
Salt-bridge mediated cooperativity and mechanical stabilization of tandem spectrin repeats†
Yanzhong
Wang‡
ab,
Yuhang
Zhang‡
 c,
Miao
Yu
d,
Peng
Xiu
b,
Yanwei
Jia
e,
Hu
Chen
c,
Shimin
Le
*c,
Jin
Qian
*b and
Jie
Yan
*af
c,
Miao
Yu
d,
Peng
Xiu
b,
Yanwei
Jia
e,
Hu
Chen
c,
Shimin
Le
*c,
Jin
Qian
*b and
Jie
Yan
*af
aMechanobiology Institute, National University of Singapore, Singapore 117411, Singapore. E-mail: phyyj@nus.edu.sg
bInstitute of Applied Mechanics, School of Aeronautics and Astronautics, Zhejiang University, Hangzhou, 310027, China. E-mail: jqian@zju.edu.cn
cDepartment of Physics, Research Institute for Biomimetics and Soft Matter, Fujian Provincial Key Lab for Soft Functional Materials Research, Xiamen University, Xiamen, 361000, China. E-mail: leshimin@xmu.edu.cn
dDepartment of Biochemistry and Division of Orthopaedic Surgery of the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China
eState Key Laboratory of Analog and Mixed-Signal VLSI, Institute of Microelectronics, University of Macau, Taipa, Macau, 999078, China
fDepartment of Physics, National University of Singapore, Singapore 117551, Singapore
First published on 3rd June 2025
Abstract
Spectrin superfamily proteins play essential roles in cells by interlinking various cytoskeletal components and bridging the cytoskeleton to both the cell membrane and the nucleus. Characterized by the spectrin repeat (SR) domain, this superfamily features a unique bundle of three antiparallel α-helices. These SRs often appear as tandem repeats linked by short segments, serving as tension-bearing structural units that support the cytoskeleton and act as signaling hubs for numerous proteins. Although the cooperative force-dependent unfolding of tandem spectrin repeats is well-documented, the precise molecular mechanisms remain unclear. In this study, we used the paradigmatic tandem SR (SR3–SR4) of α-actinin as our model system. Our results reveal that cooperativity arises from the salt bridges on the linker between the two domains. Additionally, we found that the salt bridge mechanically stabilizes the two domains, extending the lifetime of SR3–SR4 by 10 to 100 times compared to individual domains. Our full-atom MD simulations show that the linker salt bridge is a major force-bearing point, and its disruption leads to the mechanical unfolding of the domains. Finally, combining AlphaFold structural prediction and single-molecule manipulation studies of other spectrin superfamily proteins, we demonstrate that linker salt bridge-mediated cooperativity and stabilization is a potentially conserved molecular mechanism governing the mechanical responses of SRs in spectrin superfamily proteins.
New conceptsSpectrin superfamily proteins play pivotal roles within cells by cross-linking diverse cytoskeletal elements and establishing connections between the cytoskeleton, cell membrane, and nucleus. The hallmark feature of this superfamily is the spectrin repeat (SR) domain, often found as tandem repeats with short α-helical linkers. This work employs a combination of single-protein magnetic tweezers and molecular dynamics simulations to elucidate the role of the α-helical linker connecting tandem SRs in spectrin superfamily proteins. Our study reveals that this linker facilitates inter-domain salt-bridge interactions, thereby dominating the mechanical stabilization and cooperative unfolding of adjacent SRs. Our results propose a potentially conserved molecular mechanism governing the mechanical stabilization and cooperativity of neighboring SRs within spectrin superfamily proteins. |
Introduction
In cells, the actomyosin cytoskeleton consists of actin filaments, actin-binding proteins that organize the filaments into diverse structures, and myosin fibers responsible for the dynamic contraction of these filaments. The actomyosin cytoskeleton interfaces with other cytoskeletal elements such as intermediate filaments and microtubules through specific connecting proteins and associates with both the cell membrane and the nuclear membrane via sets of adaptor proteins.1–4 Actomyosin contraction or external mechanical disturbances produce dynamic tension within the filaments and related proteins, resulting in networks that are highly dynamic yet remarkably stable. These networks facilitate cells’ mechanosensing and mechanotransduction capabilities, which are vital for regulating cell–matrix and cell–cell adhesions, cell migration, differentiation, growth, tissue repair, and embryonic development.1–3The associated, tension-bearing, mechano-sensitive proteins are indispensable for this robust system. Examples include the actin filament cross-linking protein, α-actinin, which assembles the filaments into fibers; the membrane-associated actin-binding proteins, spectrin and dystrophin, which maintain the cell shape and the stability and structure of the cell membrane;5–7 the plakin family proteins, which act as linkers among actin filaments, microtubules, and intermediate filaments8 and connect intermediate filaments to the cell and nuclear membranes; and the nesprin family proteins, which link the cytoskeleton and the nuclear membrane.9 These proteins belong to the spectrin superfamily. The signature domain of this superfamily, the spectrin repeat (SR), is a characteristic bundle of three antiparallel α-helices, 106–122 amino acids long. SRs in these proteins exist as tandem repeats with short linkers, acting as tension-bearing structural modules and hubs for numerous signaling proteins.
Dynamic tension is transmitted through tandem SRs, making their tension-dependent structural dynamics crucial for their functional activities. Intriguingly, cooperative unfolding behaviors have been frequently observed among the tandem SRs of various proteins, including spectrins,10–14 dystrophin,15 utrophin,16 and α-actinin,17 as revealed by various biophysical technologies over the past few decades. Previous pioneering research on spectrins emphasized the significance of the helical nature of inter-domain linkers in maintaining cooperativity between the domains.10–14 Nonetheless, the specific molecular mechanism governing the cooperativity of the tandem SRs and their linkers remains unclear.
