
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing axial-ligand-induced switching of spin states for controllable single electron transfer-based radical initiation†
Jingyi Qina,
Yiyan Yina,
Xiaowen Guana,
Xiyang Gea,
Mengyu Caoa,
Jin Ouyang b and
Na Na*a
b and
Na Na*a
aKey Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: nana@bnu.edu.cn
bDepartment of Chemistry, Faculty of Arts and Sciences, Beijing Normal University, Zhuhai 519087, China
First published on 29th May 2025
Abstract
Radicals are highly reactive for coupling reactions while the applications are normally limited by the uncontrollable initiation and chaotic conversions. Although transition metal-based single electron transfer (SET) shows potential for controllable radical initiation, the detailed mechanism is still insufficient, especially for the roles of spin state transition in SET-based radical initiation. Herein, with an Fe(III)-catalyzed thiol–ene click (TEC) reaction as an example, the axial-ligand-induced switching of transition metals' spin states was revealed to facilitate controllable SET-based radical initiation and the subsequent coupling reactions. Given the advantages of online monitoring by ambient mass spectrometry (AMS), the short-lived radical intermediates and their dynamic changes were explored. As demonstrated, initiated by the axial coordination of sulfhydryl with Fe(III)–porphyrin, the selective generation of a thiyl radical (RS˙) via SET was achieved. Besides, as another axial-ligand, O2 in air was coordinated to Fe(III)–porphyrin, inducing the conversion of Fe(III) from a high spin (S = 5/2) to a low spin state (S = 1/2). This lowered the energy barrier for SET-based radical initiation, further facilitating the final selective coupling with the vinyl reactant. Upon revealing the axial-ligand-induced switching of the spin states by AMS and other examinations, rational design of transition metal catalysts would be promoted for efficient and highly selective radical reactions.
Introduction
Radicals have attracted much attention for their advantages of reacting with most organic molecules including sterically hindered molecules and those hard to be synthesized by catalytic reactions.1–3 In particular, with high reactivity and unique properties, radicals are expected to initiate rapid coupling to construct complex heterocyclic compounds.4,5 However, the highly active radical species would normally cause unfavourable chaotic, uncontrollable, and mysteriously baffling processes.6–8 Consequently, effective strategies for the controllable generation of radicals for chain initiation, as well as the subsequent directional conversion for chain propagation are crucial for efficient radical synthesis.For radical reactions, transition metal catalysis has been regarded as a versatile platform.9–13 Taking advantage of unpaired d-electrons, transition metals normally possess unoccupied orbitals that undergo single electron transfer (SET) to generate radicals.9,14 Significantly, upon modulating the coordination environments of transition metal sites, the spin state-related electron transfer could accelerate reaction kinetics by lowering the activation energy barrier.15–17 Predictably, the radical initiation via SET could be modulated by spin states. While the current modulation of spin states normally focuses on the rearrangement of d-electron orbitals to enhance inorganic catalytic reactions,18–21 there have been few reports revealing the role of transition metals' spin states in electron transfer-based radical initiation, thereby hindering efficient radical initiation by metals.
For instance, aryl sulfide radicals can be initiated in the metal-catalyzed thiol–ene click (TEC) reaction, which facilitates C–S coupling, exhibiting significance in pharmaceuticals and chemical engineering.22–26 Nevertheless, the initiation of various active species is usually non-selective due to the wide potential range of transition metals. Consequently, the mechanism of controllable radical reactions is worth exploring in detail.27–29 Unfortunately, although radical directional conversion (chain propagation) has been examined, a clear description of SET-based radical initiation remains lacking, let alone the roles of spin states in this initiation process (Scheme 1a).8,12,30,31 This could be largely limited by the difficulties in obtaining dynamic conversions of reactive radicals or intermediates with short lifetimes.32–35 More importantly, revealing the clear relationship between SET and spin-state switching is comprehensive and challenging, which requires detailed studies of the reaction process including the coordination behaviours of catalysts. Consequently, more efforts are needed to reveal the effect of spin states on SET-based radical initiation.
