
Effectively enhancing ion diffusion in superconcentrated ionic liquid electrolytes using co-solvent additives†
Jhonatan
Soto Puelles
ac,
Luke A.
O'Dell
 bc,
M. C. Dilusha
Cooray
ac,
Maria
Forsyth
ac and
Fangfang
Chen
*ac
bc,
M. C. Dilusha
Cooray
ac,
Maria
Forsyth
ac and
Fangfang
Chen
*ac
aInstitute for Frontier Materials, Deakin University, 221 Burwood Highway, Burwood, Victoria 3125, Australia. E-mail: fangfang.chen@deakin.edu.au
bInstitute for Frontier Materials, Deakin University, Waurn Ponds, Victoria 3216, Australia
cFuture Battery Industries Cooperative Research Centre (FBICRC), Building 220, Brand Drive, Curtin University, Bentley, WA 6102, Australia
First published on 21st March 2025
Abstract
The incorporation of high salt concentrations in ionic liquid (IL) electrolytes, forming superconcentrated ionic liquids, has been shown to improve Li-ion transference numbers and enhance cycling stability against lithium metal anodes. However, this benefit comes at the cost of significantly increased viscosity and reduced ionic conductivity due to the formation of large ion aggregates. To optimize conductivity further, a co-solvent can be introduced at an optimal concentration to enhance ion transport while preserving superior interfacial stability. The effectiveness of this approach depends on the solvent as it affects ion diffusion to varying degrees. This computational study examines how co-solvents can effectively enhance metal ion diffusion in superconcentrated ionic liquids by comparing two widely used organic solvents. We found that the key lies in their ability to effectively participate in Li solvation shells, disrupting the large Li-anion aggregates. Our results show that anion exchange in a Li(anion)x(solvent)y hybrid solvation shell occurs more rapidly than in a Li (anion)z solvation shell, facilitating Li diffusion through a structural diffusion mechanism. A co-solvent with a high donor number exhibits a stronger affinity for lithium ions, which is identified as a crucial factor in enhancing ion diffusion. This work provides valuable insights to guide the design of superconcentrated ionic liquid electrolytes for lithium–metal battery development.
Introduction
The development of future high-energy-density metal batteries relies heavily on compatible electrolytes. Room temperature ionic liquids (RILs) show great promise due to their negligible vapour pressure, good electrochemical and thermal stability, and ability to support stable cycling of alkali metal anodes.1 Typically, increasing alkali salt concentration in IL solutions leads to higher viscosity and lower ionic conductivity. However, superconcentrated ionic liquids, despite their high salt concentrations, have demonstrated enhanced alkali metal cation transport numbers and improved charge/discharge cycling stability.2–7 For example, a Li transference number exceeding 0.3 has been reported in N-propyl-N-methylpyrrolidinium bis(fluorosulfonyl)imide (C3mpyrFSI) IL with 50 mol% LiFSI8 or NaFSI,9 along with improved electrochemical performance.Chen and coworkers have conducted computational investigations to provide further insight into these electrolytes.8–10 As the salt concentration increases, large anion–alkali ion complexes (also known as aggregates) form and gradually interconnect, creating an extended network. The aggregates lead to a looser solvation shell of alkali metal ions, driving a structural Li (Na) diffusion mechanism, where Li or Na moves through anion exchange in the first solvation shell. This exchange occurs more frequently in larger ion aggregates as the salt concentration increases, leading to a smaller reduction or even an enhancement in Li or Na diffusion. In contrast, the diffusion of IL cations and anions slows down significantly,9,10 thereby increasing the metal ion transport numbers. In addition, the interfacial chemistry of superconcentrated ILs is also dominated by alkali–anion aggregates, unlike their low-concentration counterparts, which is more likely dominated by cations. This leads to an increase in inorganic products in the solid electrolyte interphase (SEI), such as LiF and Li2O, which are considered beneficial for stabilizing cycling.6,11–13
The primary drawbacks of superconcentrated ILs are the high cost of salts and the high viscosity, which negatively impact battery performance. These issues include reduced ionic conductivity and capacity,14 unstable injection and preconditioning,15 difficulty operating under extreme conditions like fast charging or cold environments, and poor wettability with the separator and high-loading cathodes.16 Therefore, future IL designs should aim to reduce the high viscosity in superconcentrated ILs or lower the salt content while preserving the interfacial feature provided by high salt concentrations to maintain the good electrochemical performance. This can be achieved through the design of new ILs, novel salts, hybrid IL systems17 with co-solvents or other additives, or exploring locally concentrated ionic liquids.18
Our previous research showed that adding 20 wt% dimethoxyethane (DME) to C3mpyrFSI improved ionic conductivity at a 50 mol% LiFSI concentration.19 The resulting electrolyte was practically non-flammable, presented enhanced electrochemical stability against high-voltage NMC cathodes,20 and had low polysulfide solubility.21 However, not all co-solvent additives are effective, necessitating further investigation into their effects on both bulk phase and interfacial properties.