In this work, we employed the paradigmatic tandem spectrin repeats (SR3–SR4) of α-actinin-1,17–19 often regarded as the ancestral protein of the family, as our model system to explore the molecular mechanism underlying the cooperativity of the tandem SRs. Using a single-protein manipulation platform with magnetic tweezers, we directly quantified the unfolding force distributions and mechanical lifetimes of the SR3–SR4 tandem repeat, as well as the individual SR3 and SR4 domains. Intriguingly, we found that SR3–SR4 not only unfolds cooperatively but also exhibits a mechanical lifetime substantially greater than that of either SR3 or SR4 alone. This observation suggests a significant mechanical stabilization effect resulting from inter-domain interactions. Our molecular dynamics simulations revealed that the linker between SR3 and SR4 plays a vital role in augmenting the mechanical stability of both SR4 and SR3 through linker-mediated salt bridges, which was experimentally confirmed by corresponding mutations that remove the salt bridges. AlphaFold3 predicts the existence of similar linker-mediated inter-domain salt bridges among spectrin superfamily proteins, suggesting inter-domain cooperativity in mechanical unfolding, supported by single-molecule mechanical unfolding experiments with dystrophin SRs. Overall, our findings uncover a potentially conserved molecular mechanism governing the mechanical stabilization and cooperativity of neighboring SRs.
Experimental section
Single-protein manipulation and analysis
A vertical magnetic tweezer setup was combined with a disturbance-free, rapid solution-exchange flow channel for conducting in vitro protein stretching experiments.20–22 All in vitro protein stretching experiments were performed in a solution containing 1× PBS, 1% BSA, 1 mM DTT, and 10 mM sodium L-ascorbate at 22 ± 1 °C. The force calibration of the magnetic tweezer setup introduces a 10% uncertainty due to the heterogeneity in the diameter of paramagnetic beads,20 while the determination of the bead height in the magnetic tweezer setup has an uncertainty of approximately 2–5 nm due to the thermal fluctuation of the tethered bead and the camera's resolution.23 Raw data obtained from magnetic tweezer experiments were analyzed using OriginPro or MATLAB software and plotted using OriginPro. Single molecular tether between the bead and the surface was ensured by monitoring microbeads’ positional changes in the x-, y-, and z-directions during stepwise unfolding or refolding events. In the case of a single tether, a discrete change in extension along the z-axis corresponding to the unfolding or refolding of a protein domain is accompanied by minimal or no displacement in the x and y directions.20In the single-molecule manipulation experiments, at least three independent individual single molecules are stretched for multiple cycles for each assay, such as the linear force-loading assay shown in Fig. 1 and the force-jump assay shown in Fig. 2. The total number of data points is indicated in each figure panel. All data points are included for the statistical analyses, such as the normalized unfolding force distribution in Fig. 1 and the characteristic lifetime fitting in Fig. 2.
 | ||
| Fig. 1 Force-dependent unfolding of SR3–SR4, SR3, and SR4 domains. (a) Top panel: Design of the single-molecule construct of the SR3–SR4 molecule. The example structure of SR3–SR4 is taken from PDB: 1SJJ,18 where blue represents SR3, green represents SR4, and red represents the linker. Bottom panel: Illustration of the single-molecule experiments where the construct is specifically tethered between a paramagnetic bead (via a DNA handle) and a coverslip surface. The illustration on the lower right showcases three experimental designs: SR3–SR4, (SR4)6, and SR3–SR4 with the vinculin-D1 domain bound to SR4, respectively. (b) Top panel: Typical force-bead height curves of the SR3–SR4 construct during the force-increase scan at a loading rate of 1 pN s−1. Five consecutive curves are depicted by colored lines, obtained by applying 10-point FFT smoothing to the raw gray data. Middle panel: Each rupture transition step in the scan is marked by a hollow circle. The dashed curve in the panel represents the theoretical estimation of the force-dependent step size for cooperative unfolding of SR3–SR4 (upper) and individual SR3/SR4 unfolding (lower). Bottom panel: Normalized histograms of rupture force distributions (N = 218). (c) Top panel: Typical force-bead height curves of the (SR4)6 construct during the force-increase scan at a loading rate of 1 pN s−1. Middle panel: Each rupture transition step in the scan is indicated by a hollow circle. Bottom panel: Normalized histograms of SR4 rupture force distributions, plotted based on N = 214 data points. (d) Top panel: Typical force-height curves of the SR3–SR4 domain in the presence of vinculin-D1 bound to SR4, obtained at a loading rate of 1 pN s−1. Middle panel: Force-dependent step sizes of SR3 unfolding observed during the force-increase scans. The dashed curve in the panel represents the theoretical estimation of the force-dependent step size for SR3 unfolding transition. Bottom panel: Normalized histograms of rupture force distributions (N = 42). | ||
 | ||
| Fig. 2 Force-dependent lifetimes of SR3–SR4, SR3, and SR4 domains. (a) A representative force-jump measurement depicting the force-dependent lifetime of the SR3–SR4 molecule. The black arrows indicate the changes in the bead height caused by the structural transitions (unfolding) of the construct. The construct was subjected to a low force of approximately 1 pN for around 10 seconds (depicted by the dark gray lines) to facilitate the formation of the SR3–SR4 complex. Subsequently, the force was rapidly increased to approximately 19.6 pN (as shown by the red lines in the example) until the complex rupture was observed. The force was then reduced to approximately 1 pN to allow the molecule to refold. (b) and (c) Representative force-jump cycles of (SR4)6 and the SR3–SR4 bound with vinculin, respectively. (d) The force-dependent lifetimes of SR3–SR4, SR3, and SR4. The hollow green/blue/black circles represent individual lifetimes measured at the corresponding forces. The solid green/blue/black squares represent the characteristic lifetimes at the respective forces. The green/blue/black lines correspond to the fitting curve based on Bell's model. The horizontal error bars reflect a force calibration uncertainty of 10 to 15%, stemming from bead heterogeneity. The vertical error bars represent the standard error of the mean. The zero-force unfolding rates k0u and transition distance Δ are as follows: 4.4 × 10−4 s−1 and 2.3 nm (SR4), 3.9 × 10−8 s−1 and 4.7 nm (SR3), and 3.0 × 10−5 s−1 and 1.7 nm (SR3–SR4), respectively. | ||
Seven plasmids were prepared for the expression of the protein constructs for single-molecule stretching experiments: three plasmids for α-actinin SR domains: (1) pET151-Avi-(ACTN1-SR3–SR4)-Spy, (2) pET151-Avi-(ACTN1-SR4)6-Spy,17 and (3) pET151-Avi-(ACTN1-SR3–SR4)Arg625Ala,Glu630Ala-Spy, and four plasmids for dystrophin SR domains as described previously.15 The DNA fragments containing the genes were synthesized by geneArt/IDTgblock/Sangon, assembled using the seamless DNA Assembly Kit, and validated through sequencing. Each plasmid was co-transformed with a BirA plasmid and expressed in Escherichia coli BL21 (DE3), cultured in LB media with D-Biotin (Sigma Aldrich), and then affinity-purified through a 6His-tag. Detailed sequence information for the constructs can be found in Text S1 (ESI†).