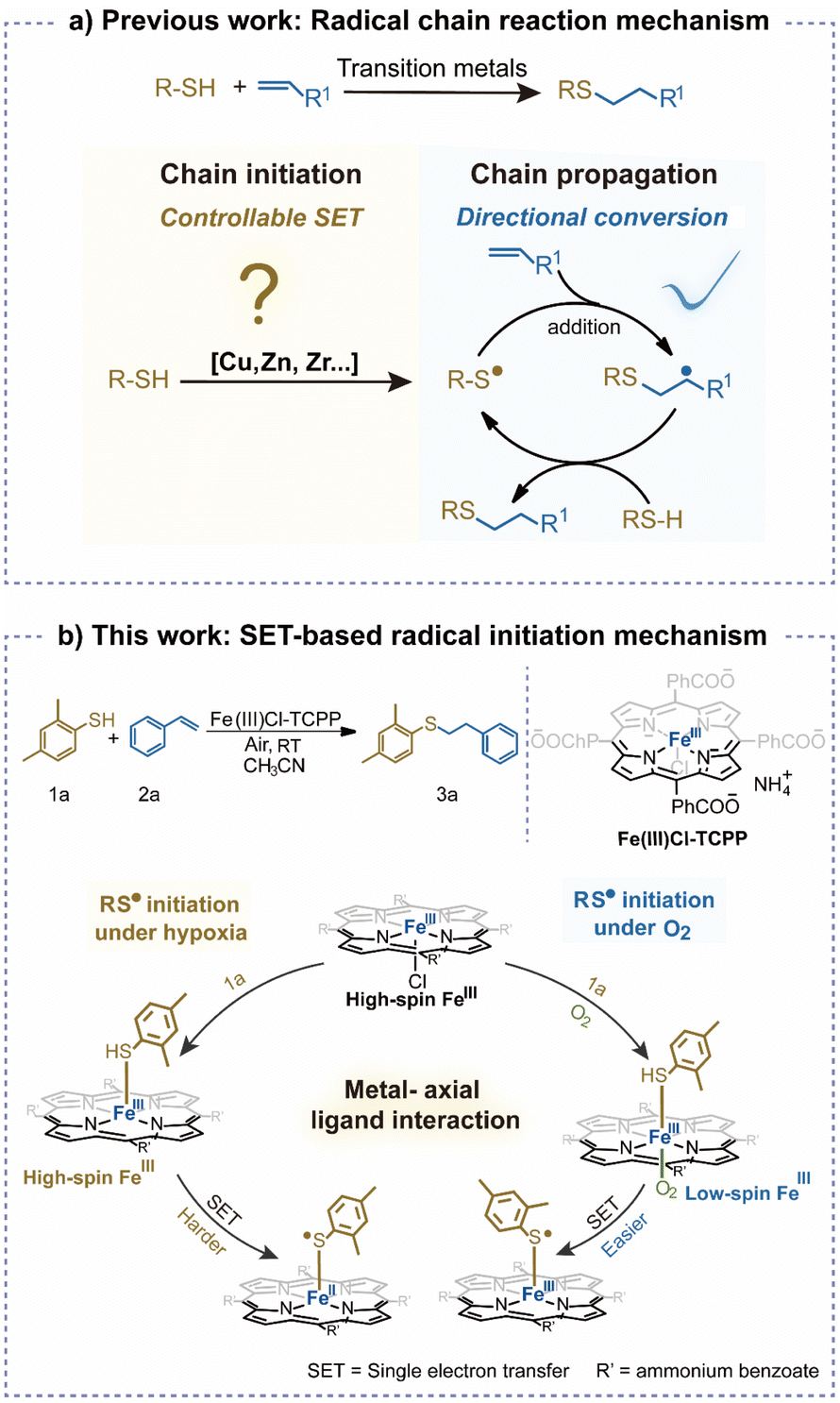 | ||
| Scheme 1 The illustration of transition metal-catalyzed thiol–ene click (TEC) reactions. (a) Radical chain reaction mechanism (previous work). (b) The mechanism of SET-based controllable radical initiation in the Fe(III)-catalyzed TEC reaction under different conditions (this work). | ||
Herein, to examine the effects of spin states on the controllable initiation of free radicals via SET, the TEC reaction of thiophenol and styrene was selected as the model reaction (Scheme 1b). Controllable generation of the thiyl radical (RS˙) was catalyzed by Fe(III)–porphyrin, which could be attributed to SET from sulfhydryl to Fe(III). To examine dynamic conversions of reactive radicals or intermediates with short lifetimes, an ambient mass spectrometry (AMS) system was constructed for online monitoring, achieving rapid structural identification without sample pretreatments.36–39 Based on the comprehensive characterizations, axial coordination of sulfhydryl with Fe(III)–porphyrin was confirmed to be crucial for the selective SET process. Moreover, O2 was revealed to act as another axial-ligand to bind with the sulfhydryl–Fe(III)–porphyrin complex, triggering the conversion of Fe(III) from a high spin (S = 5/2) to a low spin (S = 1/2) state. This lowered the energy barrier of radical generation via SET, leading to the acceleration of reaction in air. Furthermore, upon coordination with Fe(III)–porphyrin, the controllable radical initiation by SET would be facilitated by substrates with higher electron-donating abilities. This work has enabled AMS techniques for in-depth examination of controllable SET-based radical initiation, which would promote the development of transition metal catalysts for efficient and selective radical reactions.
Results and discussion
Evaluation of radical initiation in the TEC reaction
In the present TEC reaction, the C–S coupling of reactants 2,4-dimethylbenzenethiol (1a) and styrene (2a) was catalyzed by a transition metal catalyst of Fe(III)Cl–TCPP (Fig. S1†). For the rapid evaluation of TEC performance, the reaction system was directly detected by the AMS system. As shown in Fig. 1a, the reaction solution was extracted and ionized by a high-velocity (sonic) nebulizing stream of N2 gas through an external capillary. In this way, the samples in the reaction system can be directly monitored without sample pre-treatments, which enabled the determination of important short-lived intermediates. As monitored (Fig. 1b), the significant product ion of [3a + H]+ at m/z 243 was observed after reacting for 5 min. It should be noted that a relatively low abundance of disulfide at m/z 275 and sulfoxide at m/z 259 were observed, identified by collision induced dissociation (CID) analysis and HR-MS (Fig. S2 and S3†). This indicated the highly selective generation of the product of C–S coupling, in accordance with the NMR evaluations (Fig. S18-1†). | ||
Fig. 1 Evaluation of the radical initiation of the TEC reaction. (a) Schematic diagram of the AMS setup. (b) Mass spectrum of the reaction system for 5 min in air. Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol) and Fe(III)Cl–TCPP (0.25 mol%) in 3 mL solvent (CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 10:1, v:v). (c) Mass spectrum of the reaction system with the radical scavenger of TEMPO added. (d) EPR spectrum of DMPO–RS˙ in the mixture of substrate 1a and catalyst Fe(III)Cl–TCPP in air. :H2O = 10:1, v:v). (c) Mass spectrum of the reaction system with the radical scavenger of TEMPO added. (d) EPR spectrum of DMPO–RS˙ in the mixture of substrate 1a and catalyst Fe(III)Cl–TCPP in air. | ||
Subsequently, to confirm the radical initiation of the TEC reaction, the radical scavenger of 2,2,6,6-tetramethyl piperidine-1-oxyl (TEMPO) was added into the reaction system for AMS detection. As a result (Fig. 1c), no significant product signal of [3a + H]+ (m/z 243) was recorded. While the main ion peak at m/z 398 was exhibited, attributed to the coupling of the carbon radical intermediate (the precursor of product 3a) with TEMPO. The corresponding structure was confirmed by collision induced dissociation (CID) experiments (Fig. S4†). Consequently, the radical intermediate was captured by TEMPO, which greatly hindered the generation of product 3a. This indicated that the present transition metal-catalyzed TEC reaction involved stepwise radical conversions after the radical initiation.
