
Recent advances in poly(amino acids), polypeptides, and their derivatives in drug delivery
Huilin
Yuan†
a,
Mingxia
Jiang†
a,
Huapan
Fang
 *ab and
Huayu
Tian
*a
*ab and
Huayu
Tian
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Innovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM), Xiamen 361005, China. E-mail: hpfang@xmu.edu.cn; thy@xmu.edu.cn
bShenzhen Research Institute of Xiamen University, Shenzhen 518000, China
First published on 17th December 2024
Abstract
Poly(amino acids), polypeptides, and their derivatives have demonstrated significant potential as biodegradable biomaterials in the field of drug delivery. As degradable drug carriers, they can effectively load or conjugate drug molecules including small molecule drugs, nucleic acids, peptides, and protein-based drugs, enhancing the stability and targeting of the drugs in vivo. This strategy ultimately facilitates precise drug delivery and controlled release, thereby improving therapeutic efficacy and reducing side effects within the body. This review systematically describes the structural characteristics and preparation methods of poly(amino acids) and polypeptides, summarizes the advantages of poly(amino acids), polypeptides, and their derivatives in drug delivery, and detailedly introduces the latest advancements in this area. The review also discusses current challenges and opportunities associated with poly(amino acids), peptides, and their derivatives, and offers insights into the future directions for these biodegradable materials. This review aims to provide valuable references for scientific research and clinical translation of biodegradable biomaterials based on poly(amino acids) and peptides.
 Huilin Yuan | Huilin Yuan is currently a student in the College of Chemistry and Chemical Engineering, Xiamen University. She received her B.S degree from Huaqiao University in 2024, and she is now pursuing her Master's degree. Her research interests are biomedical materials and tumor therapy. |
 Mingxia Jiang | Mingxia Jiang is currently a PhD candidate majoring in Chemistry and Physics of Polymers under the supervision of Assoc. Prof. Huapan Fang at the State Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University. Currently, her research interests are biomedical materials and cancer immunotherapy. |
 Huapan Fang | Prof. Huapan Fang obtained his PhD degree from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences in 2019. From 2020 to 2022, he conducted postdoctoral research at Soochow University. In December 2022, he joined the College of Chemistry and Chemical Engineering, Xiamen University. His research topics include biomedical polymers, biomaterials, polymer nucleic acid/drug carrier, tumor immunotherapy, and protein/peptide delivery. |
 Huayu Tian | Prof. Huayu Tian obtained his PhD degree from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences in 2005. From 2006 to 2022, he conducted research at institutions including the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Korea Advanced Institute of Science and Technology, Kyushu University, and the University of Utah. In 2022, he joined the College of Chemistry and Chemical Engineering, Xiamen University. His research topics include biomedical polymers, nano gene/drug carriers, biotherapy, nucleic acid vaccines, immunotherapy, and energy polymers. |
1. Introduction
Cancer, infectious diseases, and genetic diseases pose significant threats to human lives, the rapid development of therapeutic agents including small-molecule drugs,1–4 therapeutic genes,5–7 and protein/peptide agents,8–10 has brought hope to patients. Nevertheless, these free drugs usually have poor stability and fail to effectively reach the lesion sites when administered orally or intravenously, resulting in extremely low bioavailability,11 which in turn affects the therapeutic outcomes. Moreover, free small-molecule drugs such as chemotherapeutics, often reach normal tissues or organs, leading to severe side effects within the body.12 Therefore, developing efficient and safe drug delivery systems is essential for enhancing drug efficacy and reducing side effects within the body.The traditional drug delivery systems face numerous challenges in dealing with complicated pathological environments, particularly in improving drug stability,13 enhancing targeting capability,14 and increasing bioavailability.15 Therefore, scientists are continuously exploring the development of novel drug carriers and delivery strategies to overcome these challenges.16–19 For instance, Liu et al.20 utilized fluorinated chitosan to deliver antibody drugs including anti-PD-1 and anti-CTLA-4 orally to tumor-bearing mice, effectively inhibiting tumor growth and extending the survival time of the mice. Additionally, Cheng et al.21 used phenylboronic acid-modified polyamidoamine to achieve intracellular delivery of various protein drugs. Although these biomedical carriers can enhance the delivery efficiency and contribute to therapeutic effects of drugs, they are non-biodegradable in the body, leading to a significant burden and potential side effects.
Poly(amino acids) and polypeptide-based materials have become a research focus in drug delivery due to their excellent biodegradability and safety in vivo.22 These materials are polymers or oligomers formed by the conjugation of amino acid monomers through peptide bonds. They are typically synthesized via various methods such as ring-opening polymerization of N-carboxyanhydride (NCA),23 amide condensation reaction of amino acids,24 microbial fermentation, and solid-phase peptide synthesis.25 Such carriers not only exhibit good biocompatibility and biodegradability but can also be tailored in various shapes and functions through sequence design and chemical modification. For instance, by adjusting the types and ratios of hydrophilic and hydrophobic amino acids, polypeptide molecules can self-assemble into micelles,26 fibers,2 vesicles,27 or hydrogels28 for various disease treatments. Therefore, poly(amino acids) and polypeptides have broad clinical application prospects in the field of drug delivery (Fig. 1).
 | ||
| Fig. 1 Schematic illustration of drug delivery of poly(amino acids) and peptide carrier materials in different disease types. | ||
Typically, poly(amino acids) or polypeptides can encapsulate hydrophobic small molecule drugs to form nanoparticles (NPs) through hydrophobic interactions, which eventually enhances the delivery efficiency of small molecule drugs in vivo.26 Additionally, hydrophilic small molecule drugs can be conjugated to poly(amino acids) or polypeptides via dynamic chemical bonds, thereby extending the circulation time of hydrophilic small molecule drugs in the bloodstream and their accumulation at target sites.29 With the outbreak of the COVID-19 pandemic and the rapid development of gene editing technology, nucleic acid drugs have received widespread attention. However, nucleic acid drugs such as DNA, mRNA, or siRNA are usually unstable and easily degraded by nucleases in the body.30 Furthermore, nucleic acids are often negatively charged, which hinders their uptake by target cells.31 Poly(amino acids) or polypeptides can effectively load nucleic acids via electrostatic interactions and compress them into NPs, thus improving their stability and uptake efficiency by target cells in vivo.32 Other macromolecular drugs, including protein and peptide drugs, generally have good bioactivity and are used to treat major diseases such as cancer and genetic disorders.33 However, due to their large molecular weight and poor stability, these drugs are easily degraded by proteases or peptidases in the body, ultimately resulting in poor efficacy.33 Poly(amino acid) or polypeptide carriers can effectively load protein or peptide drugs while maintaining their bioactivity, thereby enhancing the therapeutic efficacy of these macromolecular drugs in vivo.16 Moreover, a search on Web of Science for publications over the past 10 years (2014–2024) on poly(amino acids) or polypeptides for drug delivery shows that the number of papers in this field has consistently exceeded 7000 per year, with a clear upward trend, which indicated that poly(amino acid)/polypeptide-based biomaterials holds great potential for addressing human health challenges and improving public health (Fig. 2).
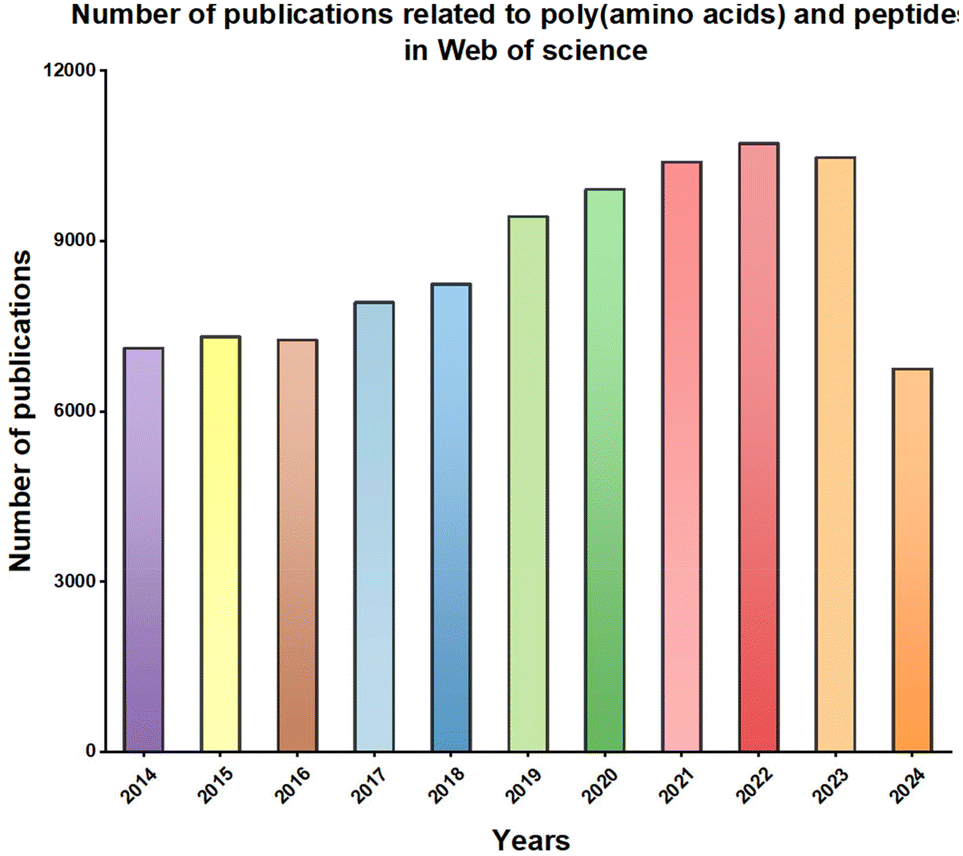 | ||
| Fig. 2 Web of science statistical chart of the number of papers published on poly(amino acids) and peptides from 2014 to August, 2024. | ||
Given the unique advantages of poly(amino acids) and polypeptides in drug delivery, this review systematically discusses the characteristics of these polymer carrier materials, common preparation methods, their applications in various diseases, and the progress of clinical research. In addition, we summarize the current challenges and opportunities faced by degradable polymer carrier materials, such as poly(amino acids) and polypeptides, and propose future directions for the development of degradable biomedical carrier materials. This review aims to provide valuable insights for both fundamental research and clinical translation of poly(amino acid)- and polypeptide-based degradable biomaterials.
2. Structure of poly(amino acids) and peptides
According to the nomenclature recommended by the International Union of Pure and Applied Chemistry (IUPAC), a polypeptide is a molecule composed of more than 20 amino acids linked by peptide bonds,34,35 and synthetic peptides are also referred to poly(amino acids). Amino acid materials exhibit excellent biocompatibility, biodegradability, and good self-assembly behavior, making them significant for biomedical applications,36 as illustrated in Fig. 3, which presents for the common structural formula of poly(amino acids) and polypeptides. The structure of poly(amino acids) consists of linear or branched polymers formed by amino acid monomers through amide bonds, similar to peptide bonds. Each amino acid monomer features a basic structural unit that include an amino group, a carboxyl group, and a side chain (R group). Poly(amino acids) are linked by repeating peptide bonds (–NH–CO–), which create the poly(amino acid) backbone, typically in a linear configuration. The diverse side chains of amino acids impart a range of chemical and physical properties to poly(amino acids), including hydrophilicity, hydrophobicity, acidity, and alkalinity. In addition, the side chains of poly(amino acid) contain numerous reactive functional groups, such as amino and carboxyl groups, which can be further modified with drugs or biologically active molecules to develop various nano-delivery systems.37–39 Furthermore, the presence of secondary structures, such as α-helices and β-folds, allows poly(amino acids) to exhibit distinct properties compared to conventional macromolecules.40 | ||
| Fig. 3 Common structural formula for poly(amino acids) and polypeptides. | ||
3. Properties of poly(amino acids), peptides and their derivatives
3.1. High drug-carrying capacity
Poly(amino acids) and peptides can bind to drug molecules through covalent or non-covalent interactions and self-assemble to form nanomicelles and NPs. These structures are utilized to encapsulate and transport drugs, enhancing their drug solubility and stability.NP systems, as modern drug delivery vehicles, significantly enhance drug solubility, improve drug stability, and promote the distribution of drugs within the body, thereby increasing clinical efficacy.41 Amphiphilic polymers can adopt various morphologies, such as micelles or vesicles, through self-assembly in specific environments. These polymers can be physically encapsulated and chemically cross-linked to achieve controlled release of model drugs. In addition, drug-filled micelles are both thermodynamically and kinetically stable, exhibiting excellent properties such as durability, prolonged effects, and safety.42 For example, glutamic acid-linked paclitaxel dimers (Glu–PTX2) can self-assemble into NPs (Glu–PTX2 NPs) in aqueous solutions, forming stable drug–polymer complexes that display a spherical morphology and good structural stability in aqueous media. These complexes exert potent cytotoxic effects in aqueous environments, thereby enhancing their bioavailability.43
3.2. Targeted delivery
Improving drug delivery to specific organs, cells, or even organelles to enhance therapeutic efficacy while minimizing dosage and side effects has long been a critical focus in the development of advanced drug delivery systems.44 Targeted drug delivery leverages polymer chains to direct drug molecules to specific regions of interest. Poly(amino acids) and peptides are particularly effective in achieving this goal, as they can bind to specific molecular targets, increasing drug concentration within target tissues or organs while reducing systemic side effects.45 Targeting peptides, in particular, exhibit strong specificity for their targets. For instance, peptides derived from cell surface protein, such as intercellular adhesion molecule-1 (ICAM-1), luteinizing hormone-releasing hormone (LHRH), bombesin, and LFA-1, have demonstrated high affinity for cell surface receptors, facilitating the selective delivery of therapeutic agents.46Several widely utilized strategies for the targeted delivery of poly(amino acid)-based therapeutics include the following. (1) Ligand modification: poly(amino acids) can be surface-functionalized with specific ligands, enabling them to recognize and bind selectively to target cells or tissues.47 This active targeting mechanism reduces non-specific cellular uptake, thereby decreasing off-target toxicity and enhancing therapeutic outcomes.48 (2) Passive targeting: by exploiting the unique size and surface characteristics of NPs, poly(amino acid) carriers can passively accumulate in tumor tissue through the enhanced permeability and retention (EPR) effect.49,50 This effect arises from the abnormal vasculature of tumors, which is characterized by increased permeability and poor lymphatic drainage, allowing NPs to preferentially localize at the tumor site. (3) Environmental responsiveness: poly(amino acid) carriers can be engineered to respond to specific physiological conditions within the tumor microenvironment, triggering localized drug release.51 For instance, polyglutamic acid NPs are designed to degrade in acidic environments, which are often found in tumors, thereby releasing their drug cargo precisely at the desired site.52 (4) Cell-penetrating peptides: a strategy to enhance transcellular drug transport involves the use of CPPs (such as TAT peptides), which facilitate the internalization of therapeutic molecules across cellular barriers.53 This approach significantly improves the delivery of small-molecule drugs into target cells, increasing intracellular drug concentrations and enhancing therapeutic efficacy.
These methods can be employed individually or in combination to enhance drug targeting and efficacy while minimizing side effects. For instance, an innovative mesoporous silica nanoparticle (MEMSN)-encapsulated cellular uptake shielding multifunctional system has demonstrated cancer-targeted triggered, triggered drug delivery to tumor cells.54 The surface of the mesoporous NPs was modified with disulfide bonds that linked β-cyclodextrin, facilitating GSH-induced intracellular drug release. Subsequently, the NP surface was further modified with the RGD peptide sequence and the matrix metalloproteinase substrate peptide Pro–Leu–Gly–Val–Arg (PLGVR) through a host–guest interaction. The modification of the NP with pAsp resulted in MEMSN that could protect the target ligand. In addition, in vitro studies confirmed that MEMSN effectively blocked its uptake by normal cells. Upon reaching the tumor cells, the pAsp protective layer was removed through the hydrolysis of PLGVR in metalloproteinase-rich cancer cells. Consequently, the targeting properties were activated, allowing the cancer cells to readily uptake the drug-coated NPs, which subsequently facilitated GSH-induced intracellular drug release (Fig. 4).