In this work, we explored the bulk phase effect, elucidating how co-solvents effectively enhance Li diffusion in superconcentrated ILs through comparing two solvents: DME and ethylene carbonate (EC). The two solvents differ in both Li affinities and dielectric constants. Li affinity can be evaluated using the donor number (DN), and a higher DN is associated with a stronger interaction with Li. DME has a higher DN (20) than EC (16.4) (Table S2†). The dielectric constant is also a key electrolyte property. Solvents with high dielectric constants reduce electrostatic interactions between oppositely charged ions, promoting salt dissociation, which is essential for achieving good ionic conductivity. EC has a much higher dielectric constant than DME (89 against 7.2). Through comparing EC and DME in a superconcentrated ionic liquid, we emphasize that a high affinity of the solvent is more effective for enhancing ion diffusion in superconcentrated ionic liquid electrolytes.
Here, the superconcentrated ionic liquid being investigated is 3.2 mol kg−1 LiFSI (about 50 mol%) in trimethylpropylammonium bis(fluorosulfonyl)imide (N1113FSI) IL, which has been recently studied experimentally.22 We replaced 20 wt% of N1113FSI with EC or DME as suggested in a previous research study.19 This gives approximately 53 mol% salt relative to the IL. For comparison, two lower co-solvent concentrations of 5 wt% and 10 wt% are also considered.
Results and discussion
The effect of co-solvents on the electrolyte bulk phase structure was investigated first. In the 50 mol% LiFSI in N1113FSI (100IL), the formation of LiFSI ion aggregates was observed. Fig. 1a illustrates the formation of these aggregates through the bridging coordination of FSI between multiple Li cations. Fig. 1b displays a snapshot of Li cations from a molecular dynamics (MD) simulation. The Li cations in the same aggregate are connected by green lines, revealing a large aggregate network in 100IL. A short movie in the ESI† shows that these aggregates are stable and long-lasting, contributing to the high viscosity of the electrolyte. | ||
| Fig. 1 (a) Simulation snapshot of LiFSI aggregation in N1113FSI with 50 mol% LiFSI salt. Li cations (green spheres) are bonded together by FSI bridging coordination. (b) A snapshot to demonstrate the extended Li–FSI network, where only green Li cations are presented. Those Li in the same aggregates were connected through green lines, representing bridging through FSI. | ||