To further confirm the generation of radical intermediates in the TEC reaction, the mixture of substrate 1a and catalyst Fe(III)Cl–TCPP was examined by EPR characterization with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the trapping agent. The presence of RS˙ was verified by the EPR signals (g = 2.08, 2.06, and 2.04) (Fig. 1d), which were attributed to the adduct of RS˙ and DMPO. This was also in accordance with the previous reports.40 Besides, to further confirm the selective radical generation, other thiophenol substrates with various substituted groups were selected for employing TEC reactions. As expected, the highest yields were obtained for the substrates with benzene-ring bearing electron-donating groups (92% of 3a for –Me, 94% of 3b for –OMe) (Fig. S5†), while the substrates with electron-withdrawing groups afforded lower product yields (65–81% of 3c–3e for –Cl, –CF3 and –NO2). These reaction products were verified by NMR characterization (Fig. S18-2 to 4†). Consequently, efficient radical initiation can be achieved with higher electron-donating abilities of substrates. In addition, the highest yield was obtained for the TEC in acetonitrile (CH3CN) and dichloromethane (DCM) (entries 1–2), much higher than in methanol (CH3OH) with the strongest coordination ability (entry 3) (Fig. S6†). It can be deduced that the coordination is crucial for radical initiation. Consequently, RS˙ was selectively generated in the TEC process, which played an important role in the subsequent C–S coupling at high yield.
Examination of radical generation upon the SET process
As proposed, RS˙ could be generated via SET between the sulfide substrate and transition metals or O2.29,41 Thus, further efforts are required to determine the real process of the present radical initiation upon electron transfer between 1a and Fe(III)Cl–TCPP or O2. To examine the generation of RS˙ upon SET, the roles of O2 and Fe(III) were firstly evaluated by EPR analysis under different conditions. For the mixture of 1a and Fe(III)Cl–TCPP, EPR signals of RS˙ were still obvious even in N2 (blue line, Fig. 2a), which were similar to that in air. While without Fe(III), no radical signal was recorded in the TCPP system (yellow line, Fig. 2a). This was in accordance with the low yield of TEC without Fe(III) active species in the catalyst (Fig. S7†). Furthermore, the highest yield was recorded in the O2 environment, a little higher than that in N2 (Fig. S7†). Consequently, it can be deduced that the transition metal of Fe(III) played an important role in the radical initiation, while O2 was not the essential factor but can facilitate the generation of RS˙. | ||
Fig. 2 Examination of radical generation upon the SET process. (a) EPR spectra of the mixture of substrate 1a and Fe(III)Cl–TCPP in N2 (blue line) or the mixture of 1a and TCPP in air (yellow line). (b) XPS spectra of Fe 2p before and after TEC reaction in air. (c) XPS spectra of Fe 2p before and after TEC reaction in N2. (d)  signals for the catalyst and reaction system. (e) UV-vis absorption of the NBT colorimetric reaction with signals for the catalyst and reaction system. (e) UV-vis absorption of the NBT colorimetric reaction with  in different systems. (f) Proposed mechanism of O2 and Fe(III)Por involving SET-based radical initiation. in different systems. (f) Proposed mechanism of O2 and Fe(III)Por involving SET-based radical initiation. | ||
To further examine the roles and changes of Fe(III)Cl–TCPP in the TEC reaction, the chemical states of the iron ion in air and N2 were evaluated by X-ray photoemission spectroscopy (XPS). In air (Fig. 2b), the XPS peaks of Fe(III) (at the binding energies of 724.3 and 711.0 eV)42,43 were observed before and after the reaction. This demonstrated the good stability of Fe(III)Cl–TCPP in the TEC reaction, which was crucial for the highly efficient catalytic reactions. However, after the reaction in N2 (Fig. 2c), significant XPS peaks of both Fe(III) (at 724.4 and 711.1 eV) and Fe(II) (at 722.6 and 709.2 eV)44 were recorded. This indicated the employment of electron transfer in the Fe(III)Cl–TCPP-catalyzed TEC. Consequently, the reduction of Fe(III) to Fe(II) could be achieved via SET from substrate 1 to Fe(III), along with the generation of RS˙. Notably, no obvious signal of Fe(II) was observed in the reaction in air. This could probably be attributed to the oxidation of the generated Fe(II) by O2 in air, which generates Fe(III) for catalytic reactions in the next TEC reactions.
Subsequently, to further examine O2 changes upon SET in the TEC reaction, the reactive oxygen species (ROS) were detected by EPR with DMPO as the radical trapping reagent. As shown in Fig. 2d, no obvious ROS signal was recorded in the system of Fe(III)Cl–TCPP, while after TEC reaction for 1 min, a significant signal of  was observed,45 demonstrating the transfer of an electron from Fe(II) to O2. In addition, the generation of
was observed,45 demonstrating the transfer of an electron from Fe(II) to O2. In addition, the generation of  was further evaluated by UV-vis absorption analysis, based on the absorption of a blue formazan deposit (∼520 nm) generated from the oxidation of nitro blue tetrazolium (NBT).46,47 As shown in Fig. 2e, strong absorption was recorded for substrate 1a with Fe(III)Cl–TCPP added (blue line) and the TEC reaction system (yellow line), while no signal was observed in substrate 2a with Fe(III)Cl–TCPP added. Consequently, it can be demonstrated that the electron transfers only occurred in the chain initiation process, rather than the chain propagation process. In fact, as shown in Fig. 2f, an electron was transferred from substrate 1 to Fe(III)Cl–TCPP, generating RS˙ and Fe(II) species. Simultaneously, the electron transfer from Fe(II) to O2 can be employed in air, along with the generation of
was further evaluated by UV-vis absorption analysis, based on the absorption of a blue formazan deposit (∼520 nm) generated from the oxidation of nitro blue tetrazolium (NBT).46,47 As shown in Fig. 2e, strong absorption was recorded for substrate 1a with Fe(III)Cl–TCPP added (blue line) and the TEC reaction system (yellow line), while no signal was observed in substrate 2a with Fe(III)Cl–TCPP added. Consequently, it can be demonstrated that the electron transfers only occurred in the chain initiation process, rather than the chain propagation process. In fact, as shown in Fig. 2f, an electron was transferred from substrate 1 to Fe(III)Cl–TCPP, generating RS˙ and Fe(II) species. Simultaneously, the electron transfer from Fe(II) to O2 can be employed in air, along with the generation of  in the TEC reaction. Consequently, this TEC reaction involved two cascade SET processes for the generation of radicals of both RS˙ and
in the TEC reaction. Consequently, this TEC reaction involved two cascade SET processes for the generation of radicals of both RS˙ and  .