Molecular dynamics simulations
Molecular dynamics (MD) simulations were carried out using the GROMACS software,24 employing the Amber99SB-ildn force field and the explicit TIP3P water model.25,26 The model for the spectrin repeats was based on the structure of Chicken Gizzard Smooth Muscle α-actinin (PDB: 1SJJ).18 The system was solvated in a 150 mM NaCl aqueous solution. To ensure overall charge neutrality, additional Na ions were added to compensate for the net negative charge of the solute. Initial energy minimization was performed using the steepest descent method. Subsequently, 100 ps of NVT (constant number of particles, volume, and temperature) followed by 1 ns of NPT (constant number of particles, pressure, and temperature) equilibration were conducted, both with harmonic positional restraints applied to the heavy atoms of the protein.A constant temperature of 300 K was maintained using a Nosé–Hoover thermostat,27 while the pressure was kept at 1 bar using isotropic coupling to a Parrinello–Rahman barostat.28 Throughout all simulations, long-range electrostatic interactions were treated with the particle mesh Ewald method.29 The constraints for heavy atom-hydrogen bonds were applied using the LINCS procedure.30
Steered molecular dynamics (SMD) simulations were performed using harmonic springs with a spring constant of k = 300 pN nm−1, and various equilibrium lengths are connected between the Cα atoms of residues at the N-terminal and the C-terminal. For each equilibrium spring length, the SMD simulation was performed for 80 ns, consisting of a 50 ns equilibration stage and a 30 ns production stage, with the total simulation time for each spectrin repeat exceeding 1 μs. The free energy and force profiles were obtained using WHAM umbrella sampling analysis.31,32 The average force values were also directly obtained based on the average spring extension ![[capital Delta, Greek, macron]](https://www.rsc.org/images/entities/char_e135.gif) x,
x, ![[F with combining macron]](https://www.rsc.org/images/entities/i_char_0046_0304.gif) = kx as well. Thus, the spring constant was determined to be sufficiently stiff to constrain the regional fluctuations, yet soft enough to allow overlapping of regional fluctuations that is needed for umbrella sampling.32
= kx as well. Thus, the spring constant was determined to be sufficiently stiff to constrain the regional fluctuations, yet soft enough to allow overlapping of regional fluctuations that is needed for umbrella sampling.32
The force differences were computed using force distribution analysis (FDA).33 For each frame, the pairwise forces ΔFij between two residues i and j can be obtained by summing up atomic forces between these two residues. To measure the force propagation through the protein, subtraction of the pairwise forces between the selected state and the reference state was performed:
| ΔFij = |Fselectedij − Freferenceij|. | (1) |
By summing up all the residue-based pairwise forces, the force of each residue was obtained:
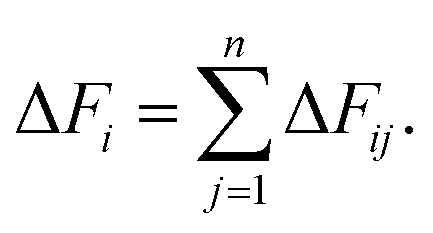 | (2) |
For each case, ΔFi was averaged over the last 30 ns simulations. The FDA was performed on each individual trajectory per condition, and the resulting average forces of the mutant were subtracted from those of the wild type. The networks shown are connected graphs of residues with force differences above a given threshold.