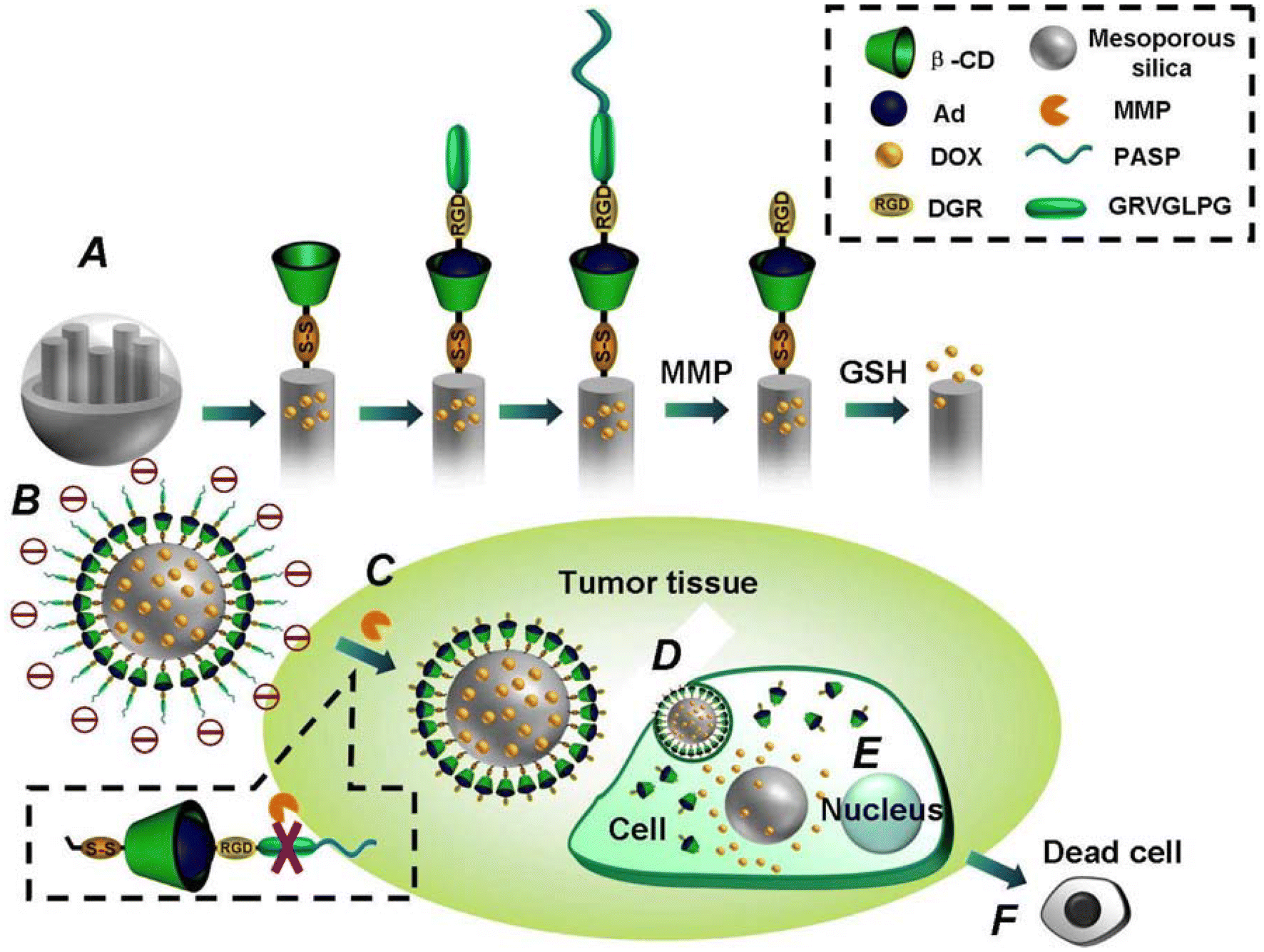 | ||
| Fig. 4 Structure of multifunctional envelope-type mesoporous silica NP and tumor-triggered targeting drug delivery.54 Copyright 2013, American Chemical Society. | ||
3.3. Low toxicity and biocompatibility
Amino acids are fundamental building blocks of biological proteins, while peptides are composed of natural amino acids that share similarities with proteins found in living organisms. As a result, peptides typically exhibit high biocompatibility, minimizing immune reactions and toxicity. These materials generally do not provoke severe toxic responses in the body, as they can be recognized and processed by the organism.38 Furthermore, peptides can be degraded into amino acids or small peptide fragments by enzymes within the body, ultimately being excreted through metabolic pathways. By modifying the amino acid sequence and structure of peptides, it is possible to design peptide materials with varying degradation rates, thereby allowing for controlled timing and rates of drug release. Table 1 presents a comparison of poly(amino acids) and polypeptides with other non-degradable biomaterials.| Polymers | Characteristic | Ref. | |
|---|---|---|---|
| Advantages | Disadvantages | ||
| Polytetrafluoroethylene (PTFE) | Chemical inertness; low friction coefficient; non-adhesive properties; thermal stability; flexible and durable | Poor tissue integration; susceptibility to deformation; limited mechanical strength; biofilm formation risk; difficult to process; expensive | 55 |
| Polyethylene (PE) | Durability and mechanical strength; chemical inertness; low friction; cost-effectiveness | Non-biodegradable; poor tissue integration; wear debris; oxidative degradation | 56 and 57 |
| Silicone | Durability and stability; flexibility and elasticity; low immune response; non-degradable; wide range of medical applications | Lack of biodegradability; tissue integration; potential for foreign body reaction; limited bioactivity; rigid manufacturing process | 58 |
| Polyurethane (PU) | Mechanical properties; biostability; versatility in formulation; hemocompatibility; cost and availability | Non-biodegradability; potential for degradation products; limited biocompatibility; surface modification required | 59 |
| Polymethyl methacrylate (PMMA) | Mechanical strength and durability; stability and long-term use; non-immunogenic; ease of shaping and processing | Lack of biodegradability; potential for biofilm formation; exothermic polymerization; brittleness | 60 |
4. Synthesis of poly(amino acids) and peptides
The synthesis of poly(amino acids) and polypeptides include chemical polymerization methods,61 enzymatic polymerization,62 biosynthesis,63 and self-assembly technology.64 Each preparation method is suited to specific application scenarios. Chemical polymerization is ideal for producing poly(amino acids) and peptides with diverse structures, while biosynthesis is more appropriate for the creating biopolymers with specific sequences. In contrast, enzymatic polymerization and self-assembly techniques prioritize gentle and environmentally friendly preparation processes. The four methods are described in detail below.4.1. Chemical polymerization
Chemical polymerization is one of the primary methods for the preparation of poly(amino acids) and currently serves as the predominant technique for both laboratory and industrial synthesis of polypeptides. This process primarily encompasses ring-opening polymerization, polycondensation reactions, and solid-phase synthesis. Among these methods, ring-opening polymerization can be conveniently conducted using α-amino acidic NCA monomers.65 These macromolecules are typically synthesized through the ring-opening polymerization of various amino anhydrides, initiated by an amine. For example, Hu et al.38 prepared triblock copolymers via ring-opening polymerization using mPEG–NH2 as the initiator, while Zhang et al.66 synthesized diblock copolymers using the macroinitiator PEG–NH2. Since NCA is a cyclic derivative of amino acids, it readily undergoes ring-opening, facilitating participation in the polymerization reaction, which makes this method widely utilized. In addition, the polycondensation reaction is a condensation process involving the carboxyl group and the amino group of an amino acid, resulting in the formation of a poly(amino acid) and a by-product, such as water or another small molecule.67 Poly(amino acids) with varying chain lengths can be produced by modifying reaction conditions, including temperature and the choice of catalyst. Furthermore, solid-phase synthesis is a stepwise technique for synthesizing poly(amino acids). This method involves immobilizing the carboxyl groups of amino acid monomers onto a solid-phase carrier, followed by the gradual addition of amino acid monomers, which are chemically linked to create a long chain. Once the reaction is complete, the poly(amino acids) are detached from the solid-phase carrier through cleavage.68 This approach allows for precise control over the sequence and molecular weight of the resulting poly(amino acids), making it suitable for synthesizing poly(amino acids) with complex structures. However, it is important to note that this method can be costly and is typically employed for small-scale or laboratory preparations.Chemical polymerization offers a degree of flexibility in the regulation of molecular weight.69 For instance, in the case of NCA ring-opening polymerization, the molecular weight of poly(amino acids) or polypeptides can be effectively modulated by altering the ratio of initiator to monomer.61 When the quantity of initiator is less than that of the monomer, each initiator molecule has increased opportunities for chain elongation, leading to the formation of polymers with higher molecular weights. Furthermore, the polymerization rate can be influenced by adjusting parameters such as reaction temperature and catalyst concentration, which in turn affects the molecular weight. Generally, elevated temperatures and higher catalyst concentrations accelerate the reaction rate, thereby facilitating an increase in the polymer's molecular weight within an optimal range.25 In addition, chemical polymerization allows for the introduction of specific stereochemical configurations through the selection of appropriate monomers and reaction conditions.61 For example, during the synthesis of poly(amino acids), ring-opening polymerization can be conducted using NCA monomers derived from amino acids with defined chirality. The utilization of L-type amino acid NCA monomers enables the synthesis of optically active poly(amino acids), with the chiral structure preserved by carefully controlling the polymerization conditions to produce materials with targeted biological activities. Moreover, chemical polymerization can yield poly(amino acids) and polypeptides with varying stereochemical structures, resulting in a diverse array of stereochemical combinations, including isotactic, syndiotactic, and atactic configurations.
4.2. Enzymatic polymerization method
Enzymatic polymerization of polymers (amino acids) and peptides is carried out under mild conditions, such as normal temperature, atmospheric pressure and near neutral pH, using enzymes as catalysts.70 In comparison to conventional chemical polymerization techniques, the utilization of mild reaction conditions necessitates less sophisticated equipment and does not require specialized reaction apparatus capable of withstanding elevated temperatures and pressures. For instance, the synthesis of poly(amino acids) and polypeptides through certain chemical polymerization methods often demands high temperatures (ranging from 100 to 200 °C) and the use of specific organic solvents. In contrast, enzymatic reactions circumvent these extreme conditions, thereby leading to reductions in production costs and energy consumption. The application of mild reaction conditions is advantageous for preserving the structural integrity of both reactants and products. Under harsh conditions, such as elevated temperatures or the presence of strong acids and bases, the structural integrity of amino acids and peptides may be compromised, resulting in denaturation or decomposition that adversely affects the quality and performance of the resulting polymer.71 Enzymatic reactions facilitate precise catalysis of the polymerization process, ensuring that the synthesized poly(amino acids) and polypeptides exhibit the desired structural and functional characteristics.In comparison to conventional chemical polymerization techniques, enzymatic polymerization exhibits notable advantages, particularly in terms of specificity. This method demonstrates a high degree of selectivity towards substrates during the synthesis of poly(amino acids) and polypeptides.72 Certain enzymes are capable of selectively polymerizing specific amino acids. For example, γ-glutamyltranspeptidase is known to catalyze the polymerization of L-glutamine and its derivatives due to the strong complementarity of its active site with the structure of glutamine.73 This allows the enzyme to accurately identify and bind glutamine molecules, thereby initiating the polymerization process. Similarly, papain preferentially recognizes peptides that contain particular amino acid sequences, as its active site can specifically bind to peptide bonds involving arginine or lysine residues. When these appropriate substrate peptides are present, papain facilitates the formation of peptide bonds, effectively linking smaller peptides into polypeptides.74 Furthermore, enzymatic polymerization exhibits regional selectivity in the growth patterns of poly(amino acid) and polypeptide chains. Enzymes can precisely dictate whether polymerization occurs at the α-amino, α-carboxyl, or side chain functional groups of amino acids or peptides. In instances where amino acids or peptides possess multiple reactive sites, the enzyme can direct the polymerization to occur preferentially at specific sites.75 For example, in the presence of lysine residues that contain multiple amino groups, the enzyme can select one amino group for polymerization based on its three-dimensional structure and the spatial orientation of its active site, resulting in a more structured poly(amino acid) or polypeptide product. Moreover, enzymatic polymerization demonstrates a pronounced ability to discriminate between stereoisomers of amino acids during the synthesis of poly(amino acids) and polypeptides.25 Most enzymes exhibit activity solely towards L-type amino acids, showing little to no reactivity with D-type amino acids or exhibiting significantly reduced efficiency.76 This selectivity arises from the specific three-dimensional spatial structure of the enzyme's active site, which closely aligns with the spatial conformation of L-amino acids. For instance, during peptide bond formation, the α-amino and α-carboxyl groups of the amino acids involved in the reaction must be precisely aligned with specific groups at the enzyme's active site. The atomic arrangement surrounding the chiral center of L-amino acids is ideally suited to meet the binding requirements of the enzyme, facilitating an efficient polymerization reaction. Conversely, D-type amino acids struggle to participate in polymerization due to the incompatibility of their spatial structure with the enzyme's active site. This stereoselectivity ultimately ensures that the resulting poly(amino acids) and polypeptides possess high optical purity and specific biological activity.
Currently, the utilization of certain enzymes for the synthesis of poly(amino acids) and polypeptides is hindered by their high cost, which contributes to elevated production expenses.77 Furthermore, these enzymes exhibit limited stability and are prone to inactivation during both storage and application. For instance, certain proteases may lose their catalytic activity when subjected to elevated temperatures, extreme pH conditions, or prolonged exposure to inhibitors. To enhance enzyme stability, it may be necessary to implement specialized preservation techniques, such as low-temperature freezing or the incorporation of stabilizing agents. Consequently, the application of enzymatic polymerization in large-scale industrial production remains constrained.78 In comparison to chemical polymerization methods, enzymatic polymerization often exhibits slower reaction rates, and maintaining uniform enzyme distribution and continuous functionality throughout large-scale reactions poses significant challenges.79 In addition, the molecular weight of products generated through enzymatic polymerization may be restricted by the specific enzyme employed and the prevailing reaction conditions, making it difficult to synthesize high molecular weight poly(amino acids) and polypeptides, as can be achieved with certain chemical polymerization techniques.
4.3. Biosynthetic methods
Biosynthesis refers to the process of synthesizing poly(amino acids) and polypeptides through the metabolic mechanisms inherent to an organism.80 This technique predominantly depends on the transfer of genetic information and the cellular systems responsible for protein synthesis, which encompass transcription and translation processes. Within cells, the genes encoded in DNA are initially transcribed to produce mRNA. Subsequently, the mRNA facilitates the assembly of amino acids in accordance with the codon sequence it contains, utilizing tRNA as a mediator, with ribosomes catalyzing the formation of polypeptide chains.81 This methodology is frequently employed for the synthesis of specific amino acid polymers and the production of naturally occurring peptides.In this case, genetic engineering is applied to synthesize poly(amino acids) by introducing genes that encode the target poly(amino acids) into microorganisms, allowing for their expression within microbial cells.62,82 The gene responsible for the target poly(amino acid) is integrated into the microorganism, facilitating the synthesis of the poly(amino acid). This process enables the production of specific sequences of poly(amino acids) that are both mass-producible and biocompatible. The method is frequently employed to synthesize biomedical materials, such as polyglutamic acid (γ-PGA).83 Furthermore, in prokaryotic expression systems, such as Escherichia coli, the degradation of short peptides by intracellular proteases poses a challenge to production efficiency. This issue can be addressed by employing protease-deficient strains or by expressing peptides as insoluble inclusions.84
The synthesis of poly(amino acids) and polypeptides utilizing transgenic organisms involves several critical steps. Initially, the design and construction of the gene must be undertaken, with the corresponding gene sequence tailored to match that of the target polypeptide. This process necessitates careful consideration of codon usage, as different organisms exhibit varying frequencies of codon preference.85 For instance, when designing genes intended for Escherichia coli, it is advisable to utilize codons that are favored by this organism to enhance the efficiency of gene expression. Subsequently, these gene fragments can be synthesized chemically or sourced from existing gene libraries, and then assembled into complete genes through genetic engineering methodologies. The constructed gene must then be introduced into a suitable host cell using an appropriate method. In the case of E. coli, a common approach is heat shock transformation, wherein the recombinant plasmid containing the target gene is mixed with E. coli cells at a low temperature, followed by a brief heat shock treatment to facilitate the uptake of the plasmid by the cells.86 For yeast, electrical transformation techniques, such as the application of high-voltage pulses, can be employed to temporarily render the cell membrane permeable, thereby allowing the entry of foreign genes.87 Finally, the expression and purification of the resultant product must be conducted. Within the host cell, the imported gene requires expression under optimal conditions. For certain genes regulated by inducible promoters, the addition of specific inducers is necessary. In E. coli, IPTG (isopropyl-beta-D-thiogalactoside) is frequently utilized to induce the expression of genes that are associated with lactose operon promoters.88 The expressed peptides can subsequently be purified using various techniques, including affinity chromatography and ion exchange chromatography.
4.4. Self-assembly technology
Under specific conditions, certain amino acids or peptides can form polymers through self-assembly.89 For instance, molecular self-assembly can lead to the formation of various types of NPs, including NPs, hydrogels, nanofibers, and nano micelles, depending on the chemical composition, molecular weight, and hydrophobicity ratio of the peptide (Fig. 5). This organization is driven by intramolecular and/or intermolecular interactions, such as hydrogen bonding, electrostatic interactions, and hydrophobic interactions, which may occur between the side functional groups of the amino acids in the peptide.40 The β-sheets and α-helices formed by the polypeptides also significantly influence the morphology and size of the peptide-based NPs.90 They are widely used in drug delivery, tissue engineering, combination therapy and other fields. Similar to NPs, nanotechnology in drug delivery can address the shortcomings of conventional methods by enabling cell-specific targeting, facilitating the transport of molecules to designated organelles, and enhancing intracellular transport. This method does not require complex chemical reactions and offer improved environmental adaptability and targeted drug delivery potential. | ||
| Fig. 5 Various types of NPs obtained from polypeptides and their derivatives and applications. | ||
Researchers have designed specific peptide sequences to facilitate the self-assembly of peptides and enhance drug loading efficiency. Initially, Zhang et al. developed and synthesized an ion-complementary peptide, EAKl6, composed of 16 amino acid residues that self-assemble in aqueous solutions to form stable hydrogel membranes visible to the naked eye.91 Furthermore, Ghadiri created a cyclic polypeptide consisting of eight amino acid residues. This peptide was self-assembled from a β-folded structure and was further highly integrated to form hollow peptide nanotubes.92 Meanwhile, with an increasing understanding of sequence–structure relationships, Woolfson et al.93 endeavored to design and explore more innovative assemblies to generate new protein functions based on α-helical helices, which are common domains in protein–protein interactions. This work opened up a potential new frontier in coiled-coil assemblies and α-helical barrels. Subsequently, Hu et al.38 prepared a series of poly(amino acid) materials through ring-opening polymerization using mPEG113–NH2 as the initiator. Cholesterol was grafted onto the side chain via an esterification reaction to render the macromolecules amphiphilic, allowing them to self-assemble in specific solvents to form NPs with distinct morphologies. At different pH values, these NPs exhibited different shapes, facilitating the study of their drug release properties.