.
Capture and detection of intermediates during SET-based radical initiation
The intermediates corresponding to both the catalyst and substrate were captured and examined by online AMS to elucidate the SET-based radical initiation for selective RS˙ generation. As detected, the characteristic ion of catalyst [Fe(III)TCPP]+ (m/z 844.5) was recorded in the reaction system at the beginning (Fig. 3a-i). With the TEC reaction proceeding (at 1 min, Fig. 3a-ii), the new ions of [Fe(III)TCPP–RS˙]+ (m/z 981.3) and [RSH–Fe(III)TCPP–O2]+ (R = 2,4-dimethylphenyl) (m/z 1014.1) were observed. The corresponding structures were confirmed by HR-MS (Fig. S8†) and CID experiments (Fig. S9†). Besides, the deuterium substituted substrate 1a was also applied to the TEC reaction to verify the structures. As a result (Fig. S10†), the ions of [Fe(III)TCPP–RS˙]+ (m/z 981.2) and [RSD–Fe(III)TCPP–O2]+ (m/z 1014.9) were observed, which further confirmed the intermediate structures. While with the TEC reaction carrying on, both intermediates of [Fe(III)TCPP–RS˙]+ (m/z 981.3) and [RSH–Fe(III)TCPP–O2]+ (m/z 1014.1) decreased (at 3 min, Fig. 3a-iii). Interestingly, the new ion of [Fe(III)TCPP–RSR1]+ (R1 = phenylethyl) (m/z 1085.9) was observed, which was attributed to the complex of product 3a and catalyst. The corresponding structure was identified by a CID experiment (Fig. S11†). Consequently, it can be deduced that the selective generation of RS˙ ([Fe(III)TCPP–RS˙]+) was related to the electron transfer within the intermediate of [RSH–Fe(III)TCPP–O2]+. Thereafter, another intermediate of [Fe(III)TCPP–RSR1]+ was obtained by the radical chain transfer along with the consumption of initial intermediates. | ||
| Fig. 3 Examination of intermediates during SET-based radical initiation. (a) Detection of the reaction system by AMS at different reaction time (R = 2,4-dimethylphenyl, R1 = phenylethyl). Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol) and Fe(III)Cl–TCPP (0.25 mol%) in 3 mL solvent (CH3CN:H2O = 10:1, v:v). (b) Dynamic monitoring of different ions. EICs of [Fe(III)TCPP]+ at m/z 844.5 (i), [1a + H]+ at m/z 139 (ii), [Fe(III)TCPP–RS˙]+ at m/z 981.3 (iii), [Fe(III)TCPP–3a]+ at m/z 1085.9 (iv) and [3a + H]+ at m/z 243 (v). (c) EPR spectra of the intermediate 1 [RSH–Fe(III)TCPP–O2]+. (d) and (e) Raman spectra of Fe(III)Cl–TCPP and intermediate 1 [RSH–Fe(III)TCPP–O2]+. (f) Two possible reaction mechanisms for the selective generation of RS˙ in different atmospheres. | ||
Subsequently, the dynamic changes of the intermediates and important species were examined by the online extracted ion chromatograms (EICs). As shown in Fig. 3b-i to ii, the reactant ion of [1a + H]+ (m/z 139) and catalyst ion of [Fe(III)TCPP]+ (m/z 844.5) decreased gradually. While the ion at m/z 981 ([Fe(III)TCPP–RS˙]+) increased gradually and reached a peak value within 1 min (Fig. 3b-iii), indicating the successful generation of RS˙. Thereafter, [Fe(III)TCPP–RS˙]+ began to decrease along with the simultaneous increase of the chain propagation intermediate of [Fe(III)TCPP–3a]+ (m/z 1086) and the final product of [3a + H]+ (m/z 243) (Fig. 3b-iv and v). Consequently, the dynamic changes of different species have confirmed the initial radical initiation, followed by chain propagation along with RS˙ consumption to obtain the final product.
To further examine the coordination between substrates and the active site of Fe(III), the reaction intermediates were characterized by EPR and Raman spectrometry. To avoid the effects of solvent coordination on the characterizations, the experiments were employed in the non-coordinating solvent of DCM. Firstly, the high-spin Fe(III) (S = 5/2) EPR signals of Fe(III)Cl–TCPP at g = 6.23, 4.29, 2.00 were detected, but these signals vanished after reaction in N2 (Fig. S12†).48,49 This confirmed the generation of Fe(II) (silent EPR signals) upon SET from substrate 1a to Fe(III), in accordance with the XPS data (Fig. 2c). Thereafter, the intermediates were examined by EPR analysis at low temperature, avoiding the rapid conversion of radical intermediates for better examinations. After adding substrate 1a to Fe(III)Cl–TCPP in air (Fig. 3c), the mixture was quickly frozen to −80 °C to obtain the significant EPR signals of low-spin Fe(III) (S = 1/2) (g = 2.38, 2.23, 1.92).50 Given that the intermediate of [RSH–Fe(III)TCPP–O2]+ (IN1) was generated by the coordination of Fe(III)–porphyrin with substrate 1a and O2 (demonstrated in Fig. 3a-ii), this intermediate could be assigned as the low-spin species.
The specific low-spin IN1 was further confirmed by low-temperature Raman spectrometry analysis. After adding substrate 1a into the catalyst system of Fe(III)Cl–TCPP in air (Fig. 3d), the oxidation and spin state marker bands of v4 and v2 exhibited a blue shift (from 1357 to 1365 and 1552 to 1561 cm−1). This indicated the formation of the low-spin structure for Fe(III)–porphyrin. In addition, some new peaks were observed in the low-frequency region (Fig. 3e), attributed to Fe(III)–S stretching vibrations (345 and 470 cm−1), Fe(III)–OO vibration (525 cm−1) and C–S stretching vibrations (619 and 774 cm−1).50,51 Briefly, the intermediate of IN1 was generated upon the coordination of Fe(III) (in iron porphyrin) with an S atom (in substrate 1) and O atom (in oxygen). This was further confirmed by the following theoretical calculations. Therefore, the thiyl radical initiation exhibited high selectivity upon the restriction of electron transfer sites from substrate 1a to Fe(III) by the coordination interaction.
Based on this, the mechanism of SET-based chain initiation is proposed in Fig. 3f. Initially, Fe(III)–porphyrin was coordinated with substrate 1a and O2 to afford a six-coordinate (6C) low-spin intermediate of [RSH–Fe(III)TCPP–O2]+ (IN1). Subsequently, upon the first SET process, the sulfhydryl of RSH transferred an electron to Fe(III) to generate the Fe(II)-complex (IN2), exhibiting the coordination of RS˙ and O2 with Fe(II)–porphyrin. Notably, the structure of IN2 was similar to the activated state of cytochrome P450 in a biological system.52 This could activate O2 to act as the final electron acceptor to oxidize Fe(II) via the second SET process. Therefore,  was generated along with the recovery of Fe(II) to Fe(III). However, in the absence of oxygen, the five-coordinate (5C) complex of [RSH–Fe(III)TCPP]+ (IN1′) was also at high spin states, formed by the interaction between thiolate (weak field ligand) and Fe(III)–porphyrin, which was further confirmed by the following theoretical calculations. Although the SET-based radical initiation was also employed, the catalyst failed to revert to its high valence state, thus impeding the continuous SET process with substrate 1a. Finally, the RS˙ was generated and coordinated with Fe(III)–porphyrin (IN3) or Fe(II)–porphyrin (IN3′) to undergo the subsequent chain propagation process.
was generated along with the recovery of Fe(II) to Fe(III). However, in the absence of oxygen, the five-coordinate (5C) complex of [RSH–Fe(III)TCPP]+ (IN1′) was also at high spin states, formed by the interaction between thiolate (weak field ligand) and Fe(III)–porphyrin, which was further confirmed by the following theoretical calculations. Although the SET-based radical initiation was also employed, the catalyst failed to revert to its high valence state, thus impeding the continuous SET process with substrate 1a. Finally, the RS˙ was generated and coordinated with Fe(III)–porphyrin (IN3) or Fe(II)–porphyrin (IN3′) to undergo the subsequent chain propagation process.
Theoretical calculations for exploring the effects of spin state switching on SET
To further examine the radical initiation to obtain RS˙ and explore the effects of spin states on electron transfer, theoretical calculations were employed using density functional theory (DFT). With an unsubstituted porphyrin and thiophenol as the models, the calculations were carried out at the BP86/D ef2-TZVP level. The geometrical parameters and electronic structures of IN1 (6C with O2) and IN1′ (5C without O2) were optimized with the Gaussian 09 program package.53 In the optimized structures of IN1 and IN1′, thiophenol and O2 acted as axial ligands to coordinate with Fe(III)–porphyrin (Fig. 4a). These respectively resulted in the adsorption energy of −1.76 and −0.85 eV, indicating their stable coordination to obtain intermediates. In addition, a difference of 0.04 Å in the Fe–S bond length resulted for optimized structures of IN1 and IN1′, which was attributed to the axial coordination of oxygen. Besides, the bond lengths of Fe–Npyr were 2.00 Å (for IN1) and 2.06 Å (for IN1′), which could be attributed to the low and high spin thiolate-bound complexes.50,54 Consequently, it can be deduced that the spin state of thiolate-coordinated Fe(III)–porphyrin was transformed to a low one with oxygen bound to the iron, which would facilitate the following reactions.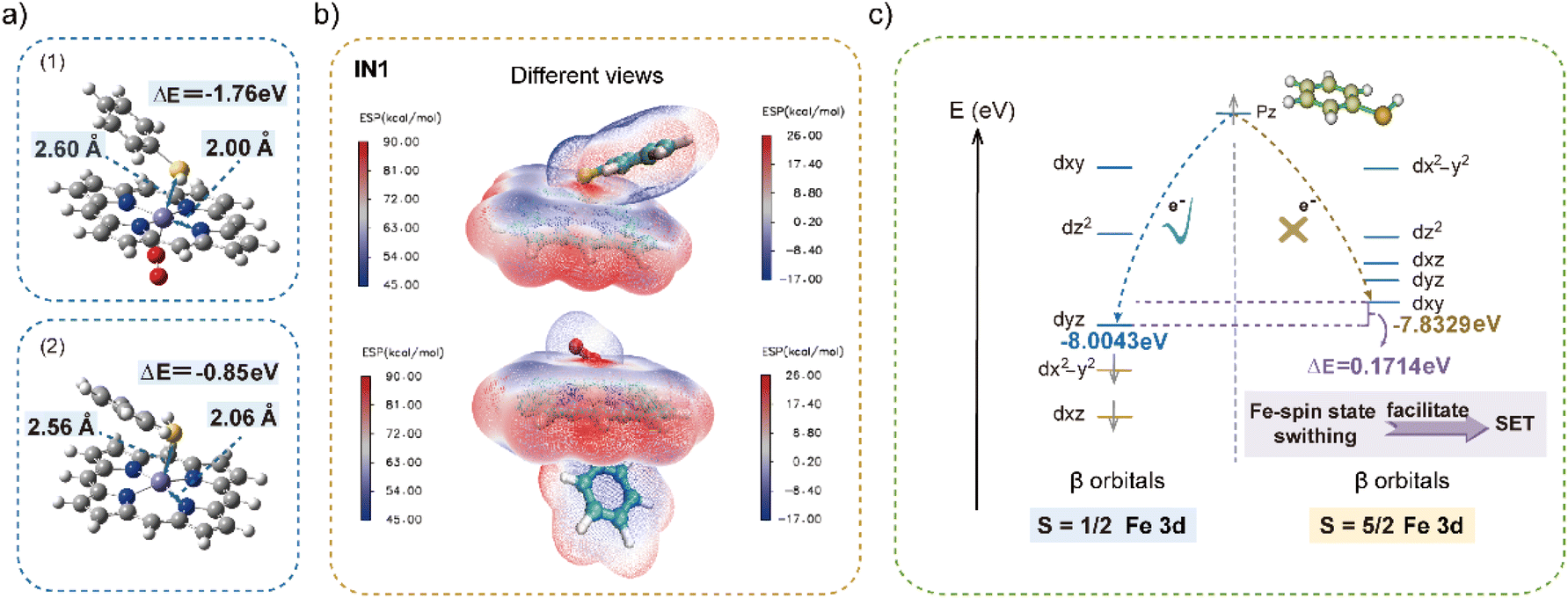 | ||
| Fig. 4 Theoretical calculations on the generation of RS˙. (a) DFT optimized structures of IN1 (1) and IN1′ (2). (b) Molecular electrostatic potentials of IN1. The left axis corresponds to the iron porphyrin and the right axis corresponds to the axial ligands. (c) Calculated Fe 3d orbitals of iron porphyrins with different spin states. The yellow lines represent the occupied 3d orbitals, and the blue lines represent the unoccupied 3d orbitals. | ||
Thereafter, to further reveal the charge distribution of intermediates, the molecular electrostatic potential (MEP) maps of IN1 were computed to examine the sites of SET.55,56 As shown in Fig. 4b and S13,† Fe(III) exhibited a positive potential with the red region, which indicated the ability of receiving electrons, whereas both regions of –SH and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in substrate 1 exhibited a negative potential (blue shed) for providing electrons. Consequently, upon the coordination between electron-rich sulfhydryl and Fe(III)–porphyrin, the electron would be transferred from the sulfhydryl group to Fe(III) for selective RS˙ generation. This was in accordance with the significantly reduced yield of TEC reaction in CH3OH, which acts as an axial ligand solvent with higher adsorption energy (−1.79 eV) than IN1 (−1.76 eV) (Fig. S14†). Therefore, CH3OH can act as a strong coordinating solvent to compete with sulfhydryl, coordinating with Fe(III)–porphyrin to impede SET. This could also be indirect proof for the SET-based radical initiation process.
C in substrate 1 exhibited a negative potential (blue shed) for providing electrons. Consequently, upon the coordination between electron-rich sulfhydryl and Fe(III)–porphyrin, the electron would be transferred from the sulfhydryl group to Fe(III) for selective RS˙ generation. This was in accordance with the significantly reduced yield of TEC reaction in CH3OH, which acts as an axial ligand solvent with higher adsorption energy (−1.79 eV) than IN1 (−1.76 eV) (Fig. S14†). Therefore, CH3OH can act as a strong coordinating solvent to compete with sulfhydryl, coordinating with Fe(III)–porphyrin to impede SET. This could also be indirect proof for the SET-based radical initiation process.
To further explore the effects of spin state and O2 coordination on SET-based radical initiation, the Fe 3d orbitals of Fe–porphyrins with different spin states and S p orbital of the substrate were calculated. Herein (Fig. 4c and S15†), the energy of the lowest unoccupied 3d orbital was calculated to evaluate the electron accepting ability in the first SET. Considering the alpha orbitals of Fe(III) with a high spin state were all occupied, the electron of sulfhydryl would be transferred into the beta orbital (E = −7.38 eV). While with O2 coordination, the energy of the lowest unoccupied beta orbital was −8.00 eV, which was lower than that of the alpha one (−7.21 eV). This induced the inclined entering of electrons into the unoccupied beta orbitals. From the thermodynamic point of view, the lower energy of the low spin state (−8.00 eV) than the high spin state (−7.38 eV) also suggested the favourable electron transfer from sulfhydryl to Fe(III). Therefore, the SET-based radical initiation of RS˙ would be further promoted in the presence of O2, facilitating the formation of 6C IN1 at a low spin state. While without O2, the 5C IN1′ in a high spin state was obtained, which limited the reaction, in accordance with the lower yield in N2. Therefore, O2 not only avoids the subsequent catalyst deactivation, but also plays a key role in the formation of IN1 at a low spin state to facilitate the SET-based initiation.
The mechanism of the SET-initiated TEC reaction
Based on the aforementioned experimental and theoretical examinations, the mechanism of SET-initiated TEC reaction in air can be proposed (Fig. 5). Initially, the axial ligands of substrate 1a and O2 were coordinated with the catalyst of Fe(III)TCPP ([Fe(III)TCPP]+ at m/z 844.5) to form IN1 at the low spin state ([RSH–Fe(III)TCPP–O2]+ at m/z 1014.1). Compared to IN1′ at the high spin state, the generated IN1 at the low spin state reduced the energy barrier of SET. In this way, the intramolecular selective SET was employed from sulfhydryl to Fe(III), which generated the intermediate complex of RS˙ and Fe(II) (IN2). Subsequently, Fe(II) in IN2 was oxidized by O2 via the second SET process, forming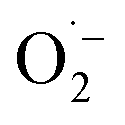 and another intermediate of IN3 ([Fe(III)TCPP–RS˙]+ at m/z 981.3). It should be noted that the generated
and another intermediate of IN3 ([Fe(III)TCPP–RS˙]+ at m/z 981.3). It should be noted that the generated  can participate in the oxidation of sulfur-containing compounds, leading to the formation of sulfoxides (Fig. 1b, m/z 259) at a relatively low abundance. Thereafter, the chain propagation would be initiated through a two-step SET-based radical initiation. This involves radical addition to the –CC bond in substrate 2a, generating a carbon-centered radical (IN4) that abstracts an H atom from another molecule of substrate 1a. This induced the generation of hydrothiolated intermediate IN5 ([Fe(III)TCPP–3a]+ at m/z 1085.9) and another equivalent of RS˙. Finally, the product ([3a + H]+ at m/z 243) was desorbed from the catalyst to complete the TEC reaction.
can participate in the oxidation of sulfur-containing compounds, leading to the formation of sulfoxides (Fig. 1b, m/z 259) at a relatively low abundance. Thereafter, the chain propagation would be initiated through a two-step SET-based radical initiation. This involves radical addition to the –CC bond in substrate 2a, generating a carbon-centered radical (IN4) that abstracts an H atom from another molecule of substrate 1a. This induced the generation of hydrothiolated intermediate IN5 ([Fe(III)TCPP–3a]+ at m/z 1085.9) and another equivalent of RS˙. Finally, the product ([3a + H]+ at m/z 243) was desorbed from the catalyst to complete the TEC reaction.
 | ||
| Fig. 5 The mechanism of the SET-initiated TEC reaction. | ||
Besides, the generated RS˙ can be directly captured by the catalyst to obtain IN3 upon the coordination between Fe(III)TCPP and RS˙, fulfilling another run of the chain propagation. Upon this sizable Fe(III)TCPP-based coordination, bulky RS˙ species with increased steric hindrance would endow active RS˙ with high stability for subsequent reactions. It should be noted that a low abundance of disulfide product was also exhibited even in an N2 atmosphere (Fig. S16†). Consequently, the homo-coupling of bulky RS˙ species (IN3) would be restricted due to the steric hindrance, which would facilitate the coupling with the vinyl reactants. Therefore, the side reactions would be avoided upon the stabilization of radicals by the Fe(III)TCPP-based coordination, which was confirmed by the decreased yield and conversion with free Fe(III) as the catalyst (Fig. S17†). Consequently, the controllable SET-based initiation of RS˙ upon axial-ligand-induced switching of spin states and the subsequent selective chain propagation were revealed.
Conclusions
In summary, axial-ligand-induced switching of spin states in an Fe(III)-catalyzed TEC reaction was revealed, which facilitated the controllable generation of RS˙ via an SET process. As demonstrated by AMS-based characterization and other examinations, RS˙ was demonstrated to be selectively and controllably generated via SET between substrate 1 and Fe(III)–porphyrin. The role of the axial ligand in inducing switching of the transition metal's spin states was revealed, which facilitated RS˙ radical initiation and subsequent reactions. With O2 as another axial ligand, the thiolate-coordinated Fe(III)–porphyrin (IN1′ S = 5/2) was transformed to a low spin state (IN1 S = 1/2), dramatically lowering the energy barrier of SET-based radical generation. This well explains the efficient and selective C–S coupling in air. Subsequently, upon coordination with the bulky Fe(III)–porphyrin, the RS˙ species (IN3) selectively coupled with the vinyl reactant for efficient TEC reactions. Consequently, based on AMS monitoring and comprehensive examinations, efficient and selective TEC reactions originated from spin-regulated SET-based RS˙ initiation. This work not only holds promise for efficient and selective radical reactions upon manipulating spin states but also broadens the applications of AMS for in-depth mechanism examinations.Data availability
All data of this study have been presented in the manuscript and ESI.†Author contributions
J. Qin and Y. Yin conceived and designed the project; X. Guan contributed to the characterization of products; J. Qin, Y. Yin and X. Ge conducted the calculations; M. Cao and J. Ouyang supported figure preparation. N. Na directed the whole project and acquired the funding. All authors have given approval to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 22474010 and 22274012), the National Key Research and Development Program of China (2024YFA1509600) and the Fundamental Research Funds for the Central Universities (No. 2233300007).Notes and references
- S. Wang, S. Tang and A. Lei, Sci. Bull., 2018, 63, 1006–1009 CrossRef CAS PubMed.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Chem. Soc. Rev., 2019, 48, 2615–2656 RSC.
- W.-C. C. Lee, D.-S. Wang, Y. Zhu and X. P. Zhang, Nat. Chem., 2023, 15, 1569–1580 CrossRef CAS PubMed.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- P. Xu, J. Xie, D.-S. Wang and X. P. Zhang, Nat. Chem., 2023, 15, 498–507 CrossRef CAS PubMed.
- Z. Huang, Y. Yang, J. Mu, G. Li, J. Han, P. Ren, J. Zhang, N. Luo, K.-L. Han and F. Wang, Chin. J. Catal., 2023, 45, 120–131 CrossRef CAS.
- H.-M. Huang, P. Bellotti and F. Glorius, Chem. Soc. Rev., 2020, 49, 6186–6197 RSC.
- Q. Zhou, M. Chin, Y. Fu, P. Liu and Y. Yang, Science, 2021, 374, 1612–1616 CrossRef CAS PubMed.
- M. Chierchia, P. Xu, G. J. Lovinger and J. P. Morken, Angew. Chem., Int. Ed., 2019, 58, 14245–14249 CrossRef CAS PubMed.
- A. Dahiya and B. K. Patel, Chem. Rec., 2021, 21, 3589–3612 CrossRef CAS PubMed.
- X. Wang, J. He, Y.-N. Wang, Z. Zhao, K. Jiang, W. Yang, T. Zhang, S. Jia, K. Zhong, L. Niu and Y. Lan, Chem. Rev., 2024, 124, 10192–10280 CrossRef CAS PubMed.
- M. Mato, D. Spinnato, M. Leutzsch, H. W. Moon, E. J. Reijerse and J. Cornella, Nat. Chem., 2023, 15, 1138–1145 CrossRef CAS PubMed.
- S. Chen, X. Liang, S. Hu, X. Li, G. Zhang, S. Wang, L. Ma, C.-M. L. Wu, C. Zhi and J. A. Zapien, Nano-Micro Lett., 2023, 15, 47 CrossRef CAS PubMed.
- C. Zhang, X. Wang, Z. Ma, H. Yao, H. Liu, C. Li, J. Zhou, R. Xu, X. Zheng, H. Wang, Q. Li, M. Gu, H. Jiang and M. Huang, Sci. Bull., 2023, 68, 2042–2053 CrossRef CAS PubMed.
- F. He, Q. Zheng, X. Yang, L. Wang, Z. Zhao, Y. Xu, L. Hu, Y. Kuang, B. Yang, Z. Li, L. Lei, M. Qiu, J. Lu and Y. Hou, Adv. Mater., 2023, 35, 2304022 CrossRef CAS PubMed.
- G.-Z. Huang, Y.-S. Xia, F. Yang, W.-J. Long, J.-J. Liu, J.-P. Liao, M. Zhang, J. Liu and Y.-Q. Lan, J. Am. Chem. Soc., 2023, 145, 26863–26870 CrossRef CAS PubMed.
- Y. Zhang, Q. Wu, J. Z. Y. Seow, Y. Jia, X. Ren and Z. J. Xu, Chem. Soc. Rev., 2024, 53, 8123–8136 RSC.
- K. Sun, Y. Huang, Q. Wang, W. Zhao, X. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2024, 146, 3241–3249 CrossRef CAS PubMed.
- Q. Zhao, M. Zhang, Y. Gao, H. Dong, L. Zheng, Y. Zhang, J. Ouyang and N. Na, J. Am. Chem. Soc., 2024, 146, 14875–14888 CrossRef CAS PubMed.
- B. H. Northrop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef CAS PubMed.
- R. Kumar, Saima, A. Shard, N. H. Andhare, Richa and A. K. Sinha, Angew. Chem., Int. Ed., 2015, 54, 828–832 CrossRef CAS PubMed.
- Q. Xiao, H. Zhang, J.-H. Li, J.-X. Jian, Q.-X. Tong and J.-J. Zhong, Org. Lett., 2021, 23, 3604–3609 Search PubMed.
- R.-J. Zhang, X.-R. Li, R.-B. Liang, Y. Xiao, Q.-X. Tong, J.-J. Zhong and L.-Z. Wu, Org. Lett., 2024, 26, 591–596 CrossRef CAS PubMed.
- Z. Wu and D. A. Pratt, Nat. Rev. Chem., 2023, 7, 573–589 CrossRef PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS PubMed.
- N. Taniguchi, ChemistrySelect, 2018, 3, 6209–6213 CrossRef CAS.
- A. K. Sinha and D. Equbal, Asian J. Org. Chem., 2019, 8, 32–47 CrossRef CAS.
- S. Mondal, F. Dumur, D. Gigmes, M. P. Sibi, M. P. Bertrand and M. Nechab, Chem. Rev., 2022, 122, 5842–5976 CrossRef CAS PubMed.
- Y. Wang, Y. Zhou, W. Sun, X. Wang, J. Yao and H. Li, Adv. Sci., 2024, 11, 2402890 CrossRef CAS PubMed.
- H. Yang, E. C. McDaniel, S. Impano, A. S. Byer, R. J. Jodts, K. Yokoyama, W. E. Broderick, J. B. Broderick and B. M. Hoffman, J. Am. Chem. Soc., 2019, 141, 12139–12146 CrossRef CAS PubMed.
- P. J. H. Williams, G. A. Boustead, D. E. Heard, P. W. Seakins, A. R. Rickard and V. Chechik, J. Am. Chem. Soc., 2022, 144, 15969–15976 CrossRef CAS PubMed.
- W. G. Walls, A. L. Vagstad, T. Delridge, J. Piel, W. E. Broderick and J. B. Broderick, J. Am. Chem. Soc., 2024, 146, 5550–5559 CrossRef CAS PubMed.
- X. Ge, Y. Yin, J. Sun, J. Ouyang and N. Na, Chem. Sci., 2023, 14, 2229–2236 RSC.
- H. Lu, Y. Yin, J. Sun, W. Li, X. Shen, X. Feng, J. Ouyang and N. Na, Chin. Chem. Lett., 2021, 32, 3457–3462 CrossRef CAS.
- S. Jin, H. Chen, X. Yuan, D. Xing, R. Wang, L. Zhao, D. Zhang, C. Gong, C. Zhu, X. Gao, Y. Chen and X. Zhang, JACS Au, 2023, 3, 1563–1571 CrossRef CAS PubMed.
- X. Li, X. Nong, C. Zhu, X. Gao, H. Chen, X. Yuan, D. Xing, L. Liu, C. Liang, D. Zang and X. Zhang, J. Am. Chem. Soc., 2024, 146, 29267–29271 CrossRef CAS PubMed.
- K. Mittra, A. Singha and A. Dey, Inorg. Chem., 2017, 56, 3916–3925 CrossRef CAS PubMed.
- N. Taniguchi and K. Kitayama, Synlett, 2018, 29, 2712–2716 CrossRef CAS.
- C. Xu, Y. Pan, G. Wan, H. Liu, L. Wang, H. Zhou, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2019, 141, 19110–19117 CrossRef CAS PubMed.
- S. Guo, G. Zhang, Y. Guo and J. C. Yu, Carbon, 2013, 60, 437–444 Search PubMed.
- L. Geng, M. Zhang, W. Zhang, M. Jia, W. Yan and G. Liu, Catal. Sci. Technol., 2015, 5, 3097–3102 RSC.
- Q. Zhao, L. Zheng, Y. Gao, J. Li, J. Wei, M. Zhang, J. Sun, J. Ouyang and N. Na, J. Am. Chem. Soc., 2023, 145, 12586–12600 CrossRef CAS PubMed.
- H. Di Wang, P. J. Pagano, Y. Du, A. J. Cayatte, M. T. Quinn, P. Brecher and R. A. Cohen, Circ. Res., 1998, 82, 810–818 CrossRef CAS PubMed.
- H.-S. Hsieh, R. Wu and C. T. Jafvert, Environ. Sci. Technol., 2014, 48, 11330–11336 CrossRef CAS PubMed.
- A. Gemenetzi, P. Stathi, Y. Deligiannakis and M. Louloudi, Chem. Phys. Lett., 2021, 764, 138282 CrossRef CAS.
- W. Shi, L. Cao, H. Zhang, X. Zhou, B. An, Z. Lin, R. Dai, J. Li, C. Wang and W. Lin, Angew. Chem., Int. Ed., 2017, 56, 9704–9709 CrossRef CAS PubMed.
- P. K. Das, S. Chatterjee, S. Samanta and A. Dey, Inorg. Chem., 2012, 51, 10704–10714 CrossRef CAS PubMed.
- S. Samanta, S. Sengupta, S. Biswas, S. Ghosh, S. Barman and A. Dey, J. Am. Chem. Soc., 2023, 145, 26477–26486 CrossRef CAS PubMed.
- N. Ueyama, N. Nishikawa, Y. Yamada, T. Okamura and A. Nakamura, J. Am. Chem. Soc., 1996, 118, 12826–12827 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- N. Suzuki, T. Higuchi, Y. Urano, K. Kikuchi, H. Uekusa, Y. Ohashi, T. Uchida, T. Kitagawa and T. Nagano, J. Am. Chem. Soc., 1999, 121, 11571–11572 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Zhang and T. Lu, Phys. Chem. Chem. Phys., 2021, 23, 20323–20328 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02194d |
| This journal is © The Royal Society of Chemistry 2025 |