Results and discussion
Enhanced mechanical stability of the SR3–SR4 tandem due to inter-domain cooperativity
In order to investigate the force-dependent unfolding behaviour of α-actinin SR3–SR4, we prepared single-molecule constructs that consist of the SR3 and SR4 as well as the linker between the domains (referred to as SR3–SR4, 500th–745th a.a., UniProt P12814), or six repeats of SR4 (621th–745th a.a., UniProt P12814, referred to as (SR4)6), spanned between an N-terminal biotinylated AviTag and a C-terminal SpyTag (Fig. 1a). The two tags enable specific tethering of the target protein between a spyCatcher-coated coverslip surface and a super-paramagnetic bead, utilizing streptavidin-labeled DNA handles as spacers between the bead and the coverslip surface.17,34 Subsequently, we applied an external force to the SR3–SR4 or (SR4)6 molecule using a magnetic tweezers setup20 and observed the structural dynamics of the domains under force. These dynamics were reflected by corresponding changes in the bead height. Here we note that the linker regions prior and after the target domain/domains are kept in the constructs to better mimic the physiological conditions. The detailed sequence information of the constructs could be found in the supporting Text S1 (ESI†).The top panels of Fig. 1b and c display the typical force–bead height curves for the SR3–SR4 and (SR4)6 tether during linear force-increase scans, respectively, with a physiologically relevant loading rate of 1 pN s−1.35,36 The middle and bottom panels of Fig. 1b and c illustrate the corresponding force-step size graph and the normalized unfolding force distribution for SR4 and SR3–SR4, obtained from N = 214 and N = 218 force-increase scan cycles, respectively. We note that bead-height changes during a force change can result from both molecular extension and bead rotation due to torque rebalance. In contrast, a stepwise bead-height change during which force remains nearly unchanged reflects a change in molecular extension, such as during protein domain unfolding or refolding.20,37
The unfolding curves of (SR4)6 show characterized step-wise unfolding of individual independent domains. The unfolding forces are peaked at around 11.0 ± 0.2 pN (mean ± standard deviation) based on Gaussian fitting. In sharp contrast, for the SR3–SR4 construct, concurrent unfolding of both domains with a single large step size that equals the sum of two domain unfolding, or nearly concurrent unfolding of the two domains with a small time interval at forces of 20.8 ± 0.2 pN was predominant. The concurrent or nearly concurrent unfolding indicates strong cooperativity between SR3 and SR4. Importantly, this also results in a strong mechanical stabilization of SR4, evidenced by an increased unfolding force of about 10 pN.
To investigate whether this cooperativity also enhances the mechanical stabilization of SR3, we utilized the D1 domain of vinculin, which is known to bind a high-affinity site in SR4 in its unfolded conformation38 and maintains SR4 in the unfolded state when the force is reduced.17 We stretched the SR3–SR4 molecule in the 200 nM vinculin-D1 domain in solution, ensuring that a vinculin-D1-bound unfolded SR4 disrupts the inter-domain cooperativity between SR3 and SR4. The top panel of Fig. 1d illustrates typical force-bead height curves for SR3–SR4 (vinculin-D1 bound) during linear force-increase scans at a loading rate of 1 pN s−1. The middle and bottom panels of Fig. 1d show the corresponding force-step size graph and the normalized unfolding force distribution of SR3, obtained from N = 42 force-increase scan cycles. The SR3 domain unfolds at forces of 15.1 ± 0.1 pN, roughly 5 pN less than the unfolding forces of SR3–SR4.
Next, we further explored the mechanical stabilization effects of the inter-domain interaction of the SR3–SR4 molecule. To directly quantify the force-dependent lifetime of the SR3–SR4 molecule, as well as that of the individual SR3 and SR4 domains, we implemented force-jump-cycle procedures (Fig. 2). Initially, the SR3–SR4 molecule was held at a small force of ∼1 pN for ∼30 seconds to allow folding. Then, we increased the force to a target level (e.g., ∼19.6 pN in Fig. 2a, shown in red) and maintained it until the unfolding events were observed. These were indicated by one large step of bead height jump or two sequential, close steps. Subsequently, we reduced the force to permit refolding of the SR3–SR4 for the next cycle. By repeating such force-jump-cycles multiple times at target forces ranging from ∼12 pN to ∼23 pN, we obtained a pool of lifetimes for each target force, leading to a force-dependent average lifetime. Similar force-jump-cycles were used for six repeats of SR4 (Fig. 2b), as well as the SR3–SR4 molecule in the presence of vinculin-D1 (Fig. 2c), to ascertain the force-dependent lifetime of individual domains. It is worth noting that, for the SR3–SR4 molecule with vinculin-D1 in solution, an additional step of force at ∼3 pN for 5 s was included to increase the exposure time of the unfolded SR4, thereby ensuring vinculin-D1 re-binding to VBS in the unfolded SR4 if the vinculin-D1 was disassociated during higher forces. For the six repeats of SR4, a pseudo-dwell-time analysis method was implemented to calculate the force-dependent lifetime of SR4.39
Fig. 2d illustrates the force-dependent lifetime of the SR3–SR4 tandem (black), SR3 (blue), and SR4 (green). The inter-domain interaction between SR3 and SR4 markedly stabilizes the domains, resulting in a significantly extended mechanical lifetime when compared to SR3 or SR4 alone. The individual SR4 domain endures ∼100 seconds at a force of ∼5 pN, and it can last for a duration in seconds at forces up to 10 pN. In sharp contrast, the force-dependent lifetime of the SR4 domain within the SR3–SR4 tandem increases by nearly two orders of magnitude. Furthermore, the differential transition distances Δ of SR3, SR4, and the SR3–SR4 tandem seem to indicate that the force-induced unfolding pathways of SR3, SR4, and the SR3–SR4 tandem are distinctly different.
Overall, the results suggest that there is an inter-domain interaction between SR3 and SR4 that results in strong cooperativity in mechanical unfolding of the two domains and contributes substantially to the mechanical stabilization of both SR3 and SR4.