So far, researchers have been combining peptide self-assembly with other functional materials to develop multifunctional systems for smart responses, precision drug delivery, and regenerative medicine. Some self-assembled peptide materials have entered preclinical studies and early clinical trials, demonstrating their promising applications. In addition, Table 2 compares the advantages and limitations of other non-degradable biomaterials with poly(amino acids) and polypeptides and their derivatives.
| Synthesis method | Characteristic | Ref. | |
|---|---|---|---|
| Advantages | Limitation | ||
| Chemical polymerization | Simple operation; low cost; high molecular weight; synthetic diversity; controlled polymerization; one-step synthesis of functional clustering peptides | Difficult monomer purification; slow polymerization speed; high environmental sensitivity; difficult to control sequence selectivity; complex initiator design and preparation; initiator residue problems; molecular weight control and distribution problems; low functional group efficiency | 61 |
| Enzymatic polymerization method | Mild reaction conditions; high selectivity; controlled polymerization; biocompatibility; functionable modification; environmental friendliness; stimulus responsiveness | Molecular weight control; polymerization activity and controllability; low functional group efficiency; high substrate concentration requirement; reversible reaction; complex functional group introduction and modification steps | 94 |
| Biosynthetic methods | Easy to operate; narrow molecular weight distribution; environmentally friendly | Slow reaction speed; difficult to prepare large molecular weight polypeptides; cumbersome steps and high cost; difficult to achieve large quantities of preparation | 95 |
| Self-assembly technology | Simple structural design; low production cost; good biocompatibility; improved cell uptake; drug targeted release; versatility; high stability and low toxicity | Low stability; easy to be affected by pH; poor water solubility; low drug load; high production cost; poor mechanical and rheological properties; self-assembly process difficult to accurately control; sensitive to environmental factors | 96 |
5. Optimization of poly(amino acids), polypeptides and their derivatives
Poly(amino acids) and peptides can be modified through derivatization via chemical alterations, binding to small molecules, or conjugation with antibodies, among other methods, to significantly enhance their functionality in drug delivery. These modifications and derivatives improve drug targeting, stability, controlled release, and biocompatibility, thereby increasing the efficiency and safety of drug delivery.5.1. Optimize the sequence and composition of poly(amino acids) or polypeptides
Different amino acids exhibit distinct side chain structures and chemical properties, allowing for the optimization of specific characteristics through the careful selection of sequences and compositions in poly(amino acids) or polypeptides. Notably, modifying the amino acid composition to increase the proportion of rigid amino acids, such as phenylalanine and tyrosine, both of which possess large and relatively rigid side chains, can enhance the interactions between poly(amino acid) or polypeptide chains, thereby improving mechanical stability.97 Furthermore, the incorporation of proline, which contains a pyrrolidine ring structure, can also contribute rigidity to the molecular chain; an appropriate increase in its content can further enhance the mechanical properties of the material.98 In addition, the design of poly(amino acids) or polypeptides with repeating units or specific sequences that promote the formation of secondary structures, such as α-helices or β-sheets, can facilitate tighter molecular arrangements and strengthen intermolecular interactions, including hydrogen bonding. This, in turn, enhances the mechanical strength and stability of the material.99 Moreover, the introduction of cross-linking groups or amino acids, such as cysteine with sulfhydryl groups, into the poly(amino acid) or polypeptide chain can lead to the formation of covalent cross-linking networks through chemical or photocross-linking methods.100 Such cross-linked structures can restrict the mobility of molecular chains and increase the material's resistance to deformation, significantly improving its mechanical stability.To regulate the rate of drug release, it is possible to systematically adjust the ratio of hydrophilic to hydrophobic amino acids. An increase in the proportion of hydrophobic amino acids facilitates the formation of a denser structure in poly(amino acids) or polypeptides, thereby reducing the diffusion rate of the drug and extending the duration of drug release.101 Conversely, a higher concentration of hydrophilic amino acids enhances the penetration of water molecules and the solubility of the drug, which accelerates its release. Additionally, the incorporation of chemical bonds or amino acid sequences that respond to specific stimuli, such as acid-sensitive hydrazone bonds or enzyme-sensitive peptide bonds, into the main or side chains of poly(amino acids) or polypeptides can also effectively modulate the drug release rate.102 Upon reaching the target site, these sensitive bonds or sequences can be cleaved under appropriate stimulus conditions, leading to the structural disintegration of the poly(amino acid) or polypeptide and facilitating rapid drug release. For instance, in the acidic environment of tumors, poly(amino acid) drug carriers containing acid-sensitive hydrazone bonds can undergo rapid degradation and drug release due to the low pH.103 Generally, carriers composed of poly(amino acids) or polypeptides with higher molecular weights and longer chain lengths exhibit a more compact structure, resulting in a longer diffusion pathway for the drug and a comparatively slower release rate.104 Therefore, the molecular weight and chain length of poly(amino acids) or polypeptides can be manipulated by adjusting polymerization conditions to regulate the drug release rate.
To enhance the solubility of the material, one can increase the content of hydrophilic amino acids, decrease the amount of hydrophobic amino acids, introduce water-soluble groups, or modify the terminal ends of poly(amino acids) or polypeptides. For example, chemical modifications such as amination at the N-terminus or acetylation at the C-terminus can alter the charge distribution and polarity of the molecule, thereby improving its solubility in water and other solvents.105
5.2. Chemical modifications
Chemical modification involves altering the structure of a poly(amino acid) or peptide molecule to enhance its suitability for specific drug delivery applications.However, PEGylation also hinders the cellular uptake of the carriers, thereby limiting their therapeutic efficacy. Consequently, numerous peptide carriers with removable PEG shells have been developed in recent years.112 Jiang et al.113 prepared two cisplatin-loaded polyglutamic acid–lysine composite nanoformulations, featuring detachable polyethylene glycol grafted onto lysine fragments through two distinct bridging chemical bonds. These formulations are responsive to specific tumor tissue microenvironments, including low pH and matrix metalloproteinases. The nanoformulations containing PEG fragments circulated in the bloodstream for a prolonged period of time with increased drug accumulation in the tumor tissue compared to those without PEG fragments. Upon reaching the tumor tissue, the nanoformulations exhibited enhanced cellular uptake and cytotoxicity due to the cleavage of the bridging chemical bond between polyethylene glycol and polylysine (Fig. 6).
 | ||
| Fig. 6 Schematic representation of PLG–CDDP nano-formulations with separable polyethylene glycol response to the tumor microenvironment for enhanced treatment of peritoneal metastases from ovarian cancer.113 Copyright 2021, Elsevier. | ||
 | ||
| Fig. 7 Liposomes were prepared by multifunctional biomaterials, responding to external trigger around the stimulated zone, rapidly releasing its contents.119 Copyright 2019, Elsevier. | ||
pH or temperature-responsive modifications are often applied to smart drug delivery systems to build pH or temperature-responsive drug delivery vectors. The drug is wrapped in a responsive poly(amino acid) or polypeptide carrier to control the release of the drug by regulating the external pH or temperature. This smart drug delivery system can improve the targeting and therapeutic effect of drugs, and can also be used to make biosensors and prepare injectable tissue engineering scaffold materials.
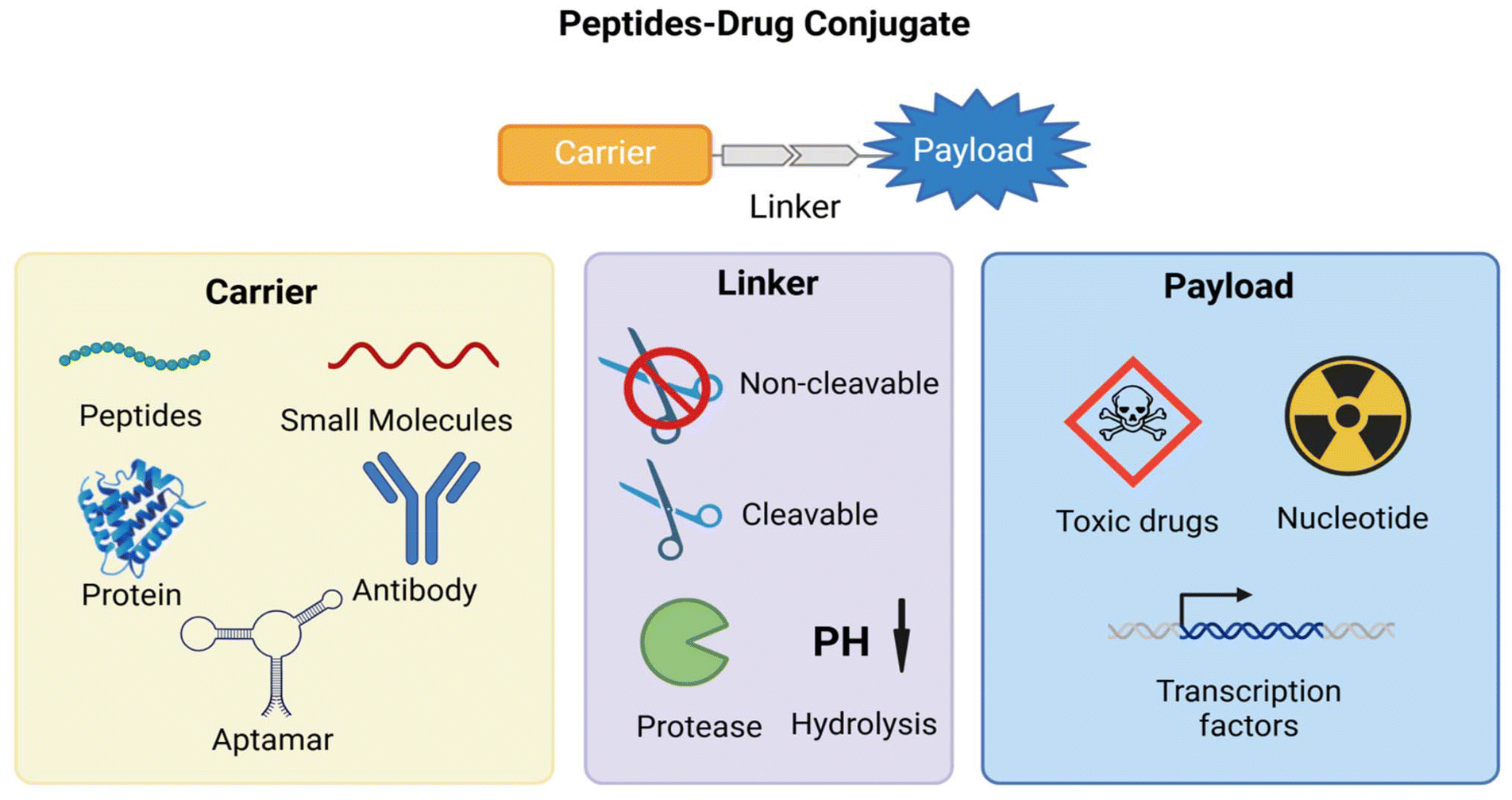 | ||
| Fig. 8 Schematic structure of peptide–drug coupling. | ||
Recently, Li et al.129 designed and synthesized a peptide–drug conjugate by linking the targeted transferrin receptor (TfR)-binding peptide analogue BP9a (CAHLHNRS) to doxorubicin (DOX) via N-succinimidyl-3-maleimidopropionate (SMP).129 They concluded that BP9a could serve as a potential TfR-targeting peptide carrier for selective drug delivery. Subsequently, Yu et al.130 investigated two new peptide–drug conjugates (PDCs), LT7-SS-DOX and DT7-SS-DOX, which were created by coupling peptide ligands composed of natural L-type amino acids. These ligands are known to suffer from issues related to enzymatic degradation and insufficient biostability. The new PDCs, LT7 (haiyprh) and its inverse analogue, DT7 (hrpyiah), were synthesized through disulfide bonding with DOX. They demonstrated enhanced serum stability, prolonged reduction-triggered drug release, and increased in vitro antiproliferative activity. Targeted peptide couplers exhibit significant potential for drug delivery and precision medicine due to their highly specific targeting capabilities and multifunctional design.
Furthermore, a pH and reducing dual-reactive peptide–dexamethasone (anti-inflammatory drug) conjugate (L-SS-DEX) was developed to improve the immunosuppressive tumor microenvironment for effective colorectal cancer therapy (Fig. 9).131 This peptide–dexamethasone conjugation ensures the stability of the dexamethasone moiety and facilitates the rapid release of dexamethasone within the tumor microenvironment. Compared to free DEX, L-SSDEX was more effective in reducing pro-tumor inflammation by inhibiting cyclooxygenase-2 (COX-2) and resulted in enhanced tumor suppression through the infiltration of CD8+ cytotoxic T cells. To achieve immune activation, a tumor-specific enhanced oxidative stress polypeptide coupler (TSEOP) was synthesized by covalently linking an enhanced oxidative stress module to a long-circulating amphiphilic peptide, which induced immunogenic cell death (ICD) in tumor cells (Fig. 10).132 In response to specific intra-tumor specific stimulation, TSEOP was degraded to generate cinnamaldehyde (CA) and the glutathione (GSH) eliminator quinone (QM). CA and QM synergistically increased oxidative stress and endoplasmic reticulum stress, leading to tumor cell death and enhanced anti-tumor immunity.
 | ||
| Fig. 9 Schematic illustration of L-SS-DEX for tumor microenvironment modulation. Reproduced with Reproduced with permission.131 Copyright 2020, Elsevier. | ||
 | ||
| Fig. 10 Schematic illustration of TSEOP for inducing ICD and boosting antitumor immunity. Reproduced with permission.132 Copyright 2020, American Chemical Society. | ||
In addition, the use of poly(amino acids) and peptides in the modification of bispecific antibodies represents a cutting-edge technology aimed at enhancing therapeutic efficacy by improving the specificity and functionality of antibodies, particularly in the fields of cancer treatment and immunotherapy. Bispecific antibodies can simultaneously bind to two different antigens, allowing for more effective targeting of cancer cells or pathogens while minimizing damage to healthy tissues.133 Modifying bispecific antibodies can result in several advantages, including increased stability of the antibody molecule, reduced immunogenicity, enhanced targeting capabilities, and prolonged half-life. This technology holds the potential to further improve the specificity and stability of antibody-based therapies, which is anticipated to lead to significant advancements in personalized medicine and targeted therapies in the future.
The hydrophobicity of cell membranes and the blood–brain barrier selectively limits the cellular uptake of exogenous molecules larger than 500 Da. However, CPPs can efficiently penetrate cell membranes and have become essential tools for delivering nucleic acids, proteins, and small molecule drugs. There are two primary pathways for CPPs to enter cells: the cytosolic pathway and the direct penetration pathway. In the cytosolic pathway, the phospholipid bilayer first invaginates and encapsulates the penetrating peptide, forming small vesicles that facilitate entry into the cell. Alternatively, this pathway may involve the formation of reverse micelles to facilitate cellular entry. In contrast, the direct penetration pathway is more straightforward. One mechanism relies on the polarity of the penetrating peptide to directly perforate the phospholipid bilayer, while another mechanism involves the peptide spreading across the surface of the bilayer like a carpet and subsequently fusing with it to gain entry into the cell.44
Early HIV transactivator (Tat) proteins represent remarkable alternative strategies for penetrating the impermeable phospholipid bilayer of cell membranes and crossing biological barriers. After being secreted by HIV-infected cells, Tat translocates into neighboring cells, altering gene transcription and facilitating the spread of the virus.135 Fawell at al. published the first report in 1994, demonstrating that Tat-derived peptides could deliver large proteins to various cell types and mammalian organs.136 Subsequently, a research team led by Bernard Lebleu eventually identified the truncated polycationic peptide GRKKRRQRRR, which contains RNA-binding and nuclear localization signaling (NLS) motifs that are sufficient for efficient translocation into cells and tissues.137 Following this, Gilles Divita's group designed a short peptide vector called MPG, the first non-covalent CPP for the delivery of nucleic acids into cultured cells.138 This was closely followed by the development of Pep-1, a peptide carrier for the cellular transfer of peptides and proteins.139 Later, peptide known as Xentry (LCLRPVG), derived from the N-terminal region of the hepatitis B virus protein X, represented a novel class of CPPs that entered cells exclusively through an energy-dependent endocytosis process, thereby expanding the range of cellular uptake pathways.140 Recent studies have demonstrated that CPPs can be chemically modified and optimized in sequence to enhance their penetration efficiency and specificity. Furthermore, they can be combined with other functional molecules to develop multifunctional vectors for imaging, diagnostic, and therapeutic purposes. CPPs have shown significant potential for preclinical and clinical research, particularly in the fields of anticancer drug delivery and gene editing. The research history of CPPs illustrates a gradual evolution from early discoveries to mechanistic studies and widespread applications. The versatility and functionality of CPPs position them as promising candidates for critical applications in biomedicine.