Adding 20 wt% EC or DME (20EC or 20DME) to the IL enhances the diffusion of all ions, as indicated by the mean square displacement (MSD) calculations in Fig. 2a–c. This enhancement is more pronounced for 20DME. The observed effect on ion diffusion is attributed to changes in ion coordination. Clearly, these co-solvent molecules coordinate with Li cations, as suggested by a prominent peak in Li-EC and Li-DME radial distribution function (RDF) profiles (Fig. 2d). The Li-EC (DME) coordination consequently reduces the number of FSI coordinated with Li, as evidenced by the decreased Li–FSI coordination number (CN) in Fig. 2e, which changes from 4.6 in 100IL to 4.0 and 3.3 in 20EC or 20DME, respectively. The co-coordination of EC or DME, therefore, interrupts Li–Li associations by breaking Li–FSI coordination.
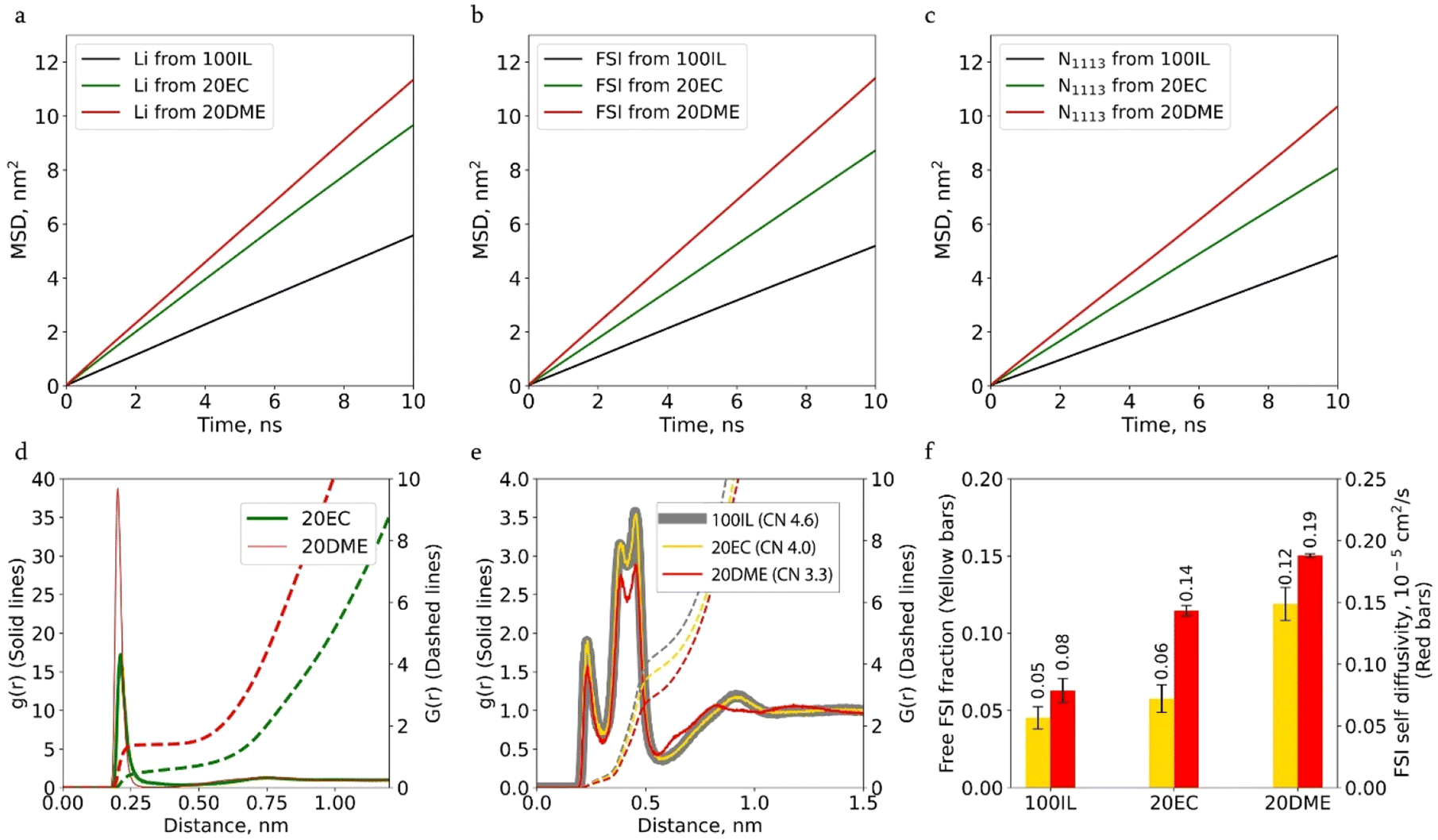 | ||
| Fig. 2 (a–c) MSD functions for Li, FSI and N1113 for 100IL, 20EC and 20DME systems. (d) RDFs between Li and the oxygen from the carbonyl group in EC (green line) and between Li and both oxygens from DME (red line), along with the coordination number profiles (dashed lines). (e) RDF between Li and the nitrogen atom from FSI in different electrolyte systems (solid lines), and the coordination number profiles (dashed lines). Between the brackets is the coordination number, which is calculated by reading the integrals at the cut-off distance of the solvation shells (the minimum near 0.6 nm). (f) Fraction of FSI that is not coordinated with Li, denominated “free” FSI (yellow bars) and FSI self-diffusivity (red bars). | ||
Replacing FSI with co-solvents in the Li-solvation shell results in an increase in ‘free FSI’, i.e. the non-Li coordinated FSI. As shown in Fig. 2f, this fraction increases from 5% in 100 IL to 6% in 20EC and 12% in 20DME. The same figure shows that the increase in the free FSI fraction is also accompanied by an increase in FSI and Li diffusivities. The higher fraction of free FSI in 20DME is also consistent with the lower Li–FSI coordination number in Fig. 2e, which is due to the more effective Li-DME coordination. DME has a higher donor number and a stronger affinity for Li than EC. According to DFT calculations, the binding energy of Li-DME (−2.65 eV) is larger than that of Li-EC (−2.07 eV) (Fig. S2†). MD simulation results suggest that almost all DME participates in Li-coordination. The fraction of Li ions in hybrid coordination structures remains higher in the presence of DME compared to that with EC (Fig. 3a), even at reduced co-solvent concentrations of 5 wt% and 10 wt%. In contrast, approximately 25% of EC molecules do not participate in Li coordination, regardless of the three EC concentrations examined (as shown in Fig. S3a†).
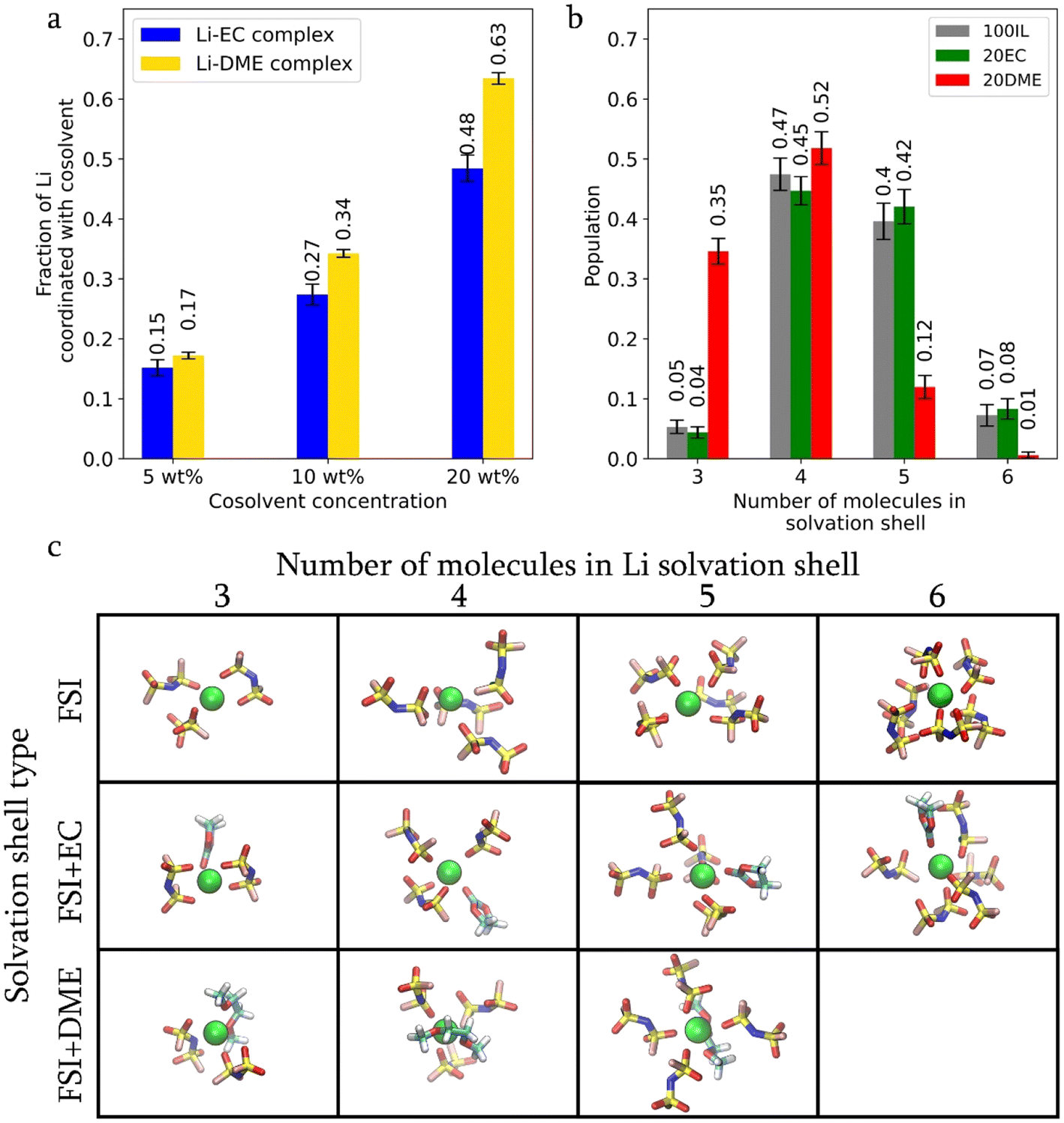 | ||
| Fig. 3 (a) Fraction of Li coordinated with EC and DME (blue and yellow bars, respectively) at different co-solvent concentrations. (b) Distribution of Li cations based on the number of molecules that form their solvation shell for the IL-in-salt system (100IL), EC system (20EC) and DME system (20DME). (c) Simulation snapshots of Li with different sizes and types of solvation shells. | ||
The size of the Li solvation shell (Fig. 3b and c) also varies. Smaller Li solvation shells, comprising three to four solvent molecules, dominate in the DME system, whereas four to five solvent molecules dominate in 20EC and 100IL cases. This is due to a decrease in the coordinated FSI when DME wraps Li with two oxygen atoms and occupies more solvation space (Fig. 3c). EC only coordinates Li through one carbonate oxygen atom, allowing more FSI to be present in the Li solvation shell. From the above results, it can be seen that DME is more effective in interrupting the formation of large Li-FSI aggregates through better Li solvation. It is worth noting that the interactions between lithium cations and various organic solvents can be detected experimentally using techniques such as Heteronuclear Overhauser Effect Spectroscopy (HOESY), a nuclear magnetic resonance (NMR) technique that measures the 1H–7Li cross-relaxation rates. Our previous research has shown that this rate is sensitive to short-range H–Li distances (<4 Å).23 However, such measurements are non-trivial and require careful analysis.24
In addition, we compared the simulated diffusivities of the electrolyte components with diffusion NMR results in 100IL, 20EC, and 20 DME in Fig. 4. Simulation results are in very good agreement with experimental results. It is clear that both EC and DME enhance the diffusion of all ions, with the effect being more pronounced for DME at higher concentrations. MD simulations qualitatively reproduce the trends in diffusivity changes across different electrolytes, consistent with experimental findings. It should be noted that classic MD normally underestimates diffusivities compared to experimental results due to the simplified treatment of the ion polarizable effect in a non-polarisable force field.25 However, in this case, the errors are within an acceptable range, from 8% to 75% (Table S1†), well below one order of magnitude. Despite these discrepancies, correctly predicting the relative diffusivity differences across various electrolytes supports the validity of the force field used in this study.
 | ||
| Fig. 4 Experimental and predicted diffusivities (black markers and yellow bars, respectively) taken at 80 °C for Li+, FSI−, N1113+ and co-solvents (a to d). | ||
To understand the effect of co-solvent on Li diffusion, the Li–FSI ion pair lifetimes and cage lifetimes were studied, following the method proposed by Zhang and coworkers.26 It is observed that the co-solvents decrease the average Li–FSI ion pair and cage lifetimes. The longest lifetime is for 100IL and the shortest lifetime is for 20DME (Fig. 5a). Thus, the structural diffusion of Li occurs more frequently in 20DME, leading to an increase in Li self-diffusivity.
 | ||
| Fig. 5 (a) Li–FSI ion pair lifetime and Li ion cage lifetime (blue and yellow bars, respectively) for the 100IL, 20EC and 20DME systems. The solid curve with markers and error bars represents the calculated Li self-diffusivity. (b) and (c) Li–FSI ion pair lifetime and Li ion cage lifetime calculated separately for Li in hybrid solvation shells and full FSI shells for the 20EC and 20DME systems. (d and e) Simulation snapshots to show the trajectory of a Li in a hybrid solvation shell (FSI + DME) and a full FSI solvation shell in a time window of 1 ns. (f) Simulation snapshot of Li in full FSI shells (green) and hybrid (FSI + DME) solvation shells (red). The fastest Li are highlighted in gray. | ||
If Li–co-solvent complexes are the main contributors to the enhanced Li diffusion, then Li in hybrid solvation shells (co-solvent + FSI) should move faster than Li in full FSI solvation shells. We prove this by comparing the ion pair lifetime, cage lifetime (Fig. 5b and c) and Li MSD (Fig. S4†) in different types of solvent shells. The analysis confirms the shorter ion pair and ion cage lifetimes for Li in hybrid shells than in the full FSI shells. The MSD results also indicate the higher diffusion of Li in hybrid shells (Fig. S4†). Two simulation snapshots in Fig. 5d and e display the moving trajectories of a Li in a DME-containing hybrid solvation shell compared to a Li in a full FSI solvation shell for 800 ps, where the former shows a longer Li travel distance.
Lastly, the fastest Li ions selected based on their MSD values are highlighted in grey in an MD snapshot (Fig. 5f). In the same snapshot, Li ions in hybrid shells and FSI shells are colored in red and green, respectively. Clearly, most of the fastest Li ions are present in hybrid shells.
We also analyzed the percentage of Li in small clusters detached from the main aggregate networks. This percentage is higher in the DME system with 22% of Li compared to 2% in the EC system (Fig. S5†) and no isolated clusters were observed in 100IL, indicating the more effective role of DME in interrupting large ion aggregates. Therefore, although the higher dielectric constant of EC was believed to help weaken ion association and was used in Li-ion batteries, in the superconcentrated ionic liquid system, the DME with a high donor number works better in disrupting Li–anion aggregates through stronger Li coordination, therefore more effectively breaking large ion-aggregates and improving overall conductivity.
Lastly, an experimental study of the effect of co-solvents on electrolyte bulk properties was conducted. Table 1 compares the ionic conductivity and Li transference numbers of three electrolyte systems. At both 50 °C and 80 °C, the addition of co-solvent significantly increases conductivity. The DME system shows the most notable enhancement by 2.6 times at 50 °C and 1.7 times at 80 °C compared to 100IL. While the Li transference number decreases slightly with the addition of co-solvents, the change remains minimal. It is 0.389 in 100IL and decreases to 0.371 in EC and 0.33 in the DME system. Therefore, adding a co-solvent can effectively increase conductivity without significantly affecting the Li transference number, making this an effective method for improving bulk phase electrolyte performance.
| Electrolyte system | Conductivity/×10−2 S cm−1 | Transference number | |
|---|---|---|---|
| 50 °C | 80 °C | 50 °C | |
| 3.2 mol kg−1 LiFSI in N1113FSI (100IL) | 2.43 | 6.22 | 0.387 ± 0.09 |
3.2 mol kg−1 LiFSI in N1113FSI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EC (80IL:20EC) :EC (80IL:20EC) |
5.67 | 9.1 | 0.371 ± 0.1 |
| 3.2 mol kg−1 LiFSI in N1113FSI:DME (80IL:20DME) |
6.2 | 10.5 | 0.33 ± 0.07 |
Conclusions
This research investigates the role of co-solvents in improving ion transport in superconcentrated ionic liquids. Two co-solvent molecules of EC and DME have been compared. It is found that the stronger affinity between the co-solvent and Li is critical and promotes the formation of more Li-solvent–FSI hybrid solvation shells, which enhance Li diffusion and reduce the large Li–FSI aggregates. DME with a higher donor number than EC works better in this role although the latter has a higher dielectric constant. This understanding provides guidance in choosing co-solvent molecules as additives to effectively enhance ion diffusion in superconcentrated ionic liquid electrolytes.Computational details
For the simulations, three electrolytes were considered. The 100IL system was an equimolar mixture of LiFSI and N1113FSI. The 20EC and 20DME systems consisted of 53 mol% LiFSI in N1113FSI with 20 wt% co-solvent. Table S3† shows the composition of the system. The composition was chosen based on a previous study with LiFSI in C3mpyrFSI, showing that the addition of 20 w% DME enhances Li anode stability in battery cycling.20 Initially, all molecules were randomly placed in a 7 nm cubic box using Packmol.27 The chemical structures of the studied species are shown in Fig. S1.†The simulations were carried out in Gromacs 2022.28 The force field parameters of Li, FSI and N1113 were taken from the CL&P forcefield,20,29,30 while EC and DME were described by the OPLS forcefield.31 A timestep of 1 fs was chosen along with the leapfrog integrator. H bonds were constrained using a Lincs algorithm. Long-range electrostatic interactions were implemented using an Ewald summation with a Fourier grid of 0.16. Short range electrostatic and van der Waals interactions were defined within a cutoff distance of 1.2 nm.
Temperature and pressure were controlled using a Nose–Hoover thermostat and a Parrinello–Rahman barostat, respectively. Three-dimensional periodic boundary conditions were considered, and a dispersion correction was used for approximating long van der Waals interactions. To correct ion–ion and dipole–dipole interactions the total charge of the ions was scaled down to ±0.7.25
First a steepest descent minimization was done on the initial systems converging to a minimum force of 1000.0 kJ/mol nm−1. Next, to improve the system dynamics, the temperature was increased from 393 K to 500 K in the first 3 ns. Then, the system was slowly quenched from 500 K to 393 K in the next 4 ns. The system temperature was kept at 393 K for an additional 3 ns. The final simulation was run in an NpT ensemble with 393 K and 1 bar for 50 ns.
For the post-simulation analysis, via Python coding, the composition and number of molecules in the solvation shell around each Li cation were analyzed for the last 5000 trajectory frames. Two approaches were used to identify Li–FSI networks. In the first one, using the data from the solvation shell analysis, a Li network was constructed by tracking FSI shared by multiple Li. In the second method, cluster analysis on Li was performed applying the DBSAN algorithm considering a maximum distance of 0.76 nm between points. Such a distance was extracted from Li–Li RDF, shown in Fig. S6.†
The Li–FSI pair and FSI cage lifetimes were calculated based on the method of Zhang and coworkers.26 First, the ion pair (IP) and ion cage (IC) functions were calculated in a time window of 1000 ps. A Li–FSI pair is formed between Li and the closest FSI anion in the solvation shell, and it is broken when another FSI coordinates Li within a closer distance. An FSI cage around Li is formed by its FSI solvation shell and it is broken when an FSI leaves or a new FSI enters. Following this, a time correlation function (TCF) (C(t)) was calculated on the corresponding IP and IC functions (eqn (1)). In eqn (1), the angle brackets represent the average and h refers to the IP or IC function. The estimated ion pair and ion cage TCFs were averaged for all the Li, obtaining a smooth decreasing curve. The resultant lifetimes were obtained by numeric integration of these curves from 0 to infinite using Simpson's rule. The algorithm was implemented with Python 3.0.
 | (1) |
Additionally, the apparent Li transference number was calculated with eqn (2), where n and D are the number of molecules and self-diffusivity, respectively.19
 | (2) |
The post-simulation analysis was carried out using Python, including MDAnalysis,32 and Gromacs post-simulation tools. The simulation snapshots were produced using VMD 1.9.4.33
Electrolyte preparation
N-Trimethyl-N-propylammonium bis(fluorosulfonyl)imide (N1113FSI, 99.5%) and lithium bis-(fluorosulfonyl)imide (LiFSI, 99.5%) were received from Solvionic (France). Dimethoxylethane (DME) and ethylene carbonate (EC) were purchased from Merck Natural Sciences Australia. All chemicals were used without further purification or drying. Appropriate amounts of LiFSI, N1113FSI and the cosovents were weighed accurately according to the different compositions used and mixed overnight at room temperature to prepare the electrolytes. All the materials were stored, and electrolytes were prepared in an argon-filled glovebox. The electrolyte compositions chosen for this work are shown in Table 2.| LiFSI-salt concentration | N1113FSI wt% | Cosolvent | Cosolvent wt% |
|---|---|---|---|
| 3.2 mol kg−1 | 100 | — | — |
| 95 | DME | 05 | |
| 80 | DME | 20 | |
| 95 | EC | 05 | |
| 80 | EC | 20 |
NMR measurements
1H, 19F and 7Li pulsed field gradient (PFG) NMR measurements were carried out to determine the diffusion coefficients of the IL cation and additive, IL anion and lithium cation, respectively. Samples were packed into 5 mm NMR tubes inside an argon glove box. A Bruker Avance III 11.7 T NMR spectrometer was used with the double stimulated echo PFG pulse sequence to eliminate convection effects. The gradient pulse length was set to 5–10 ms, the diffusion time to 50–100 ms and the maximum gradient strength was around 50 G cm−1. Sample temperatures were 80 °C with a minimum of 10 min equilibration time. Attenuation curves were fitted using the Stejskal–Tanner equation in the TopSpin software.Transference number measurement
Lithium metal foil (Gelon Energy Corp. China) of 100 μm thickness was cleaned and cut into 16 mm diameter discs. These discs were attached to two of the 0.5 mm spacers of 2032 type coin cell compartments to be used for a lithium symmetrical cell. For the separator, 16 mm diameter Celgard 3501 was dried overnight under vacuum at 40 °C and used. Different electrolyte systems were used in a volume of 80 μl for each cell. A cell-crimping machine (Hohsen, Japan) was used to crimp the coin cells. All cells were assembled in an argon filled glovebox, where H2O and O2 levels were maintained below 1 ppm. The cells were rested at 50 °C for 24 h to allow for separator wetting. The assembled cells were tested using a VMP3 (Biologic) battery cycler. These Li/Li cells were then used to measure the transference number of each electrolyte. The tLi+ value was calculated following the traditional Bruce–Vincent method.1 The equation for transference number calculation is as follows: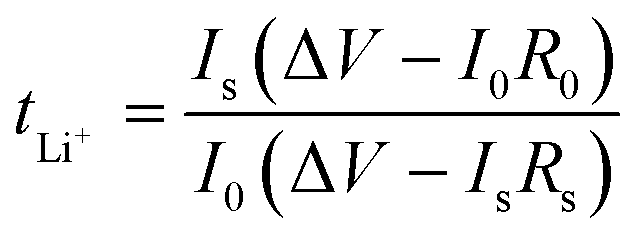
Here, ΔV is the applied voltage across the cell (10 mV), I0 and Is are initial and steady-state currents, and R0 and Rs are initial and steady state surface impedances of the measured cell.
Ionic conductivity measurements
Electrochemical impedance spectroscopy (EIS) was performed on an MTZ-35 impedance analyser to obtain the ohmic impedance (R) of the electrolytes. A dip cell with two platinum rods sheathed in glass was filled with an electrolyte. The ionic conductivity was calculated using the formula: σ = G/R, where s is ionic conductivity and G is the cell constant of the dip cell. The G value was determined by testing the calibration solution, 0.01 M KCl, at 25 °C. The conductivity was measured by applying frequency from 1 MHz to 1 Hz and an amplitude of 0.01 V. The ionic conductivity was measured at 50 and 80 °C temperatures.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Future Battery Industries Cooperative Research Centre (FBICRC) grant. The computational research was undertaken with the assistance of resources from the National Computational Infrastructure (NCI Australia) through the National Computational Merit Allocation Scheme (NCMAS), supported by the Australian Government.References
- Z. Lei, B. Chen, Y.-M. Koo and D. R. MacFarlane, Chem. Rev., 2017, 117, 6633–6635 Search PubMed.
- S. A. M. Noor, N. C. Su, L. T. Khoon, N. S. Mohamed, A. Ahmad, M. Z. A. Yahya, H. Zhu, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2017, 247, 983–993 CAS.
- H. Yoon, P. C. Howlett, A. S. Best, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2013, 160, A1629 CAS.
- G. M. Girard, M. Hilder, H. Zhu, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. MacFarlane and P. C. Howlett, Phys. Chem. Chem. Phys., 2015, 17, 8706–8713 CAS.
- T. Pathirana, R. Kerr, M. Forsyth and P. C. Howlett, Sustainable Energy Fuels, 2021, 5, 4141–4152 CAS.
- T. Pathirana, D. A. Rakov, F. Chen, M. Forsyth, R. Kerr and P. C. Howlett, ACS Appl. Energy Mater., 2021, 4, 6399–6407 CAS.
- X. Gao, F. Wu, A. Mariani and S. Passerini, ChemSusChem, 2019, 12, 4185–4193 CAS.
- F. Chen and M. Forsyth, Phys. Chem. Chem. Phys., 2016, 18, 19336–19344 CAS.