Disruption of linker-mediated salt bridges precedes the mechanical unfolding of SR3–SR4, SR3, and SR4
To investigate the molecular intricacies of the inter-domain interactions between SR3 and SR4 that contribute to the cooperative unfolding and mechanical stabilization effect of the domains, we conducted molecular dynamics (MD) simulations on the SR3–SR4 complex as well as the individual SR3 and SR4 domains. Since the X-ray structure of human α-actinin-1 has not been reported, the model of the spectrin repeats for the simulation studies was based on the structure of α-actinin-2 of smooth muscle (PDB: 1SJJ),18 which agrees well with the structural prediction using AlphaFold340 (Fig. S1, ESI†). In these simulations, harmonic spring constraints were applied to the protein domains, using the same spring constant k = 300 pN nm−1 (top panels of Fig. 3a–c). As we incrementally increased the equilibrium length of the spring, the proteins began to unfold gradually. Forces were directly obtained from the spring constraints with different equilibrium lengths (illustrated as dots in bottom panels of Fig. 3a–c). | ||
| Fig. 3 Mechanical stabilization of SR3–SR4 through the linker-mediated salt bridge. (a)–(c) Top panels: Schematic representation of molecular dynamics simulations illustrating the stretching of SR3–SR4, SR4, and SR3. Harmonic spring constraints with an identical spring constant of k = 300 pN nm−1 were applied. The equilibrium length of the spring was incrementally adjusted to unfold the proteins gradually. Bottom panels: Force versus end-to-end distance profiles for (a) SR3, (b) SR4, and (c) SR3–SR4 from MD simulations. Black dots depict the average forces directly obtained from the spring constraints at various equilibrium lengths, while the continuous curves are generated through cubic spline interpolation. The average minimum distances between SR3–SR4 residues, (d) Glu621 and Arg625, and (e) Glu630 and Arg633 were monitored during the stretching process. These distances reflect the persistence of salt bridges between the linker and SR3/SR4. Significant increases in the distance indicate the detachment of residues, corresponding to the force drops in (e). | ||
Using cubic spline interpolation, continuous force–distance curves for SR3, SR4, and the SR3–SR4 complex were generated (as shown by the curves in Fig. 3a–c). Integration of the force with respect to the end-to-end distance of the protein allowed us to derive free energy profiles (Fig. S2, blue lines, ESI†). These are in agreement with the global unbiased free energy profiles obtained via constrained local fluctuations with different spring equilibrium lengths using the standard weighted histogram analysis method (WHAM, see Fig. S2, ESI†).31 As the equilibrium length of the spring increased, the end-to-end distance of the protein extended, and the force on SR3, SR4, and the SR3–SR4 complex initially increased slowly. Upon stretching to a specific distance, a substantial drop in force was observed, signifying a partial structural transition.
During the stretching process, the microstructure of SR3/SR4/SR3–SR4 undergoes a transformation, as depicted by the representative structures in Fig. S2–S4 (ESI†). For SR3, when the end-to-end distance d = 5.1 nm, there are no significant changes in the structure of SR3. Interestingly, at d = 5.8 nm, the Lys556–Asp616 salt bridge is abolished, accompanied by a decrease in the stretching force, which corresponds to the first peak in Fig. 3a. As SR3 protein is stretched further from d = 6.2 nm to d = 6.8 nm, the Arg518–Glu565 salt bridge breaks, corresponding to the last peak in Fig. 3a (Fig. S3, ESI†). Salt-bridge residues also play a significant role during the stretching process of SR4. From d = 4.9 nm to d = 6.8 nm, the Lys633–Asp703 and Arg673–Glu733 salt bridges are abolished, and the α-helices unwind from the C terminus (Fig. S3, ESI†).
In the case of SR3–SR4, the α-helical linker partially unwinds (residues 621 to 630) at d = 12.4 nm, corresponding to the first peak in Fig. 3c. The breaking of the Glu621/Glu622–Arg625 salt bridge occurs at d = 13.6 nm, and the highest peak in Fig. 3c corresponds to the abolishment of salt bridges with Glu630, Arg633, and Asp703 (Fig. S5, ESI†). The average minimum distances between residues Glu621 and Arg625, Glu630 and Arg633 (the salt bridges between the linker and SR3–SR4) during the stretching process are recorded in Fig. 3d and e, respectively. The substantial increase in the distance signifies the unbinding of residues, which directly corresponds to the force drops in Fig. 3c. These results indicate that the Glu621/Glu622–Arg625 and Glu630–Arg633 salt bridges are crucial during the gradual partial unfolding process of SR3–SR4. Overall, the results reveal that mechanical destabilization of SR3–SR4, SR3 and SR4 starts from disruption of salt bridges on the linker between SR3 and SR4, strongly suggesting a crucial role of these linker-mediated salt bridges in their mechanical stability.