5.3. Multifunctional complexes
Poly(amino acids) and peptides can be utilized to construct multifunctional delivery systems by simultaneously incorporating multiple functionalized groups. For instance, chemotherapeutic drugs and anti-tumor immune enhancers can be combined within the same peptide or poly(amino acid) carrier to achieve synergistic therapeutic effects.141 In addition, by integrating nucleic acid drugs with chemotherapeutic drugs in poly(amino acid) carriers, the dual effects of gene silencing and chemotherapy on tumor cells can be realized concurrently.142 This multifunctional delivery system, based on poly(amino acids) and peptides, integrates various features such as targeting, stabilization, and controlled release, thereby offering new possibilities for personalized therapy and the treatment of complex diseases. Furthermore, single-function gene delivery systems often fail to meet the demands of clinical applications, making the development of safe and effective multifunctional gene delivery systems particularly crucial. Multifunctional peptide-based gene vectors have shown significant promise in clinical gene therapy. In recent years, research has concentrated on the integration of peptides with diverse functions, including CPPs, targeting ligands, chimeric peptides, and nuclear localization signals. Compared to traditional vectors and single-function peptide vectors, multifunctional peptide gene vectors can markedly enhance gene transfection efficiency and therapeutic efficacy.Researchers are developing multifunctional peptide NPs. Zhang et al.143 prepared a series of multifunctional chimeric gene vectors composed of alkyl chains, R8 transmembrane peptides, and RGD peptides. The carboxy-terminal RGD sequence enhances the specificity of target recognition for integrins αυβ3 and αυβ5, which are overexpressed on tumor cells. The hydrophobic alkyl chain at the amino terminus was utilized to improve the encapsulation of DNA and enhance the stability of the vector/DNA complex. The results demonstrated that the multifunctional peptide vector significantly increased the transfection efficiency of cancer cells compared to the peptide or RGD-modified vector with R8. Subsequently, they constructed nuclear-targeting vectors by attaching the nuclear localization signal (NLS) PKKKRKV to the R8 peptide modified with hydrophobic stearic acid. The NLS ligand can be recognized by nuclear transporter proteins (importin-α and importin-β), which help overcome the nuclear membrane barrier and promote nuclear translocation. Although peptide-based gene vectors have successfully navigated many barriers and achieved significant positive outcomes in gene therapy, balancing the complex functionality of peptides remains a challenge. Future research will focus on optimizing and coordinating the structure and function of various functional peptide vectors for more efficient and stable gene delivery.
Multi-stimulus responsive peptides can react to two or more stimuli, including pH, temperature, enzymes, light, or redox conditions. Consequently, it is feasible to develop innovative nanocarriers with enhanced responsiveness that leverage this multi-stimulus capability to efficiently release payloads at targeted sites.
5.4. Analytical technique
During the synthesis of poly(amino acids) and polypeptides, various side reactions and incomplete synthesis may occur, necessitating the development of diverse analytical techniques to characterize the resulting polymers, NMR spectroscopy serves as a valuable tool for elucidating the molecular structure of these polymers by exploiting the resonance of atomic nuclei within a magnetic field.144 This technique can provide insights into the amino acid sequence, secondary structure, and intermolecular interactions. Notably, the detailed structures of polypeptides comprising fewer than 30 amino acids can be resolved using two-dimensional and three-dimensional NMR methodologies, which are crucial for understanding the spatial conformation of polypeptides and the nature of intramolecular hydrogen bonding.145 Furthermore, by analyzing parameters such as chemical shifts and coupling constants, one can ascertain the types and sequences of amino acid residues, thereby confirming the primary structure of poly(amino acids) and polypeptides. In addition, FTIR can be used to identify various chemical bonds and functional groups in compounds, and then determine the molecular structure. In the study of poly (amino acid) and polypeptide, FTIR spectroscopy can be employed to identify various chemical bonds and functional groups within compounds based on their absorption characteristics in the infrared spectrum. In the context of poly(amino acids) and polypeptides, FTIR is primarily utilized to analyze secondary structures.146 For example, the examination of the amide I band (1620–1690 cm−1) allows for the determination of the relative proportions of α-helices, β-sheets, random coils, and turns. The overlapping vibrational peaks associated with these secondary structures within the amide I band can be distinguished through mathematical techniques such as deconvolution, facilitating the extraction of quantitative information regarding the secondary structure.GPC serves as a method for determining molecular weight by utilizing a polymer solution that is introduced into a porous filler column. The retention times of molecules with varying molecular weights differ within the column, facilitating their separation and subsequent molecular weight assessment. This technique is particularly effective for poly(amino acids) and polypeptides, allowing for rapid and precise determination of their average molecular weight and molecular weight distribution. Such measurements are crucial for investigating the degree of polymerization, chain length, and molecular uniformity, making GPC a widely employed method for characterizing the molecular weight of polymeric materials.147
DSC and TGA are instrumental in evaluating molecular stability. DSC assesses the thermal transformation behavior of substances by measuring changes in heat flow during heating or cooling, thereby providing insights into properties such as melting point, glass transition temperature, and crystallization temperature.148 For poly(amino acids) and polypeptides, DSC is utilized to evaluate thermal stability, phase transition behavior, and the strength of intermolecular interactions.149 Conversely, TGA measures the mass of a sample in relation to temperature or time under programmed conditions, enabling the analysis of thermal decomposition processes, thermal stability, and the presence of moisture and volatile substances.150 In the context of poly(amino acids) and polypeptides, TGA can ascertain decomposition temperatures and thermogravimetric conditions, thereby informing on their stability at elevated temperatures and offering guidance for material processing, storage, and usage conditions.151
Furthermore, mass spectrometry can be employed to determine the molecular weight, amino acid sequence, and modifications of poly(amino acids) and polypeptides by separating and detecting samples based on the mass-to-charge ratio of ions.152 Circular dichroism chromatography is utilized to investigate the three-dimensional structure, reaction kinetics, and conformational changes of molecules in solution.153 X-ray crystallography is a technique used to elucidate the structure of proteins, allowing for the precise determination of the spatial arrangement of atoms within crystals, thereby revealing the three-dimensional structures of proteins and peptides.154 Additionally, ultraviolet spectroscopy can provide information regarding the solution conformation of biomacromolecules, requiring only that the samples be in a solution state for analysis.155
6. Application of poly(amino acids), peptides and their derivatives in drug delivery
Poly(amino acids), peptides, and their derivative drug delivery systems can significantly enhance the stability, bioavailability, and targeting of drugs, while also reducing side effects and improving therapeutic efficacy. Below are specific application cases of poly(amino acid), peptide, and their derivative in the delivery of small molecule drugs, nucleic acid drugs, peptide drugs, and protein drugs.6.1. Types of drugs loaded
 | ||
| Fig. 11 Overview of the 2R2SP requirement for nanocarriers with high overall delivery efficiency and the 3S transition in the CAPIR cascade.157 Copyright 2017, John Wiley and Sons. | ||
The primary peptide–drug couplings utilized in anticancer therapy include peptide–DOX couplings, peptide–paclitaxel (PTX) conjugates, peptide–camptothecin (CPT) affixes, peptide–platinum (Pt) drug couplings, peptide–vessel-disrupting agent (VDA) affixes, peptide–protein conjugates, and peptide–gas molecule conjugates. For instance, Singer et al. designed poly(L-glutamic acid)–Gly–CPT conjugates to stabilize the reactive lactone form of CPT and enhance its water solubility. In addition, linking CPT to high molecular weight anionic polymers improved its solubility and increased tumor distribution through the EPR effect.158 Klein's et al.159 further developed the concept of PLG couplings of 20(S)–CPT to augment the aqueous solubility of CPT, which demonstrated stability in aqueous solutions at neutral pH and exhibited potent anti-tumor activity in vivo. They also assessed the correlation between PLG molecular weight, CPT loading, and solubility, as well as the relationship between CPT-equivalent dosage and the in vivo antitumor efficacy of various conjugates, ultimately identifying the optimal conjugate composition.
The RGD (Arg–Gly–Asp) peptide specifically recognizes and binds to integrin receptors on the surface of tumor cells. By combining the peptide with the anticancer drug paclitaxel (PTX), targeted delivery of the drug can be achieved, thereby improving therapeutic efficacy and reducing side effects on normal tissues. To address the limitations of PTX, such as solubility, membrane permeability, and non-selective cytotoxicity, Deng et al. synthesized a “smart” PDC160 (peptide–drug coupling device) by linking PTX to a multifunctional peptide that consists of a tumor-targeting peptide (TTP) and a CPP. They constructed the TTP–CPP–PTX coupling, designated LTP-1, which intelligently delivers PTX to cells overexpressing the LHRH receptor. LTP-1 demonstrates a two-fold increase in cellular uptake compared to PTX and exhibits enhanced cytotoxicity, with an IC50 of 3.8 nM (compared to 6.6 nM for PTX). Additionally, LTP-1 shows reduced cytotoxicity to normal cells and has the capability to overcome PTX resistance. Furthermore, the in vivo antitumor efficacy of LTP-1 surpasses that of PTX, without significant toxicity. Zhang et al. synthesized an amphiphilic triblock copolymer methoxy poly(ethylene glycol)-block-poly(L-glutamic acid)-block-poly-(γ-propargyl-L-glutamate) (mPEG-b-PLGA-b-PPLG). The PLGA block of mPEG-b-PLGA-b-PPLG was modified with dopamine containing catechol groups through an amidation reaction, while the PPLG block was modified with a small tertiary amine molecule via a click reaction, resulting in a novel triblock copolymer, mPEG-b-PGCA-b-PGTA. This new copolymer could simultaneously load phenylboronic acid-modified ribonuclease A (RNase A) and hydrophobic DOX through pH-reversible phenylboronic acid–catechol linkages and hydrophobic interactions, respectively. The dual-drug-loaded NPs facilitate efficient delivery of both agents to tumor sites, exhibiting high systemic stability and enabling low-pH and ROS-triggered cooperative release of DOX and RNase A within tumor cells, leading to enhanced combined anticancer efficacy (Fig. 12).161
 | ||
| Fig. 12 Schematic illustration of mPEG-b-PGCA-b-PGTA mediated intracellular codelivery of RNase A and DOX for combination cancer therapy.161 Copyright 2020, John Wiley and Sons. | ||
The design of CPP-antibiotic couplers has primarily been based mainly on HIV–TAT peptides or their analogues, the usually arginine-rich peptides with short side chains.162–164 These CPPs enter cells through a complex mechanism, which results in low membrane permeability and limited drug delivery efficacy. In response to these challenges, Jiang et al.165 developed a class of structurally simple yet highly membrane-permeable CPPs that exhibited 100-fold greater membrane permeability than conventional CPPs, such as TAT and oligoarginine, as along with an unprecedented membrane penetration mechanism. Upon approaching the cell membrane, CPPs begin to adhere to the cell membrane through interactions between their peptide surface charges and the membrane's surface charge. They gradually reorient themselves orthogonally, allowing more charged side chains to make contact with the cell membrane. Following this initial adhesion, CPPs redistribute their surface charge to one side and penetrate the membrane orthogonally, with the exposed hydrophobic side facing the lipid interior. Given that the lengths of their side chains are comparable to the thickness of the lipid bilayer, and considering the driving force provided by the negatively charged inner leaflet, the side chains can facilitate further charge redistribution by tunneling their charged groups from the outer leaflet to the inner leaflet, ultimately resulting in complete membrane penetration.165
The application of peptides in gene editing is primarily demonstrated through their uses carriers for delivering gene editing tools into target cells. One significant application of peptide drug delivery systems in the delivery of the CRISPR/Cas9. Researchers utilize peptide NPs to transport the CRISPR/Cas9 components into target cells for gene editing. For example, the delivery of Cas9 nuclease and single guide RNA (sgRNA) using peptide-modified NPs has proven effective in modifying target genes for the treatment of genetic diseases. Ramakrishna et al.170 demonstrated that a simple treatment involving recombinant Cas9 protein, coupled with CPP and CPP-complexed guide RNA, results in endogenous gene disruption in human cell lines. In this process, the Cas9 protein binds to the CPP via thioether bonding, while the guide RNA is complexed with the CPP to form cohesive, positively charged NPs.
Poly(L-lysine) (PLL) is one of the most widely utilized gene vectors. PLL is a polypeptide synthesized through ring-opening polymerization, using e-benzyloxycarbonyl-L-lysine N-carboxyanhydride (Lys(Z)–NCA) as the monomer.171 Research demonstrated that exogenous DNA could be specifically delivered into hepatocellular carcinoma cells using an asialo–orosomucoid–PLL copolymer with transfection capabilities. However, the relatively high toxicity and low transfection efficiency of PLL, particularly due to its large molecular weight, have limited its applications. The incorporation of hydrophobic groups into PLL can enhance its cellular uptake. Clements et al. developed an amphiphilic vector by substituting PLL with palmitic acid (PLL–Pa). Compared to PLL, PLL–Pa exhibited significant intracellular transport in bone marrow stromal cells (BMSC) and demonstrated markedly higher transfection efficiency.172 Kataoka et al. created redox-sensitive multi-ion complex micelles through spontaneous nuclear cross-linking of thio-PEG-b-PLL and antisense oligodeoxynucleotides (ODN). The stability of ODN against nuclease degradation within the micelles was significantly enhanced, allowing for efficient release in the intracellular environment.173 Maruyama et al. showed that by grafting PEG onto the side chain of PLL increased circulatory lifespan and tumor without compromising siRNA activity.174 On the one hand, PEGylation enhances the stability and circulation time of cationic NPs in the blood circulation. On the other hand, polyethylene glycolisation may also diminish cellular uptake and endosomal escape.175 To address these challenges, Christie et al. modified PEG–PLL with cyclic arginine–glycine–glutamate peptide (cRGD), which binds to a variety of tumor-overexpressed integrin receptors, yielding improved results.176
Glioblastoma remains the most resistant malignant brain tumor due to the lack of effective therapeutic genes and drug delivery systems, particularly outside the tumor islands.177,178 Y. S. Malik et al.179 used non-viral agents, such as polylysine-modified polyethyleneimine (PEI–PLL) copolymers, to generate genetically engineered mesenchymal stem cells (MSCs) containing suicide genes, such as HSV-TK and TRAIL. This study suggested a promising non-viral approach for developing cell-based therapeutic treatments for gliomas. Furthermore, Lin et al.180 employed methoxy polyethylene glycol (MPEG) conjugated to arginine-functionalized poly(L-lysine) dendrimer (PLLD–Arg) through a click reaction. They subsequently synthesized MPEG–PLLD–Arg interacting with α-cyclodextrin (α-CD) to form a supramolecular hydrogel via host–guest interactions. This hydrogel demonstrated highly efficient and sustained gene transfection of the cells tested, outperforming PEI-25k.
Iudin and Vasilieva et al. demonstrated the suitability of PGlu-containing NPs for capturing positively charged peptide antibiotics, such as polymyxins, through electrostatic interactions or covalent coupling.184 In all cases, the physically loaded antibiotics retained their antimicrobial properties at the free drug level, while the amide-linked conjugates exhibited reduced activity. Furthermore, all encapsulated forms of polymyxins displayed lower cytotoxicity against human embryonic kidney cells (HEK 293) compared to free cationic polymyxins. Liu et al.185 selected the less cytotoxic antimicrobial peptide MP, coupled with the CPP Antp, and observed the changes in the cell-killing activity of the fusion peptide. They found that the uncoupled antimicrobial peptide MP and the CPP Antp did not exhibit any significant killing effect on the tumor cells. In contrast, the fusion peptide MPGA, which was formed by coupling the two peptides, demonstrated a strong tumor cell-killing effect by disrupting cell membranes. This suggests that coupling antimicrobial peptides with CPPs can significantly enhance their cytotoxicity, potentially providing a promising avenue for the development of novel antitumor drugs.
Poly(amino acids), such as polyglutamic acid (PGA) and polytyrosine (PTY), have also been investigated for their potential use in vaccine delivery systems to enhance antigen stability and immunogenicity. Zhang et al.187 explored the feasibility of NP-mediated antigenic peptides to efficiently induce cytotoxic T-lymphocyte responses to tumor-associated autoantigens in a C57BL/6 mouse model. They prepared biodegradable poly(D,L-lactide-co-glycolide) nanoparticle (PLGA-NP) carrying murine melanoma antigenic peptides, hgp100 and TRP, were prepared using a double-emulsification method, demonstrating the viability of NP-mediated antigen delivery for cancer immunotherapy. M. Chiba et al.188 developed polymeric microspheres capable of controlled release of macromolecules over periods ranging from days to more than a month. These polymers are particularly effective for the controlled delivery of vaccine antigens due to the incorporation of the immune adjuvant L-tyrosine into their backbone.