- F. Chen, P. Howlett and M. Forsyth, J. Phys. Chem. C, 2018, 122, 105–114 CAS.
- M. Forsyth, H. Yoon, F. Chen, H. Zhu, D. R. MacFarlane, M. Armand and P. C. Howlett, J. Phys. Chem. C, 2016, 120, 4276–4286 CrossRef CAS.
- G. M. Hobold, C. Wang, K. Steinberg, Y. Li and B. M. Gallant, Nat. Energy, 2024, 9, 580–591 CrossRef CAS.
- H. Zeng, K. Yu, J. Li, M. Yuan, J. Wang, Q. Wang, A. Lai, Y. Jiang, X. Yan, G. Zhang, H. Xu, J. Wang, W. Huang, C. Wang, Y. Deng and S. S. Chi, ACS Nano, 2024, 18, 1969–1981 CrossRef CAS PubMed.
- D. Rakov, M. Hasanpoor, A. Baskin, J. W. Lawson, F. Chen, P. V. Cherepanov, A. N. Simonov, P. C. Howlett and M. Forsyth, Chem. Mater., 2022, 34, 165–177 CrossRef CAS.
- J. Tong, S. Wu, N. von Solms, X. Liang, F. Huo, Q. Zhou, H. He and S. Zhang, Front. Chem., 2020, 7, 945 CrossRef PubMed.
- N. Yao, L. Yu, Z. H. Fu, X. Shen, T. Z. Hou, X. Liu, Y. C. Gao, R. Zhang, C. Z. Zhao and X. Chen, Angew. Chem., Int. Ed., 2023, 62, e202305331 CrossRef CAS PubMed.
- O. Borodin, J. Self, K. A. Persson, C. Wang and K. Xu, Joule, 2020, 4, 69–100 CrossRef CAS.
- Y. Lu, Q. Cao, W. Zhang, T. Zeng, Y. Ou, S. Yan, H. Liu, X. Song, H. Zhou, W. Hou, P. Zhou, N. Hu, Q. Feng, Y. Li and K. Liu, Nat. Energy, 2024, 10, 191–204 Search PubMed.
- X. Liu, A. Mariani, H. Adenusi and S. Passerini, Angew. Chem., Int. Ed., 2023, 62, e202219318 CrossRef CAS PubMed.
- U. Pal, F. Chen, D. Gyabang, T. Pathirana, B. Roy, R. Kerr, D. R. MacFarlane, M. Armand, P. C. Howlett and M. Forsyth, J. Mater. Chem. A, 2020, 8, 18826–18839 RSC.
- U. Pal, D. Rakov, B. Lu, B. Sayahpour, F. Chen, B. Roy, D. R. MacFarlane, M. Armand, P. C. Howlett, Y. S. Meng and M. Forsyth, Energy Environ. Sci., 2022, 15, 1907–1919 RSC.
- U. Pal, G. M. A. Girard, L. A. O'Dell, B. Roy, X. Wang, M. Armand, D. R. MacFarlane, P. C. Howlett and M. Forsyth, J. Phys. Chem. C, 2018, 122, 14373–14382 CrossRef CAS.
- U. Pal, Y. Wu, K. Periyapperuma, F. Chen, A. Ziebell, S. Fantini, P. Howlett, M. Forsyth and R. Kerr, J. Phys. Chem. C, 2024, 128, 13694–13701 CrossRef CAS.
- M.-A. Sani, P.-A. Martin, R. Yunis, F. Chen, M. Forsyth, M. Deschamps and L. A. O'Dell, J. Phys. Chem. Lett., 2018, 9, 1007–1011 CrossRef CAS PubMed.
- P.-A. Martin, E. Salager, M. Forsyth, L. A. O'Dell and M. Deschamps, Phys. Chem. Chem. Phys., 2018, 20, 13357–13364 RSC.
- K. Cui, A. Yethiraj and J. R. Schmidt, J. Phys. Chem. B, 2019, 123, 9222–9229 CrossRef CAS PubMed.
- Y. Zhang and E. J. Maginn, J. Phys. Chem. Lett., 2015, 6, 700–705 CrossRef CAS PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- J. N. Canongia Lopes, J. Deschamps and A. A. H. Pádua, J. Phys. Chem. B, 2004, 108, 2038–2047 Search PubMed.
- D. A. Rakov, F. Chen, S. A. Ferdousi, H. Li, T. Pathirana, A. N. Simonov, P. C. Howlett, R. Atkin and M. Forsyth, Nat. Mater., 2020, 19, 1096–1101 CrossRef CAS PubMed.
- B. Doherty, X. Zhong, S. Gathiaka, B. Li and O. Acevedo, J. Chem. Theory Comput., 2017, 13, 6131–6145 CrossRef CAS PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05234j |
| This journal is © The Royal Society of Chemistry 2025 |