Point mutations in the salt-bridge-coordinating residues destabilize the SR3–SR4
To further evaluate the role of the linker-mediated salt bridges in modifying the mechanical stability of SR3–SR4, point mutations Arg625Ala and Glu630Ala were introduced into linker residues in MD simulations (Fig. 4). Analysis of the per residue root-mean-square-fluctuation (RMSF) revealed global differences across most residues between the wild type and the mutant (Fig. S6, ESI†), suggesting that the point mutations Arg625Ala and Glu630Ala destabilize SR3–SR4. A harmonic spring with a constant k = 300 pN nm−1 was applied to the SR3–SR4 mutant, with increasing equilibrium lengths used to stretch the protein. The force–distance curve is depicted in Fig. S7 (ESI†), displaying a force level during stretching clearly lower than that in Fig. 3c. This demonstrates that residues Arg625 and Glu630 significantly influence the unfolding force of SR3–SR4. | ||
| Fig. 4 Destabilization of SR3–SR4 due to point mutations in the linker in MD simulations. (a) Comparison of internal residue-based forces between the mutant and wild-type SR3–SR4 at different force thresholds (purple bars), highlighting the mutation residues Arg625/Glu630Ala (indicated by black arrows). (b) Comparison of salt bridges between the linker and SR3/SR4 in the wild-type SR3–SR4 (upper panel) with those in the mutant SR3–SR4 (lower panel). (c) Trajectory-averaged minimum distances between residue pairs 625 and Glu621, 625 and Glu622, 630 and Arg633, as well as Arg633 and Asp703, in both wild-type and mutant SR3–SR4. | ||
To elucidate the structural and mechanical changes in the Arg625Ala and Glu630Ala mutants, we conducted a force distribution analysis (FDA, see the Methods section) in full-atom MD simulations.33 Introducing a mutation can alter the force distribution, providing insights into how the mutation affects the force propagation pathway.33 We examined the force distribution in the mutant SR3–SR4, comparing the magnitude of the differences to the wild type to identify the allosteric pathways. Fig. 4a illustrates the network of residue pairs displaying changes in inter-residue forces exceeding a specific threshold (purple bars). The mutation notably affects the force change more in the SR4 domain than in the SR3 domain, indicating that the linker residues enhance the mechanical stability of SR4 and further stabilize SR3–SR4. Importantly, the mutations result in more than 50 pN force changes at the sites corresponding to the linker-mediated salt bridges in the wild type, suggesting that the linker-mediated salt bridges are critical force-bearing sites.
The FDA shows that force propagation began from the mutation points Arg625Ala and Glu630Ala (marked with black arrows in the bottom panel in Fig. 4a), transmitting to the SR3 and SR4 domains through salt bridges with Glu621, Glu622, and Arg625 and Glu630, Arg633 and Asp703, respectively. To identify salt bridges vital for the structural stability of SR3–SR4, we calculated the minimum distance of salt-bridge residues in the wild type and mutant, which indicates the interaction strength between two residues (Fig. 4c). In the mutant SR3–SR4, the distance between residues 625 and Glu621/Glu622 and between residues 630 and Arg633 is markedly higher than that in the wild type SR3–SR4, indicating robust interactions between these residues in wild type SR3–SR4, with point mutations leading to the abolition of the salt bridges. Additionally, the difference in the distance between Arg633 and Asp703 may suggest that the formation of the Glu630–Arg633 salt bridge may also influence residues outside the mutation points, possibly exerting an allosteric effect on SR4 to further stabilize the protein structure.
To experimentally confirm the role of the linker-mediated salt bridges in the cooperative mechanical unfolding and mechanical stabilization of the SR3–SR4, we constructed a single protein stretching construct containing the SR3–SR4 domain with the mutations on salt-bridge-coordinating residues, termed SR3–SR4Arg624Ala,Glu629Ala. Here we note that Arg624 and Glu629 of human α-actinin 1 correspond to Arg625 and Glu630 for α-actinin-2 of smooth muscle (PDB: 1SJJ),18 respectively. We then performed similar force loading experiments on the mutated SR3–SR4 (Fig. 5a). Fig. 5b–d show the representative force–bead height curves of the SR3–SR4Arg624Ala,Glu629Ala during linear force-increase scans with a loading rate of 1 pN s−1. Fig. 5c and d show the resulting force–step size graph and normalized force distributions of the unfolding events (N = 143). Clearly, the cooperative unfolding events of SR3 and SR4 at ∼22 pN were vanished. Instead, two separated unfolding events occurred with peak forces at 8.9 ± 0.3 pN and 12.5 ± 0.1 pN. In addition, we further quantified the force-dependent lifetimes of the two domains in SR3–SR4Arg624Ala,Glu629Ala (Fig. 5e and f) and showed that the force-dependent lifetimes of the mutated domains decreased by over 100–1000 times compared to those of the wild-type SR3–SR4.
 | ||
| Fig. 5 Destabilization of SR3–SR4 due to point mutations in the linker revealed by single-molecule stretching. (a) Single-molecule design of the SR3–SR4Arg624Ala,Glu629Ala construct, which breaks the linker-mediated inter-domain salt-bridges. (b) Four representative force–bead height curves of the SR3–SR4Arg624Ala,Glu629Ala during linear force-loading scans with a loading rate of 1 pN s−1. (c) The force–step size graph of the two SR domain unfoldings obtained from the linear force-loading scans (N = 143). The light gray and gray symbols indicate the first and second unfolding events during a scan. The dashed lines are theoretical calculations of the force-dependent step sizes of the SR3 and SR4, as well as the SR3 and SR4. (d) The resulting normalized distribution of the unfolding forces. The lines are Gaussian fitting of the distribution. (e). Representative time–bead height curves during force-jump scans at three targeting forces: 8 pN, 10 pN, and 12 pN. (f) The force-dependent lifetimes of the two SR domains in the SR3–SR4Arg624Ala,Glu629Ala construct. Gray symbols indicate the raw data, and the colored symbols indicate the characterized lifetime. The lines are Bell model fitting of the experimental data, which gives the zero-force unfolding rates k0u and transition distance Δ for 8.7 ± 4.6 × 10−4 s−1 and 2.3 ± 0.9 nm (red) and 4.9 ± 1.7 × 10−3 s−1 and 2.1 ± 1.3 nm (blue), respectively. | ||
Altogether, these results support the conclusion that the linker-mediated salt-bridge leads to mechanical stabilization and cooperativity of the SR3–SR4 domains, while point mutations in the salt-bridge-coordinating residues vanish the cooperativity and destabilize the SR3–SR4 domains.
In this study, using single-protein manipulation and molecular dynamics simulation technologies, we investigated the force-dependent mechanical stability of the paradigmatic tandem SR (SR3–SR4) domains from α-actinin. Our results clearly demonstrate that linker-mediated salt bridges result in inter-domain interactions, which in turn lead to strong cooperativity and stabilization during the mechanical unfolding of these domains.