6.2. Enhance the therapeutic efficacy of drugs
The first advancement involves the introduction of blood-stable yet intracellularly unstable bonds in peptide–drug couplings. This includes acid-unstable bonds, such as hydrazine or benzoic acid imine, which respond to lysosomal acidity I as well as glutathione (GSH)-sensitive disulfide bonds203,204 that react to elevated GSH levels in tumor cells.205 Hoang et al. synthesized a ROS-responsive poly(ethylene glycol)-poly(methionine) and prepared micelles through the self-assembly of the hydrophobic pro-oxidant drug piperamide.206 Increased ROS levels in cancer cells triggered a transition of the peptide from hydrophobic to hydrophilic, leading to the disassembly of the micelles and resulting in effective drug release and enhanced anticancer efficacy.
The second strategy involves the capacity of peptides and poly(amino acids) to facilitate targeted drug release in response to environmental changes or specific cellular markers. These advanced drug delivery systems can identify particular conditions within the disease microenvironment, allowing for precise drug release at the target site. This approach maximizes therapeutic efficacy while minimizing side effects.
6.2.2.1. Targeted release based on pH response. Poly(amino acids) and polypeptides are complex biological macromolecules comprised of amino acid residues, characterized by a diverse array of ionizable functional groups, including amino and carboxylic groups. The ionization state of these functional groups is influenced by the pH of the surrounding environment, which in turn affects the electrostatic interactions among molecular chains. Specifically, in polypeptides that contain histidine residues, the imidazole side chain of histidine (with a pKa of approximately 6.0) is partially protonated at physiological pH (7.4). As the ambient pH decreases towards the pKa value, the degree of protonation of histidine residues increases, resulting in altered electrostatic repulsion or attraction between polypeptide chains, which subsequently modifies the structural configuration of the carrier.207 variations in pH can induce significant structural transformations. Under acidic conditions, the protonation of amino groups enhances electrostatic repulsion between molecular chains, leading to an expansion of the carrier. Conversely, under alkaline conditions, this effect is reversed.208 Such volumetric changes can influence the encapsulation and release dynamics of drugs. For instance, if a drug is physically embedded within the carrier, the expansion of the carrier may facilitate the opening of release channels, thereby promoting drug release. Additionally, fluctuations in pH can trigger depolymerization or polymerization processes in poly(amino acids) or polypeptides. Aggregates stabilized by hydrogen bonds or electrostatic interactions may be disrupted by changes in pH, resulting in depolymerization and subsequent drug release.209 For example, poly(amino acid) complexes that rely on acid–base interactions may undergo depolymerization when the pH shifts, disrupting the acid–base equilibrium. Furthermore, certain pH-sensitive poly(amino acids) and polypeptides may experience hydrophilic transformations. In acidic environments, their hydrophilic properties are enhanced due to protonation, while in alkaline conditions, hydrophilicity diminishes and hydrophobicity increases. This alteration in hydrophilicity significantly impacts the interactions between the drug and the carrier, as well as the carrier's interactions with the surrounding environment, thereby regulating the drug release process.
Therefore, poly (amino acid) materials can be designed to be PH sensitive, allowing targeted release of drugs in specific acidic and alkaline environments. This responsiveness ensures that the drug is released at the desired site, thereby minimizing systemic side effects. For example, Shen et al.210 developed a series of poly(amino acid) polymers that achieved an accelerated cumulative release rate of NPs as the pH decreased, demonstrating a significant targeted release effect. The synthesis of these materials and the preparation of NPs offer innovative approaches in the field of sustained drug release.
The primary disadvantage of conventional chemotherapy is its inability to selectively target cancer cells, resulting in damage to normal, healthy cells. The extracellular pH is 7.4, whereas in tumor cells, due to rapid and frequent cell proliferation and lactic acid accumulation lower the pH to between 5 and 6.211 A pH-responsive carrier molecule that specifically responds to acidic conditions may provide an effective means of precisely delivering drugs within the tumor microenvironment, thereby enhancing cytotoxicity against cancer cells. This approach must also consider the varying pH levels in different regions of the gastrointestinal tract, including the stomach, duodenum, jejunum, ileum, and vagina, to ensure optimal drug uptake at the corresponding sites.212–214 The introduction of aspartic acid (Asp) or glutamic acid (Glu) into peptides, with the pKa of the side carboxyl group in the pH 4 region resulting in NPs capable of releasing drugs in the acidic extracellular environment of tumor tissue (pH 5.5–6.0).215 Zhong et al. synthesized pH-responsive block polymers composed of polyethylene glycol (mPEG) and poly(asparagyl diisopropylethylenediamine-co-phenylalanine) (P(Asp(DIP)-co-Phe)), which can self-assemble into nanovesicles that encapsulate the hydrophilic drug tirapamine and the hydrophobic photosensitizer dihydroporphyrin.216 Due to the pH sensitivity of the diisopropylethylenediamine fragments, these nanovesicles disintegrate after 24 hours of incubation in a 10 mM phosphate buffered saline (PBS) solution at pH 5.0, facilitating the release of the encapsulated active ingredients.
For example, Li et al. developed a multifunctional cationic hydrogel designed for synergistic antibacterial therapy that is responsive to cationic stimuli, pH changes, and near-infrared (NIR) light, specifically for the treatment of bacterial-infected wounds (Fig. 13).217 The hydrogel matrix was created by crosslinking quaternary ammonium/boronic acid-modified poly(aspartic acid) (QPABA) and poly(vinyl alcohol) (PVA) with the RWRWRW–NH2 peptide (MP196) linked to PDA NPs. The gelation time was recorded at 30 seconds. The incorporation of phenyl boronic ester bonds facilitated pH-triggered dissociation in the acidic environment characteristic of bacterial infections. The developed hydrogel demonstrated high in vivo antibacterial efficacy, achieving nearly 100% effectiveness when combined with NIR light exposure.
 | ||
| Fig. 13 Schematic representation of pH-responsive QPABA/PVA hydrogel with encapsulated NIRresponsive MP196@PDA NPs as a wound treatment material for antibacterial therapy. Reproduced with permission of Elsevier from ref. 217. Copyright 2023, Elsevier. | ||
6.2.2.2. Targeted release based on temperature response. Some poly(amino acids) undergo phase transitions from hydrophobic to hydrophilic or from gel state to solution state over a certain temperature range. This phase transition can facilitate the release of the drug from the carrier. For example, when the temperature rises above a certain threshold, poly(amino acid) micelles, or NPs, break down, releasing encapsulated drugs. Furthermore, alterations in temperature can induce modifications in the molecular conformation of poly(amino acids) and polypeptides.218 At lower temperatures, the molecular chains exhibit an extended configuration, creating sufficient internal space to accommodate drug molecules. As the temperature increases beyond a certain threshold, the molecular chains undergo contraction, resulting in a reduction of the internal space, which subsequently leads to the expulsion of the drug molecules.219 Additionally, the conformational changes in the molecules can influence the hydrophilicity of the carrier, thereby modulating the interactions between the drug and the carrier, as well as between the carrier and its surrounding environment, ultimately governing the drug release process.220
Since tumor tissue tends to be slightly hotter than surrounding healthy tissue, typically displaying a 1–2 °C temperature difference, this variation provides a natural targeting advantage for temperature-responsive drug release. Drug delivery systems can be designed to respond to this slight temperature increase by selectively acting on the tumor site or by external means that can precisely control the temperature increase in the tumor area. A notable example is polyn-isopropylacrylamide (PNIPAM), a classic low-critical solution temperature (LCST) type temperature-responsive polymer that often binds to poly(amino acids) or peptides. When the temperature rises to the lowest temperature (usually around 32° C), PNIPAM changes from water soluble to hydrophobic, which triggers shrinkage or aggregation of the material, leading to drug release.221 The realization of the temperature-sensitive mechanism includes physical mixing, in which drugs and temperature-sensitive poly(amino acids) or polypeptides are loaded into the carrier through physical mixing. Or the drug is chemically coupled to temperature-sensitive poly(amino acids) or polypeptides. This temperature-responsive property highlights their great potential for use in temperature-sensitive drug delivery against tumors.
6.2.2.3. Enzyme-based response release. The enzymatic sensitivity of poly(amino acids) and polypeptides is contingent upon the precise recognition between the enzyme and its substrate. In vivo, a diverse array of enzymes exists, each characterized by a unique recognition sequence and active site. For instance, matrix metalloproteinases (MMPs) possess the ability to identify and cleave specific amino acid sequences, such as glycine–proline–leucine–glycine–isoleucine–alanine (GPLGIA). When a poly(amino acid) or polypeptide drug carrier incorporates the recognition sequence of these enzymes, it becomes susceptible to enzymatic action. The enzymatic activity on poly(amino acids) or polypeptides primarily involves the hydrolysis of peptide bonds.222 Upon recognition and cleavage of a specific peptide bond within the carrier, the molecular architecture of the carrier undergoes alteration. Such structural modifications may manifest as the fragmentation of molecular chains, conformational adjustments, or the emergence of new reactive termini. For example, when an enzyme cleaves a polypeptide carrier at a central location, the previously continuous molecular chain is divided into two segments, potentially resulting in the opening of the drug delivery channel encapsulated within or altering the interaction between the carrier and the drug, thereby facilitating drug release.223 By leveraging the degradation of these polymers through specific enzymes present in the biological milieu, enzymatic reactivity governs the release of drugs from poly(amino acids). This mechanism ensures that drug release is preferentially activated at pathological sites enriched with enzymes, such as tumors or inflamed regions. This can be accomplished by integrating enzyme-sensitive segments into poly(amino acid) materials, such as polymers that contain specific amino acid sequences amenable to recognition and degradation by endogenous enzymes, thus enabling the release of encapsulated therapeutics.224 Furthermore, the design of carrier architectures, including nanoparticles, micelles, or hydrogels, can enhance the efficacy of drug release at sites of enzymatic degradation.209
In various pathological conditions, the expression and activity of enzymes in affected tissues exhibit significant deviations from those in healthy tissues. For instance, in the context of tumors, both tumor cells and adjacent stromal cells produce substantial quantities of MMPs, which facilitate the invasion and metastasis of cancer cells.225 To enhance the targeted delivery of therapeutics to tumor tissues, enzyme-sensitive poly(amino acid) or polypeptide carriers are employed. These carriers are designed to release the drug upon cleavage by specific enzymes present in the tumor microenvironment, thereby improving the drug's utilization at the tumor site while minimizing adverse effects on normal tissues.226 Within cellular compartments, various enzymes, such as acid hydrolases found in lysosomes, play a crucial role. Upon the endocytosis of the drug carrier, it is transported into endosomes and lysosomes. Enzyme-sensitive poly(amino acid) or polypeptide carriers can exploit lysosomal enzymes to facilitate drug release intracellularly. For example, a polypeptide carrier engineered to release drugs in response to lysosomal enzymes encapsulates gene therapeutics. When this carrier reaches the lysosome, it is cleaved by lysosomal enzymes, allowing the gene drug to be released, thus preventing excessive degradation by nucleases present in the lysosomal environment and enhancing the efficiency of gene transfection.227 Considering the intricate enzymatic landscape within biological systems, some drug carriers are designed to be substrates for multiple enzymes.228 This design strategy ensures that under varying physiological or pathological conditions, the presence and activity of any one of these enzymes can initiate drug release. Such a mechanism enhances the flexibility and adaptability of drug delivery systems.
6.2.2.4. Response release based on redox reactions. In vivo, different tissues, cells and cellular organelles have different redox environments. For example, the cytoplasm inside the cell is usually in a relatively reductive environment, while the extracellular environment and certain organelles (such as mitochondria) may have a high oxidation potential.229 This difference in redox environment provides a physiological basis for designing redox sensitive drug delivery vectors.
The redox sensitivity of poly(amino acids) and polypeptide drug delivery carriers is mainly achieved by introducing specific redox sensitive groups. These groups can undergo biochemical structural changes under oxidation or reduction conditions, such as disulfide bonds (–S–S–). Disulfide bond is the most common redox sensitive group. In drug delivery vehicles, disulfide bonds can link molecular chains of poly(amino acids) or polypeptides, or they can be used to link drugs to carriers.230 When the carrier enters the reducing environment within the cell (such as the cytoplasm), the disulfide bond is reduced and broken.231 For example, a polypeptide nanocaller containing disulfide bonds encapsulates an anticancer drug. In the extracellular environment, the disulfide bonds remain stable and the drug is encapsulated inside the carrier. When the nanocarriers enter the cell through endocytosis, the disulfide bond breaks, the structure of the nanocarriers disintegrates, and the drug is released under the reducing environment of the cytoplasm. In addition, polyglutamic acid and polylysine containing disulfide bonds can be rapidly degraded in an intracellular reducing environment to release active drug molecules.232 Hu et al.233 developed a series of innovative poly(amino acid) materials that create drug-carrying NPs through physical encapsulation and chemical bonding.
Redox sensitive drug delivery vectors can effectively achieve intracellular targeted delivery. Due to the differences in the redox environment inside and outside the cell, this vector can remain stable outside the cell, while releasing drugs after entering the cell, especially in the reducing environment inside the cell. This is very important for drugs that need to play a role in the cell (such as gene drugs, protein drugs, etc.), which can improve the efficiency of drug release in the cell and avoid the drug being degraded or inactivated by early release outside the cell. The redox status of tumor tissues is also different from that of normal tissues. The redox balance within tumor cells is often disrupted and often has high levels of glutathione (GSH), an important reducing agent.234 Taking advantage of this feature, redox sensitive poly(amino acids) or polypeptide carriers can target drug delivery into tumor cells for release. For example, drug carriers containing disulfide bonds are designed so that under the action of high concentration of GSH in tumor cells, disulfide bonds are broken and drugs are released, improving the killing effect of drugs on tumor cells while reducing the impact on normal tissues.235
Poly(amino acids) and polypeptides possess the ability to self-assemble into nano-sized carriers, thereby enhancing the encapsulation capacity for pharmaceuticals.237 By manipulating polymerization conditions or through strategic molecular design, the dimensions of these carriers can be accurately tailored. For instance, block copolymers derived from poly(L-glutamic acid) (PGA) and poly(L-lysine) (PLL) can spontaneously form nanomicelles in aqueous environments.238 The nanomicelle's core is hydrophobic, allowing for the effective encapsulation of hydrophobic drugs, such as paclitaxel. Typically, these carriers exhibit particle sizes ranging from 10 to 1000 nm, which not only optimizes encapsulation efficiency but also facilitates the circulation and distribution of the carriers within biological systems. Furthermore, nano-sized vectors can accumulate in tumor tissues due to the enhanced permeability and retention (EPR) effect.239 The larger vascular endothelial spaces characteristic of tumor tissues enable these nanocarriers to infiltrate more readily and persist within the tumor environment, thereby facilitating localized drug release and indirectly enhancing the effective utilization of therapeutic agents.240
The incorporation of hydrophobic groups into poly(amino acids) or polypeptides has been shown to enhance the capacity for hydrophobic drug encapsulation.241 Concurrently, the self-assembly properties of the carrier can be modified to further optimize drug encapsulation efficacy. For certain insoluble pharmaceuticals, such chemically modified carriers can significantly enhance both the solubility and stability of the drug.242 For instance, the encapsulation of the poorly soluble anticancer agent camptothecin within a modified poly(amino acid) carrier not only increases the drug's encapsulation efficiency but also mitigates the rapid precipitation and degradation of the drug in aqueous environments.243 In the case of protein therapeutics, a viable strategy involves conjugating them to polypeptide carriers via amide bonds. An illustrative example is the linkage of insulin to a poly(L-histidine)–poly(L-lysine) (PH–PLL) carrier through an amide bond, which not only safeguards insulin from proteolytic degradation but also facilitates its release in an appropriate milieu (such as an acidic intestinal environment or within cells) due to the pH-responsive characteristics of the carrier. This approach enhances both the encapsulation efficiency and stability of the drugs.
7. Pharmacological mechanisms of poly(amino acid) and peptide delivery systems
7.1. Mode of administration
Due to the specific structure of poly(amino acids) and peptides, their delivery must overcome the body's degradation and clearance mechanisms. Poly(amino acid) and peptide drug delivery systems can be utilized in various modes, including oral, intravenous, intramuscular, pulmonary, ocular, and transdermal routes. The most suitable delivery strategy is selected based on the type of disease types and therapeutic needs. The selection and optimization of each delivery method depend on the physicochemical properties of the drug as well as the requirements of the targeted therapy. When combined with the design of the carrier system, these factors can significantly enhance the drug's bioavailability and therapeutic efficacy. | ||
| Fig. 14 Biochemical and physical barriers to oral drug delivery, and intestinal mucosal structures with major intestinal cell types.248 Copyright 2022, Theranostics. | ||
7.2. Cellular uptake mechanisms
In poly(amino acids) and peptide-based drug delivery systems, the cellular uptake mechanism is a pivotal factor influencing the overall effectiveness and therapeutic efficacy of drug delivery. The uptake mechanism of the drug carrier plays a key role in determining whether the drug can successfully enter target cells and thereby exert its intended therapeutic effect. | ||
| Fig. 15 Common uptake pathways of NP. (a) After interacting with the cell surface, NPs is absorbed through various endocytosis or pinocytosis. (b) Most NPs must escape from various vesicle cavities or endosomes before acidification.265 Copyright 2021, Springer Nature. | ||
One of the common pathways for cellular uptake of external substances is cytophagy, which is mediated by lattice proteins. This process involves the formation of vesicles enveloped in lattice proteins at the cell membrane. Poly(amino acids) and peptide drug carriers can efficiently enter the cell through this pathway. When these drug carriers bind to receptors or membrane proteins on the cell surface, the lattice proteins aggregate to form membrane-coated vesicles, which are subsequently internalized into the cell.263 After entering the cell, these endocytic vesicles are transported via endosomes and can eventually be degraded or translocated to other organelles. Grid protein-mediated cytophagy is highly selective and regulated, making it suitable for targeted drug delivery.264 This mechanism is particularly effective in delivering therapies against cancer cells. By designing peptide or poly(amino acids) drug carriers that specifically bind to cancer cell receptors, efficient intracellular entry can be achieved.
