
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnique selectivity of rare-earth metal ambiphilic carbenes towards organic molecules and novel reactivity patterns with isonitriles†
Fuxiang Chaia,
Weikang Wua,
Thayalan Rajeshkumard,
Zeming Huang b,
Qingbing Yuana,
Yun Weia,
Laurent Maron*d and
Shaowu Wang*bc
b,
Qingbing Yuana,
Yun Weia,
Laurent Maron*d and
Shaowu Wang*bc
aKey Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecule-Based Materials, College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, Anhui, P. R. China. E-mail: swwang@mail.ahnu.edu.cn
bAnhui Laboratory of Clean Catalytic Engineering, Anhui Laboratory of Functional Coordinated Complexes for Materials Chemistry and Application, College of Chemical and Environmental Engineering, Anhui Polytechnic University, Wuhu 241000, Anhui, P. R. China. E-mail: wsw@ahpu.edu.cn
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China
dLPCNO, CNRS & INSA, Université Paul Sabatier, 135 Avenue de Rangueil, 31077 Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 23rd May 2025
Abstract
Studies on the chemistry of highly active rare-earth (RE) metal ambiphilic carbenes face challenges due to the lack of appropriate model molecular platforms because the corresponding ambiphilic carbenes have empty p-orbitals. Here, the synthesis of novel multi-chelated amidate rare-earth metal pincer complexes bearing ambiphilic carbenes is realized by using indol-2-yl-based ligands. The ambiphilic carbene (indol-2-yl carbon) of these complexes is extremely active and shows unique selective reactivity towards various organic molecules including carbodiimides, imine, diphenyldiazomethane, aluminum alkyls, 9-BBN and isonitriles, which demonstrate a range of unprecedented reactivity patterns such as formal [2 + 2] cyclometallation, [3 + 3] annulation, and aza-[4 + 1] annulation, while unprecedented aza-[4 + 1 + 1] annulation products were isolated from the reactions of the rare-earth metal complexes bearing electrophilic carbenes and strongly polarized metallacyclopropanes with 2,6-dimethylphenylisonitrile. Such reactivities differ sharply from those of common transition-metal complexes with the corresponding substrates. The electronic and steric effects of the ligands and the effects of central metal ions on the reactivity patterns were investigated both computationally and experimentally.
Introduction
The design and synthesis of new ligands for catalysis is among the ever-green research areas in modern chemistry.1 Particularly, organometallic complexes containing pincer ligands are well known for their exceptional thermal stability, distinctive reactivity and wide applicability in catalysis.2 The reactivity of pincer complexes can be manipulated through modulations of substituents on the donor groups or introduction of diverse connections between the central backbone and donor groups.3 Thus, various symmetric pincer ligands bearing different backbones such as phenyl,4 pyridyl,5 pyrrolyl,6 carbazolyl,7 carbon or heteroatom-bridged diaryl8 and all-carbon carbolong pincer ligands9 have been developed. Our group has been engaged in the development of pincer-type rare-earth metal complexes featuring non-symmetric indol-2-yl-based ligands with development of new catalysis for the transformations of various organic molecules.10 On the other hand, amidate metal complexes demonstrated distinctive reactivity and catalytic performance,11 with applications in olefin polymerization,12 copolymerization,13 ester polymerization,14 and alkene hydroamination.15 Despite these advances, multi-chelated amidate pincer ligated complexes bearing both ambiphilic carbenes and nucleophilic alkyls have so far been unreported and their reactivity remains elusive.Singlet carbenes in late transition-metal complexes are known to have lone pair electrons donated to the metal center and an empty p-orbital to accept d-electrons from the metal to form π-backbonding (Scheme 1).16 A recent study indicated that the p-orbital receptor properties of these carbenes can be effectively harnessed.17 Given that rare-earth metal ions have no d-electron to form π-backbonding, rare-earth metal complexes bearing such electrophilic carbenes might possess unique reactivity for application in catalysis and activation of small molecules. However, the synthesis of rare-earth metal electrophilic carbene complexes has just been realized until very recently, and 1,1-alkyl or 1,1-H(D) migratory reactions, and unique reactivity patterns towards pyridine derivatives were found with these complexes (Scheme 1).11g,18,19 The chemistry of rare-earth metal complexes bearing an electrophilic carbene is far less developed in comparison with those of late transition metals.
 | ||
| Scheme 1 Bonding and reactivity of the RE complexes bearing indol-2-yl ligands. | ||
N-Heterocyclic carbenes (NHCs) are one of the most widely used ligands and catalysts for their strong electron-donating and Lewis basic properties, as well as stable bonding with metals, and easy adjustment.20 Cyclic (alkyl)(amino)carbenes (CAACs) are receiving extensive attention due to their ambiphilic character.21 CAACs have also been applied in various fields such as transition-metal catalysis,22 and light emitting materials as CAAC–Cu complexes.23 Additionally, isolated CAACs have high thermal stability that can be utilized in stabilizing paramagnetic substances and activating small molecules (such as CO,24 P4,25 H2 (ref. 26) and NH3 (ref. 26)). Following these pioneering studies of the CAACs, other types of ambiphilic carbenes have been developed and applied in catalysis or activation of small molecules.27 However, these carbenes have only been used as supporting ligands in rare-earth metal chemistry, and studies on the chemistry of rare-earth metal complexes bearing both ambiphilic carbene and nucleophilic alkyl have to overcome some challenges including the lack of a model molecular platform due to their high activity and instability.
The reactions of isonitriles with transition metal compounds have been extensively studied with findings of cyclization reactions, migration insertion and multi-component reactions.28 The insertion of isonitriles into the metal-element σ bond will result in the formation of complexes having η1- or η2-imidoyl (iminoacyl) (Scheme 2);29 or the η1 or η2 formyl complexes;30 or the bridged µ2-CNR ligands. The insertion of isonitriles into the multiple M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C carbon bond will form metallacyclopropanes (η2 ketenimine) or κ1 ketenimine;31,32 insertion of isonitrile into the Sc–C bond of aryl rare-earth metal complexes generates imidoyls, these complexes participate in further transformation reactions and then form an unusual indoline scandium with another isonitrile molecule.32,33 These findings enrich the methods for constructing chemical bonds in organic synthesis. However, the reactivity pattern of rare-earth metal complexes having both ambiphilic carbenes and nucleophilic carbons with isonitriles remains elusive.
C carbon bond will form metallacyclopropanes (η2 ketenimine) or κ1 ketenimine;31,32 insertion of isonitrile into the Sc–C bond of aryl rare-earth metal complexes generates imidoyls, these complexes participate in further transformation reactions and then form an unusual indoline scandium with another isonitrile molecule.32,33 These findings enrich the methods for constructing chemical bonds in organic synthesis. However, the reactivity pattern of rare-earth metal complexes having both ambiphilic carbenes and nucleophilic carbons with isonitriles remains elusive.
 | ||
| Scheme 2 Reaction modes of isonitrile with metal complexes. | ||
Herein, we report the design, synthesis and reactivity study of homodinuclear pincer-type rare-earth metal alkyl complexes featuring novel multi-chelated 1-amidate-3-imino functionalized indol-2-yl ligands, and reactivity study of rare-earth metal complexes bearing electrophilic carbon and strongly polarized metallacyclopropanes with 2,6-dimethylphenylisonitrile. The RE–Ccarbene bonds of these complexes exhibit unique selective reactivity towards electrophiles and nucleophiles, which could be associated with the ambiphilic carbene nature of the indol-2-yl carbon as revealed by DFT calculations and experimental results.
Results and discussion
Synthesis and characterization of multi-chelated amidate homodinuclear pincer rare-earth metal complexes bearing ambiphilic carbene and nucleophilic alkyl
The reactions of the ligand precursors H2LAd [LAd = 1-[(N-2,6-iPr2C6H3)NCOCH2]-3-(C10H15NCH)–C8H4N, Ad = adamantyl, C10H15]; H2LDipp [LDipp = 1-[(N-2,6-iPr2C6H3)NCOCH2]-3-(2,6-iPr2C6H3NCH)–C8H4N; Dipp = 2,6-iPr2C6H3]; H2LDMA [LDMA = 1-[(N,N-Me2)NCOCH]-3-(2,6-iPr2C6H3NCH)–C8H4N] (see the ESI†) with RE(CH2SiMe)3(THF)n (RE = Gd, Dy, Y, Er, Yb, Lu) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio at room temperature in tetrahydrofuran (THF) or toluene for 3 h produced the corresponding novel multi-chelated amidate homodinuclear pincer rare-earth metal complexes [η1:η1:(µ-η1:η2)-LAdRE(CH2SiMe3)(THF)]2 [RE = Gd(1a), Dy(1b), Y(1c), Er(1d), Yb(1e), Lu(1f)]; [η1:η1:(µ-η1:η2)-LDippRE(CH2SiMe3)(THF)]2 [RE = Y(2a), Er(2b), Yb(2c), Lu(2d)]; [η1:η1:(µ-η1:η2)-LDMARE(CH2SiMe3)(THF)]2 [RE = Y(3a), Er(3b), Yb(3c), Lu(3d)] in high yields (Scheme 3, Fig. 1 and S52–S65 in the ESI†). All complexes were fully characterized by spectroscopic methods and elemental analyses, and their structures were confirmed by single-crystal X-ray diffraction.
:1 stoichiometric ratio at room temperature in tetrahydrofuran (THF) or toluene for 3 h produced the corresponding novel multi-chelated amidate homodinuclear pincer rare-earth metal complexes [η1:η1:(µ-η1:η2)-LAdRE(CH2SiMe3)(THF)]2 [RE = Gd(1a), Dy(1b), Y(1c), Er(1d), Yb(1e), Lu(1f)]; [η1:η1:(µ-η1:η2)-LDippRE(CH2SiMe3)(THF)]2 [RE = Y(2a), Er(2b), Yb(2c), Lu(2d)]; [η1:η1:(µ-η1:η2)-LDMARE(CH2SiMe3)(THF)]2 [RE = Y(3a), Er(3b), Yb(3c), Lu(3d)] in high yields (Scheme 3, Fig. 1 and S52–S65 in the ESI†). All complexes were fully characterized by spectroscopic methods and elemental analyses, and their structures were confirmed by single-crystal X-ray diffraction.
 | ||
| Scheme 3 Synthesis of the homodinuclear rare-earth metal alkyl complexes 1–3. | ||
 | ||
| Fig. 1 Representative diagram for the complexes 1 (left), 2 (middle) and 3 (right) with thermal ellipsoids set at 30% probability level. All the hydrogen atoms are omitted, the diisopropylphenyl (Dipp) group and adamantyl (Ad) group are drawn in wireframe style for clarity. Selected bond lengths (Å) and selected bond angles (deg) can be read in the ESI.† | ||
The diamagnetic complexes 1c, 1f, 2a, 2d, 3a, and 3d were further characterized by NMR spectroscopy. In the 1H NMR spectrum of complex 1c, the signal centred at −0.23 ppm could be assigned to the resonances of the methylene protons of the CH2SiMe3 group, and is comparable with the resonance at −0.34 ppm (s, 2H) attributed to the protons of the methylene of the CH2SiMe3 in the 2-amidate indolyl yttrium complex.4b Unlike complex 1c, the methylene protons of the Y–CH2SiMe3 in complex 2a gave doublet resonances centred at −0.16 ppm (2JY–H = 8.0 Hz). Complex 3a exhibits more pronounced coupling of yttrium ion nucleus with the protons of the CH2SiMe3, resulting in doublet resonances at −0.06 ppm (2JY–H = 15.0 Hz) and −0.16 ppm (2JY–H = 15.0 Hz). In addition, the corresponding methylene protons of the lutetium complexes were found to resonate at −0.37 ppm, −0.66 ppm (1f); −0.22 ppm, −0.37 ppm (2d); and −0.24 ppm, −0.32 ppm (3d), respectively, suggesting that the substituents of the ligands and coordinated THF might prevent the Lu–CCH2SiMe3 bonds from free rotation, thus showing different proton resonances for the alkyls (–CH2SiMe3). The signal centred at 200.4 ppm in the 13C{1H} NMR spectrum of 1c is attributed to the resonance of the carbon of the indol-2-yl coupled with the yttrium ion nucleus with 1JY–C = 40.0 Hz, which can be compared with previously reported data.10a,11g,18,19 The signal centred at 201.8 ppm assigned to the indol-2-yl carbon (C2-ind) in the 13C{1H} spectrum of 2a is coupled to the yttrium ion nucleus (1JY–C = 50.0 Hz), moving downfield as compared to that in 1c. The signal centred at 201.3 ppm (1JY–C = 50.0 Hz) in the 13C{1H} spectrum of 3a can be compared with the literature data (indol-2-yl carbon of L*RE(CH2SiMe3)2(THF)x (L* = 1-(2-C4H7OCH2)-3-(2,6-iPr2C6H3NCH)C8H4N) at 201.8 ppm).18 While the corresponding carbon resonances in the lutetium complexes 1f, 2d, 3d shift even more downfield to 206.7, 209.2, and 210.9 ppm, respectively, these resonances for the indol-2-yl carbon atoms fall in the range of singlet metal carbenes.34a
X-ray analysis reveals that the amidates in complexes 1, 2 and 3 adopt the µ-η1:η2 mode with the oxygen atoms of the amidates bridging two rare-earth metal ions; the amidates bond with one of the metal ions in η2 mode, and the imino-functionalized indol-2-yl motifs bond with the central metal ions in a manner of η1:η1. Thus, the bonding modes of the ligands with the central metal ions can be described as η1:η1:(µ-η1:η2) on the basis of analysis of the structural bond lengths (Tables S1–S3 in the ESI†). The indol-2-yl carbon atoms, imino, amidate groups and central metals are nearly coplanar in these rare-earth metal complexes. Obviously, two alkyl groups and two coordinated THF are present in the trans-form in complexes 1 and 2. In the formation of complexes 1 and 2, a sequence of indol-2-yl C–H activation and deprotonation is involved, while in the formation of complexes 3, the activation of the indol-2-yl C–H and α C–H of the amide is involved to generate an amino-functionalized enolate anion bonding with the central metal ions in µ-η1:η2 modes. This difference highlights the influence of substituents on the reactivity of the ligand precursors with RE(CH2SiMe)3(THF)2. It is found that the imino groups did not insert into the RE–Calkyl bonds, which was different from previous results.14 It's probably due to the stability of the multiple-chelated amidate pincer structure, the steric hindrance of the adamantyl or the aryl groups and electron isomerization resulting in the enamido motifs (Scheme 3). Unlike complexes 1 and 2, the central metal ions in complexes 3 have six-coordination-number due to the absence of THF coordination, and the two alkyl groups present in the trans-form.