We propose that linker-mediated salt bridges may represent a conserved molecular mechanism underlying the frequently observed cooperative mechanical unfolding of SR tandems. Indeed, analysis of AlphaFold3-predicted structures of various spectrin superfamily proteins, including α-actinin, α-spectrin, β-spectrin, dystrophin, and dystonin (Fig. S8, ESI†),40 reveals linker-mediated bridges in over 50% of the neighboring SR domains (Fig. 6a). Our previous studies on the force-dependent unfolding dynamics of all 24 SR domains in the dystrophin protein15 demonstrated multiple concurrent unfolding events between neighboring SR domains. This finding is consistent with the mechanism based on linker-mediated salt bridges analyzed from structures predicted by AlphaFold3 (Fig. 6b–e).40 Altogether, short helical linker-mediated inter-domain salt bridges may be a conserved molecular mechanism utilized by tandem SRs for mechanical stabilization and cooperativity of neighboring domains.
 | ||
| Fig. 6 Highly conserved linker-mediated inter-domain salt-bridges among the spectrin superfamily. (a) The percentages of linker-mediated inter-domain salt-bridges among the spectrin superfamily. The example protein sequences are based on the UniProt database (dystonin: Q03001; dystrophin: P11532; α-actinin: P12814; β-spectrin: Q9H254; α-spectrin: P02549); the structures are predicted by AlphaFold3.40 (b)–(e) Comparison of the predicted linker inter-domain salt bridges in dystrophin's 24 SR domains and the force-dependent unfolding dynamics of dystrophin. The single-molecule stretching experiments on dystrophin are repeating experiments consistent with our previous publication.15 The potential assignments of the predicted structure and the unfolding events are indicated by arrows in the panels. | ||
Cooperative mechanical unfolding of multiple domains concurrently or nearly concurrently has a significant impact on tension transmission. This process releases tension when pulling a tandem of domains over a large tension magnitude due to the sudden release of a long flexible polypeptide polymer, suggesting a potential role in the mechanical protection of force-transmission linkages. This is particularly important given the large and rapid extension ranges of spectrin superfamily proteins, such as spectrin networks, nesprins, and dystrophin, during large-scale dynamic cellular deformation and muscle contraction.41–43
Cooperative mechanical unfolding of multiple domains concurrently or nearly concurrently also leads to strong mechanical stabilization of SRs, which may play a significant role in mechanotransduction. Some SRs contain signaling sites buried within the folded domain.17,38 The mechanical enhancement results in auto-inhibition of these signaling sites, requiring a much higher tension threshold to activate the signaling pathway. Using α-actinin SRs as an example, the SR4 domain contains a vinculin binding site.17,38 The salt-bridge mediated increase in mechanical stability results in a higher activation force threshold to expose the buried binding site for vinculin binding.46 Additionally, some SRs may bind their partners in their folded state.44 For such SRs, the enhanced mechanical stability offers higher resilience against mechanical stress for such binding partners.
Salt-bridge mediated mechanical stabilization might be a conserved molecular mechanism not only for SRs but also for other protein domain types to regulate their mechanical behaviors.45,46 Interestingly, it has been recently reported that the salt-bridge formed within the modulation domains (M1–M3) of the C. elegans α-catenin leads to strong mechanical stabilization of the M1 domain, which is mechanically weak by itself. This results in a high activation force threshold to expose the buried signaling protein binding site within the M1.46
An insightful finding from this study is that it is possible to introduce mechanical stability at a specific site within a domain or at the domain–domain interface, which additionally leads to domain–domain cooperativity during mechanical unfolding. This can be utilized as a guiding principle in bioengineering to tune the mechanical properties of various protein domains or protein–protein interactions, promising useful applications in the development of future protein based functional materials.
Author contributions
S. L., J. Y., and J. Q. conceived the study. S. L., Y. W., M. Y. and Y. Z. designed and performed the single-molecule experiments and analyzed the data; Y. W. and Y. Z. performed the MD simulation; Y. J. and P. X. provided technological support. J. Y., S. L., Y. W., Y. Z., M. Y., J. Q., and H. C. interpreted the data; S. L. and J. Y. wrote the paper with inputs from all authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research in China was funded by the National Natural Science Foundation of China (NSFC Grant No. 32271367 and 12204389 to S. L., 12125205 to J. Q., and 32301094 to M. Y.) and the Fundamental Research Funds for the Central Universities (No. 20720220029 to S. L.). The research in Singapore was supported by the National Research Foundation (NRF), Singapore, the Mechanobiology Institute under its MID-SIZED GRANT (MSG) (NRF-MSG-2023-0001 to J. Y.), and the Singapore Ministry of Education Academic Research Funds Tier 2 (MOE-T2EP50123-0008 to J. Y.).Notes and references
- D. A. Fletcher and R. D. Mullins, Nature, 2010, 463, 485–492 CrossRef CAS PubMed.
- L. Blanchoin, R. Boujemaa-Paterski, C. Sykes and J. Plastino, Physiol. Rev., 2014, 94, 235–263 CrossRef CAS PubMed.
- R. K. H. Liem, Cold Spring Harbor Perspect. Biol., 2016, 8, a018259 CrossRef PubMed.
- S. Le, M. Yu and J. Yan, Curr. Opin. Solid State Mater. Sci., 2021, 25, 100895 CrossRef CAS.
- M. Edlund, M. A. Lotano and C. A. Otey, Cell Motil. Cytoskeleton, 2001, 48, 190–200 CrossRef CAS PubMed.