8. Progress in the clinical use of poly(amino acids) and peptides
The application of poly(amino acids) and peptides in clinical medicine is rapidly advancing and is increasingly recognized as a promising approach for treating a wide range of diseases.271 The high specificity and low toxicity of these biomolecules demonstrate significant potential in drug development and therapeutic applications.Antimicrobial peptides (AMPs) exhibit potent antimicrobial activity as integral components of the natural immune system.272 In contrast to conventional antibiotics, antimicrobial peptides are less prone to drug resistance and can swiftly eliminate bacteria, fungi, and viruses.273 In recent years, significant advancements have been made in the prevention and treatment of infections using antimicrobial peptides. For example, gramicidin274 and polymyxin B275 have been extensively utilized to treat ocular and dermal infections. Additionally, research is ongoing to develop novel antimicrobial peptides to address the challenges posed by multi-drug resistant strains.
Progress has also been made in the application of peptides for metabolic diseases. Insulin, a classical peptide hormone, is widely in the treatment of diabetes. Ongoing research has led to the development of novel long-acting insulin analogs that enhance the stability of glycemic control and improve patient compliance.259 GLP-1 receptor agonists, representing a new generation of antidiabetic medications, have demonstrated promising glucose-lowering effects as well as cardiovascular protective benefits.276 In the treatment of neurological disorders, peptides have also shown significant potential. For example, neuropeptide Y (NPY) and its receptor antagonists are currently being investigated for their efficacy in treating anxiety and depression.277 In addition, Aβ peptide vaccines and inhibitors for Alzheimer's disease are undergoing clinical trials aimed at slowing or halting the progression of the disease.278
In conclusion, advancements in the clinical application of poly(amino acids) and peptides indicate that these compounds are poised to become significant therapeutic options for a wide range of diseases.279 Although challenges such as stability, delivery systems, and production costs remain, the clinical use of poly(amino acids) and peptide drugs is expected to become increasingly promising as technology progresses and research efforts intensify.
9. Outlook and conclusions
With the continuous advancement of biomedical and materials science, poly(amino acids), peptides, and their derivatives have demonstrated significant potential in the field of drug delivery. Future research will further explore the ability of these materials to enhance drug efficacy, reduce side effects, and achieve precision therapy.One critical area of future development lies in improving the targeting specificity and controlled release capabilities of poly(amino acid) and peptide-based drug carriers. While these carriers have made progress in achieving targeted delivery and controlled release through the incorporation of targeting ligands and responsive design elements, there remains a need for greater precision and efficiency. By optimizing the selection of ligands and improving the structural design of the carriers, it is possible to develop more effective systems that achieve enhanced targeted delivery and more precise release profiles, thereby improving therapeutic outcomes.
Another promising direction for future research is the development of multifunctional carriers. These carriers not only transport and release drugs but also integrate additional functions such as diagnosis, imaging, and therapy. For instance, smart, responsive carriers can be designed to respond to physiological changes in the body, such as pH, temperature, or enzyme activity, allowing real-time monitoring of drug release and therapeutic efficacy. Additionally, these multifunctional systems may enable the combination of various therapeutic modalities, such as photothermal therapy or immunotherapy, offering synergistic effects and improving treatment efficiency.
Despite the favorable biocompatibility of poly(amino acids) and peptides, their in vivo metabolic pathways and long-term safety profiles need further investigation. Ensuring that these drug delivery systems are non-toxic and exhibit low immunogenicity is crucial for their successful clinical application. Therefore, future studies should focus on understanding the impact of carrier degradation products on biological systems and on optimizing the chemical structure of these materials to ensure their safety and degradability in vivo.
Scaling up the production of poly(amino acid) and peptide-based carriers for clinical use presents another significant challenge. Much of the current research is still at the laboratory stage, and achieving large-scale production with standardized preparation methods will be essential for clinical translation. Developing efficient, cost-effective synthesis and production processes, while ensuring consistent and reproducible product quality, will be critical. Furthermore, extensive preclinical and clinical studies are needed to validate the safety and efficacy of these systems.
Personalized and precision medicine represents an exciting frontier for the application of poly(amino acids) and peptides in drug delivery. By integrating patient-specific genomic, proteomic, and other big data analyses, drug delivery systems can be tailored to meet individual patient needs, enabling precision therapies. This approach would allow customization of the type of drug carrier, dosage, and release method based on the patient's specific condition and physiological state, thus improving therapeutic outcomes and minimizing adverse effects. In addition to the current materials, there is a need to explore new materials and structures to meet the diverse requirements of drug delivery. Advances in nanotechnology, supramolecular chemistry, and biomaterials science provide novel ideas for designing and synthesizing drug carriers with unique properties. For example, self-assembly techniques and nanomaterials may be employed to construct carriers with higher drug-loading capacities and improved stability.280,281
Although significant progress has been made in the field of drug delivery, poly(amino acids), peptides, and their derivatives still face several challenges. Drug delivery systems must remain stable in complex in vivo environments and release their cargo under specific conditions, which presents a major challenge for the design and optimization of carriers. Additionally, overcoming biological barriers, such as the blood–brain barrier, is crucial for enhancing the effectiveness of drug delivery systems. Ensuring the long-term safety and minimizing the immune responses of these carriers is also a critical issue that requires further investigation.
In conclusion, while there are many challenges ahead, poly(amino acids), peptides, and their derivatives hold great promise for advancing drug delivery technologies. Continued research and innovation will help overcome existing technological barriers, leading to more efficient and safer drug delivery systems that can provide new solutions for clinical treatment. The future of this field will rely on interdisciplinary collaboration, with synergistic contributions from materials science, pharmacy, biology, and clinical medicine.
Author contributions
Huilin Yuan and Mingxia Jiang wrote the original draft, Huapan Fang and Huayu Tian designed the project, reviewed and revised the original manuscript. All authors have read and agreed to the published version of the manuscript.Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this study.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are thankful to National Key Research and Development Program of China (2021YFB3800900); Natural Science Foundation of Xiamen, China (3502Z202371004); National Natural Science Foundation of China (52473150, 51925305, 51873208, 51833010, and 52203183); Shenzhen Science and Technology Program (JCYJ20240813145515020); Fundamental Research Funds for the Central Universities (20720230004); and the talent cultivation project Funds for the Innovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (HRTP-[2022]52).References
- S. Hoelder, P. A. Clarke and P. Workman, Mol. Oncol., 2012, 6, 155–176 CrossRef CAS
.
- H. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2010, 94, 1–18 CrossRef CAS
- M. Jiang, W. Chen, W. Yu, Z. Xu, X. Liu, Q. Jia, X. Guan and W. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 43963–43974 CrossRef CAS
- A. Sood and R. Panchagnula, Chem. Rev., 2001, 101, 3275–3304 CrossRef CAS PubMed
- H. Li, Y. Yang, W. Hong, M. Huang, M. Wu and X. Zhao, Signal Transduction Targeted Ther., 2020, 5, 1 CrossRef
- H. Fang, Z. Guo, L. Lin, J. Chen, P. Sun, J. Wu, C. Xu, H. Tian and X. Chen, J. Am. Chem. Soc., 2018, 140, 11992–12000 CrossRef CAS
- L. Xu, Z. Shao, X. Fang, Z. Xin, S. Zhao, H. Zhang, Y. Zhang, W. Zheng, X. Yu, Z. Zhang and L. Sun, Exploration, 2024, 20230165 CrossRef
- X. Liu, F. Wu, Y. Ji and L. Yin, Bioconjugate Chem., 2018, 30, 305–324 CrossRef
- H. Fang, L. Chen, Z. Deng, Y. Gao, Y. Yang, Q. Chen and Z. Liu, ACS Nano, 2023, 17, 17595–17595 CrossRef CAS PubMed
- H. Fang, Z. Guo, J. Chen, L. Lin, Y. Hu, Y. Li, H. Tian and X. Chen, Nat. Commun., 2021, 12, 6742 CrossRef CAS PubMed
- S. Antimisiaris, A. Marazioti, M. Kannavou, E. Natsaridis, F. Gkartziou, G. Kogkos and S. Mourtas, Adv. Drug Delivery Rev., 2021, 174, 53–86 CrossRef CAS PubMed
- S. Dragojevic, J. S. Ryu and D. Raucher, Molecules, 2015, 20, 21750–21769 CrossRef CAS PubMed
- R. Liu, C. Luo, Z. Pang, J. Zhang, S. Ruan, M. Wu, L. Wang, T. Sun, N. Li and L. Han, Chin. Chem. Lett., 2023, 34, 107518 CrossRef CAS
- S. Hossen, M. K. Hossain, M. Basher, M. Mia, M. Rahman and M. J. Uddin, J. Adv. Res., 2019, 15, 1–18 CrossRef CAS PubMed
- A. Akhtar, A. Andleeb, T. S. Waris, M. Bazzar, A.-R. Moradi, N. R. Awan and M. Yar, J. Controlled Release, 2021, 330, 1152–1167 CrossRef CAS
- H. Fang, Y. Wu, L. Chen, Z. Cao, Z. Deng, R. Zhao, L. Zhang, Y. Yang, Z. Liu and Q. Chen, ACS Nano, 2023, 17, 4748–4763 CrossRef CAS PubMed
- H. Fang, L. Chen, Z. Deng, Y. Gao, Y. Yang, Q. Chen and Z. Liu, ACS Nano, 2023, 17, 1128–1143 CrossRef CAS PubMed
- M. Jiang, W. Chen, Y. Sun, J. Zeng, L. Ma, J. Gong, X. Guan, K. Lu and W. Zhang, Int. J. Biol. Macromol., 2023, 242, 125223 CrossRef CAS
- H. Fang, L. Zhang, Y. Wu, L. Chen, Z. Deng, Z. Zheng, Y. Wang, Y. Yang and Q. Chen, Chem. Eng. J., 2024, 498, 155781 CrossRef CAS
- L. Liu, Y. Pan, C. Zhao, P. Huang, X. Chen and L. Rao, ACS Nano, 2023, 17, 3225–3258 CrossRef CAS PubMed
- J. Lv, C. Liu, K. Lv, H. Wang and Y. Cheng, Sci. China Mater., 2020, 63, 620–628 CrossRef
- J. V. González-Aramundiz, M. V. Lozano, A. Sousa-Herves, E. Fernandez-Megia and N. Csaba, Expert Opin. Drug Delivery, 2012, 9, 183–201 CrossRef
- E. Liarou, S. Varlas, D. Skoulas, C. Tsimblouli, E. Sereti, K. Dimas and H. Iatrou, Prog. Polym. Sci., 2018, 83, 28–78 CrossRef CAS
- J. Martin, A. Desfoux, J. Martinez, M. Amblard, A. Mehdi, L. Vezenkov and G. Subra, Prog. Polym. Sci., 2021, 115, 101377 CrossRef CAS
- S. Mallakpour and M. Dinari, J. Macromol. Sci., Part A:Pure Appl. Chem., 2011, 48, 644–679 CrossRef CAS
- H. Xu, Q. Yao, C. Cai, J. Gou, Y. Zhang, H. Zhong and X. Tang, J. Controlled Release, 2015, 199, 84–97 CrossRef CAS
- L. Zhao, N. Li, K. Wang, C. Shi, L. Zhang and Y. Luan, Biomaterials, 2014, 35, 1284–1301 CrossRef CAS
- M. J. Webber and E. T. Pashuck, Adv. Drug Delivery Rev., 2021, 172, 275–295 CrossRef CAS PubMed
- A. W. Du and M. H. Stenzel, Biomacromolecules, 2014, 15, 1097–1114 CrossRef CAS PubMed
- B. B. Mendes, J. Conniot, A. Avital, D. Yao, X. Jiang, X. Zhou, N. Sharf-Pauker, Y. Xiao, O. Adir and H. Liang, Nat. Rev. Methods Primers, 2022, 2, 24 CrossRef CAS PubMed
- H. J. Vaughan, J. J. Green and S. Y. Tzeng, Adv. Mater., 2020, 32, 1901081 CrossRef CAS
- Y. Liu, C.-F. Xu, S. Iqbal, X.-Z. Yang and J. Wang, Adv. Drug Delivery Rev., 2017, 115, 98–114 CrossRef CAS
- T. D. Brown, K. A. Whitehead and S. Mitragotri, Nat. Rev. Mater., 2020, 5, 127–148 CrossRef
- H. B. Gdr, N. Sharon and E. Australia, Pure Appl. Chem., 1984, 56, 595–624 CrossRef
- R. Augustine, N. Kalva, H. A. Kim, Y. Zhang and I. Kim, Molecules, 2019, 24, 2961 CrossRef CAS
- K. Bauri, S. G. Roy, S. Pant and P. De, Langmuir, 2013, 29, 2764–2774 CrossRef CAS
- J. Rodríguez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef PubMed
- Z. Hu, J. Wang, S. Han, J. Hu and A. Reheman, New J. Chem., 2022, 46, 19888–19899 RSC
- P. Wu, H. Zhao, X. Gou, X. Wu, S. Zhang, G. Deng and Q. Chen, Int. J. Nanomed., 2019, 4059–4069 CrossRef CAS
- C. Bonduelle, Polym. Chem., 2018, 9, 1517–1529 RSC