The bond lengths of RE1–C2-ind, RE1–O1, RE1–N1, RE1–N3 and RE1–O1A in complexes 1a–1e, 2a–2e and 3a–3d decrease with the decrease of ionic radius from Gd3+ to Lu3+ and Y3+ to Lu3+ (see Tables S1–S3 in the ESI†), which is consistent with the lanthanide contraction.34b The distances of RE1–C2-ind [2.388(5) Å for 1c; 2.348(7) Å for 1d; 2.351(6) Å for 1e; 2.320(6) Å for 1f] in complexes 1c–1f are shorter than those of RE1–C2-ind [2.396(5) Å for 2a; 2.367(3) Å for 2b; 2.357(3) Å for 2c; 2.335(3) Å for 2d] of complexes 2a–2d, probably due to the steric effects of the substituents on the ligands. The RE–C2-ind distances in 1 and 2 are shorter than those found in L*RE(CH2SiMe3)2(THF)x [(RE = Y, x = 1, 2.452(6) Å; RE = Er, x = 0, 2.387(4) Å; RE = Dy, x = 0, 2.396(3) Å; {L* = 1-(2-C4H7OCH2)-3-(2,6-iPr2C6H3NCH)C8H4N}],18 which can be attributed to coordination number and steric differences.
The O–C–N angles of the amidates range from 115.0(3)° to 117.3(6)° in complexes 1 and from 116.3(3)° to 116.8(4)° in complexes 2. These data are very close to those (115.3(9)° to 119.3(11)°) in the amidate-functionalized N-heterocyclic carbene (NHC) rare-earth metal amido complexes.34c The N–C–O angles of the amidates (in the range of 107.7(4)° to 109.7(11)°) in complexes 3 are significantly smaller than the corresponding angles in complexes 1 and 2, which can be attributed to the bonding differences between the amidate in 1 or 2 and the amino-functionalized enolate in 3.
DFT calculations show that the indol-2-yl carbon atoms have a natural charge of −0.253 in 1c and −0.260 in 2a, which are substantially smaller than those of carbon atoms of the alkyl (–CH2SiMe3) (natural charge of −1.711 in 1c and −1.716 in 2a) in the corresponding complexes (Table 1). The natural charge of the indol-2-yl carbon atoms in complexes 1 and 2 can be compared with that of −0.228 found in 8a, which bears strongly polarized carboryne-based metallacyclopropanes.19b This suggests some difference in reactivity and selectivity between the RE–C2-ind and RE–CCH2SiMe3 bonds. The natural charges of the indol-2-yl carbon atoms in the complexes 1c, 2a and 8a are also smaller than those of the sp2 carbanion (natural charge of −0.58) bonding with lutetium ion in the pentamethylcyclopentadienyl supported lutetacyclopropene, which displayed nucleophilic property according to the DFT calculations and experimental results.35 The natural charges of the indol-2-yl carbon atoms in complexes 1c, 2a and 8a are comparable to those in five-membered organometallic carbenes (ranging from −0.17 to −0.29 for different metals), which exhibit ambiphilic reactivity as indicated by the high values of protons and hydride affinities based on DFT calculations,36 while the resonances for the indol-2-yl carbon atoms (see above) of these complexes can be compared with the resonances (∼190–210 ppm) of the carbonyl carbon atoms of the ketones and aldehydes, which were generally accepted as electrophiles, demonstrating the electrophilic character of the indol-2-yl carbon atoms. Taken together, the indol-2-yl carbon atoms in these complexes can be described as ambiphilic carbon atoms.
| Atom label | Natural charges in 1c (Y) | Natural charges in 2a (Y) | Natural charges in 8a (Y)19b |
|---|---|---|---|
| Y1 | 2.054 | 2.164 | 2.007 |
| C1 | −0.253 | −0.260 | −0.228 |
| O1 | −0.867 | −0.901 | −0.622(imino N) |
| O1A | −0.867 | −0.901 | −0.819(carboryne C) |
| N2A | −0.662 | −0.681 | −0.816(carboryne C) |
| N3 | −0.654 | −0.667 | _ |
| O2 | −0.636 | −0.665 | _ |
| C34 or C36 | −1.711(C34 in 1c) | −1.716(C36 in 2a) | _ |
Selective reactivity of the RE–C2-ind bond towards organic compounds containing electrophilic carbon or polar N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) N double bond. Unusual [2 + 2] cyclometallation and [3 + 3] annulation
N double bond. Unusual [2 + 2] cyclometallation and [3 + 3] annulation
DFT calculations reveal that the natural charge of the indol-2-yl carbon of the complex 1c is −0.25 (Table 1) and the natural charges of the carbon atoms of the alkyls (–CH2SiMe3) connected to the metal center are around −1.71, implying that these different RE–C bonds (RE–Ccarbene and RE–CCH2SiMe3) may show different reactivities towards small organic molecules. To this end, the above-synthesized complexes 1 were subjected to reactions with a range of electrophiles (Scheme 4). Complexes 4a–4d (Fig. S66–S69 in the ESI†) were crystallized from different solvents when 0.5 equiv. of complexes 1 were treated with carbodiimides or an aldimine. To our surprise, the RE–Ccarbene bonds selectively reacted with these electrophiles to generate novel [2 + 2] cyclometallation complexes 4a–4d, in which the indol-2-yl carbon bonds with the central metal ions in η1 mode (RE–C2-ind bond lengths: 2.803(4) Å in 4a, 3.050(3) Å in 4b, 3.062(3) Å in 4c, 2.833(5) Å in 4d, Scheme 4) due to the π-electron moving towards indol-2-yl carbon atoms after the reaction. In contrast, the RE–CCH2SiMe3 bonds of the complexes 1, which have the more nucleophilic carbon atoms as demonstrated by DFT calculations, did not react with the corresponding substrates, but reacted with the α C–H bond of the amidate to produce a novel amido-functionalized enolate functionality chelated with the central metal ions in µ-η1:η2 mode (complexes 4a–4d in Scheme 4). These results are completely different from those of previous studies on the reactivities of rare-earth metal alkyls with carbodiimides and imine, which only produced the carbodiimides or imine insertion products with amidinate or amido being bonded with central metal ions in η2 or σ bond ways.37
 | ||
| Scheme 4 The reactions of complexes 1 with electrophiles. | ||
When complexes 1b and 1e were reacted with 4 equiv. of diphenyldiazomethane (Ph2CN2) at room temperature in THF for 0.5 h, complexes 4e and 4f were isolated (Fig. S70 and S71 in the ESI†). While treatment of 1b with 4 equiv. of diphenyldiazomethane in THF overnight or standing the THF solution of 4e overnight generated the product 4g (Fig. S72 in the ESI†) via a rare formal [3 + 3] annulation (Schemes 3, 5 and Fig. 2). The formation of complex 4g might involve a sequence of insertion of polar NN bonds of the Ph2CN2 into RE–Ccarbene and RE–Calkyl bonds to afford 4e, activation of the α C–H bond of the amidate by the anionic [PhCNNCH2SiMe3]− to produce the amido-functionalized enolate and ring closure with CN bond. The final step is accompanied by transformation of the amidate functionality into an imine functionalized alkoxide type motif.