- V. Bennett and D. M. Gilligan, Annu. Rev. Cell Biol., 1993, 9, 27–66 CrossRef CAS PubMed.
- T. Haenggi and J. M. Fritschy, Cell. Mol. Life Sci., 2006, 63, 1614–1631 CrossRef CAS PubMed.
- G. Wiche, J. Cell Sci., 1998, 111, 2477–2486 CrossRef CAS PubMed.
- Q. Zhang, J. N. Skepper, F. Yang, J. D. Davies, L. Hegyi, R. G. Roberts, P. L. Weissberg, J. A. Ellis and C. M. Shanahan, J. Cell Sci., 2001, 114, 4485–4498 CrossRef CAS PubMed.
- R. Law, P. Carl, S. Harper, P. Dalhaimer, D. W. Speicher and D. E. Discher, Biophys. J., 2003, 84, 533–544 CrossRef CAS PubMed.
- R. Law, G. Liao, S. Harper, G. Yang, D. W. Speicher and D. E. Discher, Biophys. J., 2003, 85, 3286–3293 CrossRef CAS PubMed.
- R. I. MacDonald and J. A. Cummings, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1502–1507 CrossRef CAS PubMed.
- S. Batey, L. G. Randles, A. Steward and J. Clarke, J. Mol. Biol., 2005, 349, 1045–1059 CrossRef CAS PubMed.
- L. G. Randles, R. W. S. Rounsevell and J. Clarke, Biophys. J., 2007, 92, 571–577 CrossRef CAS PubMed.
- S. Le, M. Yu, L. Hovan, Z. Zhao, J. Ervasti and J. Yan, ACS Nano, 2018, 12, 12140–12148 CrossRef CAS PubMed.
- S. Rajaganapathy, J. L. McCourt, S. Ghosal, A. Lindsay, P. M. McCourt, D. A. Lowe, J. M. Ervasti and M. V. Salapaka, Sci. Rep., 2019, 9, 5210 CrossRef PubMed.
- S. Le, X. Hu, M. Yao, H. Chen, M. Yu, X. Xu, N. Nakazawa, F. M. Margadant, M. P. Sheetz and J. Yan, Cell Rep., 2017, 21, 2714–2723 CrossRef CAS PubMed.
- J. Liu, D. W. Taylor and K. A. Taylor, J. Mol. Biol., 2004, 338, 115–125 CrossRef CAS PubMed.
- J. Ylänne, K. Scheffzek, P. Young and M. Saraste, Structure, 2001, 9, 597–604 CrossRef PubMed.
- H. Chen, H. Fu, X. Zhu, P. Cong, F. Nakamura and J. Yan, Biophys. J., 2011, 100, 517–523 CrossRef CAS PubMed.
- S. Le, R. Liu, C. T. Lim and J. Yan, Methods, 2016, 94, 13–18 CrossRef CAS PubMed.
- S. Le, M. Yao, J. Chen, A. K. Efremov, S. Azimi and J. Yan, Nucleic Acids Res., 2015, 43, e113 CrossRef PubMed.
- S. Le, M. Yu and J. Yan, Angew. Chem., Int. Ed., 2019, 58, 18663–18669 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950 CrossRef CAS PubMed.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- P. Cong, L. Dai, H. Chen, J. R. van der Maarel, P. S. Doyle and J. Yan, Biophys. J., 2015, 109, 2338–2351 CrossRef CAS PubMed.
- W. Stacklies, C. Seifert and F. Graeter, BMC Bioinf., 2011, 12, 101 CrossRef PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed.
- M. H. Jo, P. Meneses, O. Yang, C. C. Carcamo, S. Pangeni and T. Ha, Science, 2024, 383, 1374–1379 CrossRef CAS PubMed.
- Y. Hu, H. Li, C. Zhang, J. Feng, W. Wang, W. Chen, M. Yu, X. Liu, X. Zhang and Z. Liu, Cell, 2024, 187, 3445–3459 CrossRef CAS PubMed.
- X. Zhao, X. Zeng, C. Lu and J. Yan, Nanotechnology, 2017, 28, 414002 CrossRef PubMed.
- P. R. J. Bois, R. A. Borgon, C. Vonrhein and T. Izard, Mol. Cell. Biol., 2005, 25, 6112–6122 CrossRef CAS PubMed.
- Y. Cao, R. Kuske and H. Li, Biophys. J., 2008, 95, 782–788 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- C. M. Denais, R. M. Gilbert, P. Isermann, A. L. McGregor, M. te Lindert, B. Weigelin, P. M. Davidson, P. Friedl, K. Wolf and J. Lammerding, Science, 2016, 352, 353–358 CrossRef CAS PubMed.
- Y. Kalukula, A. D. Stephens, J. Lammerding and S. Gabriele, Nat. Rev. Mol. Cell Biol., 2022, 23, 583–602 CrossRef CAS PubMed.
- A. Ghisleni, C. Galli, P. Monzo, F. Ascione, M.-A. Fardin, G. Scita, Q. Li, P. Maiuri and N. C. Gauthier, Nat. Commun., 2020, 11, 5108 CrossRef CAS PubMed.
- M. Reinhard, J. Zumbrunn, D. Jaquemar, M. Kuhn, U. Walter and B. Trueb, J. Biol. Chem., 1999, 274, 13410–13418 CrossRef CAS PubMed.
- S. Barrick, J. Li, X. Kong, A. Ray, E. Tajkhorshid and D. Leckband, Mol. Cell. Biol., 2018, 29, 111–122 CrossRef CAS PubMed.
- S. Le, M. Yu, C. Fu, J. A. Heier, S. Martin, J. Hardin and J. Yan, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2400654121 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nh00112a |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |