
Commercial kitchen operations produce a diverse range of gas-phase reactive nitrogen species†
Leigh R.
Crilley‡
 a,
Jenna C.
Ditto§
bc,
Melodie
Lao
a,
Zilin
Zhou¶
b,
Jonathan P. D.
Abbatt
c,
Arthur W. H.
Chan
bc and
Trevor C.
VandenBoer
*a
a,
Jenna C.
Ditto§
bc,
Melodie
Lao
a,
Zilin
Zhou¶
b,
Jonathan P. D.
Abbatt
c,
Arthur W. H.
Chan
bc and
Trevor C.
VandenBoer
*a
aDepartment of Chemistry, York University, Canada. E-mail: tvandenb@yorku.ca
bDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, Canada
cDepartment of Chemistry, University of Toronto, Canada
First published on 23rd October 2024
Abstract
Gas-phase reactive nitrogen species (Nr) are important drivers of indoor air quality. Cooking and cleaning are significant direct sources indoors, whose emissions will vary depending on activity and materials used. Commercial kitchens experience regular high volumes of both cooking and cleaning, making them ideal study locations for exploring emission factors from these sources. Here, we present a total Nr (tNr) budget and contributions of key species NO, NO2, acidic Nr (primarily HONO) and basic Nr (primarily NH3) using novel instrumentation in a commercial kitchen over a two-week period. In general, highest tNr was observed in the morning and driven compositionally by NO, indicative of cooking events in the kitchen. The observed HONO and basic Nr levels were unexpectedly stable throughout the day, despite the dynamic and high air change rate in the kitchen. After summing the measured NOx, HONO and Nr,base fractions, there was on average 5 ppbv of Nr unaccounted for, expected to be dominated by neutral Nr species. Using co-located measurements from a proton transfer reaction mass spectrometer (PTR-MS), we propose the identities for these major Nr species from cooking and cleaning that contributed to Nr,base and the neutral fraction of tNr. When focused specifically on cooking events in the kitchen, a vast array of N-containing species was observed by the PTR-MS. Reproducibly, oxygenated N-containing class ions (C1–12H3–24O1–4N1–3), consistent with the known formulae of amides, were observed during meat cooking and may be good cooking tracers. During cleaning, an unexpectedly high level of chloramines was observed, with monochloramine dominating the profile, as emitted directly from HOCl based cleaners or through surface reactions with reduced-N species. For many species within the tNr budget, including HONO, acetonitrile and basic Nr species, we observed stable levels day and night despite the high air change rate during the day (>27 h−1). The stable levels for these species point to large surface reservoirs which act as a significant indoor source, that will be transported outdoors with ventilation.
Environmental significanceCooking and cleaning are significant sources of reactive nitrogen species, which are key drivers of the multiphase chemical processes that can lead to poor indoor air quality. Here we present measurements in a commercial kitchen of the total gaseous budget of nitrogenous molecules and demonstrate that a chemically diverse array of nitrogenous species make up about 10% of the total budget. Both cooking and cleaning were found to emit these complex mixtures, while surface reservoirs have a dominant role in controlling their levels. Our observations show that emissions from both sources may be driving complex multiphase chemistry indoors that extends beyond the criterion pollutants and can impact urban air quality. |
1. Introduction
Indoor air pollutants are produced, removed, and transformed by many indoor processes including direct emissions, indoor–outdoor air exchange, multiphase chemistry, and surface deposition.1 Reactive nitrogen (Nr) species, defined here as all N-containing compounds except N2 and N2O,2 are important drivers of indoor air quality due to their chemical activity. Key Nr species include nitrogen oxides (NOx = NO + NO2), nitrous acid (HONO), and ammonia (NH3), which are all known to have detrimental health effects and can be present indoors at levels which exceed outdoor levels by several orders of magnitude.1 As people spend up to 90% of their time indoors, encompassing time at home and work, a better understanding of the sources and processes driving poor indoor air quality across a diverse range of environments is required.Cooking and cleaning are known to be significant direct sources of Nr indoors, whose emissions vary depending on the activity type and materials used. Cooking with gas appliances can result in very high direct emissions of NOx and HONO (mixing ratios up to 400 and 100 parts per billion (ppbv), respectively).3–8 These sources are especially pertinent in homes that have low ventilation rates or are without a range hood. Under these non-ideal conditions, in a few minutes of use, a natural gas stove can increase NO2 levels to exceed the 1 h Canadian maximum residential exposure limit (90 ppbv).9 Transportation of NO2 indoors is also possible if outdoor levels are high.
The dominant loss of NO2 from indoor environments can vary, depending on the rate of air exchange. When ventilation is sufficiently high it dominates the removal of NO2 by transport to the outdoors. The next most important loss is reactive uptake of NO2 onto indoor surfaces through which HONO will readily form. This HONO can then build up in areas with low levels of indoor lighting (in terms of photon flux and/or energy) and away from windows that generate sunlit areas.10–12 While the mechanism for surface uptake of NO2 is debated under typical atmospheric NOx regimes, the reported association between NO2 and HONO driven by surface chemistry is strong. The best-described heterogeneous mechanism remains NO2 hydrolysis, which proceeds through the formation of an N2O4 intermediate, ultimately forming HONO and nitric acid (HNO3) (R1).12
 | (R1) |
The last indoor sink for NO2 is through reaction with ozone (O3). Loss of NO2 has recently been shown for O3 in domestic settings where experimental additions have been performed,13–16 and the reaction is capable of forming the ambient nocturnal pollutants NO3, N2O5, and ClNO2. This has been observed without O3 addition being required in an athletic facility impacted by polluted outdoor air.17 Outside of these unique situations, it is not yet clear the extent to which this chemical sink for NO2 prevails indoors, particularly in domestic settings where the O3 mixing ratio is known to be low.
Through the loss of NO2, an abundance of HONO is produced, with studies consistently reporting stable indoor mixing ratios on the order of a few ppbv,4,18,19 when point sources are not also present. This observation has been explained through theoretical models that use a surface reservoir for HONO,4 drawing off recent evidence for such outdoors.18,19 More recently, a study by Wang et al.20 performed ventilation experiments that observed HONO levels rapidly decreasing with increased ventilation (e.g. opening doors and windows), followed by a return to the original levels when the ventilation was decreased to the initial state. These observations are consistent with reversible equilibrium partitioning of HONO from a surface reservoir into the overlying air (R2). In this work by Wang and co-workers, surface nitrite (NO2−) was also found to be present on the surfaces of deployed glass plates, providing clear evidence from the surface to support this proposed partitioning behaviour.
| HONO(g) ⇌ HONO(surf) ⇌ H(aq)+ + NO2(aq)− | (R2) |
As a result, the equilibrium of HONO from a sufficiently large surface reservoir should always obtain steady state levels under any ventilation regime until the reservoir is depleted. The capacity of such reservoirs, which depends on the effective pH of the surface environment (shown for bulk aqueous exchange in (R2)), and the controls on their dynamics remain open questions.15 In addition to production from NO2, other indoor sources of HONO are all thought to be direct emissions and include gas stoves, cigarettes or smoking of other materials, and candles.11,21 Laboratory experiments have suggested other secondary HONO production routes, such as photo-enhanced reduction of NO2 on window films containing PAHs,22 but remain unconstrained for their importance across indoor environments more generally from field observations. Indoor losses of HONO include ventilation, photolysis (R3), and deposition/uptake to surfaces (R2). In all cases, these losses are of concern because of the potential release of the hydroxyl radical (OH) from HONO photolysis, where OH could initiate oxidation chemistry in the presence of organic molecules to form secondary pollutants like O3 and respirable fine airborne particles (PM2.5) indoors and outdoors.19,23–25
 | (R3) |
In addition to combustion products, cooking of protein rich food is an important source of N-containing gases and particles indoors, thought to be primarily formed via the reaction of amino acids and cooking oils.26,27 The specific nitrogenous species emitted will vary depending on the fuel, cooking oil, food/ingredients, style (e.g., frying, grilling, baking, etc.), temperature, and ventilation.28 As such, the literature reports a diverse range of gas- and particle-phase nitrogenous species emitted during cooking, and includes NH3,29 amides,28 pyrazine and pyridine derivatives,30 nitrophenol and nitrobenzenes,31 heterocyclic nitrogen species, and isocyanic acid (HNCO).32,33 Overall, cooking is a source of a diverse range of Nr species, where the concentrations of individual compounds are typically low28,32 and with unknown exposure impact, with the exception of small highly reduced or oxidised Nr species (e.g. NH3, NO).
Cleaning is another primary source of Nr indoors. Elevated levels of chloramines (NH2Cl, NHCl2 and NCl3) have been observed indoors after use of chlorinated products, such as bleach-based cleaners that contain HOCl.17,34–37 The levels of chloramines produced during cleaning can be high, with estimated upper limits of 60, 0.5, and 6.8 ppbv for NH2Cl, NHCl2 and NCl3, respectively,34 which is concerning due to their well-established toxicity. Cleaning is also a source of reduced Nr species, particularly when using ammonium hydroxide-based cleaning products. Ampollini et al.29 demonstrated that using an ammonium hydroxide cleaner before vinegar (or dilute acetic acid)-based solution resulted in very high NH3 emissions (>1 ppmv), likely due to acid–base neutralisation on the surface. These studies highlight that surface reactions between cleaning products and/or other sorbed compounds can lead to the release of toxic compounds like NH3 and chloramines. In addition, other Nr species such as amines or HNCO that are not traditionally measured may be important chemical actors for indoor transformations.32 Therefore, a budget of total reactive nitrogen is important to ascertain the key species contributing to this chemical pool of species indoors, but one has yet to be established.
Regular high volumes of cooking and cleaning are typical in commercial kitchens (e.g. restaurants, catering, etc.) and as such are excellent study locations to characterize emissions from these two sources and their interactions,38 as the rate of these activities is much higher than in residences, which have already been identified for potential impacts on urban air quality.39 Commercial kitchens are chemically complex spaces influenced by high ventilation rates and are potentially significant sources of indoor and outdoor air pollutants, and yet they are understudied, especially in relation to Nr species. In our companion paper,40 we explored the indoor exposure and outdoor air transport of cooking and cleaning-derived particulate matter, as well as the unexpected presence of cleaning-derived chlorinated reaction products. The current work presents a time-resolved total gas-phase reactive nitrogen (tNr) budget in a commercial kitchen, along with contributions from the key species NOx, HONO and basic Nr (e.g. NH3 and amines) to determine the extent that the budget can be closed. Using supporting measurements from a co-located Vocus proton transfer reaction mass spectrometer (PTR-MS), we seek to identify key Nr species from cooking and cleaning that contribute to the behaviour of the basic fraction of tNr, as well as the contributors to tNr that have not been previously speciated; we consider this as a ‘missing’ fraction in the collective knowledge of the indoor chemistry community. Finally, the role of surfaces as reservoirs that control Nr species capable of partitioning, such as HONO, NH3, acetonitrile, triethylamine, and monochloramine among many other organic nitrogen-bearing species, will be examined in this space which has long operated as a commercial kitchen where very large surface reservoirs have developed over time.
2. Method
2.1 KOCENA campaign
Full details of the Kitchen Organic Carbon, Emissions, Nitrogen, and Aerosol (KOCENA) campaign will be presented in an overview paper reporting a wider range of observations. Briefly, this study was performed in a commercial kitchen on a university campus in Toronto, Canada from Sept 1–17, 2021. The space is located partially below ground, without any exterior windows to allow sunlight to penetrate in to it. Instead, it is constantly illuminated by light emitting diode fixtures (LEDs). During this sampling period, the kitchen activities ramped up from being closed during lockdown to preparing meals to support on average 300 transactions per day by mid-September, alongside catered orders which could span a few to tens of meals. The kitchen consisted of a large commercial cooking space (henceforth called “main kitchen”), where meals were prepared from the raw ingredients, and a customer-facing food sales area, where meals were ordered, heated, and served. Operating hours were between 8:00–22:00 local time, during which both cooking and cleaning occurred according to health and safety guidelines. Cooking in the main kitchen typically began around 8:00 and concluded around 17:00 between Monday and Friday, while the customer-facing area operated daily. The bulk of cleaning began around 17:00 and continued until closing at 22:00, but was an ongoing periodic activity throughout the day as required by health and safety guidelines. Food sales in the customer-facing area also continued until closing at 22:00. Cooking times for meals or dishes prepared, or cleaning conducted, in the main kitchen were recorded daily by kitchen staff.2.2 Instrumentation
All instruments were housed in a climate-controlled room and sampled from a common inlet that was located near the highest density of cooking appliances in the main kitchen. The common inlet was 1/4 inch O.D. (0.64 cm) perfluoroalkoxy alkane (PFA) tubing that was 6.6 m long and had a combined total flow of 2.1 litres per minute. The tip of the inlet was positioned to sample from the main cooking area, which included equipment such as ovens, gas stoves, and dishwashers in close proximity – approximately one to three metres away. A polytetrafluoroethylene (PTFE) filter was installed at the entrance of the inlet to prevent intrusion of particles.For the tNr pathway, air is directed to a custom-built Pt catalytic oven held at 800 °C to convert all Nr species to NOx and it is referred to as the tNr oven from here onward. Basic species within the tNr pathway (e.g., NH3 and amines) are similarly determined by selective scrubbing of the sample gas flow with a phosphorous acid (H3PO3) coated annular denuder prior to the sample flow entering the tNr oven, which will be referred to collectively as Nr,base. In all cases, NOx is the measured analyte – as the final combustion product(s) – determined using a commercially available Mo-catalyst chemiluminescent analyser (Serinus 40, American Ecotech LLC, Warren, RI, USA). Automated flow control of the 3-way solenoid valves was achieved with a LabJack microcontroller and custom LabView program. Calibration, QA/QC, and maintenance activities are provided in Section S1.1 of the ESI.
2.3 Data analysis
All data analysis was performed in R (3.6.3) using R studio. Diurnal trends were calculated using the openair package.42 As the PTR-MS measured at higher time resolution compared to the tNr instrument, when calculating diurnal averages, we only included PTR-MS measurements if there was a corresponding tNr measurement at 1 minute resolution to avoid biasing the mean diurnal trends for comparison. Furthermore, there needed to be at least 25% of tNr data coverage within any given one-hour period for it to be included in the calculated mean diurnal trends.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cubic feet per minute (CFM) of ventilation flow. The geometric main kitchen volume was determined to be 617 m3 by measurement of the dimensions of the walls around the perimeter and throughout the room. These three values were then used to calculate the kitchen air change rate (eqn (1); ACR, h−1).
000 cubic feet per minute (CFM) of ventilation flow. The geometric main kitchen volume was determined to be 617 m3 by measurement of the dimensions of the walls around the perimeter and throughout the room. These three values were then used to calculate the kitchen air change rate (eqn (1); ACR, h−1). | (1) |
3. Results and discussion
3.1 Measured tNr budget in a commercial kitchen
The time series of the tNr and its components (NOx, HONO and Nr,base) shows episodic peaks throughout the campaign. In general, spikes in tNr were observed in the morning and were driven by NO and NO2, indicating likely cooking events with gas appliances in the kitchen. The short duration of these peaks results from the kitchen ventilation system rapidly removing the emissions.40 Outside of these peaks, the measured tNr levels were generally constant over time (Fig. 1). The mean diurnal trends in tNr also indicate that levels peaked during the morning (6–10 am) and were relatively stable outside of these times (Fig. 2). Overall, the largest single contributor to the tNr budget was NO2 (Fig. 2C), followed by Nr,base species, NO, then HONO. While the instrument measures the acidic fraction of indoor air (Nr,acid), this is present dominantly as HONO since prior reports on HNO3 in indoor air have found very low levels.47,48 | ||
| Fig. 1 Time series of (top) the fractions of HONO (yellow) and tNr,base (green) and (bottom) measured mixing ratios of tNr (black), NO (blue), and NO2 (orange) at a 1 minute time resolution over the whole campaign. Large tick marks denote midnight of each observation day and small ticks mark noon. | ||
 | ||
| Fig. 2 (A) Mean cumulative diurnal trends of measured tNr and key species quantified by the tNr instrument over the whole campaign. Pie charts present the average gaseous Nr budget (B) when operations and occupancy ceased at night (10 pm to 6 am) and (C) during daytime occupancy when cooking was active (9 am to 8 pm). | ||
The average contributions of measured tNr fractions were fairly consistent between day and night, in the absence of cooking (10 pm to 6 am, Fig. 2B) and when cooking occurred (9 am to 8 pm; Fig. 2C). The clear exception was NO, which accounted for a larger fraction of the tNr during the day (11%, Fig. 2C), consistent with emissions from gas stove combustion during cooking.9,49 The observed increase in NO around the typical start of staff-logged cooking activities (6–7 am) was driven in part by intrusion of polluted outdoor air during the start-up period for the ventilation system (Fig. S2†). The HONO and Nr,base levels were unexpectedly stable throughout the day (Fig. 2), despite the dynamic and high ACR in the kitchen. This is not driven by inlet effects, as the inlet was shared by the PTR-MS which observed rapid increases and decreases of many polar and oxygenated species related to both cooking and cleaning, and others that demonstrated similar stability.40 These HONO and Nr,base observations point to a significant surface reservoir that has rapid air-surface partitioning20 and we explore this in more detail below (Sections 3.3.1 and 3.3.5). After summing the measured NOx, HONO and Nr,base fractions, there was on average 5 ppbv of Nr unaccounted for (Fig. 2). This missing fraction of Nr (grey shading in Fig. 2) is expected to be dominated by neutral Nr species based on the sampling strategy of the instrument and is explored further via the PTR-MS data in the sections that follow.
 | ||
| Fig. 3 Mean diurnal trends of measured Nr,base and Nr,neutral measured by tNr instrument along with NH2Cl measured by the PTR-MS. The range of PTR-MS sensitives provides an estimate of upper and lower limits for NH2Cl mixing ratios as outlined in Ditto et al.40 | ||
A vast number of N-containing ions was detected by PTR-MS (see Section 3.2) but here we focus on the basic Nr species class of amines for which the PTR-MS was calibrated using standards of known concentration, namely triethylamine (TEA) and monochloramine (NH2Cl). Amides, while having known cooking sources,28 are not expected to be captured by the H3PO3 denuder as they are typically very weak bases or even very weak acids. As a strong base that is also volatile, TEA has previously been found to be emitted during fish cooking,50,51 some instances of which occurred during the measurement period for this campaign. The ambient levels were below the co-deployed passive sampler detection limits for TEA (LOD = 2.59 ppbv for 5 days of collection52), while the PTR-MS detected it at levels above its detection limits (on average 25 pptv), with a more detailed discussion on these differences in Section S1.5.†
A major nitrogen-containing compound detected by PTR-MS was monochloramine, NH2Cl, primarily observed in the afternoon at ppbv levels and in the low pptv range outside of these times (Fig. 3). In the companion paper to this work, cleaning was demonstrated to be the likely source of NH2Cl at this time40 consistent with previous reports.34 Calibration of PTR-MS for NH2Cl has proven problematic17,34,36 and in the current work we present sensitivities for NH2Cl using different approaches, as outlined in Ditto et al.40 Briefly, the sensitivity was determined experimentally,54 theoretically,53 and also constrained to measured Nr,base levels, in order give a range of possible values (Fig. 3). The three approaches agreed to within a factor of 2, thus the true NH2Cl mixing ratio likely lies within this range. However, this also suggests that NH2Cl would have accounted for a significant fraction of the Nr,base (42–88%) during the afternoon when levels peaked despite our knowledge that the NH3 levels agreed well through the independent verification by passive samplers, but which do not provide time-resolved insight.
Our calculation assumes that NH2Cl was quantitively converted to NOx in the tNr oven and/or scrubbed with the H3PO3 denuder. These have not been experimentally determined to be true due to the difficulty in generating stable gas-phase levels of NH2Cl.34,55 This assumption may not be valid, at least for complete (i.e. 100%) conversion or scrubbing of NH2Cl, as we did not observe a notable increase in Nr,base during the afternoon when NH2Cl levels peaked for extended periods of time (Fig. 3). While the estimated range of NH2Cl levels were generally within the total amount of measured Nr,base (Fig. 3), it seems unlikely that NH2Cl would account for the vast majority Nr,base in the afternoon. For this to be true, the levels of other Nr,base species (e.g. NH3) would have had to correspondingly decrease, which is unlikely as NH3 is emitted during cleaning,29 and cooking was still occurring at this time. Fig. S3† demonstrates that when there was a peak in NH2Cl (up to 80 ppbv, 9:25–9:45 pm), the measured tNr, and the Nr,neutral fraction in particular (Fig. 2), did increase relative to previous or later measurements when NH2Cl was notably lower. The magnitude of the missing fraction in Nr,neutral (Fig. 3, black line) would suggest that the tNr instrument is responding to NH2Cl because it is passing through the H3PO3 denuder either entirely or in part. This is possible, as these molecules are less polarizable and weaker bases than NH3. Similarly, NH2Cl is less likely to adsorb or ionize on surfaces conditioned with atmospheric water vapour and be less subject to inlet effects compared to NH3, which transfers readily from our instrument characterization.41 The conversion efficiency of NH2Cl in the catalytic oven should be substantial, based on the oven being characterized to do this to alkylamines, but whether the conversion is quantitative is not currently known. Comparing the magnitude of our estimated mixing ratios of NH2Cl to the PTR-MS theoretical calibration factor, a lower limit, there is good agreement (3–4 ppbv) and some consistent features in the time trends. At the experimental calibration factor limit, however, a substantial disagreement emerges (4–8 ppbv difference). The observed high levels of NH2Cl in this study were a surprise and future work will seek to improve the tNr instrument performance with respect to chloramines, as these are increasingly being recognized as compounds of interest in both indoor and outdoor environments.17,34,36,54 These improvements would likely be by increasing the combustion temperature to 825 °C or greater, in line with our related work on total chlorine detection.56
 | ||
| Fig. 4 (A) Cumulative mean diurnal trends of quantified tNr and sub-species along with calibrated N-containing ions thought to arise from neutral molecules detected by PTR-MS. Pie charts represent the average gaseous Nr budget (B) at night when operations ceased (10 pm to 6 am), and (C) during occupation for daytime cooking (9 am to 8 pm). | ||
It is important to note that the levels reported in the previous paragraph are mean diurnal levels over the whole campaign, and the contributions from N-containing species during cooking events may be higher,40 which is explored in detail in the next section. Overall, with the addition of these four neutral N-containing species, roughly 3 ppbv of the tNr was still unaccounted for by mass balance, with the variability in mean tNr levels on the order of ±4 ppbv. Therefore, much of the missing fraction could reside within the tNr instrument uncertainty (20%, (ref. 41)), yet 3 ppbv is also not a negligible amount. We suggest that this points to contributions from other N-containing species that were detected, but that the PTR-MS was not directly calibrated for. One such species could be isocyanic acid (HNCO), shown in recent work to be present in the ppbv range after cooking and cleaning activities in a house.32
3.2 Cooking and cleaning as a source of multiple gas-phase N-containing species
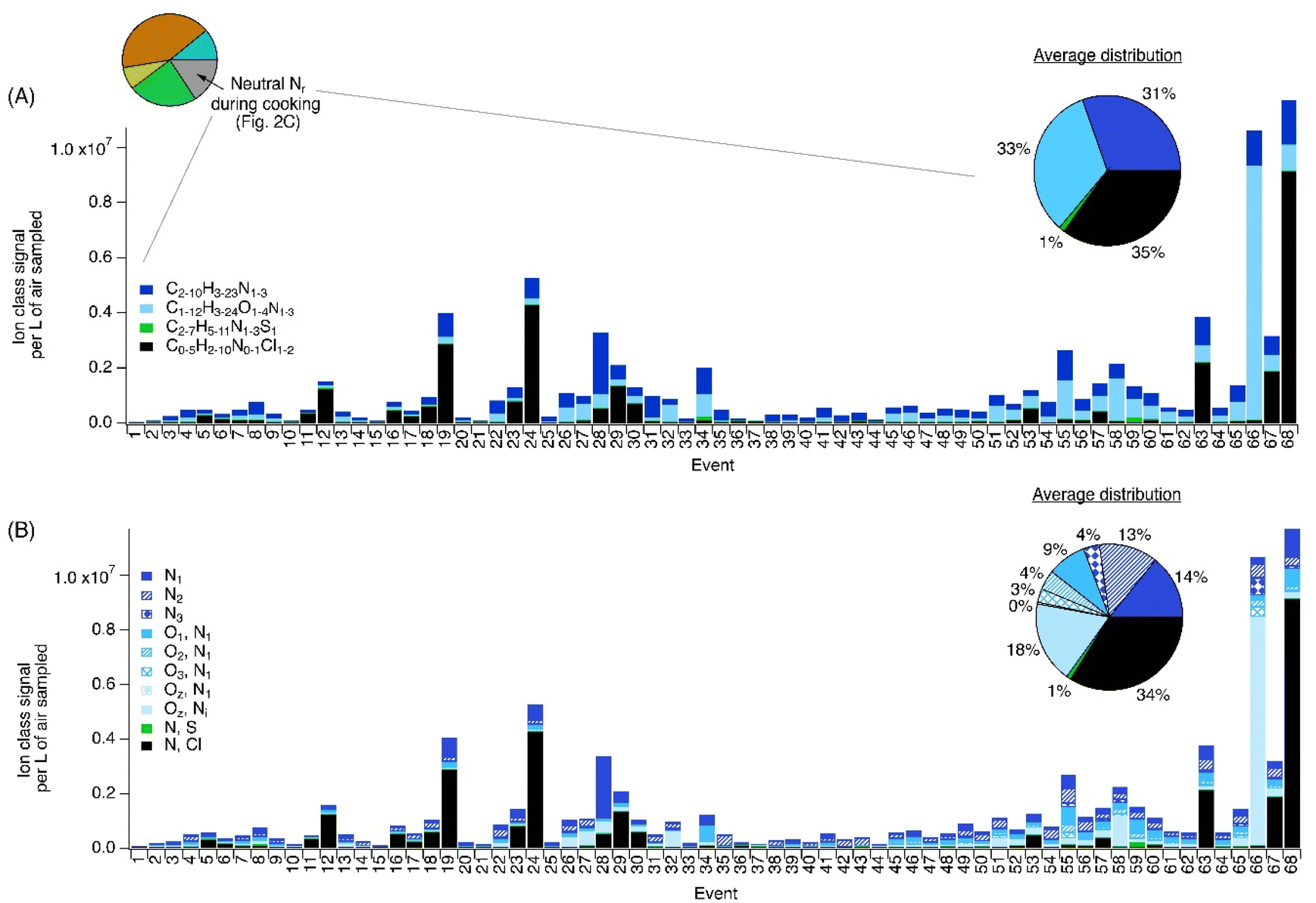 | ||
| Fig. 5 (A) Nitrogen containing ion signal fractionation for each of 68 distinct cooking events throughout the sampling campaign. (B) Nitrogen specific and associated heteroatom signal groupings. We note that the data shown in (B) is the same as the data shown in (A), but further broken down by the number of N and O atoms to describe the formulas in additional detail. A description of the cooking events is available in Ditto et al.40 The pie charts illustrate the average ion class distribution across all events. In panel B, i and z subscripts are used to reflect the number of N atoms being >1 and the number of O atoms being >3, respectively. For comparison with the tNr data, we also included the daytime pie chart from Fig. 2C here, to illustrate the further chemical speciation of neutral Nr segment that we explored here with the PTR-MS during cooking events. | ||
The time series of these ions revealed episodic peaks and higher levels during the morning to early afternoon (Fig. S4 and S5†), consistent with a period of expected cooking-related emissions in the kitchen and outdoor surveys conducted on commercial kitchen plumes.38 Other uncalibrated amine ions consistently detected at high levels with similar temporal and diurnal trends were possible cyclic amines C5H5NH+ (suggested to be pyridine, Fig. S6†) and C6H8N2H+ (suggested to be alkylpyrazine, Fig. S7†). Pyridine is known to be emitted during meat roasting,63 while alkylpyrazine has been associated with the aroma of foods, including coffee64 and baked potato.65 The observations of these ions combined with the observed temporal patterns, points to a cooking-related source for C5H5NH+ and C6H8N2H+. Cooking was not the only likely source of ions in the C2–10H3–23N1–3 class, with the ion C5H9NH+ having very similar diurnal trends to NH2ClH+ (Fig. S8†) which points to cleaning as another source of this nitrogenous ion (Section 3.2.4).
These dishes were also cooked at other times (e.g. events 27, 32 and 58 for butter chicken, tagine and lentils, respectively) and while these events also had high contributions from C1–12H3–24O1–4N1–3 ions and specifically C4H8N2O3H+ (Fig. 5A), the levels were not as high and may be because of the location of preparation or the procedure used (e.g. extent of covering, duration, maximum temperature, etc.). Other cooking related ions were observed in the C1–7H5–11N1–3S1 ions class, which only accounted for a small fraction (1%, Fig. 5) of nitrogen containing ions detected across all the events. The major ion in this class was possibly a thiozoline (C5H8N2OSH+), previously associated with meat aromas67 and could originate from a cooking-related source.
As cooking is demonstrated here to be a likely major source of the C2–10H3–23N1–3 and C1–12H3–24O1–4N1–3 classes of ions, we compared the ions detected during selected cooking events with those emitted during a controlled laboratory study that simulated cooking processes by heating amino acids with triglycerides (cooking oil).28 During this work, Ditto et al.40 observed that the reactions between thermal degradation products of triglycerides and amino acids (forming NH3 upon heating) resulted in the emissions of numerous amides. Fig. S10† demonstrates that there was 15% overlap with ions from simulated cooking emissions and a preparation of chicken, salmon, and vegetable in the kitchen cooking logs. A similar comparison showed this overlap rose to 30% for a pork and vegetable cooking event (Fig. S9†). Some of the major N-containing ions that were observed to overlap were C2H3NH+ (acetonitrile) and C4H7N3H+ as well as oxygenated N-containing ions such as CH3NOH+, suggesting that meat cooking is a source of molecules that are precursors to these ions. The oxygenated N-containing ions observed in both studies, such as CH3NOH+ were probably amides, which are known to be toxic.28 We cannot definitively assign these ions to amides from these field measurements, as structures cannot be unambiguously ascertained by PTR-MS. Interestingly, many N-containing ions in this class designation (85 and 70%, by ion intensity; Fig. S9 and S10†) were not observed to overlap with the simulated meat cooking emissions study,28 suggesting that cooking related sources of this compound class are highly variable under realistic conditions where many protein sources and combinations of oils may be used simultaneously, similar to the observations reported by Coggon et al. in Las Vegas.68 This highlights the complexity of Nr species emitted by cooking, as the vast majority were not emitted during the controlled lab experiments of meat cooking in heated oil, which could be one major cooking source of amides.69,70
The complexity of cooking emissions in the kitchen, due to the mixture of two different cooking areas, combinations of different ingredients, overlapping preparations, and potentially different procedures and appliances being used on any given day, makes it challenging to isolate useful tracer ions for a particular dish or even a single form of food. In general, oxygenated N-containing class ions (C1–12H3–24O1–4N1–3), consistent with the known formulae of amides or combinations of reduced nitrogen groups (e.g., amines) with oxygenated groups, appear to be good tracers for meat cooking although identifying all tracer ions was not possible and could be the focus of future targeted research under controlled lab conditions.
The levels of NH2ClH+ maximized in the afternoon (Fig. 3) and in our companion paper,40 where cleaning emissions were determined to be the dominant source of this species. Specifically, the use of cleaning products containing sodium hypochlorite in the kitchen were likely primary drivers, either through direct emissions37 or via surface reactions with N-containing compounds such as amines (e.g. ammonium salts, long chain alkyl amines, monoethanolamine) that were also present on surfaces or in other cleaning solutions. In contrast to previous reports of bleach cleaning in a residential house,34,37 we did not observe elevated ion counts that could be assigned to NHCl2H+ or NCl3H+ by PTR-MS during cleaning activities in the kitchen.40 This was likely due to the inability of this PTR to detect these compounds with more chlorine atoms.54 Regardless, our observed NH2ClH+ during cleaning is similar to a recent report from indoor sports centres36 and may reflect the prevalence of substantial cleaning emissions of this molecule from large-scale/commercial environments compared to domestic settings. Other prominent chlorinated N-bearing ions with greater hydrocarbon character were readily observed and included C5H10ClNH+, C3H6ClNH+, C2H4ClNH+ and CH4ClNH+, which are thought to originate from unique parent compounds (i.e. not as a result of fragmentation in the PTR-MS40). The C1–5H4–10ClNH+ ion series were likely N-chloraldimines, which are formed from the reaction of HOCl and amino acids on surfaces.35 Since amino acids are expected to be ubiquitous on kitchen work surfaces from food material transfer as a source, the observed ions for these reaction products would be ever-present due to the stringent cleaning procedures to maintain health and safety in a commercial kitchen work environment.
3.3 Surfaces are significant reservoirs of Nr in the kitchen
A well-recognized precursor to HONO in any environment is NO2, so the interplay of these two species was explored first. Heterogeneous surface uptake is expected to be an important loss process of indoor NO2 as these environments have high surface area to volume ratios (S/V), with a lower limit value of 3.2 m−1 calculated for this commercial kitchen, ignoring the surfaces provided by appliances, shelving and materials thereon, as well as their porosity.12,72–74 Outdoors, the conversion of NO2 to HONO on surfaces has been implied through the ratio of HONO/NO2 relative to NOx emitted directly from vehicle combustion (HONO/NOx = 0.5–2%).75–78 Indoors, in the absence of a direct HONO source, it can be used similarly to infer the potential for this heterogeneous NO2 chemistry as a source of HONO.79 Here, the background HONO/NO2 ratio in the absence of direct sources of either molecule in the kitchen was on average 0.19 ± 0.03, with little diurnal variation observed, despite the difference in ACR during operating hours and at night. The consistently high HONO/NO2 therefore points to heterogeneous reactions on surfaces acting as an source of gas phase and reservoir HONO.20 This ratio is also lower than that reported for direct HONO emissions from gas stoves, which have found ranges of HONO/NO2 spanning 0.5 to 1.0 depending on the duration of gas stove operation, with 1.0 being the most commonly observed.4,5 Therefore, if cooking emissions were not rapidly ventilated, it might be reasonable to expect that HONO was deposited to surfaces in addition to the heterogeneous reaction of NO2, prior to the upgraded HVAC being installed in this space a few years prior to this study.
Competing with the heterogeneous surface source of HONO (Fig. 6, top) are losses by air exchange and photolysis (Fig. 6, middle). The measured spectral irradiance in this space, despite lighting being on 24 hours per day, 7 days per week, was generated from LEDs overhead in place of traditional fluorescent tubes,84 where the new lighting (λ > 405 nm) did not provide photons with high enough energy to facilitate meaningful photolytic loss of HONO. As a result, the majority of HONO loss within the kitchen was driven by ventilation to the outdoor environment, which was then rapidly replaced from the surface reservoir on the timescale of our observations (∼1–5 min). From this simple box model, the predicted HONO was found to be substantially lower than the measured HONO and generally followed the same trends, but inverted in sign, due to the strong impact of the ventilation on the fate of molecules in this space (Fig. 6, bottom). Additional sources of HONO are required to reconcile the difference between the model and observations and was true for both boundary condition scenarios for this model (Table S2†).
 | ||
| Fig. 6 Mean diurnal predicted HONO levels calculated from production by NO2 uptake on surfaces (top) and loss by photolysis and ventilation (middle) compared to measured HONO by day of week (bottom), using a γNO2 of 2 × 10−6 and a nighttime ACR of 0.5 h−1. The shaded areas represent the 95% confidence interval of the mean. Ventilation dominates the fate of HONO leading to predicted concentrations lower than measured, implicating the presence of additional sources. | ||
Comparing the measurements to the production of HONO from NO2 in the model, outdoor to indoor transport could partially reconcile the observations (Section S2.1†), but the discrepancy persists throughout the day regardless of whether the ventilation is active (Fig. 6). As a result, we conclude that outdoor HONO transported indoors was unlikely to have played a substantial part in the overall temporal dynamics or magnitude of HONO in this space, but it may contribute in the morning when the probability was highest. Similarly, due to the low energy LED lighting and absence of windows, we are able to rule out photochemical contributions. Overall, comparing the predicted production rate of HONO and the measured values confirms that, throughout all hours, HONO in the commercial kitchen is overwhelmingly produced by release from a surface reservoir, followed by heterogeneous uptake of NO2 (Fig. 6). Even when the ACR is high, the indoor HONO concentration remains constant despite being removed in abundance (Fig. S12–S14), such that the high rate of transfer is most likely explained by sustained surface emissions. With all seven days showing the same trends in Fig. 6, it is not surprising to find a dynamic situation from the measured to predicted ratio for HONO chemistry in this space. This is because the cooking activities and ACR were similarly dynamic. The overall diurnal campaign averages provide broader insight into the general drivers of HONO chemistry in this busy indoor space (Fig. 7). From this, we consider the relative roles of NO2 and surfaces in the two disparate daytime and nighttime ACR regimes in more detail to investigate their dynamics.
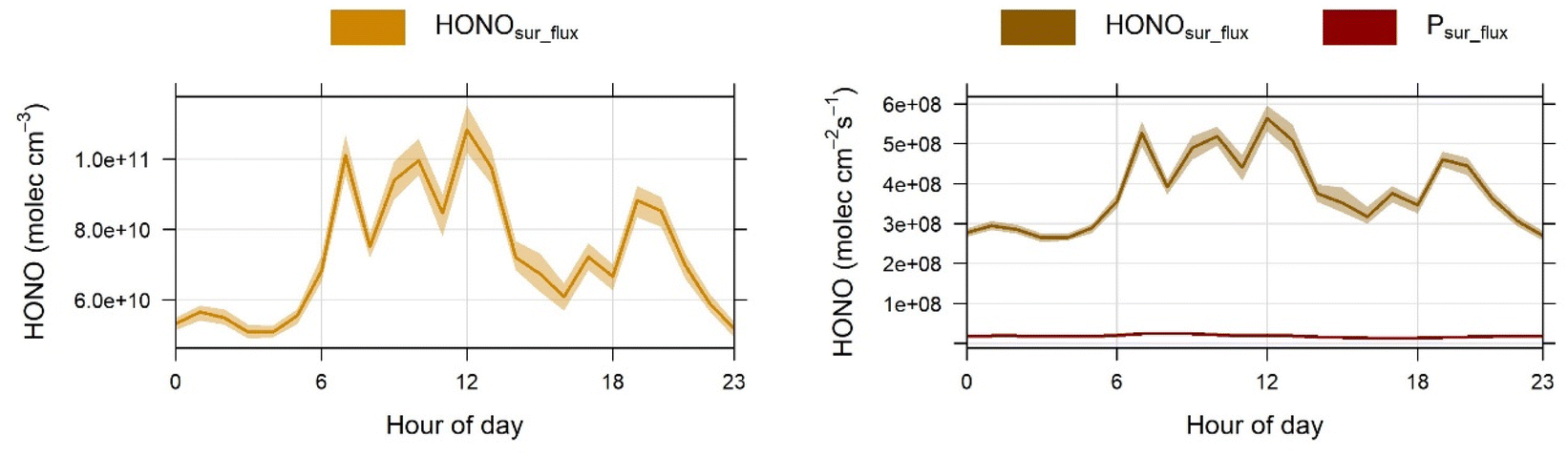 | ||
| Fig. 7 (Left) Mean diurnal trends of HONO levels originating from surface emissions (HONOsur_flux) using E7, and (Right) the calculated fluxes required to maintain these levels from surface emissions (HONOsur_flux) and NO2 uptake (Psur_flux). The shaded areas represent the 95% confidence interval of the mean. Note the different y-axis scales and units. | ||
A mean surface emission flux of 7.4 × 108 molecules cm−2 s−1 exists during the high ACR operating hours (8 am to 10 pm). In comparison both Wang et al.49 and Collins et al.4 observed fluxes in the range of 2.2 to 2.9 × 107 molecules cm−2 s−1 for ACRs from 0.0 to 1.0 h−1 during rapid ventilation experiments, where time constants to recover from the perturbation were used to determine HONO fluxes. Our fluxes, to maintain similar indoor HONO mixing ratios, are 20 times larger or more, consistent with the potentially higher surface loadings, S/V and/or porosity of the kitchen materials in which a reservoir has been built up historically with low ventilation, and/or the observed higher NO2 reactivity. Overall, residential and commercial kitchen space controls over HONO mixing ratios are similar in mechanism but occurring at a larger magnitude in this commercial space with a potential legacy of less well-ventilated operation conditions to facilitate large persistent pollutant reservoirs. The limited information on air infiltration rates in commercial buildings, as well as the uncertainty in the conversion of NO2 on the heavily impacted kitchen surfaces, can lead to outcomes that are somewhat different from those depicted from our best estimates discussed here (Table S2†). Given the strong departure from prior reports in our observed NO2 loss rates, the NO2-dominant scenario is an upper limit without precedent and should be treated as such in terms of its explanatory power. This is the only model scenario where the nighttime HONO levels can be reasonably captured by NO2 heterogeneous reactions. Regardless, our more modest model runs and this upper limit all agree on the presence of a substantial and sustained HONO surface source to reconcile the observations when this kitchen space is actively ventilated.
The similar levels between day and night for the majority of Nr ions, despite the difference in ACR, also point to a very fast and strong source to compete with the very high ACR in the kitchen. Otherwise, signal increases would have been observed overnight, when there were no cooking emissions present, and these would have dropped dramatically upon ventilation power up in the morning. Taken together, these observations suggest a significant surface reservoir for these species also with rapid equilibrium control on indoor air composition. These observations also appear to reflect the decades-long legacy of surface contamination built up in the ventilation system and kitchen, such as from use of cooking oils.84 Similar findings have been reported for the persistence of SVOCs in the indoor air of an office,85 and the accumulation of gases like HONO in a test house.49 Surface fluxes for a subset of VOCs measured by the PTR-MS are shown in Table S3.† Fluxes were estimated by focusing on measured air concentrations from 6–7 am, while ventilation in the kitchen was powered on but no other sources except surfaces existed in the kitchen and no changes to VOC signals were observed. This calculation assumes constant VOC surface flux rate throughout the ventilated period in the kitchen (6 am to 10 pm) and represents the net effect of surface emissions and deposition.
Compared to past floor area specific VOC emission factors from a newly constructed residential test facility with low VOC-emitting materials, and comparisons to other occupied or unoccupied homes therein,86 surface fluxes from this commercial kitchen generally fell within similar orders of magnitude but are compound-dependent. For example, ethanol fluxes were about 6 times higher in the kitchen than in Poppendieck et al.,86 which is consistent with cooking activity in the kitchen being a major source. Acetone, acetic acid, toluene, and terpene fluxes were similar in the commercial kitchen and residential environments. Hexanal fluxes in the kitchen were about 1 order of magnitude lower than the homes, while D5-siloxane fluxes were 2 orders of magnitude greater in the kitchen which is consistent with its high occupancy with sometimes more than ten staff, relative to when unoccupied.
Surface fluxes of nitrogen containing species have been observed indoors in other work, albeit they seldom have a decades-long legacy of intense use for 10 hours per day.87 For example, Wang et al.20 studied indoor surfaces as dynamic temporary reservoirs for volatile species at the HOMEChem test house and observed that ammonia participated in rapid gas–surface exchange, with a response time to return to steady state mixing ratios after enhanced ventilation periods ranging from 500–2500 s. This response time was longer than that for many of the other VOCs studied, suggesting that NH3 could perhaps be more strongly sorbed to surfaces. Similar observations were also made by Wang et al.32 for isocyanic acid (HNCO) during the same field campaign, with mixing ratios increasing as a function of temperature, with increased evaporation from surfaces occurring at higher air and surface temperatures. Surface reservoirs of acetonitrile indoors were observed by Arata et al.88 who reported stable levels of <1 ppb yet calculated a net loss of 0.09 mg h−1 indoors during the HOMEChem campaign based on the indoor to outdoor ratio decreasing over the day. In contrast, the majority of the other VOCs reported by Arata et al.88 exhibited a net gain indoors.
4. Conclusions
High time resolution measurements of the first gas phase tNr budget in a commercial kitchen identified the contributions of a variety of key Nr species and determined the extent that this budget can be closed with modern high time resolution analytical instrumentation. The mean diurnal tNr budget was not closed using the measured levels of NO, NO2, HONO and basic Nr (e.g. NH3 and amines); a substantial missing fraction (ca. 5 ppbv) was found. Using supporting PTR-MS measurements of calibrated N-containing species, the missing fraction of Nr was found to be partially closed by the neutral organic species acetonitrile, propionamide, pyrrole and acrylonitrile, but with roughly 3 ppbv of the missing fraction remaining unaccounted for and composed of a wide variety of chemical structures that were not calibrated on the PTR-MS. That these neutral species could not fully account for the observed missing fraction may point to there being notable contributions from unheralded N-containing species indoors.Cooking events in the kitchen produced a diverse and numerous array of N-containing species detected by the PTR-MS. This variety was due to the complexity of cooking occurring in the kitchen with different cooking areas, combinations of different ingredients and appliances, as well as overlapping meal preparations and techniques. This made identifying specific tracers for cooking a particular dish or single form of food challenging. When grouped by their broad molecular formulae, oxygenated N-containing class ions (C1–12H3–24O1–4N1–3), consistent with the known formulae of amides or other combinations of reduced nitrogen groups with oxygenated groups, appear to be good tracers for meat cooking. This could be the focus of future targeted research under controlled lab conditions and from surveys of urban air impacted by cooking to explore if specific amides could be used as such tracers.
Many of the highest abundance N-containing ions measured by the PTR-MS were chlorinated, with monochloramine the dominant ion in this class. The observed elevated levels of monochloramine were surprising and found to be emitted from use of HOCl based cleaners in the kitchen via direct emissions37 or surface reactions with reduced N-containing compounds. This is concerning due to the established toxicity of NH2Cl, and therefore emphasizes an emerging need to understand worker exposure levels with more accurate measurements in the future. Unfortunately, quantification of NH2Cl with PTR-MS remains challenging and will need improvement to obtain quantification via both the PTR-MS and tNr instruments, as such chlorinated anthropogenic compounds are being recognized to have increasing importance for both indoor and outdoor atmospheric chemistry.36,54 As this particular PTR-MS is not sensitive to higher order chloramines, differences in cleaning practises in a commercial kitchen compared to domestic settings (e.g. cleaners employed or limiting reagents with respect to surface composition) and changes in the relative emissions of these molecules remains an open line of inquiry. The discrepancy between these observations and those made prior is also related to differences in the sensitivity and selectivity of different atmospheric chemical ionisation mass spectrometers to these compounds, based on the reagent ion, and warrants further investigation and optimization.
Throughout the measurement period, levels of HONO were relatively stable day and night, despite the very large changes in ACR, pointing to gas-phase HONO being in equilibrium with a large reservoir present on the surfaces in the kitchen. We used a simple chemical model constructed with production and loss processes bounded by our observations or those from others. It was found that the predicted HONO was always lower than the measured HONO under the best estimates for NO2 heterogeneous uptake. Overall, HONO in the commercial kitchen was found to be mostly produced from surfaces fluxes, while NO2 reaction played a minor supporting role in controlling HONO, where only the most extreme NO2 reactivity conditions put the two processes on parity during the day or dominated explanatory power under low ventilation conditions. A similar lack of diurnal patterns as seen for HONO were observed for the neutral and basic Nr species acetonitrile and triethylamine, as well as many other N-containing species. Thus, the stable levels despite high ACR points to large reservoirs of Nr present on the surfaces in this commercial kitchen and was a surprising finding in this study. The magnitude of the reservoir in the indoor space, by mass, and how one might conduct its amelioration warrants further research.
Data availability
The data from this study are available upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the entire research team who contributed to this field campaign: beyond the authors listed here, we extend our thanks to C. J. Young and L. Salehpoor (York University), T. Kahan (University of Saskatchewan). We thank C. J. Young for the loan of the EC9841 and Serinus 10 during the kitchen observations. We also wish to thank the building operators, facilities managers, and kitchen staff for access to the observation space, mechanical rooms, operations and sales logs, and ventilation data. ML acknowledges support through the Harold I. Schiff award in atmospheric chemistry. JCD and AWHC acknowledge funding from the Canada Research Chairs program (CRC-2019-00028); TV, ML, LRC, JCD and JPDA acknowledge funding from the Alfred P. Sloan Foundation CIE Program (G-2019-11404, G-2018-11062, G-2018-11051), and NSERC Discovery Grants and Discovery Launch Supplement (RGPIN-2020-06166 and DGECR-2020-00186).References
- J. P. D. Abbatt and C. Wang, The atmospheric chemistry of indoor environments, Environ. Sci.: Process. Impacts, 2020, 22, 25–48 CAS.
- J. M. Roberts, C. E. Stockwell, R. J. Yokelson, J. de Gouw, Y. Liu, V. Selimovic, A. R. Koss, K. Sekimoto, M. M. Coggon, B. Yuan, K. J. Zarzana, S. S. Brown, C. Santin, S. H. Doerr and C. Warneke, The nitrogen budget of laboratory-simulated western US wildfires during the FIREX 2016 Fire Lab study, Atmos. Chem. Phys., 2020, 20, 8807–8826 CrossRef CAS.
- V. Bartolomei, E. Gomez Alvarez, J. Wittmer, S. Tlili, R. Strekowski, B. Temime-Roussel, E. Quivet, H. Wortham, C. Zetzsch, J. Kleffmann and S. Gligorovski, Combustion Processes as a Source of High Levels of Indoor Hydroxyl Radicals through the Photolysis of Nitrous Acid, Environ. Sci. Technol., 2015, 49, 6599–6607 CrossRef CAS.
- D. B. Collins, R. F. Hems, S. Zhou, C. Wang, E. Grignon, M. Alavy, J. A. Siegel and J. P. D. Abbatt, Evidence for Gas-Surface Equilibrium Control of Indoor Nitrous Acid, Environ. Sci. Technol., 2018, 52, 12419–12427 CrossRef CAS PubMed.
- S. Zhou, C. J. Young, T. C. VandenBoer, S. F. Kowal and T. F. Kahan, Time-Resolved Measurements of Nitric Oxide, Nitrogen Dioxide, and Nitrous Acid in an Occupied New York Home, Environ. Sci. Technol., 2018, 52, 8355–8364 CrossRef CAS.
- M. Dennekamp, S. Howarth, C. A. J. Dick, J. W. Cherrie, K. Donaldson and A. Seaton, Ultrafine particles and nitrogen oxides generated by gas and electric cooking, Occup. Environ. Med., 2001, 58, 511–516 CrossRef CAS PubMed.
- L. Kiyoung, X. Jianping, S. G. Alison, O. Halûk, P. L. Brian, J. W. Charles and D. S. John, Nitrous acid, nitrogen dioxide, and ozone concentrations in residential environments, Environ. Health Perspect., 2002, 110, 145–150 Search PubMed.
- N. A. Mullen, J. Li, M. L. Russell, M. Spears, B. D. Less and B. C. Singer, Results of the California Healthy Homes Indoor Air Quality Study of 2011-2013: impact of natural gas appliances on air pollutant concentrations, Indoor Air, 2016, 26, 231–245 CrossRef CAS.
- E. D. Lebel, C. J. Finnegan, Z. Ouyang and R. B. Jackson, Methane and NOx Emissions from Natural Gas Stoves, Cooktops, and Ovens in Residential Homes, Environ. Sci. Technol., 2022, 56, 2529–2539 CrossRef CAS.
- S. F. Kowal, S. R. Allen and T. F. Kahan, Wavelength-Resolved Photon Fluxes of Indoor Light Sources: Implications for HOx Production, Environ. Sci. Technol., 2017, 51, 10423–10430 CrossRef CAS PubMed.
- B. Bottorff, C. Wang, E. Reidy, C. Rosales, D. K. Farmer, M. E. Vance, J. P. D. Abbatt and P. S. Stevens, Comparison of Simultaneous Measurements of Indoor Nitrous Acid: Implications for the Spatial Distribution of Indoor HONO Emissions, Environ. Sci. Technol., 2022, 56(19), 13573–13583 CrossRef CAS.
- B. J. Finlayson-Pitts, L. M. Wingen, A. L. Sumner, D. Syomin and K. A. Ramazan, The heterogeneous hydrolysis of NO2 in laboratory systems and in outdoor and indoor atmospheres: an integrated mechanism, Phys. Chem. Chem. Phys., 2003, 5, 223–242 RSC.
- S. Zhou, C. J. Young, T. C. VandenBoer and T. F. Kahan, Role of location, season, occupant activity, and chemistry in indoor ozone and nitrogen oxide mixing ratios, Environ. Sci.: Process. Impacts, 2019, 21, 1374–1383 CAS.
- C. Arata, K. J. Zarzana, P. K. Misztal, Y. Liu, S. S. Brown, W. W. Nazaroff and A. H. Goldstein, Measurement of NO3 and N2O5 in a Residential Kitchen, Environ. Sci. Technol. Lett., 2018, 5, 595–599 CrossRef CAS.
- D. K. Farmer, M. E. Vance, D. Poppendieck, J. Abbatt, M. R. Alves, K. C. Dannemiller, C. Deeleepojananan, J. Ditto, B. Dougherty and O. R. Farinas, The chemical assessment of surfaces and air (CASA) study: using chemical and physical perturbations in a test house to investigate indoor processes, Environ. Sci. Process. Impacts., 2024 10.1039/D4EM00209A.
- M. F. Link, J. Li, J. C. Ditto, H. Huynh, J. Yu, S. M. Zimmerman, K. L. Rediger, A. Shore, J. P. D. Abbatt and L. A. Garofalo, Ventilation in a Residential Building Brings Outdoor NOx Indoors with Limited Implications for VOC Oxidation from NO3 Radicals, Environ. Sci. Technol., 2023, 57, 16446–16455 CrossRef CAS.
- A. Moravek, T. C. Vandenboer, Z. A. Finewax, D. Pagonis, B. Nault, W. L. Brown, D. A. Day, A. V. Handshy, H. Stark, P. Ziemann, J. L. Jimenez, J. de Gouw and C. J. Young, Reactive Chlorine Emissions from Cleaning and Reactive Nitrogen Chemistry in an Indoor Athletic Facility, Environ. Sci. Technol., 2022, 56(22), 15408–15416 CrossRef CAS PubMed.
- T. C. VandenBoer, S. S. Brown, J. G. Murphy, W. C. Keene, C. J. Young, A. A. P. Pszenny, S. Kim, C. Warneke, J. A. de Gouw and J. R. Maben, Understanding the role of the ground surface in HONO vertical structure: high resolution vertical profiles during NACHTT-11, J. Geophys. Res. Atmos., 2013, 118, 10–155 Search PubMed.
- T. C. VandenBoer, M. Z. Markovic, J. E. Sanders, X. Ren, S. E. Pusede, E. C. Browne, R. C. Cohen, L. Zhang, J. Thomas and W. H. Brune, Evidence for a nitrous acid (HONO) reservoir at the ground surface in Bakersfield, CA, during CalNex 2010, J. Geophys. Res. Atmos., 2014, 119, 9093–9106 CrossRef CAS.
- C. Wang, D. B. Collins, C. Arata, A. H. Goldstein, J. M. Mattila, D. K. Farmer, L. Ampollini, P. F. DeCarlo, A. Novoselac, M. E. Vance, W. W. Nazaroff and J. P. D. Abbatt, Surface reservoirs dominate dynamic gas-surface partitioning of many indoor air constituents, Sci. Adv., 2020, 6, eaay8973 CrossRef CAS.
- A. Klosterköther, R. Kurtenbach, P. Wiesen and J. Kleffmann, Determination of the emission indices for NO, NO2, HONO, HCHO, CO, and particles emitted from candles, Indoor Air, 2021, 31, 116–127 CrossRef.
- J. Liu, H. Deng, P. S. J. Lakey, H. Jiang, M. Mekic, X. Wang, M. Shiraiwa and S. Gligorovski, Unexpectedly High Indoor HONO Concentrations Associated with Photochemical NO2 Transformation on Glass Windows, Environ. Sci. Technol., 2020, 54, 15680–15688 CrossRef CAS PubMed.
- C. J. Young, S. Zhou, J. A. Siegel and T. F. Kahan, Illuminating the dark side of indoor oxidants, Environ. Sci.: Process. Impacts, 2019, 21, 1229–1239 CAS.
- R. Sommariva, L. R. Crilley, S. M. Ball, R. L. Cordell, L. D. J. Hollis, W. J. Bloss and P. S. Monks, Enhanced wintertime oxidation of VOCs via sustained radical sources in the urban atmosphere, Environ. Pollut., 2021, 274, 116563 CrossRef CAS PubMed.
- T. F. Kahan, C. J. Young and S. Zhou, Indoor photochemistry, in, Handbook of Indoor Air Quality, ed. Y. Zhang, P. K. Hopke and C. Mandin, Springer Singapore, Singapore, 2021, pp. 1–30 Search PubMed.
- Y. Zhao and B. Zhao, Emissions of air pollutants from Chinese cooking: a literature review, Build. Simul., 2018, 11, 977–995 CrossRef.
- K. L. Abdullahi, J. M. Delgado-Saborit and R. M. Harrison, Emissions and indoor concentrations of particulate matter and its specific chemical components from cooking: a review, Atmos. Environ., 2013, 71, 260–294 CrossRef CAS.
- J. C. Ditto, J. P. D. Abbatt and A. W. H. Chan, Gas- and Particle-Phase Amide Emissions from Cooking: Mechanisms and Air Quality Impacts, Environ. Sci. Technol., 2022, 56, 7741–7750 CrossRef CAS.
- L. Ampollini, E. F. Katz, S. Bourne, Y. Tian, A. Novoselac, A. H. Goldstein, G. Lucic, M. S. Waring and P. F. DeCarlo, Observations and Contributions of Real-Time Indoor Ammonia Concentrations during HOMEChem, Environ. Sci. Technol., 2019, 53, 8591–8598 CrossRef CAS PubMed.
- J. Zeng, Z. Yu, M. Mekic, J. Liu, S. Li, G. Loisel, W. Gao, A. Gandolfo, Z. Zhou, X. Wang, H. Herrmann, S. Gligorovski and X. Li, Evolution of Indoor Cooking Emissions Captured by Using Secondary Electrospray Ionization High-Resolution Mass Spectrometry, Environ. Sci. Technol. Lett., 2020, 7, 76–81 CrossRef CAS.
- E. Reyes-Villegas, T. Bannan, M. Le Breton, A. Mehra, M. Priestley, C. Percival, H. Coe and J. D. Allan, Online Chemical Characterization of Food-Cooking Organic Aerosols: Implications for Source Apportionment, Environ. Sci. Technol., 2018, 52, 5308–5318 CrossRef CAS PubMed.
- C. Wang, J. M. Mattila, D. K. Farmer, C. Arata, A. H. Goldstein and J. P. D. Abbatt, Behavior of Isocyanic Acid and Other Nitrogen-Containing Volatile Organic Compounds in The Indoor Environment, Environ. Sci. Technol., 2022, 56, 7598–7607 CrossRef CAS.
- R. F. Hems, C. Wang, D. B. Collins, S. Zhou, N. Borduas-Dedekind, J. A. Siegel and J. P. D. Abbatt, Sources of isocyanic acid (HNCO) indoors: a focus on cigarette smoke, Environ. Sci.: Process. Impacts, 2019, 21, 1334–1341 CAS.
- J. M. Mattila, P. S. J. Lakey, M. Shiraiwa, C. Wang, J. P. D. Abbatt, C. Arata, A. H. Goldstein, L. Ampollini, E. F. Katz, P. F. DeCarlo, S. Zhou, T. F. Kahan, F. J. Cardoso-Saldaña, L. H. Ruiz, A. Abeleira, E. K. Boedicker, M. E. Vance and D. K. Farmer, Multiphase Chemistry Controls Inorganic Chlorinated and Nitrogenated Compounds in Indoor Air during Bleach Cleaning, Environ. Sci. Technol., 2020, 54, 1730–1739 CrossRef CAS PubMed.
- Z. Finewax, D. Pagonis, M. S. Claflin, A. V Handschy, W. L. Brown, O. Jenks, B. A. Nault, D. A. Day, B. M. Lerner, J. L. Jimenez, P. J. Ziemann and J. A. de Gouw, Quantification and source characterization of volatile organic compounds from exercising and application of chlorine-based cleaning products in a university athletic center, Indoor Air, 2021, 31, 1323–1339 CrossRef CAS.
- A. A. Angelucci, L. R. Crilley, R. Richardson, T. S. E. Valkenburg, P. S. Monks, J. M. Roberts, R. Sommariva and T. C. VandenBoer, Elevated levels of chloramines and chlorine detected near an indoor sports complex, Environ. Sci.: Process. Impacts, 2023, 25, 304–313 CAS.
- J. P. S. Wong, N. Carslaw, R. Zhao, S. Zhou and J. P. D. Abbatt, Observations and impacts of bleach washing on indoor chlorine chemistry, Indoor Air, 2017, 27, 1082–1090 CrossRef CAS.
- S. Kim, J. Machesky, D. R. Gentner and A. A. Presto, Real-world observations of reduced nitrogen and ultrafine particles in commercial cooking organic aerosol emissions, Atmos. Chem. Phys., 2024, 24, 1281–1298 CrossRef CAS.
- T. J. Carter, D. R. Shaw, D. C. Carslaw and N. Carslaw, Indoor cooking and cleaning as a source of outdoor air pollution in urban environments, Environ. Sci.: Process. Impacts, 2024, 26, 975–990 CAS.
- J. C. Ditto, L. R. Crilley, M. Lao, T. C. VandenBoer, J. P. D. Abbatt and A. W. H. Chan, Indoor and outdoor air quality impacts of cooking and cleaning emissions from a commercial kitchen, Environ. Sci.: Process. Impacts, 2023, 25, 964–979 CAS.
- L. R. Crilley, M. Lao, L. Salehpoor and T. C. VandenBoer, Emerging investigator series: an instrument to measure and speciate the total reactive nitrogen budget indoors: description and field measurements, Environ. Sci.: Process. Impacts, 2023, 25, 389–404 CAS.
- D. C. Carslaw and K. Ropkins, openair — an R package for air quality data analysis, Environ. Model. Softw., 2012, 27–28, 52–61 CrossRef.
- M. Mendez, N. Blond, D. Amedro, D. A. Hauglustaine, P. Blondeau, C. Afif, C. Fittschen and C. Schoemaecker, Assessment of indoor HONO formation mechanisms based on in situ measurements and modeling, Indoor Air, 2017, 27, 443–451 CrossRef CAS PubMed.
- T. C. VandenBoer, C. J. Young, R. K. Talukdar, M. Z. Markovic, S. S. Brown, J. M. Roberts and J. G. Murphy, Nocturnal loss and daytime source of nitrous acid through reactive uptake and displacement, Nat. Geosci., 2015, 8, 55–60 CrossRef CAS.
- M. E. Monge, B. D'Anna and C. George, Nitrogen dioxide removal and nitrous acid formation on titanium oxide surfaces-an air quality remediation process?, Phys. Chem. Chem. Phys., 2010, 12, 8991–8998 RSC.
- R. Ammar, M. E. Monge, C. George and B. D’Anna, Photoenhanced NO2 Loss on Simulated Urban Grime, ChemPhysChem, 2010, 11, 3956–3961 CrossRef CAS PubMed.
- M. Brauer, P. B. Ryan, H. H. Suh, P. Koutrakis, J. D. Spengler, N. P. Leslie and I. H. Billick, Measurements of nitrous acid inside two research houses, Environ. Sci. Technol., 1990, 24, 1521–1527 CrossRef CAS.
- F. Vichi, L. Mašková, M. Frattoni, A. Imperiali and J. Smolík, Simultaneous measurement of nitrous acid, nitric acid, and nitrogen dioxide by means of a novel multipollutant diffusive sampler in libraries and archives, Herit. Sci., 2016, 4, 4 CrossRef.
- C. Wang, B. Bottorff, E. Reidy, C. M. F. Rosales, D. B. Collins, A. Novoselac, D. K. Farmer, M. E. Vance, P. S. Stevens and J. P. D. Abbatt, Cooking, Bleach Cleaning, and Air Conditioning Strongly Impact Levels of HONO in a House, Environ. Sci. Technol., 2020, 54, 13488–13497 CrossRef CAS.
- S. Robbana-Barnat, M. Rabache, E. Rialland and J. Fradin, Heterocyclic amines: occurrence and prevention in cooked food, Environ. Health Perspect., 1996, 104, 280–288 CrossRef CAS PubMed.
- K. Kikugawa, Formation of mutagens, 2-amino-3, 8-dimethylimidazo [4, 5-# f] quinoxaline (MeIQx) and 2-amino-3, 4, 8-trimethylimidazo [4, 5-# f] quinoxaline (4, 8-DiMeIQx), in heated fish meats, Mutat. Res. Mol. Mech. Mutagen., 1987, 179, 5–14 CrossRef CAS PubMed.
- L. Salehpoor and T. C. VandenBoer, Suppressor and calibration standard limitations in cation chromatography of ammonium and 10 alkylamines in atmospheric samples, Anal. Methods, 2023, 15, 3822–3842 RSC.
- K. Sekimoto, S.-M. Li, B. Yuan, A. Koss, M. Coggon, C. Warneke and J. de Gouw, Calculation of the sensitivity of proton-transfer-reaction mass spectrometry (PTR-MS) for organic trace gases using molecular properties, Int. J. Mass Spectrom., 2017, 421, 71–94 CrossRef CAS.
- C. Wang, J. Liggio, J. J. B. Wentzell, S. Jorga, A. Folkerson and J. P. D. Abbatt, Chloramines as an important photochemical source of chlorine atoms in the urban atmosphere, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220889120 CrossRef CAS.
- T. Wu, T. Földes, L. T. Lee, D. N. Wagner, J. Jiang, A. Tasoglou, B. E. Boor and E. R. I. I. I. Blatchley, Real-Time Measurements of Gas-Phase Trichloramine (NCl3) in an Indoor Aquatic Center, Environ. Sci. Technol., 2021, 55, 8097–8107 CrossRef CAS PubMed.
- T. C. Furlani, R. Ye, J. Stewart, L. R. Crilley, P. M. Edwards, T. F. Kahan and C. J. Young, Development and validation of a new in situ technique to measure total gaseous chlorine in air, Atmos. Meas. Tech., 2023, 16, 181–193 CrossRef CAS.
- M. A. Bari, W. B. Kindzierski, A. J. Wheeler, M.-È. Héroux and L. A. Wallace, Source apportionment of indoor and outdoor volatile organic compounds at homes in Edmonton, Canada, Build. Environ., 2015, 90, 114–124 CrossRef.
- R. Sheu, C. Stönner, J. C. Ditto, T. Klüpfel, J. Williams and D. R. Gentner, Human transport of thirdhand tobacco smoke: a prominent source of hazardous air pollutants into indoor nonsmoking environments, Sci. Adv., 2022, 6, eaay4109 CrossRef.
- A. Arı, P. Ertürk Arı, S. Yen
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) soy-Karakaş and E. O. Gaga, Source characterization and risk assessment of occupational exposure to volatile organic compounds (VOCs) in a barbecue restaurant, Build. Environ., 2020, 174, 106791 CrossRef.
soy-Karakaş and E. O. Gaga, Source characterization and risk assessment of occupational exposure to volatile organic compounds (VOCs) in a barbecue restaurant, Build. Environ., 2020, 174, 106791 CrossRef. - X. Wang, Y. Wang, G. Hu, D. Hong, T. Guo, J. Li, Z. Li and M. Qiu, Review on factors affecting coffee volatiles: from seed to cup, J. Sci. Food Agric., 2022, 102, 1341–1352 CrossRef CAS.
- Y. Liu, P. K. Misztal, J. Xiong, Y. Tian, C. Arata, R. J. Weber, W. W. Nazaroff and A. H. Goldstein, Characterizing sources and emissions of volatile organic compounds in a northern California residence using space- and time-resolved measurements, Indoor Air, 2019, 29, 630–644 CAS.
- C. E. Stockwell, P. R. Veres, J. Williams and R. J. Yokelson, Characterization of biomass burning emissions from cooking fires, peat, crop residue, and other fuels with high-resolution proton-transfer-reaction time-of-flight mass spectrometry, Atmos. Chem. Phys., 2015, 15, 845–865 CrossRef.
- S. A. O. Adeyeye, Heterocyclic Amines and Polycyclic Aromatic Hydrocarbons in Cooked Meat Products: A Review, Polycycl. Aromat. Compd., 2020, 40, 1557–1567 CrossRef CAS.
- L. Hashim and H. Chaveron, Use of methylpyrazine ratios to monitor the coffee roasting, Food Res. Int., 1995, 28, 619–623 CrossRef CAS.
- E. C. Coleman and C.-T. Ho, Chemistry of baked potato flavor. 1. Pyrazines and thiazoles identified in the volatile flavor of baked potato, J. Agric. Food Chem., 1980, 28, 66–68 CrossRef CAS.
- I. M. Weiss, C. Muth, R. Drumm and H. O. K. Kirchner, Thermal decomposition of the amino acids glycine, cysteine, aspartic acid, asparagine, glutamic acid, glutamine, arginine and histidine, BMC Biophys., 2018, 11, 2 CrossRef.
- E. G. Vilar, M. G. O'Sullivan, J. P. Kerry and K. N. Kilcawley, Volatile organic compounds in beef and pork by gas chromatography-mass spectrometry: a review, Sep. Sci., 2022, 5, 482–512 CAS.
- M. M. Coggon, C. E. Stockwell, L. Xu, J. Peischl, J. B. Gilman, A. Lamplugh, H. J. Bowman, K. Aikin, C. Harkins and Q. Zhu, Contribution of cooking emissions to the urban volatile organic compounds in Las Vegas, NV, Atmos. Chem. Phys., 2024, 24, 4289–4304 CrossRef CAS.
- F. Klein, S. M. Platt, N. J. Farren, A. Detournay, E. A. Bruns, C. Bozzetti, K. R. Daellenbach, D. Kilic, N. K. Kumar, S. M. Pieber, J. G. Slowik, B. Temime-Roussel, N. Marchand, J. F. Hamilton, U. Baltensperger, A. S. H. Prévôt and I. El Haddad, Characterization of Gas-Phase Organics Using Proton Transfer Reaction Time-of-Flight Mass Spectrometry: Cooking Emissions, Environ. Sci. Technol., 2016, 50, 1243–1250 CrossRef CAS PubMed.
- J. J. Schauer, M. J. Kleeman, G. R. Cass and B. R. T. Simoneit, Measurement of Emissions from Air Pollution Sources. 4. C1−C27 Organic Compounds from Cooking with Seed Oils, Environ. Sci. Technol., 2002, 36, 567–575 CrossRef CAS PubMed.
- W. W. Nazaroff, Residential air-change rates: a critical review, Indoor Air, 2021, 31, 282–313 CrossRef PubMed.
- A. Gandolfo, L. Rouyer, H. Wortham and S. Gligorovski, The influence of wall temperature on NO2 removal and HONO levels released by indoor photocatalytic paints, Appl. Catal., B, 2017, 209, 429–436 CrossRef CAS.
- E. G. Alvarez, M. Sörgel, S. Gligorovski, S. Bassil, V. Bartolomei, B. Coulomb, C. Zetzsch and H. Wortham, Light-induced nitrous acid (HONO) production from NO2 heterogeneous reactions on household chemicals, Atmos. Environ., 2014, 95, 391–399 CrossRef.
- T. Wainman, C. J. Weschler, P. J. Lioy and J. Zhang, Effects of surface type and relative humidity on the production and concentration of nitrous acid in a model indoor environment, Environ. Sci. Technol., 2001, 35, 2200–2206 CrossRef CAS.
- R. Kurtenbach, K. H. Becker, J. A. G. Gomes, J. Kleffmann, J. C. Lörzer, M. Spittler, P. Wiesen, R. Ackermann, A. Geyer and U. Platt, Investigations of emissions and heterogeneous formation of HONO in a road traffic tunnel, Atmos. Environ., 2001, 35, 3385–3394 CrossRef CAS.
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, J. D. Surratt, N. M. Donahue, J. T. Jayne and D. R. Worsnop, Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef.
- L. J. Kramer, L. R. Crilley, T. J. Adams, S. M. Ball, F. D. Pope and W. J. Bloss, Nitrous acid (HONO) emissions under real-world driving conditions from vehicles in a UK road tunnel, Atmos. Chem. Phys., 2020, 20, 5231–5248 CrossRef CAS.
- Y. Liu, K. Lu, Y. Ma, X. Yang, W. Zhang, Y. Wu, J. Peng, S. Shuai, M. Hu and Y. Zhang, Direct emission of nitrous acid (HONO) from gasoline cars in China determined by vehicle chassis dynamometer experiments, Atmos. Environ., 2017, 169, 89–96 CrossRef CAS.
- F. Spataro and A. Ianniello, Sources of atmospheric nitrous acid: state of the science, current research needs, and future prospects, J. Air Waste Manage. Assoc., 2014, 64, 1232–1250 CrossRef CAS PubMed.
- R. Bröske, J. Kleffmann and P. Wiesen, Heterogeneous conversion of NO2 on secondary organic aerosol surfaces: a possible source of nitrous acid (HONO) in the atmosphere?, Atmos. Chem. Phys., 2003, 3, 469–474 CrossRef.
- J. Kleffmann, K. H. Becker and P. Wiesen, Heterogeneous NO2 conversion processes on acid surfaces: possible atmospheric implications, Atmos. Environ., 1998, 32, 2721–2729 CrossRef CAS.
- M. A. Donaldson, A. E. Berke and J. D. Raff, Uptake of Gas Phase Nitrous Acid onto Boundary Layer Soil Surfaces, Environ. Sci. Technol., 2014, 48, 375–383 CrossRef CAS PubMed.
- M. A. Donaldson, D. L. Bish and J. D. Raff, Soil surface acidity plays a determining role in the atmospheric-terrestrial exchange of nitrous acid, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18472–18477 CrossRef CAS.
- Z. Zhou, L. R. Crilley, J. C. Ditto, T. C. VandenBoer and J. P. D. Abbatt, Chemical Fate of Oils on Indoor Surfaces: Ozonolysis and Peroxidation, Environ. Sci. Technol., 2023, 57, 15546–15557 CrossRef CAS.
- Y. Li, L. He, D. Xie, A. Zhao, L. Wang, N. M. Kreisberg, J. Jayne and Y. Liu, Strong temperature influence and indiscernible ventilation effect on dynamics of some semivolatile organic compounds in the indoor air of an office, Environ. Int., 2022, 165, 107305 CrossRef CAS PubMed.
- D. G. Poppendieck, L. C. Ng, A. K. Persily and A. T. Hodgson, Long term air quality monitoring in a net-zero energy residence designed with low emitting interior products, Build. Environ., 2015, 94, 33–42 CrossRef.
- J. C. Ditto, M. Webb, H. N. Huynh, J. Yu, G. C. Morrison, B. J. Turpin, M. R. Alves, K. Mayer, M. F. Link, A. H. Goldstein, D. Poppendieck, M. E. Vance, D. K. Farmer, A. W. H. Chan and J. P. D. Abbatt, The Role of Indoor Surface pH in Controlling the Fate of Acids and Bases in an Unoccupied Residence, ACS ES&T Air, 2024, 1(9), 1015–1027, DOI:10.1021/acsestair.4c00044.
- C. Arata, P. K. Misztal, Y. Tian, D. M. Lunderberg, K. Kristensen, A. Novoselac, M. E. Vance, D. K. Farmer, W. W. Nazaroff and A. H. Goldstein, Volatile organic compound emissions during HOMEChem, Indoor Air, 2021, 31, 2099–2117 CrossRef CAS PubMed.
- W. D. Fahy, F. Wania and J. P. D. Abbatt, When Does Multiphase Chemistry Influence Indoor Chemical Fate?, Environ. Sci. Technol., 2024, 58, 4257–4267 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00491d |
| ‡ Current address: Atmospheric Services, WSP Australia, Brisbane. |
| § Current address: Department of Energy, Environmental, and Chemical Engineering, Washington University in St. Louis. |
| ¶ Current address: Exposure and Biomonitoring Division, Environmental Health Science and Research Bureau, Health Canada. |
| This journal is © The Royal Society of Chemistry 2025 |