 | ||
| Scheme 5 A tentative pathway to complex 4g. | ||
 | ||
| Fig. 2 Molecular structure of 4g (left), 5c (middle) and 5e (right) with the thermal ellipsoid at 30% probability level. All hydrogen atoms were omitted and the diisopropylphenyl (Dipp) group was drawn in wireframe style for clarity. Selected bond lengths (Å) and selected bond angles (deg) can be read in the ESI.† | ||
Selective reactivity of complexes 1 with Lewis acids
To further probe the selective reactivity of the RE–Ccarbene bonds of complexes 1 towards electrophiles, we then tested their reactions with Lewis acidic compounds such as aluminum alkyls and 9-BBN (9-borabicyclo[3.3.1]nonane). It is interesting to find that the RE–Ccarbene bonds instead of the RE–CCH2SiMe3 bonds, whose carbon has larger natural charge, of complex 1e selectively reacted with the aluminum alkyls AlMe3 and AlEt3 to generate novel complexes 5a and 5b (Scheme 6, Fig. S73 and S74 in the ESI†). These complexes bear indol-2-Alalkyl motifs with two sp3 C–H bonds being σ donated to the ytterbium center, which is reduced to ytterbium(II) from ytterbium(III). Such a reduction is believed to proceed via the homolysis of the Yb–CCH2SiMe3 bond11g,38 due to its lower redox couple (−1.15 E295/V) of Yb3+/Yb2+, which is supported by the identification of the coupling product Me3SiCH2–CH2SiMe3 (m/z = 174.126) from the reaction mixture. The reduction of ytterbium(III) to ytterbium(II) by the aluminum alkyls is occasionally proposed in the polymerization of olefins with mixed ytterbium complexes and aluminum alkyl catalytic systems to explain their poor catalytic activity compared with other rare-earth metal catalysts.11d,f,39 However, the proposed ytterbium(II) intermediate has seldom been isolated and characterized. It is surprising to find that either the AlMe3 or the AlEt3 exists on the same side to the ytterbium metal in the solid state of the complexes 5a and 5b. Furthermore, the distances of 2.718(8) Å in 5a, and 2.733(4) Å in 5b of the sp3 C–H interaction with the Yb metal are significantly shorter than that of 2.86 Å found in SmII–Me interaction in the bis(Me3Si-fluorene-AlMe3)Sm complex,40 indicating strong σ donation in complexes 5a and 5b. This reactivity pattern is different from the reactions of L′Ln(CH2C6H4NMe2-o)2 (L′ = (PhCH2)2NC(NC6H3iPr2-2,6)2) with AlMe3 or AlEt3 producing the methyl bridged rare-earth metal–aluminum mixed metal complexes41 or the ethylene and ethyne complexes.42 The Al–C2-ind bond length (2.071(9) Å in 5a, and 2.086(5) in 5b) is within the expected range compared with Al–Cfluorenyl (2.101(7) Å).40 The average YbII–C(CH2) bond length of 2.711 Å in 7-coordinate 5b is longer than that found in 6-coordinate YbIIAl2Et8(THF)2 (2.633(2) Å),43 which may be due to an electrostatic interaction.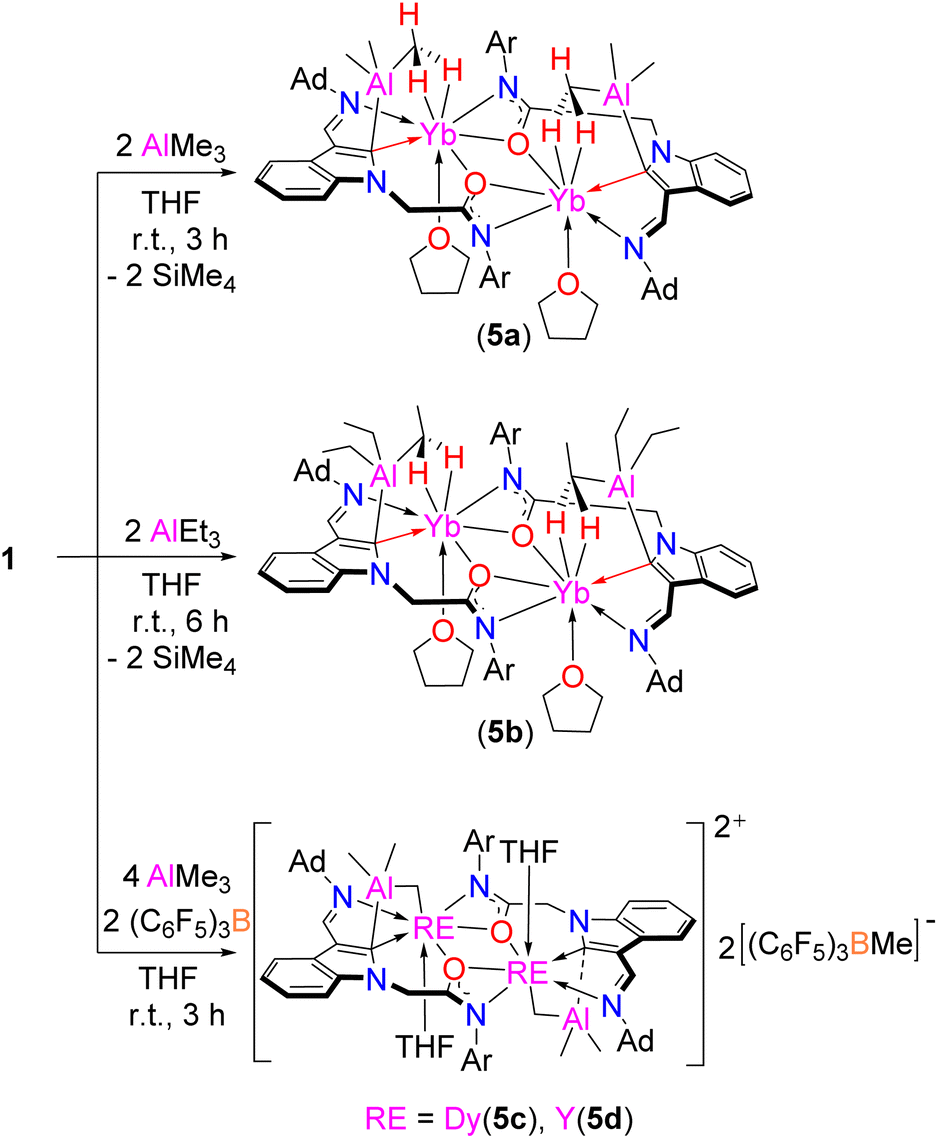 | ||
| Scheme 6 The reactions of complexes 1 with AlR3. | ||
When the rare-earth metal complexes 1b and 1c with the central metal having higher redox potential in comparison with Yb3+/Yb2+ were treated sequentially with AlMe3 and (C6F5)3B, the cationic rare-earth metal complexes 5c and 5d were isolated from a mixed solution of n-hexane and chlorobenzene (Scheme 5, Fig. S75 and S76 in the ESI†). In this process, the RE–Ccarbene bond selectively reacts with AlMe3 to deliver the indol-2-yl carbon connected aluminum intermediate, followed by activation of the C–H bond of the AlMe3 to generate the rare hetero-rare-earth metal–aluminum four-membered metallacycles 5c and 5d. The RE–C(CH2) bond lengths of 2.353(6) Å in 5c and 2.356(5) Å in 5d are comparable to the σ bond length of the Y–CCH2SiMe3 in 1c, and the Al–C(CH2) bond lengths of 1.811(6) Å in 5c and 1.809(5) Å in 5d are shorter than that of 1.986(19) Å in 5a. This reactivity pattern is different from those of the reactions of L′Ln(CH2C6H4NMe2-o)2 (L′ = (PhCH2)2NC(NC6H3iPr2-2,6)2) with AlMe3 producing the carbyne complex [(PhCH2)2NC(NC6H3iPr2-2,6)2]2Y2(µ2-Me)(AlMe3)2(µ4-CH) and alkyl abstraction product AlMe2(CH2C6H4NMe2-o) upon allowing the reaction mixture to stand for 7 days or running the reaction at 60 °C for 18 h.41 In the present case, one methyl group was transferred to (C6F5)3B to produce the anionic [(C6F5)3BMe]− part of the complexes, which are also different from the result of the reaction of [(C5Me5)La(AlMe4)2] with (C6F5)3B, which afforded a CH3/C6F5 exchange product [{[(C5Me5)La{(µ-Me)2AlMe(C6F5)}][Me2Al(C6F5)2]}2].44 The present results are also different from those of the reactions of aluminum alkyls with B(C6F5)3 in ether solvent producing the borate [Me2Al(THF)2]+[MeB(C6F5)3]−,45 indicating electronic and steric effects of the ligands on the reactivity patterns.
Encouraged by the above results, the reactivity of complexes 1 with Lewis acidic 9-BBN has been investigated. A rare nonsymmetric dinuclear yttrium complex 5e (Schemes 6 and 7, Fig. 2 and S77 in the ESI†) was obtained when complex 1c was treated with 1 equiv. of 9-BBN at room temperature in THF. X-ray analysis reveals that complex 5e is composed of a rare Y–C2–ind–B–H four-membered bora-metallacycle with the indol-2-yl carbon bonding to the metal in the η1 way. The formation of 5e might involve selective reaction of the Y–Ccarbene bond with 9-BBN, while the Y–CH2SiMe3 bond of which the carbon has larger natural charge remains intact. Complex 5e contains a unique multi-chelate amidate pincer ligand bearing a four-membered bora-metallacycle with the indol-2-yl carbon bonding to the metal centre in the η1 way in one part, and a multi-chelate amido-functionalized enolate pincer ligand in the other part (Schemes 7 and 8). The Y(2)–C(42)2-ind bond length (2.741(9) Å) is significantly longer than Y(1)–C(1)2-ind (2.381(10) Å). This suggests that the RE–C(42)2-ind bond might have transformed from the σ bond into the π bond (η1) way due to the electron-deficient bora functionality to make the π-electron of the ligand move towards the indol-2-yl carbon (Scheme 1). The Y–H bond length (2.3817(11) Å) was slightly long with those of the Sc–H (borate bridged) (Sc–H = 2.01(3) Å and 2.11(2) Å) when the ionic radii differences were taken into account.46 The reaction of complex 1e with 2 equiv. of 9-BBN in THF leads to the isolation of the central symmetric dinuclear complex 5f bearing a four-membered bora-metallacycle with the indol-2-yl carbon being η1 bonded (Yb–C2-ind bond length 2.650(7) Å) to the metal centres (Scheme 7 and Fig. S78 in the ESI†). The formation of 5f is believed to go through selective reaction of the Yb–C2-ind bond with 9-BBN to afford the 2-borate indolyl, and the Yb–CH2SiMe3 bond activates the α C–H of the amidate group to generate the multi-chelate amido-functionalized enolate 2-borate indol-2-yl ligated dinuclear complex. It is noted that while the four-membered bora-metallacycle has been proposed as an intermediate in the metathesis reactions of the RE–C bond with hydroborate or 9-BBN in the catalytic hydroboration of unsaturated compounds,47 to date, structural information about this kind of four-membered ring is rare.
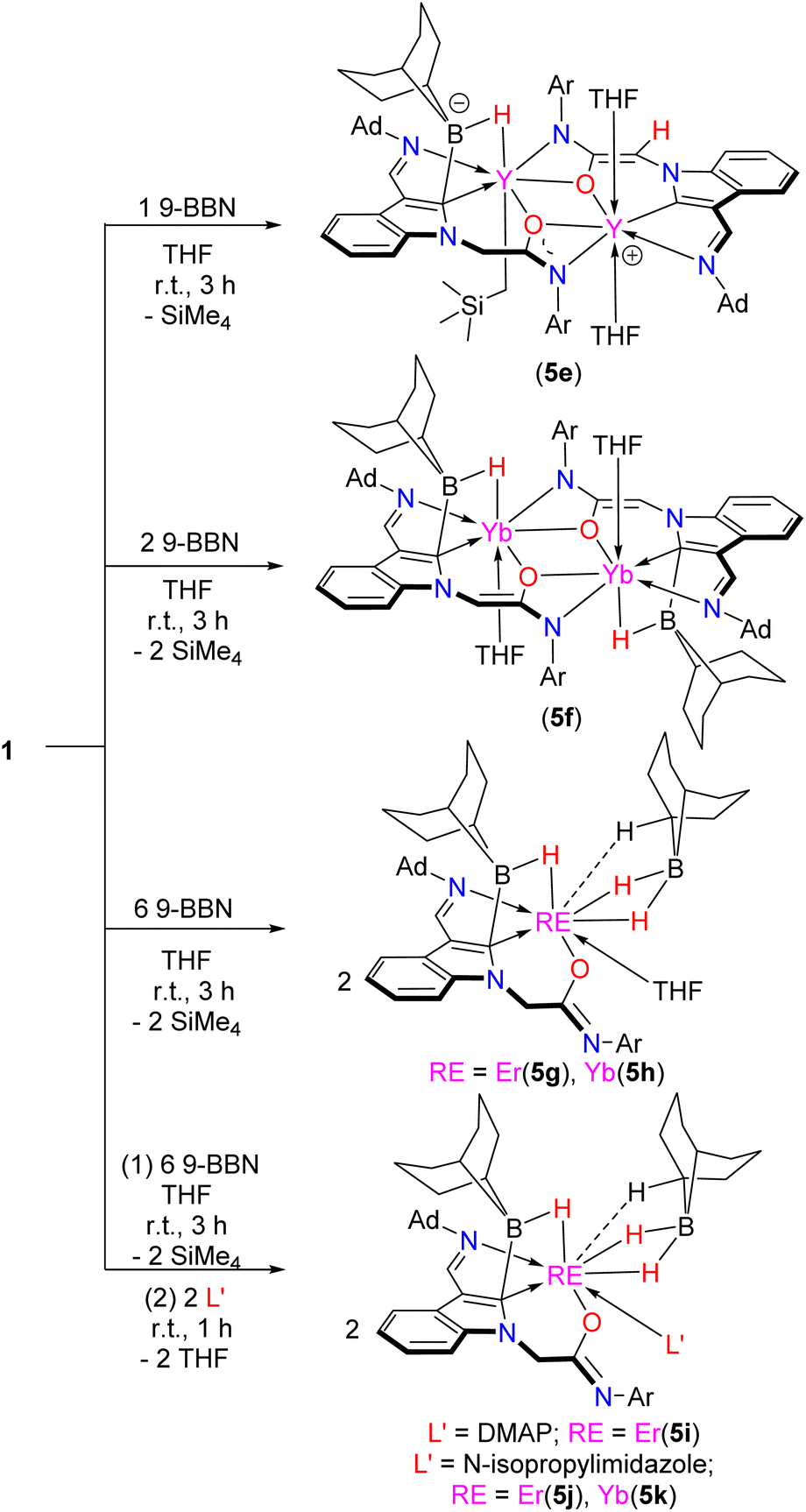 | ||
| Scheme 7 The reactions of complexes 1 with 9-BBN. | ||
 | ||
| Scheme 8 The formation process of complex 5e. | ||
Interestingly, when complexes 1 were treated with excess 9-BBN (6 equiv.) in THF, the THF coordinated mononuclear 2-borate indolyl supported and 9-BBN bridged rare-earth metal hydrides 5g and 5h were isolated (Scheme 7 and Fig. S79–S80 in the ESI†). Stepwise reactions of complexes 1 with excess 9-BBN in THF followed by treatment with donor molecules such as 4-N,N-dimethylaminopyridine (DMAP) or N-isopropylimidazole (IPIMD) provide DMAP- and IPIMD-coordinated complexes 5i–5k (Scheme 7 and Fig. S81–S83 in the ESI†). Similar to the formation of complexes 5e and 5f, the RE–Ccarbene bond selectively reacted with 9-BBN first, and then the RE–Calkyl bond reacted with another equivalent of 9-BBN to split the dinuclear complexes into mononuclear ones. An electrostatic C–H interaction (RE–H bond distances 1.976(13) Å to 2.439(18) Å) of the 9-BBN with the rare-earth metal center was found. It is interesting to find that the amidate transforms into the imino-functionalized alkoxide form. This prevents the α C–H of the original amidate from being activated by either the RE–CH2SiMe3 or the newly generated RE–H bonds to generate the amido-functinonalized enolate, which is different from the formation of 5e and 5f. The indol-2-yl carbon bonds with the central metal ion in the η1 way due to the electron-deficient bora functionality to make the π-electron of the ligand move towards the carbon. The RE–H bond distances found in 5g and 5h (Scheme 7) can be compared to those found in the divalent ytterbium and europium 9-BBN complexes, (THF)4Yb[(µ-H)2BC8H14]2, and (THF)4Eu[(µ-H)2BC8H14]2,48a the trivalent yttrium tetrahydroborate, (MeOCH2CH2C5H4)2Y[(µ-H)2BH2],48b and (C5Me5)2Y(µ-H)2BC8H14.48c It is noted that the α C–H activation has not been found in the formation of 5i–5k with the addition of 4-N,N-dimethylaminopyridine (DMAP) or N-isopropylimidazole (IPIMD), which is different from the previous reports.11g
Selective reactions of the complexes with isonitriles. Unique selectivity and ligand effects on the reactivity patterns
The above results of reactions of the synthesized complexes with electrophiles prompted us to investigate the reactivity of these complexes towards nucleophiles such as isonitriles. It is unexpectedly found that the tert-butylisonitrile (tBuNC, 2 equiv.) is selectively reacted with carbene carbon atoms (indol-2-yl carbon atoms) of the complexes 1 in THF at ambient temperature, complexes 6a–6d are generated in good yields, and the highly polarized RE–CCH2SiMe3 bonds remain intact (Scheme 9, Fig. 3 and Fig. S84–S87 in the ESI†). X-ray crystallography confirms that complexes 6a–6d contain the aza-metallacyclobutene motifs with the Y–C2-ind distance of 2.757(6) Å, C2-ind–Cimino bond length of 1.510(10) Å, and Y–N4 bond length of 2.459(6) Å. The N4–C38 distance of 1.385(10) Å reveals a CN double bond character. Complexes 6a–6d cannot undergo further insertion reaction or C–C coupling in the presence of an excess of tBuNC (>2 equiv.) under the reaction conditions. When other isonitriles such as tert-octylisonitile and cyclohexylisonitrile were used, the corresponding rare-earth metal complexes 6e and 6f (Scheme 9, Fig. S88 and S89 in the ESI†) containing the aza-metallacyclobutene moiety were also isolated, and the RE–CCH2SiMe3 bonds remained unreacted. This reactivity pattern is consistent with the electrophilic nature of the indol-2-yl carbon as revealed by DFT calculations (Table 1). However, aryl isonitriles seem incompatible in this reaction system in that the reaction of 2,6-dimethylphenylisonitrile (XylNC) with complex 1f gave an unidentifiable mixture.
 | ||
| Scheme 9 The reactions of complexes 1 with isonitriles. | ||
 | ||
| Fig. 3 Molecular structure of 6b (left), 7d (middle) and 9a (right) with the thermal ellipsoid at 30% probability level. All hydrogen atoms were omitted and the diisopropylphenyl (Dipp) group was drawn in wireframe style for clarity. Selected bond lengths (Å) and selected bond angles (deg) can be read in the ESI.† | ||
It is proposed that the formation of complexes 6 involves coordination of isonitrile to the electrophilic metal centre with the release of THF to afford the intermediate AdY1, which then undergoes 1,1-migratory insertion to the electrophilic indol-2-yl carbene carbon18,19a,c to produce the intermediate AdY2. The Y–C2-ind bond cleavage occurs with intramolecular redox to afford the intermediate AdY3, which then abstracts the proton of the α C–H of the amidate group to deliver the final product (Scheme S30 in the ESI†). This reactivity pattern is different from the known reaction of a free singlet carbene with an isonitrile to generate a ketenimine.49 To the best of our knowledge, the selectivity and reactivity found in the above reactions have so far been unreported.50 It is noteworthy that the Y–N4 bonds (2.459(6) Å) in 6b are significantly longer than those in the [(C5H5)Y(µ,η2-HCNCMe3)]2 dimer (2.325(4) Å),51a and in the Cp*2Zr(iminoacyl)CH3(Cp* = C10H15) monomer (2.368(2) Å)51b respectively, due to different coordination numbers and/or different central metal ions.
Next, taking the reaction of 1c with tBuNC to produce 6b as an example, DFT calculations were performed to investigate the possible reaction pathways and reasonable energy profiles (see the ESI† for details). The results showed that the total reaction was exergonic −42.4 kcal mol−1, and the reaction barriers were consistent with room temperature conditions. The formation of complex 6b might proceed via a stepwise process at each metal centre to accomplish the formation of the aza-metallacyclobutene moiety. After coordination of the tBuNC, the 1,1-tBuNC migratory insertion accompanied by Y–C2-ind cleavage leads to the reduction of isonitrile C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond to the imidoyl anion (intramolecular redox). This process is preferred to the 1,1-CH2SiMe3 migratory insertion with a formation of relatively stable intermediate AdB (−6.3 kcal mol−1) vs. AdB′ (8.4 kcal mol−1). The intermediate AdB transforms into the intermediate AdC (5.6 kcal mol−1), which then undergoes another 1,1-tBuNC migratory insertion accompanied by Y–C2-ind cleavage leading to reduction of isonitrile CN triple bond to generate the intermediate AdD (−12.9 kcal mol−1). The process of abstraction of the α C–H of the amidate group to form intermediate AdE (−27.0 kcal mol−1) is favourable to the cycloaddition of the imino nitrogen of the ligand to the ketenimine carbon. The AdE then abstracts another proton of the α C–H of the amidate group to deliver the final product 6b (−42.4 kcal mol−1) (Scheme 10).
N triple bond to the imidoyl anion (intramolecular redox). This process is preferred to the 1,1-CH2SiMe3 migratory insertion with a formation of relatively stable intermediate AdB (−6.3 kcal mol−1) vs. AdB′ (8.4 kcal mol−1). The intermediate AdB transforms into the intermediate AdC (5.6 kcal mol−1), which then undergoes another 1,1-tBuNC migratory insertion accompanied by Y–C2-ind cleavage leading to reduction of isonitrile CN triple bond to generate the intermediate AdD (−12.9 kcal mol−1). The process of abstraction of the α C–H of the amidate group to form intermediate AdE (−27.0 kcal mol−1) is favourable to the cycloaddition of the imino nitrogen of the ligand to the ketenimine carbon. The AdE then abstracts another proton of the α C–H of the amidate group to deliver the final product 6b (−42.4 kcal mol−1) (Scheme 10).
 | ||
| Scheme 10 Computed enthalpy profile for the formation of 6b from 1c. The enthalpy is given in kcal mol−1. | ||
To get more direct information of the substituent effects on the reactivity, we then investigated the reactivity of different complexes towards various isonitriles. It is surprising to find that when complexes 2 bearing the ligand with a diisopropylphenyl substituent instead of the adamantyl substituent in 1 were treated with different isonitriles (R–NC, R = tBu, tOctyl, Bn, Xyl) under the otherwise same conditions, novel [4 + 1] annulation products of 1-amido-functionalized enolate indolo-fused 2-amido-pyrrolyl complexes 7 (Scheme 11, Fig. 3, and S90–S95 in the ESI†) were isolated with moderate yields. In this process, the carbene carbon atoms (indol-2-yl carbon atoms) have displayed unexpected selective reactivity towards the nucleophilic isonitriles, which is completely different from that found in reactions of 1 with isonitriles. This suggests that the substituents on the ligand, which may lead to changes in both steric hindrance and electronic nature of complexes 1 and 2 (different natural charges of the central metal ion and the bonding atoms, see Table 1), have profound effects on the reaction pathways. The current results with isonitriles are also completely different from those in recently reported reactions of rare-earth metal monoalkyl complexes with isonitriles, which provided multi-substituted β-diketiminato or multi-substituted imidazolyl complexes.52 On the basis of the DFT calculation results, it is proposed that coordination of the isonitrile to complex 7a (as an example) gives the intermediate DippY1, which then undergoes 1,1-migratory insertion to generate DippY2. Isomerization of DippY2 gives the ketenimine49 intermediate DippY3. Then the amido nitrogen of the ligand attacks the central carbon of the ketenimine to finish the cyclization to form the [4 + 1] annulation products of indolo-fused 2-amido pyrrolyl fragments DippY4. The RE–CCH2SiMe3 bonds activate the α C–H bonds of the amidate to deliver the final 1-amido functionalized enolate chelate indolo-fused 2-amido-pyrrolyl ligated complexes (Schemes 12 and S31 in the ESI†).
 | ||
| Scheme 11 The reactions of complexes 2 with isonitriles. | ||
 | ||
| Scheme 12 Computed enthalpy profile for the formation of 7a from 2a. The enthalpy is given in kcal mol−1. | ||
It should be noted that when complexes 2 were directly treated with excess tert-octylisonitrile (tOctylNC) (4 equiv.), the C–C coupling or C–N coupling products through insertion of RNC to the RE–CCH2SiMe3 bonds or through insertion of RNC to the RE–N bonds as those reported in the literature were not obtained,51,53 while the tOctylNC coordinated complex 7c was isolated instead. Also of note is that the original bonding modes of the ligand change from η1:η1:(µ-η1:η2) to σ:η1:(µ-η1:η2) after the cyclization, that is, the newly generated amido-functionalized pyrrolyls bond with the central metal in σ:η1 (RE–C2-pyr: 2.669(13) Å to 2.793(13) Å) and the amido-functionalized enolates bond with the central metal in µ-η1:η2 ways in complexes 7a–7c. When the substituents of isonitriles are benzyl or xyl (7d–7f), the reactions are compatible with these substrates, which are different from that of reaction of 1 with the xyl substituted isonitrile. It is found that the CC double bonds of the newly formed amido-functionalized enolates in 7d–7f have bonding interaction with the central metal as indicated by the RE–C bond distances of 2.685(7) Å to 2.715(5) Å.
In order to further understand the reaction process, the formation of complex 7a from the reaction of complex 2a with tBuNC was calculated by DFT (Scheme 12, see the ESI† for details). It reveals that the replacement of THF by tBuNC, followed by a stepwise 1,1-migratory insertion on each metal centre to generate the intermediate DippD are parallel to those for the formation of 6b as discussed above. The transformation of DippD into the indolo-fused amido-functionalized pyrrolyl motif (DippE) is favoured over the transformation of DippD into aza-metallacyclobutene (DippE′) (4.0 kcal mol−1 vs. 4.6 kcal mol−1). The intermediate DippE then undergoes a stepwise C–H activation with the RE–CH2SiMe3 bond followed by coordination of THF to deliver the final products. These results suggest that the substituents on the ligands have influence on the electronic properties of the complexes and stability of the transition state and intermediates, thereby determining the reaction pathways of the complexes.
To prove the above results that ligands have profound influences on the reactivity patterns of rare-earth metal complexes with isonitriles, we then studied the reactivity of the rare-earth metal complexes bearing both electrophilic carbene and strongly polarized three-membered metallacycles with isonitrile. Treatment of the carboryne-based strongly polarized metallacyclopropane (κ2-L1)RE(η2-C2B10H10)(THF)3 [L1 = 1-(2-N-C5H10NCH2CH2)-3-(2,6-iPr2C6H3NCH)–C8H4N, RE = Y(8a), Er(8b), Lu(8c)]19b with 3 equiv. of XylNC afforded the unprecedented selective reaction complexes 9 bearing an α,β-di(2,6-dimethylphenyl)amido-γ-(N-2,6-diisopropylphenyl)carboline (or indolo-fused multi-functionalized pyridine)-based ligand (Scheme 13, Fig. 3 and S96–S98 in the ESI†). It is surprising to find that the highly strained and polarized metallacyclopropane moiety remains intact in the process, while the formation of the indolo-fused multi-functionalized pyridine moiety involves the coupling of the electrophilic carbene carbon and the nucleophilic nitrogen atom of the ligand (1-(2-C5H10NCH2CH2)-3-(2,6-iPr2C6H3NCH)–C8H4N)-2-yl with two molecules of 2,6-dimethylphenylisonitrile via an unexpected aza-[4 + 1 + 1] annulation, which is obviously different from the reactivity of the zirconium–carboryne complex with isonitrile to produce the zirconium complex having the [tBuNCC]− ketenimine moiety functionalized carborane anion through the insertion of isonitrile into the Zr–Ccage bond accompanied by the Zr–Ccage bond cleavage.19b The result is also different from those of the reactions of lutetium metallacyclopropene with phenylnitrile followed by treatment with 2,6-dimethylphenylisonitrile to produce the functionalized pyrrolyl moiety complex.35 This result is also obviously different from the reactions of the rare-earth metal complexes 1 and 2 with isonitriles to generate the multi-substituted aza-[4 + 1] annulation and the [2 + 2] cyclometallation products as discussed above. These differences in reactivity patterns towards isonitriles may probably be due to ligands, central metal ions, coordination environments around the central metals, and electrophilic property of the indol-2-yl carbon atoms (comparison of the natural charge of the carbon about −0.253 in 1c and −0.260 in 2a with −0.228 in the corresponding carboryne-based yttrium metallacycle 8a, see Table 1). It is found that the average lengths of the RE–Ccage bond in 9 are shorter than those of the corresponding rare-earth metallacyclopropane complexes (κ2-L1)RE(η2-C2B10H10)(THF)3, which may be attributed to the difference in ionic radii between the different coordination numbers in complexes (κ2-L1)RE(η2-C2B10H10)(THF)3. It is noted that the coupling of the imino nitrogen atom with unsaturated substrates is only involved in the aza-Diels–Alder reactions, where the imine nitrogen generally bears electron-withdrawing groups.
 | ||
| Scheme 13 Reactions of the indol-2-yl rare-earth metal carboryne-based metallacyclopropanes with XylNC. | ||
DFT calculations of complexes (κ2-L1)RE(η2-C2B10H10)(THF)3 (RE = Y, Lu) showed that the natural charges of the indol-2-yl carbon are −0.228 in yttrium and −0.213 in lutetium complexes,19b which are comparable with those found in complexes 1 and 2, indicating that the indol-2-yl carbon is an electrophilic carbon because the corresponding p-orbital lacks electrons. These results are parallel to our previous findings of the indol-2-yl carbon which exhibited electrophilic property to make the 1,1-alkyl and 1,1-H (or D) migratory insertion possible,18,19a and findings of unique reactivity patterns towards pyridine derivatives.19c Based on these results, the formation of the γ-carboline-based α,β-diamido moiety functionalized complexes could be described as follows: complex (κ2-L1)Y(η2-C2B10H10)(THF)3 was transformed into the intermediate A through ligand substitution, in which the indol-2-yl carbon is more electrophilic than that in (κ2-L1)Y(η2-C2B10H10)(THF)3 (natural charge: −0.132 in A vs. −0.228 in (κ2-L1)Y(η2-C2B10H10)(THF)3, Table S29 in the ESI†). The migratory insertion of the polarized coordinated isonitrile to the enamido-functionalized carbene with Y–Ccarbene bond cleavage produced the ketenimine intermediate B, which has been found in the reaction of free singlet carbene with isonitrile,49 and is parallel to the above results of reactions of complexes 2 with isonitriles delivering intermediate dippD. Then B underwent isomerization from cis-coordinated isonitrile to trans-coordinated isonitrile to give the ketenimine intermediate C, which then reacted with the coordinated isonitrile through insertion to afford the ketenimine intermediate D of the five-membered metallacycle. The difference in reactivity between the above formed rare-earth metal ketenimine intermediate and the present rare-earth metal ketenimine intermediate in the reactions with isonitriles can be attributed to the existence of the electrophilic carboryne motif, which may make the central carbon of the ketenimine more electrophilic, thereby allowing the coordinated isonitrile to attach to it easily. The amido group of the enamido functionality attached the carbon of the CN double bond to produce the γ-carboline-based α,β-amidoimino moiety E, which then coordinates with two molecules of THF with release of one isonitrile to produce the final product 9a (Scheme 14).
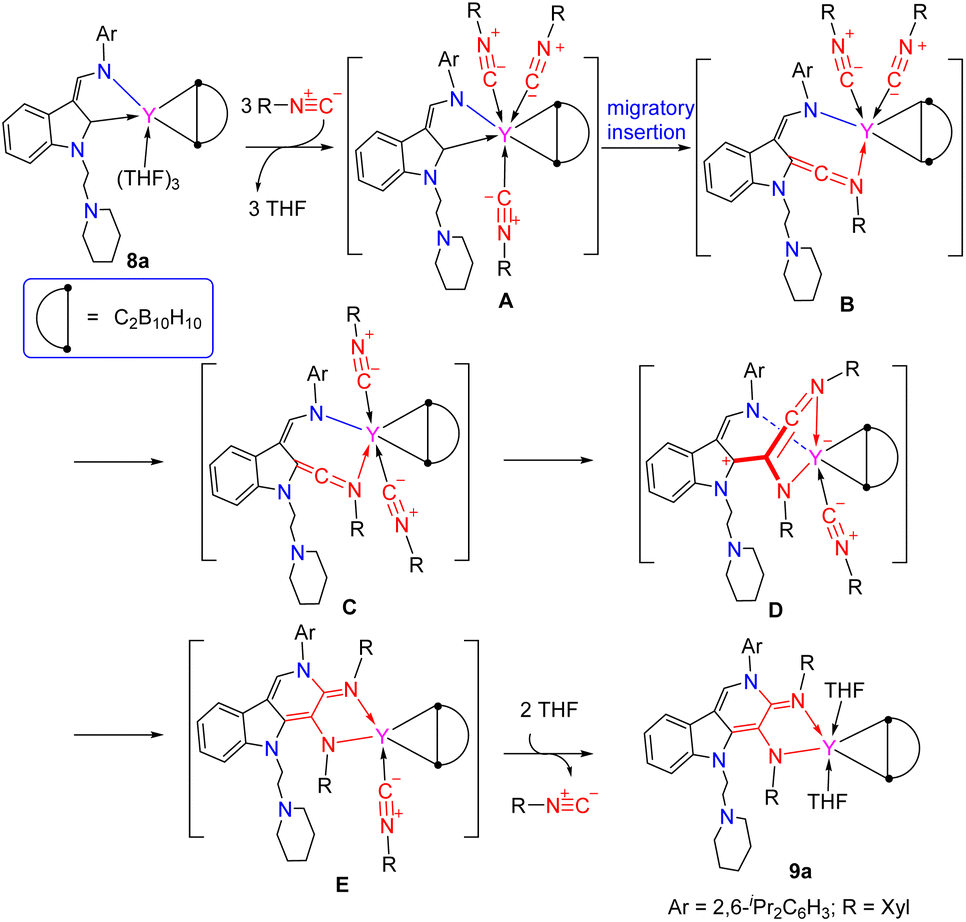 | ||
| Scheme 14 A possible reaction pathway for the reaction of indolyl rare-earth carboryne-based metallacyclopropanes with XylNC. | ||
To gain some insights into the formation of complex 9a from complex 8a, the computational studies were carried out at the level of B3PW91 including dispersion corrections (see the ESI† for details). The first step of the reaction is the formation of an yttrium centre with three coordinated isonitrile molecules, which will give a more stable A with corresponding indol-2-yl carbon which is more electrophilic (natural charge from −0.228 to −0.132). Two isomers were found (namely A and A′ in Scheme S32 in the ESI†) that would allow two different reactivities. The most stable isomer is A which is 4.8 kcal mol−1 more stable than A′, so that the latter will never be formed, preventing the red pathway in Scheme S32 in the ESI.† The red pathway, although kinetically accessible (barrier of 18.1 kcal mol−1), is the insertion of isonitrile into the Y–Ccage bond, while the black pathway, which is kinetically competitive (within the precision of the computational method) with the red pathway (barrier of 23.3 kcal mol−1), is the insertion into the Y–Ccarbene bond. Following the intrinsic reaction coordinates for both pathways, it yields complex B′ and C where the latter is more stable than the former by 2.7 kcal mol−1 so that only complex C will be observed. This completely ruled out the reaction of isonitrile with the Y–Ccage bond. Complex C is an amido complex bearing a CCN group which is reminiscent of an anionic aza-allyl group. This CCN cumulenic ketenimine system contains a central sp-hybridized carbon atom that is electron-deficient. Thus, a second molecule of isonitrile undergoes a nucleophilic addition to the central carbon of the ketenimine at TS2 with an associated barrier of 20.8 kcal mol−1. Following the intrinsic reaction coordinate, it yields another stable ketenimine intermediate D (−23.9 kcal mol−1), which readily isomerizes through a ring closure reaction of the nucleophilic addition of the Y–N bond to the central carbon of the ketenimine (TS3). The associated barrier of this last step is 16.4 kcal mol−1 and leads to the stable formation of intermediate E (−64.9 kcal mol−1). The latter exchanges an isonitrile with two THF molecules to yield the experimentally observed complex 9a. This ligand exchange is very exothermic in line with the crystallization of complex 9a (Scheme S32 in the ESI†).
Our attempts to isolate the isonitrile coordinated intermediates to prove the coordinative (or nucleophilic) ability of the nitriles in the formation of complexes 6, 7 and 9 are not successful. Then, the non-THF coordinated complexes 3 bearing the amino-functionalized enolate chelate ligands were synthesized, and their reactivity towards isonitriles was studied. To our delight, the tOctylNC coordinated (Yb–CCNtOctyl bond lengths of 2.656(9) Å) complex 10a (Scheme 15 and Fig. S99 in the ESI†) was separated in a moderate yield from the reaction of the dinuclear ytterbium complex 3c with 2 equiv. of tOctylNC in toluene at ambient temperature. This result supports the proposed coordination of isonitrile with Lewis acidic rare-earth metal centers and provides insights into the proposed intermediate equivalents AdA, DippA and A in the formation of complexes 6, 7 and 9. Complex 10a does not further react with excess tOctylNC on either the synthetic or NMR scale, indicating ligand effects on the reactivity. However, when dinuclear complex 3a was treated with XylNC, complex 10b (Scheme 15, Fig. 4 and S100 in the ESI†) bearing an aza-metallacyclobutene moiety was isolated. In 10b, the CC double bond (C2-ind–Cisonitrile = 1.383(6) Å, and Cisonitrile–Nisonitrile = 1.388(6) Å) is different from the aza-metallacyclobutene having a CN double bond (C2–ind–Cisonitrile = 1.510(10) Å, Cisonitrile–Nisonitrile = 1.385(10) Å) in the four-membered ring in complexes 6. It is proposed that the formation of 10b involves coordination of the isonitrile to the metal centre to give the intermediate 10a, which then undergoes 1,1-migratory insertion to generate the ketenimine intermediate. The ketenimine inserts into the RE–CCH2SiMe3 bonds to deliver the final products, which is also different from that in the formation of 6 (Scheme S33 in the ESI†).
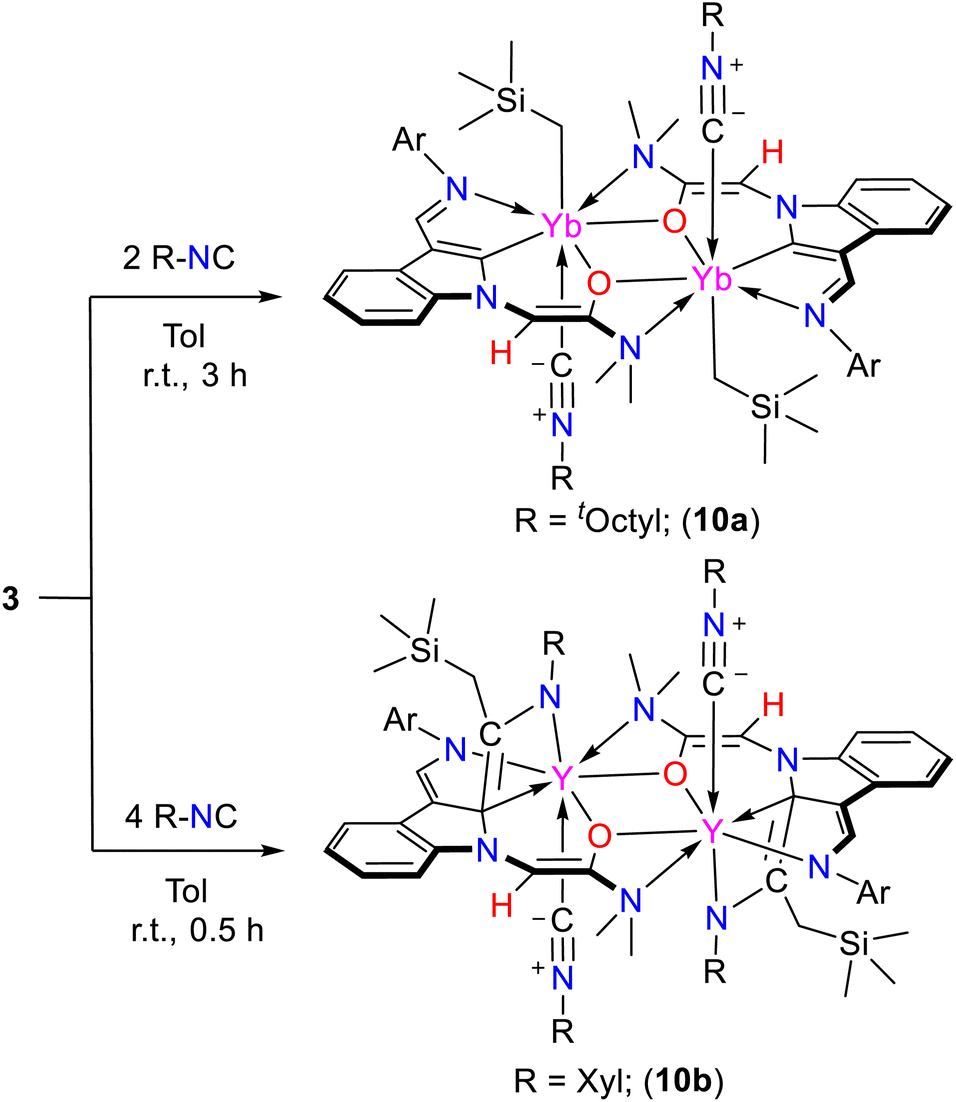 | ||
| Scheme 15 The reactions of complexes 3 with isonitriles. | ||
 | ||
| Fig. 4 Molecular structure of 10b with the thermal ellipsoid at 30% probability level. All hydrogen atoms were omitted and the diisopropylphenyl (Dipp) group was drawn in wireframe style for clarity. Selected bond lengths (Å) and selected bond angles (deg) can be read in the ESI.† | ||
This reactivity pattern is different from those in the formation of complexes 6, 7 and 9 highlighting again the influence of ligands on the reactivity patterns. Thus, four different reactivity patterns were found in the reactions of the synthesized rare-earth metal complexes with isonitriles: (1) replacement of THF of complexes 1 by isonitriles affords the isonitrile coordinated intermediates, which then undergo a stepwise 1,1-isonitrile migratory insertion to the carbene carbon to generate the intermediates having imidoyl anion attached to the indol-2-yl carbon via intramolecular redox. The imidoyl anion abstracts proton of the α C–H of the amidate to produce the final complexes 6 containing the aza-metallacyclobutene with a CN double bond. (2) The stepwise coordination and 1,1-isonitrile migratory insertion with subsequent isomerization with complexes 2 produce the ketenimine intermediate, which then was attached by the amido nitrogen to generate [4 + 1] annulation products of the indolo-fused 2-amido-functionalized pyrrolyl ligated complexes 7. (3) Coordination of the polarized isonitrile to the metal centre to produce a more electrophilic indol-2-yl carbon followed by 1,1-isonitrile migratory insertion to give the ketenimine intermediate. A second molecule of isonitrile undergoes a nucleophilic addition to the central carbon of the ketenimine yielding another ketenimine intermediate which is attached by the amido nitrogen followed by coordination with two molecules of THF to form the unprecedented aza-[4 + 1 + 1] annulation product 9. (4) Coordination of the isonitrile to the metal centre followed by 1,1-isonitrile migratory insertion to give the ketenimine intermediate, which then inserts into the RE–CCH2SiMe3 bond to produce the complexes 10 with aza-metallacyclobutene motifs having a CC double bond.
Conclusions
In summary, three different types of novel rare-earth metal complexes bearing 1-multi-chelated amidate-3-imino functionalized indol-2-yl pincer ligands (complexes 1 and 2) or 1-multi-chelated amino-functionalized enolate-3-imino functionalized indol-2-yl pincer ligand (complexes 3) were synthesized with rationally designed ligands. DFT calculations and experimental results reveal that the indol-2-yl carbon atoms of the ligands in the complexes can be described as ambiphilic carbenes, which display unprecedented selective reactivity towards electrophiles and nucleophiles. The RE–C2-ind bonds (or RE–Ccarbene bonds) of these complexes exhibit unexpected selective reactivity towards electrophiles like carbodiimides or imine to generate unusual [2 + 2] cyclometallation products of the aza-metallacyclobutane moiety. The reactions of these complexes with diphenyldiazomethane Ph2CN2 produce either insertion complexes or a rare complex bearing an indolo-fused six-membered ring via [3 + 3] annulation, depending on the amount of Ph2CN2. The RE–Ccarbene bonds also show selective reactivity towards Lewis acidic compounds such as AlR3 (R = Me, Et) and 9-BBN to produce alumina- or bora-four-membered metallacycles with short RE–H–C interactions. The reactions of the newly synthesized complexes and complexes bearing electrophilic carbene and metallacyclopropane with isonitriles display four types of unique selective reactivity patterns of the carbenes: (1) the replacement of coordinated THF by isonitriles followed by 1,1-isonitrile migratory insertion to the carbene carbon with subsequent intramolecular redox and abstraction of proton of the α C–H of the amidate to generate the formal [2 + 2] cyclometallation products of aza-metallacyclobutene having a CN double bond; (2) the substitution of THF by isonitriles followed by 1,1-isonitrile migratory insertion to the carbene carbon and isomerization to the ketenimine intermediate, which was then attacked by the nitrogen of the ligands with subsequent α C–H activation by the RE–CH2SiMe3 to deliver the novel aza-[4 + 1] annulation products of 1-(amido-functionalized enolate)-indolo-fused-2-amido-pyrrolyl ligated complexes; (3) coordination of the polarized isonitrile to the metal centre to produce a more electrophilic indol-2-yl carbon followed by 1,1-isonitrile migratory insertion to give the ketenimine intermediate. A second molecule of isonitrile undergoes a nucleophilic addition to the central carbon of the ketenimine yielding another ketenimine intermediate which is attached by amido nitrogen followed by coordination with two molecules THF to form the unprecedented aza-[4 + 1 + 1] annulation products; (4) the coordination of the isonitrile with subsequent 1,1-isonitrile migratory insertion into the carbene carbon and isomerization to a ketenimine intermediate, which then gets inserted into the RE–CCH2SiMe3 bond to generate the multi-substituted [2 + 2] cyclometallation products of aza-metallacyclobutene having a CC double bond. This work provides plenty of insights into the reactivity of rare-earth metal complexes bearing both ambiphilic carbene and nucleophilic carbon atoms, and demonstrates unique selective reactivity patterns of the rare-earth metal ambiphilic carbenes, which may be applicable in understanding and designing new catalytic reactions. Further work in this field is in progress.
Data availability
Experimental procedures, X-ray crystallographic details, NMR spectroscopy data, and computational details are available in the ESI.† CCDC 2258652–2258653, 2386247–2386289, 2258655, 2278784, 2278786 and 2278788 contain the supplementary crystallographic data for this paper.Author contributions
F. Chai led and performed the synthesis, reactivity study of the amido-functionalized indol-2-yl rare-earth metal complexes and experiments for the characterization of the complexes, and manuscript draft writing. W. Wu led and performed the synthesis, reactivity study of the indol-2-yl rare-earth carboryne-based metallacyclopropanes and experiments for the characterization of the complexes. T. Rajeshkumar performed the DFT calculations and analyse the results under the supervision of L. Maron, Z. Huang, Q. Yuan and Y. Wei assisted in the analysis of experimental data. L. Maron supervised and participated in the discussion on DFT calculation results. S. Wang is responsible for the project design, discussion, manuscript writing, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (No. 22031001, U23A2079 and 22301005). The authors are thankful for the grants from the Anhui Province (202305a12020018, GXXT-2021-052) and the Wuhu Science and Technology project (2023jc16). We appreciate the help of Dongjing Hong (Anhui Normal University) in the crystal data process and structure solution and refinement.Notes and references
- (a) H. Nishiyama, Chem. Soc. Rev., 2007, 36, 1133–1141 RSC; (b) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed; (c) N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed.
- (a) C. Allais, J.-M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829–10868 CrossRef CAS PubMed; (b) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (c) A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed; (d) E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 Search PubMed; (e) A. Haque, L. Xu, R. A. Al-Balushi, M. K. Al-Suti, R. Ilmi, Z. L. Guo, M. S. Khan, W.-Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2019, 48, 5547–5563 Search PubMed; (f) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed; (g) G. Kleinhans, A. J. Karhu, H. Boddaert, S. Tanweer, D. Wunderlin and D. I. Bezuidenhout, Chem. Rev., 2023, 123, 8781–8858 CrossRef CAS PubMed.
- M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11–27 Search PubMed.
- (a) Y. Pan, T. Xu, G.-W. Yang, K. Jin and X.-B. Lu, Inorg. Chem., 2013, 52, 2802–2808 CrossRef CAS PubMed; (b) J. Zhang, H. Yao, Y. Zhang, H. Sun and Q. Shen, Organometallics, 2008, 27, 2672–2675 CrossRef CAS; (c) A. Adhikary, S. Saha, N. S. Kumar, A. G. Oliver, J. A. Krause and H. Guan, Organometallics, 2023, 42, 1525–1537 Search PubMed.
- (a) V. Rad’kov, V. Dorcet, J.-F. Carpentier, A. Trifonov and E. Kirillov, Organometallics, 2013, 32, 1517–1527 CrossRef; (b) D. Sengupta, R. Bhattacharjee, R. Pramanick, S. P. Rath, N. S. Chowdhury, A. Datta and S. Goswami, Inorg. Chem., 2016, 55, 9602–9610 CrossRef CAS PubMed; (c) R. Pramanick, R. Bhattacharjee, D. Sengupta, A. Datta and S. Goswami, Inorg. Chem., 2018, 57, 6816–6824 CrossRef CAS PubMed.
- (a) L. Wang, D. Liu and D. Cui, Organometallics, 2012, 31, 6014–6021 CrossRef CAS; (b) P. Zhang, H. Liao, H. Wang, X. Li, F. Yang and S. Zhang, Organometallics, 2017, 36, 2446–2451 CrossRef CAS; (c) M.-C. Chang, A. J. McNeece, E. A. Hill, A. S. Filatov and J. S. Anderson, Chem. Eur. J., 2018, 24, 8001–8008 Search PubMed.
- (a) K. R. D. Johnson, B. L. Kamenz and P. G. Hayes, Organometallics, 2014, 33, 3005–3011 CrossRef CAS; (b) K. R. D. Johnson and P. G. Hayes, Organometallics, 2013, 32, 4046–4049 Search PubMed.
- (a) D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed; (b) E. A. LaPierre, W. E. Piers, D. M. Spasyuk and D. W. Bi, Chem. Commun., 2016, 52, 1361–1364 RSC; (c) W. Ren, H. Liu, F. You, P. Mao, Y.-M. So, X. Kang and X. Shi, Dalton Trans., 2021, 50, 1334–1343 Search PubMed; (d) Q. Zhuo, J. Yang, X. Zhou, T. Shima, Y. Luo and Z. Hou, J. Am. Chem. Soc, 2023, 145, 22803–22813 (J. Am. Chem. Soc., 2024, 146, 10984–10992) CrossRef CAS PubMed; (e) T. Shima, Q. Zhuo, X. Zhou, P. Wu, R. Owada, G. Luo and Z. Hou, Nature, 2024, 632, 307–312 CrossRef CAS PubMed.
- (a) X.-Y. Cao, Q. Zhao, Z. Lin and H. Xia, Acc. Chem. Res., 2014, 47, 341–354 CrossRef CAS PubMed; (b) D. Chen, Y. Hua and H. Xia, Chem. Rev., 2020, 120, 12994–13086 CrossRef CAS PubMed; (c) Q. Li, Y. Hua, C. Tang, D. Chen, M. Luo and H. Xia, J. Am. Chem. Soc., 2023, 145, 7580–7591 Search PubMed.
- (a) S. Zhu, W. Xu, D. Hong, W. Wu, F. Chai, X. Zhu, S. Zhou and S. Wang, Inorg. Chem., 2023, 62, 381–391 CrossRef CAS PubMed; (b) Z. Huang, S. Wang, X. Zhu, Y. Wei, Q. Yuan, S. Zhou, X. Mu and H. Wang, Chin. J. Chem., 2021, 39, 3360–3368 Search PubMed; (c) Y. Wei, Q. Bao, L. Song, D. Hong, J. Gao, S. Wang, X. Zhu, S. Zhou and X. Mu, Dalton Trans., 2022, 51, 2953–2961 RSC; (d) Z. Zhao, Y. Tong, C. Liu, Z. Huang, S. Wang and F. Wang, New J. Chem., 2024, 48, 14690–14696 Search PubMed.
- (a) W. J. Evans, J. C. Brady and J. W. Ziller, Inorg. Chem., 2002, 41, 3340–3346 CrossRef CAS PubMed; (b) X. Zhu, S. Wang, S. Zhou, Y. Wei, L. Zhang, F. Wang, Z. Feng, L. Guo and X. Mu, Inorg. Chem., 2012, 51, 7134–7143 CrossRef CAS PubMed; (c) Z. Feng, X. Zhu, S. Wang, S. Zhou, Y. Wei, G. Zhang, B. Deng and X. Mu, Inorg. Chem., 2013, 52, 9549–9556 CrossRef CAS PubMed; (d) L. Guo, X. Zhu, G. Zhang, Y. Wei, L. Ning, S. Zhou, Z. Feng, S. Wang, X. Mu, J. Chen and Y. Jiang, Inorg. Chem., 2015, 54, 5725–5731 CrossRef CAS PubMed; (e) L. Guo, S. Wang, Y. Wei, S. Zhou, X. Zhu and X. Mu, Inorg. Chem., 2017, 56, 6197–6207 CrossRef CAS PubMed; (f) D. Hong, X. Zhu, S. Wang, Y. Wei, S. Zhou, Z. Huang, S. Zhu, R. Wang, W. Yue and X. Mu, Dalton Trans., 2019, 48, 5230–5242 RSC; (g) S. Zhu, W. Wu, D. Hong, F. Chai, Z. Huang, X. Zhu, S. Zhou and S. Wang, Inorg. Chem., 2024, 63, 14860–14875 Search PubMed.
- S. A. Ryken and L. L. Schafer, Acc. Chem. Res., 2015, 48, 2576–2586 CrossRef CAS PubMed.
- S. Liu, W. Sun, Y. Zeng, D. Wang, W. Zhang and Y. Li, Organometallics, 2010, 29, 2459–2464 CrossRef CAS.
- (a) L. J. E. Stanlake, J. D. Beard and L. L. Schafer, Inorg. Chem., 2008, 47, 8062–8068 CrossRef CAS PubMed; (b) W. Zhang, Y. Wang, J. Cao, L. Wang, Y. Pan, C. Redshaw and W.-H. Sun, Organometallics, 2011, 30, 6253–6261 CrossRef CAS.
- (a) Z. Zhang and L. L. Schafer, Org. Lett., 2003, 5, 4733–4736 CrossRef CAS PubMed; (b) R. K. Thomson, J. A. Bexrud and L. L. Schafer, Organometallics, 2006, 25, 4069–4071 CrossRef CAS; (c) L. J. E. Stanlake and L. L. Schafer, Organometallics, 2009, 28, 3990–3998 CrossRef CAS; (d) E. Chong and L. L. Schafer, Org. Lett., 2013, 15, 6002–6005 CrossRef CAS PubMed; (e) D. C. Leitch, R. H. Platel and L. L. Schafer, J. Am. Chem. Soc., 2011, 133, 15453–15463 CrossRef CAS PubMed; (f) E. Chong, J. W. Brandt and L. L. Schafer, J. Am. Chem. Soc., 2014, 136, 10898–10901 CrossRef CAS PubMed; (g) J. W. Brandt, E. Chong and L. L. Schafer, ACS Catal., 2017, 7, 6323–6330 CrossRef CAS; (h) H. Hao and L. L. Schafer, ACS Catal., 2020, 10, 7100–7111 CrossRef CAS; (i) H. Hao, T. Bagnol, M. Pucheault and L. L. Schafer, Org. Lett., 2021, 23, 1974–1979 CrossRef CAS PubMed.
- (a) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS PubMed; (b) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed; (c) K. H. Dötz and J. Stendel, Chem. Rev., 2009, 109, 3227–3274 CrossRef PubMed; (d) A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed; (e) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 Search PubMed; (f) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed; (g) H. Amouri, Chem. Rev., 2023, 123, 230–270 CrossRef CAS PubMed.
- (a) A. A. Danopoulos, N. Tsoureas, J. C. Green and M. B. Hursthous, Chem. Commun., 2003, 756–757 RSC; (b) T. Steninke, B. K. Shaw, H. Jong, B. O. Patrick, M. D. Fryzuk and J. C. Green, J. Am. Chem. Soc., 2009, 131, 10461–10466 CrossRef PubMed; (c) C. Romain, K. Miqueu, J.-M. Sotiropoulos, S. Bellemin-Laponnaz and S. Dagorne, Angew. Chem., Int. Ed., 2010, 49, 2198–2201 CrossRef CAS PubMed; (d) E. Despagnet-Ayoub, L. M. Henling, J. A. Labinger and J. E. Bercaw, Organometallics, 2013, 32, 2934–2938 CrossRef CAS; (e) T. Hatanaka, Y. Ohki and K. Tatsumi, Angew. Chem., Int. Ed., 2014, 53, 2727–2729 Search PubMed; (f) E. Despagnet-Ayoub, M. K. Takase, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2015, 137, 10500–10503 Search PubMed; (g) C. Romain, D. Specklin, K. Miqueu, J.-M. Sotiropoulos, C. Fliedel, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2015, 34, 4854–4863 CrossRef CAS; (h) R. M. Brown, J. B. Garcia, J. Valjus, C. J. Roberts, H. M. Tuononen, M. Parvez and R. Roesler, Angew. Chem., Int. Ed., 2015, 54, 6274–6277 CrossRef CAS PubMed; (i) X. Ren, C. Gourlaouen, M. Wesolek and P. Braunstein, Angew. Chem., Int. Ed., 2017, 56, 12557–12560 Search PubMed; (j) C. C. Quadri, R. Lalrempuia, J. Hessevik, K. W. Törnroos and E. L. Roux, Organometallics, 2017, 36, 4477–4489 CrossRef CAS; (k) M. T. Nguyen, D. Gusev, A. Dmitrienko, B. M. Gabidullin, D. Spasyuk, M. Pilkington and G. I. Nikonov, J. Am. Chem. Soc., 2020, 142, 5852–5861 CrossRef CAS PubMed; (l) K. Lubitz and U. Radius, Organometallics, 2019, 38, 2558–2572 CrossRef CAS; (m) F. He, C. Gourlaouen, H. Pang and P. Braunstein, Chem. Eur. J., 2022, 28, e202104234 CrossRef CAS PubMed.
- Z. Huang, R. Wang, T. Sheng, X. Zhong, S. Wang, X. Zhu, Q. Yuan, Y. Wei and S. Zhou, Inorg. Chem., 2021, 60, 18843–18853 CrossRef CAS PubMed.
- (a) D. Hong, T. Rajeshkumar, S. Zhu, Z. Huang, S. Zhou, X. Zhu, L. Maron and S. Wang, Sci. China Chem., 2023, 66, 117–126 CrossRef CAS; (b) W. Wu, T. Rajeshkumar, D. Hong, S. Zhu, Z. Huang, F. Chai, W. Wang, Q. Yuan, Y. Wei, Z. Xie, L. Maron and S. Wang, Inorg. Chem., 2024, 63, 18365–18378 CrossRef CAS PubMed; (c) W. Wu, T. Rajeshkumar, S. Zhu, F. Chai, D. Hong, Z. Huang, Q. Yuan, L. Maron and S. Wang, Chem. Sci., 2024, 15, 20315–20327 RSC.
- (a) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed; (b) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (c) N. I. Saper and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 17667–17676 CrossRef CAS PubMed; (d) H. M. O'Brien, M. Manzotti, R. D. Abrams, D. Elorriaga, H. A. Sparkes, S. A. Davis and R. B. Bedford, Nat. Catal., 2018, 1, 429–437 CrossRef; (e) Y. Tang, I. Benaissa, M. Huynh, L. Vendier, N. Lugan, S. Bastin, P. Belmont, V. César and V. Michelet, Angew. Chem., Int. Ed., 2019, 58, 7977–7981 CrossRef CAS PubMed; (f) J. Sun, L. Luo, Y. Luo and L. Deng, Angew. Chem., Int. Ed., 2017, 56, 2720–2724 CrossRef CAS PubMed; (g) J. Cheng, L. J. Wang, P. Wang and L. Deng, Chem. Rev., 2018, 118, 9930–9987 CrossRef CAS PubMed; (h) M. Iglesias and L. A. Oro, Chem. Soc. Rev., 2018, 47, 2772–2808 RSC; (i) A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed; (j) Q. Zhao, G. R. Meng, M. Szostak and S. P. Nolan, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed; (k) K. Balayan, H. Sharma, K. Vsnks, S. Ravindranathan, R. G. Gonnade and S. S. Sen, Chem. Commun., 2023, 59, 8540–8543 RSC; (l) P. Parvathy and P. Parameswaran, Chem. Eur. J., 2023, 29, e202300582 CrossRef CAS PubMed.
- (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed; (b) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed; (c) S. Termühlen, J. Blumenberg, A. Hepp, C. G. Daniliuc and F. E. Hahn, Angew. Chem., Int. Ed., 2021, 60, 2599–2602 CrossRef PubMed.
- (a) V. Lavallo, G. D. Frey, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 5224–5228 CrossRef CAS PubMed; (b) V. M. Marx, A. H. Sullivan, M. Melaimi, S. C. Virgil, B. K. Keitz, D. S. Weinberger, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 1919–1923 CrossRef CAS PubMed; (c) G. Ung and J. C. Peters, Angew. Chem., Int. Ed., 2015, 54, 532–535 CrossRef CAS PubMed; (d) Y. Wei, B. Rao, X. Cong and X. Zeng, J. Am. Chem. Soc., 2015, 137, 9250–9253 CrossRef CAS PubMed; (e) M. P. Wiesenfeldt, Z. Nairoukh, W. Li and F. Glorius, Science, 2017, 357, 908–912 CrossRef CAS PubMed; (f) D. L. Nascimento and D. E. Fogg, J. Am. Chem. Soc., 2019, 141, 19236–19240 CrossRef CAS PubMed.
- (a) D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. A. Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed; (b) A. S. Romanov, C. R. Becker, C. E. James, D. Di, D. Credgington, M. Linnolahti and M. Bochmann, Chem. Eur. J., 2017, 23, 4625–4637 Search PubMed; (c) R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed; (d) M. Deng, N. F. M. Mukthar, N. D. Schley and G. Ung, Angew. Chem., Int. Ed., 2020, 59, 1228–1231 CrossRef CAS PubMed.
- V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2006, 45, 3488–3491 CrossRef CAS PubMed.
- J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052–7055 CrossRef CAS PubMed.
- (a) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 Search PubMed; (b) G. D. Frey, J. D. Masuda, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 9444–9447 Search PubMed; (c) A. V. Zhukhovitskiy, M. G. Mavros, K. T. Queeney, T. Wu, T. V. Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 8639–8652 CrossRef CAS PubMed; (d) S. Würtemberger-Pietsch, H. Schneider, T. B. Marder and U. Radius, Chem. Eur. J., 2016, 22, 13032–13036 Search PubMed; (e) A. Bakker, M. Freitag, E. Kolodzeiski, P. Bellotti, A. Timmer, J. Ren, B. S. Lammers, D. Moock, H. W. Roesky, H. Mönig, S. Amirjalayer, H. Fuchs and F. Glorius, Angew. Chem. Int. Ed., 2020, 59, 13643–13646 CrossRef CAS PubMed; (f) G. Kaur, R. L. Thimes, J. P. Camden and D. M. Jenkins, Chem. Commun., 2022, 58, 13188–13197 Search PubMed; (g) A. V. Zhukhovitskiy, M. G. Mavros, K. T. Queeney, T. Wu, T. V. Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 8639–8652 CrossRef CAS PubMed; (h) C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- (a) T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039–16041 CrossRef CAS PubMed; (b) M. Braun, W. Frank, G. J. Reiss and C. Ganter, Organometallics, 2010, 29, 4418–4420 CrossRef CAS; (c) Z. R. McCarty, D. N. Lastovickova and C. W. Bielawski, Chem. Commun., 2016, 52, 5447–5450 RSC; (d) M. B. Gildner and T. W. Hudnall, Chem. Commun., 2019, 55, 12300–12303 Search PubMed; (e) P. R. Sultane, G. Ahumada, D. Janssen-Müller and C. W. Bielawski, Angew. Chem. Int. Ed., 2019, 58, 16320–16325 Search PubMed; (f) M. Košutić, S. Neuner, A. Ren, S. Flür, C. Wunderlich, E. Mairhofer, N. Vušurović, J. Seikowski, K. Breuker, C. Höbartner, D. J. Patel, C. Kreutz and R. Micura, Angew. Chem. Int. Ed., 2015, 54, 15128–15133 CrossRef PubMed; (g) H. Song, H. Kim and E. Lee, Angew. Chem. Int. Ed., 2018, 57, 8603–8607 CrossRef CAS PubMed; (h) D. Martin, N. Lassauque, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2012, 51, 6172–6175 CrossRef CAS PubMed; (i) M. J. López-Gómez, D. Martin and G. Bertrand, Chem. Commun., 2013, 49, 4483–4485 RSC; (j) E. Tomás-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753–7756 Search PubMed; (k) C. M. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255–9260 CrossRef CAS PubMed; (l) Z. R. Turner, Chem. Eur. J., 2016, 22, 11461–11468 CrossRef CAS PubMed.
- (a) A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed; (b) C. D. Graaff, E. Ruijter and R. V. A. Orru, Chem. Soc. Rev., 2012, 41, 3969–4009 RSC; (c) A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- (a) J. Fernández-Gallardo, L. Bellarosa, G. Ujaque, A. Lledós, M. J. Ruiz, R. Fandos and A. Otero, Organometallics, 2012, 31, 7052–7062 CrossRef; (b) Y. Zhang, R. J. Keaton and L. R. Sita, J. Am. Chem. Soc., 2003, 125, 8746–8747 CrossRef CAS PubMed; (c) E. Barnea, T. Andrea, J.-C. Berthet, M. Ephritikhine and M. S. Eisen, Organometallics, 2008, 27, 3103–3112 CrossRef CAS; (d) L. Pauly, A. K. Adegboyega, C. A. Brown, D. E. Pérez and E. A. Ison, Organometallics, 2024, 43, 3002–3012 CrossRef CAS.
- (a) E. Lu, Y. Chen and X. Leng, Organometallics, 2011, 30, 5433–5441 CrossRef CAS; (b) J. Zhang, W. Yi, Z. Zhang, Z. Chen and X. Zhou, Organometallics, 2011, 30, 4320–4324 CrossRef CAS; (c) G. P. McGovern, F. Hung-Low, J. W. Tye and C. A. Bradley, Organometallics, 2012, 31, 3865–3879 CrossRef CAS; (d) C. S. Weinert, P. E. Fanwick and I. P. Rothwell, Organometallics, 2005, 24, 5759–5766 CrossRef CAS; (e) R. Fandos, J. Fernández-Gallardo, A. Otero, A. Rodríguez and M. J. Ruiz, Organometallics, 2011, 30, 1551–1557 CrossRef CAS; (f) J. Fernández-Gallardo, A. Bajo, R. Fandos, A. Otero, A. Rodrıguez and M. J. Ruiz, Organometallics, 2013, 32, 1752–1758 CrossRef.
- (a) S. N. MacMillan, J. M. Tanski and R. Waterman, Chem. Commun., 2007, 4172–4174 RSC; (b) A. J. Roering, J. J. Davidson, S. N. MacMillan, J. M. Tanski and R. Waterman, Dalton Trans., 2008, 4488–4498 RSC; (c) A. F. Maddox, J. J. Davidson, T. Shalumova, J. M. Tanski and R. Waterman, Inorg. Chem., 2013, 52, 7811–7816 CrossRef CAS PubMed; (d) S. Clément, L. Guyard, M. Knorr, S. Dilsky, C. Strohmann and M. Arroyo, J. Organomet. Chem., 2007, 692, 839–850 CrossRef; (e) S. Clément, S. M. Aly, D. Bellows, D. Fortin, C. Strohmann, L. Guyard, A. S. Abd-El-Aziz, M. Knorr and P. D. Harvey, Inorg. Chem., 2009, 48, 4118–4133 CrossRef PubMed.
- (a) M. D. Fryzuk, P. B. Duval, S. S. S. H. Mao, S. J. Rettig, M. J. Zaworotko and L. R. MacGillivray, J. Am. Chem. Soc., 1999, 121, 1707–1716 CrossRef CAS; (b) F. Basuli, B. C. Bailey, L. A. Watson, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Organometallics, 2005, 24, 1886–1906 CrossRef CAS; (c) D. J. Yarrow, J. A. Ibers, Y. Tatsuno and S. Otsuka, J. Am. Chem. Soc., 1973, 95, 8590–8597 CrossRef CAS; (d) T. Sielisch and U. Behrens, J. Organomet. Chem., 1986, 310, 179–187 CrossRef CAS; (e) R. Aumann and H. Heinen, Chem. Ber., 1988, 121, 1085–1091 CrossRef CAS; (f) I. Fernández, F. P. Cossío and M. A. Sierra, Organometallics, 2007, 26, 3010–3017 CrossRef; (g) K. G. Moloy, P. J. Fagan, J. M. Manriquez and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 56–67 CrossRef CAS; (h) F. Basuli, B. C. Bailey, D. Brown, J. Tomaszewski, J. C. Huffman, M. H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2004, 126, 10506–10507 CrossRef CAS PubMed; (i) J. B. Russell, M. G. Jafari, J. H. Kim, B. Pudasaini, A. Ozarowski, J. Telser, M. H. Baik and D. J. Mindiola, Angew. Chem., Int. Ed., 2024, 63, e202401433 CrossRef CAS PubMed.
- B. F. Wicker, M. Pink and D. J. Mindiola, Dalton Trans., 2011, 40, 9020–9025 RSC.
- (a) P. S. Pregosin, NMR in Organometallic Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed; (b) R. B. Jordan, Inorg. Chem., 2023, 62, 3715–3721 CrossRef CAS PubMed; (c) Z. Huang, S. Wang, X. Zhu, Q. Yuan, Y. Wei, S. Zhou and X. Mu, Inorg. Chem., 2018, 57, 15069–15078 CrossRef CAS PubMed.
- Z.-J. Lv, Z. Chai, M. Zhu, J. Wei and W. Zhang, J. Am. Chem. Soc., 2021, 143, 9151–9161 CrossRef CAS PubMed.
- J. S. Gopinath and P. Parameswaran, Organometallics, 2024, 43, 1411–1424 CrossRef CAS.
- (a) S. Li, J. Cheng, Y. Chen, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 6360–6363 CrossRef CAS PubMed; (b) W. Yi, J. Zhang, F. Zhang, Y. Zhang, Z. Chen and X. Zhou, Chem. Eur. J., 2013, 19, 11975–11983 CrossRef CAS PubMed; (c) Y. Cao, Z. Du, W. Li, J. Li, Y. Zhang, F. Xu and Q. Shen, Inorg. Chem., 2011, 50, 3729–3737 CrossRef CAS PubMed; (d) K. C. Casey, J. K. Appiah and J. R. Robinson, Inorg. Chem., 2020, 59, 14827–14837 CrossRef CAS PubMed.
- G. Zhang, S. Wang, X. Zhu, S. Zhou, Y. Wei, Z. Huang and X. Mu, Organometallics, 2017, 36, 3812–3822 CrossRef CAS.
- (a) G. Zhang, S. Wang, S. Zhou, Y. Wei, L. Guo, X. Zhu, L. Zhang, X. Gu and X. Mu, Organometallics, 2015, 34, 4251–4261 CrossRef CAS; (b) X. Zhu, Y. Li, Y. Wei, S. Wang, S. Zhou and L. Zhang, Organometallics, 2016, 35, 1838–1846 CrossRef CAS; (c) G. Zhang, B. Deng, S. Wang, Y. Wei, S. Zhou, X. Zhu, Z. Huang and X. Mu, Dalton Trans., 2016, 45, 15445–15456 RSC; (d) L. Guo, X. Zhu, G. Zhang, Y. Wei, L. Ning, S. Zhou, Z. Feng, S. Wang, X. Mu, J. Chen and Y. Jiang, Inorg. Chem., 2015, 54, 5725–5731 CrossRef CAS PubMed; (e) G. Zhang, S. Wang, X. Zhu, S. Zhou, Y. Wei, Z. Huang and X. Mu, Organometallics, 2017, 36, 3812–3822 CrossRef CAS; (f) X. Zhu, Y. Jiang, J. Chen, S. Wang, Z. Huang, S. Zhu, X. Zhao, W. Yue, J. Zhang, W. Wu and X. Zhong, Chin. J. Chem., 2020, 38, 478–488 CrossRef CAS; (g) D. Diether, M. Meermann-Zimmermann, K. W. Törnroos, C. Maichle-Mössmer and R. Anwander, Dalton Trans., 2020, 49, 2004–2013 RSC; (h) M. M. Katzenmayer, B. M. Wolf, A. Mortis, C. Maichle-Mössmer and R. Anwander, Chem. Commun., 2021, 57, 243–246 RSC; (i) E. C. Moinet, B. M. Wolf, O. Tardif, C. Maichle-Mössmer and R. Anwander, Angew. Chem., Int. Ed., 2023, 62, e202219316 CrossRef CAS PubMed.
- H. Nakamura, Y. Nakayama, H. Yasuda, T. Maruo, N. Kanehisa and Y. Kai, Organometallics, 2000, 19, 5392–5399 CrossRef CAS.
- W. Jiang, F. Kong, I. D. Rosal, M. Li, K. Wang, L. Maron and L. Zhang, Chem. Sci., 2023, 14, 9154–9160 RSC.
- W. Jiang, T. Rajeshkumar, M. Guo, L. Maron and L. Zhang, Chem. Sci., 2024, 15, 3495–3501 RSC.
- M. G. Schrems, H. M. Dietrich, K. W. Tornroos and R. Anwander, Chem. Commun., 2005, 5922–5924 RSC.
- M. Zimmermann, K. W. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2008, 47, 775–778 CrossRef CAS PubMed.
- J. Klosin, G. R. Roof and E. Y. Chen, Organometallics, 2000, 19, 4684–4686 CrossRef CAS.
- J. Chu, C. Wang, L. Xiang, X. Leng and Y. Chen, Organometallics, 2017, 36, 4620–4625 CrossRef CAS.
- (a) R. D. Dicken, A. Motta and T. J. Marks, ACS Catal., 2021, 11, 2715–2734 CrossRef CAS; (b) M. A. Iqbal, X. Yan, R. Li, F. Zhijia, S. Zhang and X. Li, New J. Chem., 2024, 48, 3149–3155 RSC.
- (a) X. Chen, S. Lim, C. E. Plečnik, S. Liu, B. Du, E. A. Meyers and S. G. Shore, Inorg. Chem., 2004, 43, 692–698 CrossRef CAS PubMed; (b) D. A. Laske, R. Duchateau, J. H. Teuben and A. L. Spek, J. Organomet. Chem., 1993, 462, 149–153 CrossRef CAS; (c) W. J. Evans, S. E. Lorenz and J. W. Ziller, Chem. Commun., 2007, 4662–4664 RSC.
- D. Magis, J. J. Cabrera-Trujillo, J. Vignolle, J.-M. Sotiropoulos, D. Taton, K. Miqueu and Y. Landais, J. Am. Chem. Soc., 2024, 146, 16802–16813 CrossRef CAS PubMed.
- J. W. Collet, T. R. Roose, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Angew. Chem., Int. Ed., 2020, 59, 540–558 CrossRef CAS PubMed.
- (a) W. J. Evans, J. H. Meadows, W. E. Hunter and J. L. Atwood, Organometallics, 1983, 2, 1252–1254 CrossRef CAS; (b) J. Chen, T. Chen, J. R. Norton and M. Rauch, Organometallics, 2018, 37, 4424–4430 CrossRef CAS.
- D. Guo, D. Hong, Z. Huang, S. Zhou, X. Zhu and S. Wang, Inorg. Chem., 2024, 63, 9539–9551 CrossRef CAS PubMed.
- (a) C. Wang, W. Mao, L. Xiang, Y. Yang, J. Fang, L. Maron, X. Leng and Y. Chen, Chem. Eur. J., 2018, 24, 13903–13917 CrossRef CAS PubMed; (b) J. Zhang, W. Yi, Z. Zhang, Z. Chen and X. Zhou, Organometallics, 2011, 30, 4320–4324 CrossRef CAS; (c) E. Lu, Y. Chen and X. Leng, Organometallics, 2011, 30, 5433–5441 CrossRef CAS; (d) N. Obata, H. Mizuno, T. Koitabashi and T. Takizawa, B. Chem. Soc. Jpn., 1975, 48, 2287–2293 CrossRef CAS; (e) M. Pérez-Gómez, S. Hernández-Ponte, J. A. García-López, R. Frutos-Pedreño, D. Bautista, I. Saura-Llamas and J. Vicente, Organometallics, 2018, 37, 4648–4663 CrossRef; (f) A. Obanda, K. Valerius, J. T. Mague, S. Sproules and J. P. Donahue, Organometallics, 2020, 39, 2854–2870 CrossRef CAS; (g) Z. Chai, J. Wei and W. Zhang, Organometallics, 2023, 42, 2736–2741 CrossRef CAS; (h) Y. Lv, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2013, 135, 14784–14796 CrossRef CAS PubMed; (i) J. Chen, N. Yassin, T. Gunasekara, J. R. Norton and M. Rauch, J. Am. Chem. Soc., 2018, 140, 8980–8989 CrossRef CAS PubMed; (j) Q. Zhuo, J. Yang, X. Zhou, T. Shima, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2024, 146, 10984–10992 CrossRef CAS PubMed; (k) C. Saviozzi, L. Biancalana, T. Funaioli, M. Bortoluzzi, M. D. Franco, M. Guelfi, V. Gandin and F. Marchetti, Inorg. Chem., 2024, 63, 1054–1067 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2258652–2258653, 2386247–2386289, 2258655, 2278784, 2278786 and 2278788. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01502b |
| This journal is © The Royal Society of Chemistry 2025 |