- E. M. Cagil, J. Mol. Struct., 2020, 1217, 128382 CrossRef CAS
- R.-S. Lee, K.-Y. Peng, S.-W. Wang and Y.-Z. Li, Polym. J., 2014, 46, 710–721 CrossRef CAS
- Z. Wang, M. Zhuang, T. Sun, X. Wang and Z. Xie, Bioorg. Med. Chem. Lett., 2017, 27, 2493–2496 CrossRef CAS PubMed
- Y.-J. Li, X.-F. Gong, D. Wang, Z. Yang, H. Cao and L. Wang, Acta Polym. Sin., 2022, 53, 445–456 CAS
- S. Guo, J. Wang, Q. Wang, J. Wang, S. Qin and W. Li, Heliyon, 2024, 10, e26009 CrossRef CAS PubMed
- S. Majumdar and T. J. Siahaan, Med. Res. Rev., 2012, 32, 637–658 CrossRef CAS PubMed
- A. David, Adv. Drug Delivery Rev., 2017, 119, 120–142 CrossRef CAS
- M. F. Attia, N. Anton, J. Wallyn, Z. Omran and T. F. Vandamme, J. Pharm. Pharmacol., 2019, 71, 1185–1198 CrossRef CAS
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed
- Y. Wang, T. Deng, X. Liu, X. Fang, Y. Mo, N. Xie, G. Nie, B. Zhang and X. Fan, Int. J. Nanomed., 2024, 6253–6277 CrossRef
- P. Mi, Theranostics, 2020, 10, 4557 CrossRef CAS
- A. Komin, L. Russell, K. Hristova and P. Searson, Adv. Drug Delivery Rev., 2017, 110, 52–64 CrossRef
- J. Zhang, Z.-F. Yuan, Y. Wang, W.-H. Chen, G.-F. Luo, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, J. Am. Chem. Soc., 2013, 135, 5068–5073 CrossRef CAS PubMed
- V. F. Cardoso, D. M. Correia, C. Ribeiro, M. M. Fernandes and S. Lanceros-Méndez, Polymers, 2018, 10, 161 CrossRef PubMed
- M. Hussain, R. A. Naqvi, N. Abbas, S. M. Khan, S. Nawaz, A. Hussain, N. Zahra and M. W. Khalid, Polymers, 2020, 12, 323 CrossRef CAS PubMed
- N. C. Paxton, M. C. Allenby, P. M. Lewis and M. A. Woodruff, Eur. Polym. J., 2019, 118, 412–428 CrossRef CAS
- M. Zare, E. R. Ghomi, P. D. Venkatraman and S. Ramakrishna, J. Appl. Polym. Sci., 2021, 138, 50969 CrossRef CAS
- S. Wendels and L. Avérous, Bioact. Mater., 2021, 6, 1083–1106 CAS
- U. Ali, K. J. B. A. Karim and N. A. Buang, Polym. Rev., 2015, 55, 678–705 CrossRef CAS
- A. R. Mazo, S. Allison-Logan, F. Karimi, N. J.-A. Chan, W. Qiu, W. Duan, N. M. O'Brien-Simpson and G. G. Qiao, Chem. Soc. Rev., 2020, 49, 4737–4834 RSC
- K. Numata, Polym. J., 2015, 47, 537–545 CrossRef CAS
- M. Kunioka, Appl. Microbiol. Biotechnol., 1997, 47, 469–475 CrossRef CAS
- Z. Li, Y. Zheng, J. Yan, Y. Yan, C. Peng, Z. Wang, H. Liu, Y. Liu, Y. Zhou and M. Ding, ChemBioChem, 2023, 24, e202300132 CrossRef CAS
- H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Chem. Commun., 2014, 50, 139–155 RSC
- S. Zhang, W. Fu and Z. Li, Polym. Chem., 2014, 5, 3346–3351 RSC
- S. S. Gupta, V. Mishra, M. D. Mukherjee, P. Saini and K. R. Ranjan, Int. J. Biol. Macromol., 2021, 188, 542–567 CrossRef CAS
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373–7390 RSC
- M. Thompson and C. Scholz, Nanomaterials, 2021, 11, 1119 CrossRef CAS
- A. Douka, S. Vouyiouka, L.-M. Papaspyridi and C. D. Papaspyrides, Prog. Polym. Sci., 2018, 79, 1–25 CrossRef CAS
-
S. P. Schwendeman, M. Cardamone, A. Klibanov, R. Langer and M. R. Brandon, in Microparticulate systems for the delivery of proteins and vaccines, CRC Press, 2020, pp. 1–49 Search PubMed
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS
- M. Saini, A. Kashyap, S. Bindal, K. Saini and R. Gupta, Front. Microbiol., 2021, 12, 641251 CrossRef PubMed
- K. Yazawa and K. Numata, Molecules, 2014, 19, 13755–13774 CrossRef PubMed
- A. Boto, C. C. González, D. Hernández, I. Romero-Estudillo and C. J. Saavedra, Org. Chem. Front., 2021, 8, 6720–6759 RSC
- Y. Qiu, D. Xu, P. Lei, S. Li and H. Xu, Trends Biotechnol., 2024, 42, 310–325 CrossRef CAS PubMed
- F. Guzmán, S. Barberis and A. Illanes, Electron. J. Biotechnol., 2007, 10, 279–314 Search PubMed
- S. Thapa, H. Li, J. Ohair, S. Bhatti, F.-C. Chen, K. A. Nasr, T. Johnson and S. Zhou, Mol. Biotechnol., 2019, 61, 579–601 CrossRef CAS
- J. Chapman, A. E. Ismail and C. Z. Dinu, Catalysts, 2018, 8, 238 CrossRef
- F. B. Oppermann-Sanio and A. Steinbüchel, Naturwissenschaften, 2002, 89, 11–22 CrossRef
- S. Chatterjee and S. Yadav, Life, 2019, 9, 25 CrossRef CAS PubMed
- M. Mindt, T. Walter, P. Kugler and V. F. Wendisch, Biotechnol. J., 2020, 15, 1900451 CrossRef CAS
- Z. Luo, Y. Guo, J. Liu, H. Qiu, M. Zhao, W. Zou and S. Li, Biotechnol. Biofuels, 2016, 9, 1–12 CrossRef
-
D. Kour, K. L. Rana, S. Thakur, S. Sharma, N. Yadav, A. A. Rastegari, A. N. Yadav and A. K. Saxena, in New and future developments in microbial biotechnology and bioengineering, Elsevier, 2019, pp. 35–75 Search PubMed
- H. Goodarzi, N. Torabi, H. S. Najafabadi and M. Archetti, Gene, 2008, 407, 30–41 CrossRef CAS
- A. Berlec and B. Štrukelj, J. Ind. Microbiol. Biotechnol., 2013, 40, 257–274 CrossRef CAS
- T. Kotnik, W. Frey, M. Sack, S. H. Meglič, M. Peterka and D. Miklavčič, Trends Biotechnol., 2015, 33, 480–488 CrossRef CAS
- D. G. Yansura and D. J. Henner, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 439–443 CrossRef CAS
- N. Stephanopoulos, J. H. Ortony and S. I. Stupp, Acta Mater., 2013, 61, 912–930 CrossRef CAS PubMed
- E. De Santis and M. G. Ryadnov, Chem. Soc. Rev., 2015, 44, 8288–8300 RSC
- S. Zhang, Biotechnol. Adv., 2002, 20, 321–339 CrossRef CAS
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed
- D. N. Woolfson, G. J. Bartlett, M. Bruning and A. R. Thomson, Curr. Opin. Struct. Biol., 2012, 22, 432–441 CrossRef CAS PubMed
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2017, 17, 1700177 CrossRef PubMed
-
S. B. Bankar, P. R. Nimbalkar, P. V. Chavan and R. S. Singhal, Role of materials science in food bioengineering, 2018, 381–412 Search PubMed
- C. Cai, J. Lin, Y. Lu, Q. Zhang and L. Wang, Chem. Soc. Rev., 2016, 45, 5985–6012 RSC
- K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935–3953 RSC
- J. C. Jiménez-Cruz, R. Guzmán-Mejía, P. Navarro-Santos, S. García-Zavala, R. Herrera-Bucio, H. A. García-Gutiérrez and J. A. Aviña-Verduzco, J. Mol. Struct., 2023, 1294, 136354 CrossRef
- J. Shen, Y. Chen, X. Li, X. Zhou and Y. Ding, Int. J. Biol. Macromol., 2024, 270, 131758 CrossRef CAS
- T. Miao, J. Wang, Y. Zeng, G. Liu and X. Chen, Adv. Sci., 2018, 5, 1700513 CrossRef PubMed
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS PubMed
- G. Liu, J. F. Lovell, L. Zhang and Y. Zhang, Int. J. Mol. Sci., 2020, 21, 6380 CrossRef CAS PubMed
- M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani and M. Bozorgomid, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC
- A. Varanko, S. Saha and A. Chilkoti, Adv. Drug Delivery Rev., 2020, 156, 133–187 CrossRef CAS
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS
- F. Oroojalian, F. Charbgoo, M. Hashemi, A. Amani, R. Yazdian-Robati, A. Mokhtarzadeh, M. Ramezani and M. R. Hamblin, J. Controlled Release, 2020, 321, 442–462 CrossRef CAS
-
D. Agyei, K.-X. Tan, S. Pan, C. C. Udenigwe and M. K. Danquah, in Peptide applications in biomedicine, biotechnology and bioengineering, Elsevier, 2018, pp. 231–251 Search PubMed
- L. Diao and B. Meibohm, Clin. Pharmacokinet., 2013, 52, 855–868 CrossRef CAS
- J. Thundimadathil, Drug Discovery, 2019, 503–530 CAS
- I. W. Hamley, Biomacromolecules, 2014, 15, 1543–1559 CrossRef CAS
- Y. Wu, D. Zhong, Y. Li, H. Wu, H. Zhang, H. Mao, J. Yang, K. Luo, Q. Gong and Z. Gu, Nanoscale, 2021, 13, 4887–4898 RSC
- Z. Jiang, X. Feng, H. Zou, W. Xu and X. Zhuang, Bioact. Mater., 2021, 6, 2688–2697 CAS
- S. Cavalli, F. Albericio and A. Kros, Chem. Soc. Rev., 2010, 39, 241–263 RSC
- Y. Shi, A. Lu, X. Wang, Z. Belhadj, J. Wang and Q. Zhang, Acta Pharm. Sin. B, 2021, 11, 2396–2415 CrossRef CAS PubMed
- D. Zhi, Y. Bai, J. Yang, S. Cui, Y. Zhao, H. Chen and S. Zhang, Adv. Colloid Interface Sci., 2018, 253, 117–140 CrossRef CAS
- B. Yang, A. Gomes Dos Santos, S. Puri, A. Bak and L. Zhou, Expert Opin. Drug Delivery, 2022, 19, 1233–1245 CrossRef CAS
- W. Li, F. Separovic, N. M. O'Brien-Simpson and J. D. Wade, Chem. Soc. Rev., 2021, 50, 4932–4973 RSC
- H. Bi, J. Xue, H. Jiang, S. Gao, D. Yang, Y. Fang and K. Shi, Asian J. Pharm. Sci., 2019, 14, 365–379 CrossRef
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS PubMed
- L. J. Abbott, A. K. Tucker and M. J. Stevens, J. Phys. Chem. B, 2015, 119, 3837–3845 CrossRef CAS PubMed
- M. Karimi, P. Sahandi Zangabad, A. Ghasemi, M. Amiri, M. Bahrami, H. Malekzad, H. Ghahramanzadeh Asl, Z. Mahdieh, M. Bozorgomid and A. Ghasemi, ACS Appl. Mater. Interfaces, 2016, 8, 21107–21133 CrossRef CAS
- Y. Zou, L. Zhang, L. Yang, F. Zhu, M. Ding, F. Lin, Z. Wang and Y. Li, J. Controlled Release, 2018, 273, 160–179 CrossRef CAS
- V. Sinha and A. Trehan, J. Controlled Release, 2003, 90, 261–280 CrossRef CAS
- R. He, B. Finan, J. P. Mayer and R. D. DiMarchi, Molecules, 2019, 24, 1855 CrossRef CAS
- T. Yang, J. Zhai, D. Hu, R. Yang, G. Wang, Y. Li and G. Liang, Pharmaceutics, 2022, 14, 1919 CrossRef CAS PubMed
- J. Yoo, C. Park, G. Yi, D. Lee and H. Koo, Cancers, 2019, 11, 640 CrossRef CAS
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC
- S. Li, H. Zhao, Y. Fan, G. Zhao, R. Wang, F. Wen, J. Wang, X. Wang, Y. Wang and Y. Gao, Chem. Biol. Drug Des., 2020, 95, 58–65 CrossRef CAS PubMed
- J. Yu, X. Mao, X. Yang, G. Zhao and S. Li, Molecules, 2024, 29, 1758 CrossRef CAS
- S. Ma, W. Song, Y. Xu, X. Si, D. Zhang, S. Lv, C. Yang, L. Ma, Z. Tang and X. Chen, Biomaterials, 2020, 232, 119676 CrossRef CAS PubMed
- S. Ma, W. Song, Y. Xu, X. Si, S. Lv, Y. Zhang, Z. Tang and X. Chen, Nano Lett., 2020, 20, 2514–2521 CrossRef CAS PubMed
- J. M. Lambert and R. V. Chari, J. Med. Chem., 2014, 57, 6949–6964 CrossRef CAS PubMed
- S. Ali, C. Dussouillez, B. Padilla, B. Frisch, A. J. Mason and A. Kichler, J. Gene Med., 2022, 24, e3401 CrossRef CAS
- A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS
- S. Fawell, J. Seery, Y. Daikh, C. Moore, L. L. Chen, B. Pepinsky and J. Barsoum, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 664–668 CrossRef CAS
- E. Vives, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef CAS
- M. C. Morris, P. Vidal, L. Chaloin, F. Heitz and G. Divita, Nucleic Acids Res., 1997, 25, 2730–2736 CrossRef CAS
- M. C. Morris, J. Depollier, J. Mery, F. Heitz and G. Divita, Nat. Biotechnol., 2001, 19, 1173–1176 CrossRef CAS
- K. Montrose, Y. Yang, X. Sun, S. Wiles and G. W. Krissansen, Sci. Rep., 2013, 3, 1661 CrossRef PubMed
- Y. Zhang, W. Song, Y. Lu, Y. Xu, C. Wang, D.-G. Yu and I. Kim, Biomolecules, 2022, 12, 636 CrossRef CAS
- C. Qiu, Y. Wu, Q. Shi, Q. Guo, J. Zhang, Y. Meng, C. Wang, F. Xia, J. Wang and C. Xu, Int. J. Biol. Sci., 2023, 19, 789 CrossRef CAS PubMed
- J.-X. Chen, H.-Y. Wang, C.-Y. Quan, X.-D. Xu, X.-Z. Zhang and R.-X. Zhuo, Org. Biomol. Chem., 2010, 8, 3142–3148 RSC
- R. Fu and T. A. Cross, Annu. Rev. Biophys. Biomol. Struct., 1999, 28, 235–268 CrossRef CAS
- S. P. Brown and H. W. Spiess, Chem. Rev., 2001, 101, 4125–4156 CrossRef CAS PubMed
- H. Yang, S. Yang, J. Kong, A. Dong and S. Yu, Nat. Protoc., 2015, 10, 382–396 CrossRef CAS PubMed
- H. Sun, F. Meng, A. A. Dias, M. Hendriks, J. Feijen and Z. Zhong, Biomacromolecules, 2011, 12, 1937–1955 CrossRef CAS PubMed
-
A. Fortunato, in Drug-biomembrane interaction studies, Elsevier, 2013, pp. 169–212 Search PubMed
- B. Mahesh, D. Kathyayani, D. C. Gowda, A. Sionkowska and S. Ramakrishna, Mater. Chem. Phys., 2022, 281, 125847 CrossRef CAS
- N. Saadatkhah, A. Carillo Garcia, S. Ackermann, P. Leclerc, M. Latifi, S. Samih, G. S. Patience and J. Chaouki, Can. J. Chem. Eng., 2020, 98, 34–43 CrossRef CAS
- I. M. Weiss, C. Muth, R. Drumm and H. O. Kirchner, BMC Biophys., 2018, 11, 1–15 CrossRef
-
M. Kinter and N. E. Sherman, Protein sequencing and identification using tandem mass spectrometry, John Wiley & Sons, 2005 Search PubMed
- B. Ranjbar and P. Gill, Chem. Biol. Drug Des., 2009, 74, 101–120 CrossRef CAS PubMed
- L. Maveyraud and L. Mourey, Molecules, 2020, 25, 1030 CrossRef CAS PubMed
- K. Wang, D.-W. Sun, H. Pu and Q. Wei, Trends Food Sci. Technol., 2017, 67, 207–219 CrossRef CAS
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed
- Q. Sun, Z. Zhou, N. Qiu and Y. Shen, Adv. Mater., 2017, 29, 1606628 CrossRef PubMed
- J. W. Singer, R. Bhatt, J. Tulinsky, K. R. Buhler, E. Heasley, P. Klein and P. de Vries, J. Controlled Release, 2001, 74, 243–247 CrossRef CAS PubMed
- R. Bhatt, P. de Vries, J. Tulinsky, G. Bellamy, B. Baker, J. W. Singer and P. Klein, J. Med. Chem., 2003, 46, 190–193 CrossRef CAS
- X. Deng, R. Mai, C. Zhang, D. Yu, Y. Ren, G. Li, B. Cheng, L. Li, Z. Yu and J. Chen, Eur. J. Med. Chem., 2021, 213, 113050 CrossRef CAS PubMed
- P. Zhang, Y. Zhang, X. Ding, W. Shen, M. Li, E. Wagner, C. Xiao and X. Chen, Adv. Mater., 2020, 32, 2000013 CrossRef CAS
- A. Brezden, M. F. Mohamed, M. Nepal, J. S. Harwood, J. Kuriakose, M. N. Seleem and J. Chmielewski, J. Am. Chem. Soc., 2016, 138, 10945–10949 CrossRef CAS PubMed
- E. K. Lei, M. P. Pereira and S. O. Kelley, Angew. Chem., Int. Ed., 2013, 52, 9660–9663 CrossRef CAS PubMed
- M. P. Pereira, J. Shi and S. O. Kelley, ACS Infect. Dis., 2015, 1, 586–592 CrossRef CAS
- Y. Jiang, M. Han, Y. Bo, Y. Feng, W. Li, J. R. Wu, Z. Song, Z. Zhao, Z. Tan and Y. Chen, ACS Cent. Sci., 2020, 6, 2267–2276 CrossRef CAS
- K. Bulaklak and C. A. Gersbach, Nat. Commun., 2020, 11, 1–4 CrossRef
- S. Mali, Indian J. Hum. Genet., 2013, 19, 3 CrossRef CAS PubMed
- K. Tatiparti, S. Sau, S. K. Kashaw and A. K. Iyer, Nanomaterials, 2017, 7, 77 CrossRef
- S. Biswas, N. Yadav, P. Juneja, A. K. Mourya, S. Kaur, D. M. Tripathi and V. S. Chauhan, ACS Omega, 2022, 7, 36811–36824 CrossRef CAS PubMed
- S. Ramakrishna, A.-B. K. Dad, J. Beloor, R. Gopalappa, S.-K. Lee and H. Kim, Genome Res., 2014, 24, 1020–1027 CrossRef CAS PubMed
- J. Chen, X. Guan, Y. Hu, H. Tian and X. Chen, Top. Curr. Chem., 2017, 375, 32 CrossRef PubMed
- B. A. Clements, V. Incani, C. Kucharski, A. Lavasanifar, B. Ritchie and H. Uludağ, Biomaterials, 2007, 28, 4693–4704 CrossRef CAS
- Y. Kakizawa, A. Harada and K. Kataoka, Biomacromolecules, 2001, 2, 491–497 CrossRef CAS PubMed
- A. Kano, K. Moriyama, T. Yamano, I. Nakamura, N. Shimada and A. Maruyama, J. Controlled Release, 2011, 149, 2–7 CrossRef CAS PubMed
- H. Hatakeyama, H. Akita and H. Harashima, Adv. Drug Delivery Rev., 2011, 63, 152–160 CrossRef CAS PubMed
- R. J. Christie, Y. Matsumoto, K. Miyata, T. Nomoto, S. Fukushima, K. Osada, J. Halnaut, F. Pittella, H. J. Kim and N. Nishiyama, ACS Nano, 2012, 6, 5174–5189 CrossRef CAS PubMed
- H. Fang, Y. Feng, J. Chen, H. Tian and X. Chen, Mater. Today Chem., 2019, 11, 269–282 CrossRef CAS
- H. Fang, H. Tian and X. Chen, J. Controlled Release, 2017, 259, e47 CrossRef
- Y. S. Malik, M. A. Sheikh, Z. Xing, Z. Guo, X. Zhu, H. Tian and X. Chen, Acta Biomater., 2018, 80, 144–153 CrossRef CAS PubMed
- Q. Lin, Y. Yang, Q. Hu, Z. Guo, T. Liu, J. Xu, J. Wu, T. B. Kirk, D. Ma and W. Xue, Acta Biomater., 2017, 49, 456–471 CrossRef CAS PubMed
- X. S. Sun, M.-S. Jang, Y. Fu, J. H. Lee, D. S. Lee, Y. Li and H. Y. Yang, Mater. Sci. Eng., C, 2020, 114, 111069 CrossRef CAS
- Y. Jeong, D. Lee, K. Choe, H. Ahn, P. Kim, J.-H. Park and Y.-C. Kim, J. Ind. Eng. Chem., 2017, 48, 79–87 CrossRef CAS
- G. Del Rio, M. A. Trejo Perez and C. A. Brizuela, Biosci. Rep., 2022, 42, BSR20221789 CrossRef
- D. Iudin, M. Vasilieva, E. Knyazeva, V. Korzhikov-Vlakh, E. Demyanova, A. Lavrentieva, Y. Skorik and E. Korzhikova-Vlakh, Int. J. Mol. Sci., 2022, 23, 2771 CrossRef CAS PubMed
- S. Liu, H. Yang, L. Wan, J. Cheng and X. Lu, Cancer Biother. Radiopharm., 2013, 28, 289–297 CrossRef CAS
- J. Li, Y. Zhou, Z. Su, X. Li, L. Zhang and S. Li, Molecules, 2024, 29, 1247 CrossRef CAS PubMed
- Z. Zhang, S. Tongchusak, Y. Mizukami, Y. J. Kang, T. Ioji, M. Touma, B. Reinhold, D. B. Keskin, E. L. Reinherz and T. Sasada, Biomaterials, 2011, 32, 3666–3678 CrossRef CAS
- M. Chiba, J. Hanes and R. Langer, Biomaterials, 1997, 18, 893–901 CrossRef CAS PubMed
- S. H. Kelly, L. S. Shores, N. L. Votaw and J. H. Collier, Adv. Drug Delivery Rev., 2017, 114, 3–18 CrossRef CAS PubMed
- L. Shi, J. Zhang, M. Zhao, S. Tang, X. Cheng, W. Zhang, W. Li, X. Liu, H. Peng and Q. Wang, Nanoscale, 2021, 13, 10748–10764 RSC
- S. Y. Fam, C. F. Chee, C. Y. Yong, K. L. Ho, A. R. Mariatulqabtiah and W. S. Tan, Nanomaterials, 2020, 10, 787 CrossRef CAS
-
P. Fabbri and M. Messori, in Modification of polymer properties, Elsevier, 2017, pp. 109–130 Search PubMed
- R. J. Nevagi, Z. G. Khalil, W. M. Hussein, J. Powell, M. R. Batzloff, R. J. Capon, M. F. Good, M. Skwarczynski and I. Toth, Acta Biomater., 2018, 80, 278–287 CrossRef CAS
- V. R. de la Rosa, J. Mater. Sci.: Mater. Med., 2014, 25, 1211–1225 CrossRef CAS PubMed
- S. N. Mahand, S. Aliakbarzadeh, A. Moghaddam, A. S. Moghaddam, B. Kruppke, M. Nasrollahzadeh and H. A. Khonakdar, Eur. Polym. J., 2022, 178, 111484 CrossRef
- O. Sedlacek and R. Hoogenboom, Adv. Ther., 2020, 3, 1900168 CrossRef
- N. Škalko-Basnet, Biol.: Targets Ther., 2014, 107–114 Search PubMed
- N. Alharbi, M. Skwarczynski and I. Toth, Biotechnol. Adv., 2022, 60, 108029 CrossRef CAS PubMed
- S. D'Souza, Adv. Pharm., 2014, 2014, 304757 Search PubMed
- T. J. Deming, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 283–297 CAS
- Z. Li, J. Huang and J. Wu, Biomater. Sci., 2021, 9, 574–589 RSC
- P. Zhang, H. Yang, W. Shen, W. Liu, L. Chen and C. Xiao, ACS Biomater. Sci. Eng., 2020, 6, 2167–2174 CrossRef CAS PubMed
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., 2003, 115, 4788–4791 CrossRef
- J. Li, L. Zhang, Y. Lin, H. Xiao, M. Zuo, D. Cheng and X. Shuai, RSC Adv., 2016, 6, 9160–9163 RSC
- W. Teng, F. Jia, H. Han, Z. Qin, Q. Jin and J. Ji, Polym. Chem., 2017, 8, 2490–2498 RSC
- Q. T. Hoang, D. Lee, D. G. Choi, Y.-C. Kim and M. S. Shim, J. Ind. Eng. Chem., 2021, 95, 101–108 CrossRef
- F. A. de Oliveira, L. J. Albuquerque, G. Delecourt, V. Bennevault, P. Guégan and F. C. Giacomelli, Curr. Gene Ther., 2021, 21, 431–451 CrossRef CAS
- F. Ofridam, M. Tarhini, N. Lebaz, É. Gagnière, D. Mangin and A. Elaissari, Polym. Adv. Technol., 2021, 32, 1455–1484 CrossRef CAS
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS
- C. Shen, J. Wang, X. Wu, J. Xu, J. Hu and A. Reheman, J. Drug Delivery Sci. Technol., 2023, 87, 104827 CrossRef CAS
- T. Volk, E. Jähde, H. Fortmeyer, K. Glüsenkamp and M. Rajewsky, Br. J. Cancer, 1993, 68, 492–500 CrossRef CAS
- B. Singh, S. Maharjan, T. Jiang, S.-K. Kang, Y.-J. Choi and C.-S. Cho, Biomaterials, 2015, 59, 144–159 CrossRef CAS
- T. Woraphatphadung, W. Sajomsang, P. Gonil, S. Saesoo and P. Opanasopit, Carbohydr. Polym., 2015, 121, 99–106 CrossRef CAS
- J. M. Knipe, F. Chen and N. A. Peppas, Biomacromolecules, 2015, 16, 962–972 CrossRef CAS PubMed
- C. R. Justus, L. Dong and L. V. Yang, Front. Physiol., 2013, 4, 354 Search PubMed
- Y. Zhong, S. Huang, C. Zheng, J. Huang, B. Li, S. Han, H. Xiao, Y. Wang and X. Shuai, Biomater. Sci., 2021, 9, 5218–5226 RSC
- W. Li, J. Cai, W. Zhou, X. Zhao, M. Wang, X. Zhou and L. Ren, Colloids Surf., B, 2023, 221, 112982 CrossRef CAS PubMed
-
S. Damodaran and K. L. Parkin, in Fennema's food chemistry, CRC Press, 2017, pp. 235–356 Search PubMed
- Z. Kang, Y. Peng, L. Zhou, Z. Li, T. Wang, Z. Zhang, Q. Liao, J. Gao, Y. Li and Y. Zhang, Mater. Chem. Front., 2018, 2, 1609–1617 RSC
- K. Zhang, X. Tang, J. Zhang, W. Lu, X. Lin, Y. Zhang, B. Tian, H. Yang and H. He, J. Controlled Release, 2014, 183, 77–86 CrossRef CAS
- G. Pasparakis and C. Tsitsilianis, Polymer, 2020, 211, 123146 CrossRef CAS
- F. Rypáček, J. Pytela, R. Kotva, V. Škarda and I. Cífková, Macromol. Symp., 2011, 123, 9–24 CrossRef
- C. Ding, L. Tong, J. Feng and J. Fu, Molecules, 2016, 21, 1715 CrossRef
- Y. Li, D. Maciel, J. Rodrigues, X. Shi and H. Tomás, Chem. Rev., 2015, 115, 8564–8608 CrossRef CAS
- G. A. Conlon and G. I. Murray, J. Pathol., 2019, 247, 629–640 CrossRef PubMed
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed
- Y. Sun, Y. Sha, G. Cui, F. Meng and Z. Zhong, Adv. Drug Delivery Rev., 2023, 192, 114624 CrossRef CAS
- M. Chourasia and S. Jain, J. Pharm. Pharm. Sci., 2003, 6, 33–66 CAS
- Y.-M. Go and D. P. Jones, Biochim. Biophys. Acta, Gen. Subj., 2008, 1780, 1273–1290 CrossRef CAS
- M. Gongora-Benitez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef CAS
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed
- D.-X. Ren, P.-C. Chen, P. Zheng and Z.-N. Xu, Colloids Surf., A, 2019, 577, 412–420 CrossRef CAS
- Z. Hu, G. Wang, R. Zhang, L. Wang, J. Wang, J. Hu and A. Reheman, Colloids Surf., B, 2023, 224, 113232 CrossRef CAS PubMed
- B. Niu, K. Liao, Y. Zhou, T. Wen, G. Quan, X. Pan and C. Wu, Biomaterials, 2021, 277, 121110 CrossRef CAS PubMed
- M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935–2946 CrossRef CAS PubMed
- K. K. Jain, Drug Delivery Syst., 2020, 1–54 Search PubMed
- M. Stepanova, A. Nikiforov, T. Tennikova and E. Korzhikova-Vlakh, Pharmaceutics, 2023, 15, 2641 CrossRef CAS
- M. Byrne, R. Murphy, A. Kapetanakis, J. Ramsey, S. A. Cryan and A. Heise, Macromol. Rapid Commun., 2015, 36, 1862–1876 CrossRef CAS
- M. A. Subhan, F. Parveen, N. Filipczak, S. S. K. Yalamarty and V. P. Torchilin, J. Pers. Med., 2023, 13, 389 CrossRef PubMed
- J. L.-S. Au, B. Z. Yeung, M. G. Wientjes, Z. Lu and M. G. Wientjes, Adv. Drug Delivery Rev., 2016, 97, 280–301 CrossRef CAS PubMed
- K. N. Sill, B. Sullivan, A. Carie and J. E. Semple, Biomacromolecules, 2017, 18, 1874–1884 CrossRef CAS PubMed
- K. T. Savjani, A. K. Gajjar and J. K. Savjani, Int. Scholarly Res. Not., 2012, 2012, 195727 Search PubMed
- P. Botella and E. Rivero-Buceta, J. Controlled Release, 2017, 247, 28–54 CrossRef CAS PubMed
- M. Plaza-Oliver, M. J. Santander-Ortega and M. V. Lozano, Drug Delivery Transl. Res., 2021, 11, 471–497 CrossRef CAS
- C. Li, L. Yuan, X. Zhang, A. Zhang, Y. Pan, Y. Wang, W. Qu, H. Hao, S. A. Algharib and D. Chen, J. Controlled Release, 2022, 352, 540–555 CrossRef CAS PubMed
- M. Azman, A. H. Sabri, Q. K. Anjani, M. F. Mustaffa and K. A. Hamid, Pharmaceutics, 2022, 15, 975 CrossRef CAS
- A. Abdel-Moneim and H. Ramadan, Drug Dev. Res., 2022, 83, 301–316 CrossRef CAS
- G. Chen, W. Kang, W. Li, S. Chen and Y. Gao, Theranostics, 2022, 12, 1419 CrossRef CAS PubMed
- S.-J. Cao, Z.-Q. Lv, S. Guo, G.-P. Jiang and H.-L. Liu, J. Drug Delivery Sci. Technol., 2021, 61, 102124 CrossRef CAS
- A. Gonella, S. Grizot, F. Liu, A. López Noriega and J. Richard, Expert Opin. Drug Delivery, 2022, 19, 927–944 CrossRef CAS
- W. Li, J. Tang, D. Lee, T. R. Tice, S. P. Schwendeman and M. R. Prausnitz, Nat. Rev. Mater., 2022, 7, 406–420 CrossRef
- B. Tyler, D. Gullotti, A. Mangraviti, T. Utsuki and H. Brem, Adv. Drug Delivery Rev., 2016, 107, 163–175 CrossRef CAS
- H. Liu, X. Shan, J. Yu, X. Li and L. Hu, Curr. Pharm. Biotechnol., 2020, 21, 180–193 CAS
- R. Gaudana, H. K. Ananthula, A. Parenky and A. K. Mitra, AAPS J., 2010, 12, 348–360 CrossRef CAS
- B. C. Ilochonwu, A. Urtti, W. E. Hennink and T. Vermonden, J. Controlled Release, 2020, 326, 419–441 CrossRef CAS
- B. Grassiri, Y. Zambito and A. Bernkop-Schnürch, Adv. Colloid Interface Sci., 2021, 288, 102342 CrossRef CAS PubMed
- N. T. T. Nhàn, D. E. Maidana and K. H. Yamada, Cells, 2023, 12, 1071 CrossRef
- R. Ruan, M. Chen, L. Zou, P. Wei, J. Liu, W. Ding and L. Wen, Ther. Delivery, 2016, 7, 89–100 CrossRef CAS PubMed
- H. Li, Y. Shi, X. Ding, C. Zhen, G. Lin, F. Wang, B. Tang and X. Li, Int. J. Biol. Macromol., 2024, 133452 CrossRef CAS
- E. Dahlén, N. Veitonmäki and P. Norlén, Ther. Adv. Vaccines Immunother., 2018, 6, 3–17 CrossRef PubMed
- S. Volpi, U. Cancelli, M. Neri and R. Corradini, Pharmaceutics, 2020, 14, 14 CrossRef PubMed
- R. Kumar, D. S. Dkhar, R. Kumari, Divya, S. Mahapatra, A. Srivastava, V. K. Dubey and P. Chandra, Biotechnol. Bioeng., 2022, 119, 3022–3043 CrossRef CAS PubMed
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS PubMed
- M. Mettlen, P.-H. Chen, S. Srinivasan, G. Danuser and S. L. Schmid, Annu. Rev. Biochem., 2018, 87, 871–896 CrossRef CAS
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2020, 20, 101–124 CrossRef PubMed
- I. Szabó, M. A. Yousef, D. Soltész, C. Bató, G. Mező and Z. Bánóczi, Pharmaceutics, 2022, 14, 907 CrossRef
- M. V. Chiarpotti, G. S. Longo and M. G. Del Pópolo, Colloids Surf., B, 2021, 197, 111373 CrossRef CAS PubMed
- Z. Zhao, Y. Wang, J. Han, K. Wang, D. Yang, Y. Yang, Q. Du, Y. Song and X. Yin, Int. J. Nanomed., 2014, 5849–5862 CrossRef
- P. Lajoie and I. R. Nabi, Int. Rev. Cell Mol. Biol., 2010, 282, 135–163 CAS
- A. Filippini and A. D'Alessio, Biomolecules, 2020, 10, 1218 CrossRef CAS PubMed
- L. B. Vong, N.-T. Trinh and Y. Nagasaki, J. Controlled Release, 2020, 326, 140–149 CrossRef CAS
- R. Seyfi, F. A. Kahaki, T. Ebrahimi, S. Montazersaheb, S. Eyvazi, V. Babaeipour and V. Tarhriz, Int. J. Pept. Res. Ther., 2020, 26, 1451–1463 CrossRef CAS
- J. Lei, L. Sun, S. Huang, C. Zhu, P. Li, J. He, V. Mackey, D. H. Coy and Q. He, Am. J. Transl. Res., 2019, 11, 3919 CAS
- K. Agam and D. K. Arya, Int. J. Pharm. Prof. Res., 2023, 14, 49–72 Search PubMed
-
M. H. Rigatto, D. R. Falci and A. P. Zavascki, Polymyxin Antibiotics: From Laboratory Bench to Bedside, 2019, pp. 197–218 Search PubMed
- X. Ma, Z. Liu, I. Ilyas, P. J. Little, D. Kamato, A. Sahebka, Z. Chen, S. Luo, X. Zheng and J. Weng, Int. J. Biol. Sci., 2021, 17, 2050 CrossRef CAS
- T. Rana, T. Behl, A. Sehgal, S. Singh, N. Sharma, A. Abdeen, S. F. Ibrahim, V. Mani, M. S. Iqbal and S. Bhatia, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2022, 114, 110478 CrossRef CAS
- L. Pinheiro and C. Faustino, Curr. Alzheimer Res., 2019, 16, 418–452 CrossRef CAS
- L. Chen, L. Zhang, R. Zhao, J. Shen, Y. Wang, J. Zhu, H. Fang, N. Liu, C. Wang and T. Wei, Nano Today, 2023, 50, 101834 CrossRef CAS
- M. Jiang, L. Zhao, X. Cui, X. Wu, Y. Zhang, X. Guan, J. Ma and W. Zhang, J. Adv. Res., 2022, 35, 49–60 CrossRef CAS PubMed
- Q. Cao, H. Fang and H. Tian, Biomaterials, 2024, 310, 122628 CrossRef CAS
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |