
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic Ni-doped ZrO2 with subnanometric Fe clusters for tandem C–C bond cleavage and coupling†
Xin Zhaoa,
Jie Wena,
Qian Qiangbc,
Dawang Tanga,
Fengliang Wanga,
Ruiqi Fang *a,
Changzhi Libc and
Yingwei Li*a
*a,
Changzhi Libc and
Yingwei Li*a
aState Key Laboratory of Pulp and Paper Engineering, Guangdong Provincial Key Laboratory of Fuel Cell Technology, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: fangrq@scut.edu.cn; liyw@scut.edu.cn
bCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 14th May 2025
Abstract
The sustainable valorization of lignin β-O-4 compounds into high-value natural products through one-pot tandem catalysis presents an urgent yet scientifically challenging frontier in biomass conversion. Herein, we report a mesoporous Fe3@Ni1-ZrO2 catalyst featuring subnanometric Fe–O clusters anchored on atomic Ni-doped ZrO2 nanosheets. This engineered architecture enables the one-pot tandem conversion of lignin β-O-4 segments to flavones under aerobic and base-free conditions, delivering 56.2% yield with a space-time yield (STY) of 3.3 g gcat−1 h−1 in continuous flow operation. Moreover, the system demonstrates exceptional substrate versatility through efficient conversion of diverse lignin β-O-4 dimers and substituted 2′-phenoxyacetophenones into bioactive flavones. Mechanistic investigations combining controlled experiments and density functional theory (DFT) calculations reveal a cooperative catalytic mechanism, i.e., ZrO2 nanosheets mediate selective oxidative cleavage of C–C bonds in β-O-4 segments, and subnanometric Fe3 clusters activate aldol condensation of cleavage intermediates, while atomic Ni sites suppress competing pathways to govern the selectivity. This synergistic interplay within the Fe3@Ni1-ZrO2 framework establishes a robust catalytic microenvironment to enable a high-efficiency tandem process.
Introduction
Lignin is a major component of lignocellulosic biomass, constituting approximately 30% of organic carbon reserves on Earth.1–3 As the largest and only renewable aromatic feedstock,4–7 it features a complex and recalcitrant polymeric structure consisting of multiple methoxylated phenylpropane subunits interconnected by diverse C–O and C–C bonds (β-O-4, 5–5 linkages, etc.).8–12 Typical lignin valorization strategies prioritize C–O bond cleavage to acquire various phenolic chemicals,13–18 whereas C–C bond scission remains a formidable challenge due to high bond dissociation energies. In order to improve the overall atomic utilization efficiency and biorefinery economics, selective cleavage of the C–C bond has emerged as a critical frontier. The β-O-4 linkage is the most frequent motif in lignin and integrates both C–O and C–C bonds,19–23 making it an ideal model system for elucidating bond cleavage mechanisms and guiding lignin valorization, as demonstrated by recent elegant progress.24–27Flavonoids are a class of plant-derived phenolic metabolites that exhibit potent antioxidant properties and versatile bioactivities against cancer, cardiovascular diseases, and aging.28–31 Currently, flavonoid production relies predominantly on plant extraction,32 yet low biosynthesis rates and limited natural abundance fail to meet escalating pharmaceutical demands. Consequently, efficient artificial synthesis of flavonoids from renewable resources is urgently needed but remains largely unexplored.33 On account of the functionalized aromatic skeleton of lignin β-O-4 segments in the structure, they offer a structurally congruent feedstock for flavonoid synthesis, which is promising to achieve a value-added biorefinery concept. In order to realize this promising transformation, a multi-step process including two sections is required, i.e., β-O-4 segment depolymerization to aromatic monomers and a coupling reaction to assemble flavonoid oxygen-containing heterocycles. To optimize synthetic efficiency, process economics and product quality, the development of one-pot systems catalyzed by a robust catalyst featuring multifunctionality and high atom utilization is undoubtedly the most promising choice.
In this work, we present a three-dimensional (3D) superstructure catalyst featuring subnanometric Fe–O clusters anchored on atomically dispersed Ni-doped ZrO2 mesoporous nanosheets (denoted as Fe3@Ni1-ZrO2). This precisely engineered configuration achieves one-pot multistep conversion of lignin β-O-4 segments into flavones under an air atmosphere without requiring acidic or alkaline additives. Mechanistic investigations combining controlled experiments with density functional theory (DFT) calculations reveal the unique compositional features and structural advantages of Fe3@Ni1-ZrO2. As illustrated in Scheme 1, the reaction initiates with ZrO2 nanosheets mediating selective C–C bond oxidative cleavage in β-O-4 segments (e.g., 2-phenoxy-1-phenylethanol), generating the benzaldehyde intermediate. Subsequently, the subnanometric Fe3 clusters activate aldol condensation between the cleavage intermediate and 2′-hydroxyacetophenone to form unsaturated carbonyl intermediates, while atomic Ni sites suppress competing pathways to govern the selectivity. Finally, a concerted cyclization-dehydrogenation process affords the synthesis of flavone. Fe3@Ni1-ZrO2 achieves 56.2% flavone yield with approximately 100% C–C bond cleavage selectivity while demonstrating broad substrate compatibility across diverse β-O-4 dimers and substituted 2′-phenoxyacetophenones, producing various flavonoids in 43.3–60.2% yields.
 | ||
| Scheme 1 Schematic illustration of the one-pot tandem reaction for upgrading lignin β-O-4 segments into flavones and the application of flavonoids. | ||
Results and discussion
Synthesis and characterization of Fe3@Ni1-ZrO2
Fig. 1a schematically illustrates the synthesis routes of Fe3@Ni1-ZrO2. PCN-222(Fe/Ni) was first synthesized via a solvothermal method. The X-ray diffraction (XRD) patterns of the as-synthesized PCN-222(Fe/Ni), PCN-222(Fe), PCN-222(Ni), and pristine PCN-222 exhibit high crystallinity, in good accordance with the literature (Fig. S1†).34 Field-emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) images reveal that the PCN-222(Fe/Ni) crystals possess a uniform spindle morphology with a size of ca. 1 μm, a solid structure, and smooth external surfaces (Fig. S2†). Then, PCN-222(Fe/Ni) was pyrolyzed at 600 °C (based on thermogravimetric analysis (TGA) results, Fig. S3†) to synthesize Fe3@Ni1-ZrO2. | ||
| Fig. 1 (a) Schematic illustration of the synthesis route of Fe3@Ni1-ZrO2. (b and c) TEM, (d) corresponding element mapping, (e and f) HAADF-STEM, (g) SAED pattern, (h and i) AC HAADF-STEM, and (j) element mapping in high-resolution scale images of Fe3@Ni1-ZrO2. | ||
SEM (Fig. S4a†) and TEM (Fig. 1b and c) images of Fe3@Ni1-ZrO2 reveal a well-preserved spindle morphology with uniform size but rough surfaces, attributed to the penetration of random nanosheets into both the interior and surface. The aberration-corrected high-angle annular dark-field scanning TEM (HAADF-STEM) image (Fig. S4b†) and the corresponding energy-dispersive X-ray spectroscopy (EDS) elemental mappings demonstrate the homogeneous distribution of Zr, O, Fe, and Ni elements throughout the architecture, with no obvious agglomeration (Fig. 1d). This observation is further supported by elemental line-scan profiles (Fig. S4c and d†). HAADF-STEM images of localized regions (Fig. 1e and f) unambiguously uncover the mesoporous structure of the ZrO2 nanosheets. Additionally, N2 adsorption–desorption isotherms (Fig. S5†) of Fe3@Ni1-ZrO2 exhibit a specific surface area of 377.8 m2 g−1, while pore size distribution curves indicate the presence of mesopores ranging predominantly from 2 to 40 nm. The selected-area electron diffraction (SAED) patterns (Fig. 1g) display distinct spotty diffraction rings, indicating the single-crystalline nature of the ZrO2 nanosheets. The spherically aberration-corrected HAADF-STEM (AC HAADF-STEM) image (Fig. 1h) resolves atomically dispersed Ni atoms (isolated dark dots) within the ZrO2 lattice and sub-nanometer Fe clusters (Fig. 1i). Moreover, high-resolution elemental mappings (Fig. 1j) corroborate the atomic dispersion of Ni species and the localized Fe clusters. Finally, characterization of the counterparts (Fe3@ZrO2, Ni1-ZrO2, and ZrO2) confirms their analogous structural features (Fig. S6–S8†).
The XRD patterns of Fe3@Ni1-ZrO2 (Fig. 2a) exhibit only weak crystalline phases of monoclinic ZrO2 (m-ZrO2), which can be attributed to its two-dimensional (2D) sheet-like morphology. Furthermore, no characteristic peaks corresponding to metallic Fe or Ni are detected, possibly due to their atomic-scale dispersion. Inductively coupled plasma optical emission spectrometer (ICP-OES) analysis (Table S1†) reveals that the Zr, Fe, and Ni contents in Fe3@Ni1-ZrO2 are 26.8, 4.4, and 0.8 wt%, respectively. In addition, the XRD patterns (Fig. 2a) do not show clear characteristic peaks of carbon due to its amorphous nature in Fe3@Ni1-ZrO2. X-ray photoelectron spectroscopy (XPS) analysis of C 1s (Fig. S9†) indicates that carbon species predominantly exist as C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, accompanied by a small amount of O–CO groups. In the Zr 3d region (Fig. 2b), the characteristic doublet peaks of Zr4+ are observed at binding energies of 183.16 and 185.52 eV. Notably, the Zr4+ peaks in Fe3@Ni1-ZrO2 exhibit a distinct shift toward higher binding energy (ca. 0.49 eV), suggesting electron transfer from Zr to Fe or Ni atoms. A similar shift is observed for Zr4+ in both Fe3@ZrO2 and Ni1-ZrO2, with the latter showing a more pronounced shift, possibly due to the doping effect of Ni atoms in the ZrO2 lattice. The high-resolution O 1s XPS spectrum (Fig. 2c) is deconvoluted into three components at 530.4, 531.83, and 533.0 eV, corresponding to lattice oxygen (metal–O bonds), oxygen vacancies, and surface-adsorbed oxygen species, respectively.35–37
O bonds, accompanied by a small amount of O–CO groups. In the Zr 3d region (Fig. 2b), the characteristic doublet peaks of Zr4+ are observed at binding energies of 183.16 and 185.52 eV. Notably, the Zr4+ peaks in Fe3@Ni1-ZrO2 exhibit a distinct shift toward higher binding energy (ca. 0.49 eV), suggesting electron transfer from Zr to Fe or Ni atoms. A similar shift is observed for Zr4+ in both Fe3@ZrO2 and Ni1-ZrO2, with the latter showing a more pronounced shift, possibly due to the doping effect of Ni atoms in the ZrO2 lattice. The high-resolution O 1s XPS spectrum (Fig. 2c) is deconvoluted into three components at 530.4, 531.83, and 533.0 eV, corresponding to lattice oxygen (metal–O bonds), oxygen vacancies, and surface-adsorbed oxygen species, respectively.35–37
 | ||
| Fig. 2 (a) XRD patterns and XPS spectra in (b) the Zr 3d region and (c) the O 1s region of Fe3@Ni1-ZrO2, Fe3@ZrO2, Ni1-ZrO2, and ZrO2. (d) XANES spectra at the Fe K-edge, (e) Fourier transform (FT) at the Fe K-edge, (f) corresponding EXAFS fitting curves of Fe3@ZrO2 (Fe) and Fe3@Ni1-ZrO2 (Fe) in R-space, (g) XANES spectra at the Ni K-edge, (h) Fourier transform (FT) at the Ni K-edge, (i) corresponding EXAFS fitting curves of Ni1-ZrO2 (Ni) and Fe3@Ni1-ZrO2 (Ni) in R-space, and (j) WT k3-weighted EXAFS spectra. | ||
The chemical states and coordination environments of atomic Fe and Ni species in Fe3@Ni1-ZrO2 and the counterparts (Fe3@ZrO2 and Ni1-ZrO2) were further investigated using XPS and X-ray absorption fine spectroscopy (XAFS, Fig. 2d–j, S10–S15, and Tables S2 and S3†).38 The Fe K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. 2d, S10a, b and S11a–c†) show that the absorption threshold position is located between FeO and Fe2O3, indicating an intermediate oxidation state of Fe between +2 and +3 in both Fe3@Ni1-ZrO2 and Fe3@ZrO2. This observation is corroborated by high-resolution Fe 2p XPS spectra (Fig. S12a†), and the Fe 2p3/2 peak at 711.04 eV corresponds to positively charged Fe species (close to Fe2+). Fourier transformed (FT) Fe K-edge extended X-ray absorption fine structure (EXAFS) analysis of k3-weighted spectra in R-space (Fig. 2e) reveals a dominant peak centered at 1.46 Å, assigned to the first-shell Fe–O coordination. This suggests that the local Fe coordination in Fe3@Ni1-ZrO2 and Fe3@ZrO2 is similar to that of the Fe2O3 reference. A minor second-shell Fe–Fe correlation peak is observed at 2.61 Å for Fe3@Ni1-ZrO2 and 2.66 Å for Fe3@ZrO2, with the shorter distance in the former likely attributable to enhanced electron transfer between Fe3 and Ni1-ZrO2 after Ni doping. Quantitative fitting (Fig. 2f and Table S2†) further confirms that Fe3@Ni1-ZrO2 exhibits an Fe–Fe coordination number of ca. 3 (lower than the Fe2O3 reference), an average Fe–Fe bond length of 3.08 Å (much shorter than the Fe2O3 reference), and an average Fe–O bond length of 1.98 Å (comparable to the Fe2O3 reference).39 These findings are consistent with wavelet transform (WT) analysis (Fig. 2j and S13a†), which identifies Fe–O and Fe–Fe bonding features, consistent with the EXAFS results.
The Ni K-edge XANES spectra of Fe3@Ni1-ZrO2 and Ni1-ZrO2 (Fig. 2g, S10c, d and S11d–f†) disclose a +2 oxidation state for Ni. The high-resolution Ni 2p XPS spectra (Fig. S12b†) show that the Ni 2p3/2 peaks of both Fe3@Ni1-ZrO2 and Ni1-ZrO2 are located at 855.11 eV, which is assigned to Ni2+. In the Ni K-edge FT-EXAFS spectra (Fig. 2h), one dominant peak at 1.31 Å (phase-uncorrected) is observed in both Fe3@Ni1-ZrO2 and Ni1-ZrO2, attributed to the Ni–O coordination shell. The quantitative least-squares EXAFS curve-fitting analysis (Fig. 2i) reveals a phase-corrected Ni–O bond length of 1.92 Å in both Fe3@Ni1-ZrO2 and Ni1-ZrO2, which is shorter than that of the NiO reference (2.09 Å), indicative of lattice distortion due to Ni2+ doping into ZrO2. Moreover, the absence of peaks near ca. 2.13 Å (metallic Ni–Ni in Ni foil) or 2.95 Å (NiO reference second shell) confirms the atomic dispersion of Ni species in Fe3@Ni1-ZrO2 and Ni1-ZrO2. Furthermore, the WT of Ni K-edge EXAFS oscillations (Fig. 2j and S13b†) displays a prominent intensity maximum at approximately 3.9 Å−1, corresponding to the Ni–O coordination environment, which further excludes metallic Ni contributions.
Based on these results, optimized structural models of Fe3@Ni1-ZrO2 and related counterparts (i.e., Fe3@ZrO2, Ni1-ZrO2, and ZrO2) were constructed (Fig. S14†). Charge density difference plots reveal distinct electronic redistributions, i.e., in pristine ZrO2 (Fig. S15d†), electron accumulation around O atoms and depletion around Zr atoms. After Ni-doping to obtain Ni1-ZrO2, the charge density around O atoms reduces slightly, while mild electron depletion occurs around Ni atoms (Fig. S15c†). Bader charge analysis indicates that each Ni atom carries an average charge of +0.361|e|, whereas the charges on Zr (+2.52|e| vs. 2.42|e| in pristine ZrO2) and O (−1.36|e| vs. −1.38|e| in pristine ZrO2) atoms increase, confirming electron transfer from Ni to ZrO2. For Fe3@Ni1-ZrO2 (Fig. S15a†), the average charge on each Ni atom decreases significantly to +0.027|e| (compared to +0.361|e| in Ni1-ZrO2), while each Fe atom carries an average charge of +0.262|e|. Specifically, individual Fe atoms exhibit charges of +0.556|e| (1-Fe), −0.112|e| (2-Fe), and +0.333|e| (3-Fe), respectively. In contrast, Fe atoms in Fe3@ZrO2 (Fig. S15b†) carry charges of −0.29|e| (1-Fe), −0.877|e| (2-Fe), and −0.492|e| (3-Fe), respectively, with an average of −0.553|e|. These results suggest charge transfer from the Fe3 cluster to the Ni atom in Fe3@Ni1-ZrO2. Furthermore, the projected density-of-states (pDOS) demonstrates enhanced electronic states near the Fermi level for all models, particularly in Fe3@Ni1-ZrO2 (Fig. S16†).
Catalytic performance of Fe3@Ni1-ZrO2
Next, the as-prepared Fe3@Ni1-ZrO2 was subsequently employed for upgrading lignin β-O-4 segments into flavonoid natural products. A one-pot cascade conversion of 2-phenoxy-1-phenylethanol (1a) and 2′-hydroxyacetophenone (1b) to flavone (6a) was investigated as a model reaction (Fig. 3a). The proposed mechanism involves four key steps. Specifically, 1a first undergoes oxidation to 2′-phenoxyacetophenone (2a) and subsequent C–C bond cleavage to generate the key intermediate, i.e., benzaldehyde (3a). Another product is anisole that consistently exists in the catalytic system and is not involved in any subsequent reactions, whose yield is shown in Table S4.† Next, 3a reacts with 1b to obtain 2′-hydroxychalcone (4a) through a C–C coupling reaction, which then undergoes intramolecular self-cyclization to form a flavanone (5a). Finally, 5a evolves into the target product flavone (6a) through oxidative dehydrogenation. | ||
| Fig. 3 (a) Possible reaction route. (b) Catalytic performance of different catalysts. Reaction conditions: 2-phenoxy-1-phenylethanol (1a, 1 mmol), 2′-hydroxyacetophenone (1b, 1.2 mmol), catalyst (0.05 g), toluene (3 mL), 150 °C, 30 h, and air (0.5 MPa). (c) Conversion and yield as a function of reaction time in the proposed one-pot cascade catalytic system over Fe3@Ni1-ZrO2. Reaction conditions: 2-phenoxy-1-phenylethanol (1a, 1 mmol), 2′-hydroxyacetophenone (1b, 1.2 mmol), catalyst (0.05 g), toluene (3 mL), 150 °C, and air (0.5 MPa). (d) Control experiments using 2-phenoxy-1-phenylethanol (1a) as the only substrate. Reaction conditions: 2-phenoxy-1-phenylethanol (1a, 1 mmol), catalyst (0.05 g), toluene (3 mL), 150 °C, 30 h, and air (0.5 MPa). (e) Control experiments using 3a and 1b as the substrates over Fe3@Ni1-ZrO2. (f) Control experiments using 2b and 1b as the substrates over Fe3@Ni1-ZrO2. (g) Control experiments using 3a and 2b as the substrates. | ||
Under optimized conditions (150 °C, 0.5 MPa air, and 30 h), Fe3@Ni1-ZrO2 affords 56.2% yield of flavone (6a) at 78.1% conversion of 1a (Fig. 3b and Table S4†), with a turnover number (TON) of 4.04 (Table S4†). The time-tracking experiment (Fig. 3c and Table S5†) revealed sequential formation and consumption of intermediates 2a (maximum yield at 18 h) and 3a (maximum yield at 24 h), consistent with a stepwise pathway involving oxidation of 1a to 2a, followed by C–C cleavage to 3a. Notably, no C–O cleavage by-products were detected. Control experiments (Fig. 3b and Table S4†) confirmed the necessity of both the catalyst and air (as the terminal oxidant), with reactive oxygen species (ROS) identified as superoxide radicals (˙O2−) via electron paramagnetic resonance (EPR) analysis (Fig. S17†).
To benchmark the performance of Fe3@Ni1-ZrO2, control experiments were conducted with its counterparts (Fe3@ZrO2, Ni1-ZrO2, and ZrO2) under identical reaction conditions (Fig. 3b and Table S4†). Fe3@ZrO2 achieved 53.6% conversion of 2-phenoxy-1-phenylethanol (1a) with 85.0% selectivity toward C–C bond cleavage, yielding only 6.3% flavone (6a) alongside 13.9% benzyl benzoate (3b). The latter by-product is probably formed via esterification of 2b with benzoic acid, generated through oxidation of intermediate 3a (dashed pathway in Fig. 3a). Ni1-ZrO2 and ZrO2 afforded 62.1% and 33.6% conversion of 2-phenoxy-1-phenylethanol (1a), respectively, but failed to produce flavone (6a). Notably, Ni1-ZrO2 exhibited 100% selectivity toward C–C bond cleavage, whereas ZrO2 showed 37.3% selectivity. These results highlight the critical role of both sub-nanometric Fe3 clusters and atomically dispersed Ni species in enhancing the catalytic activity of ZrO2 (validated via control experiments and DFT calculations; Fig. 3d and 4). Specifically, sub-nanometric Fe3 clusters uniquely drive flavone synthesis, while atomic Ni sites optimize selectivity by suppressing side reactions (further supported by control experiments and DFT calculations; Fig. 3d and 4). In addition, commercially available bulk ZrO2 and impregnation-calcination-derived Ni/Fe-ZrO2 (see the ESI† for synthesis details) were tested but showed negligible activity (Fig. 3b), underscoring the superiority of the elaborately engineered Fe3@Ni1-ZrO2.
 | ||
| Fig. 4 (a–c) Free energy diagram of the one-pot oxidative tandem reaction for synthesis of flavone from 2-phenoxy-1-phenylethanol (1a), and (d) the simplified surface structures of various reaction species along the reaction pathway over Fe3@Ni1-ZrO2. Fe (green), Ni (pink), Zr (yellow), O (blue), C (gray), and H (pink white). | ||
To further probe the reaction mechanism, the oxidative cleavage of 2-phenoxy-1-phenylethanol (1a) was independently investigated (Fig. 3d and S18†). The results show that 1a conversion (54.2%) in this isolated cleavage system was notably lower than that in the one-pot tandem system (78.1%) under identical reaction conditions, demonstrating that the tandem process enhances oxidative cleavage efficiency by continuously removing intermediates (e.g., benzaldehyde (3a) and benzoic acid) through downstream reactions. These intermediates also rationalize the formation of esterification by-products (e.g., benzyl benzoate) observed in the one-pot system. Additionally, the results once again confirm that the dual-active-site catalyst Fe3@Ni1-ZrO2 (containing atomically dispersed Fe and Ni species) exhibits superior activity compared to single-active-site analogues (Fe3@ZrO2 and Ni1-ZrO2).
Next, reactions using 2′-hydroxyacetophenone (1b) and benzaldehyde (3a) or benzyl alcohol (2b) as substrates were systematically investigated (Fig. 3e and f). The synthesis of flavone (6a) proceeds predominantly via aldol condensation between intermediates 1b and 3a, rather than between 1b and 2b. The formation of benzyl benzoate (3b) as a by-product in the reaction of 1b and 2b suggests competing esterification alongside the aldol condensation pathway in systems containing 2b (Fig. 3f). Mechanistic investigation of the reaction between 3a and 2b (Fig. 3g) uncovered critical catalyst specificity, i.e., Ni1-ZrO2 exhibited negligible oxidative esterification activity, aligning with the absence of both flavone (6a) and benzyl benzoate (3b) production in the one-pot tandem system (Fig. 3b). In contrast, Fe3@ZrO2 afforded 55.0% yield of 3b, while Fe3@Ni1-ZrO2 only gives 31.5% 3b yield, demonstrating that atomic Ni-doping could suppress oxidative esterification partly, thereby optimizing one-pot tandem reaction selectivity. Control experiments confirmed the reaction pathway, i.e., 1a is first oxidized to 2a, which undergoes C–C cleavage to generate 3a. Subsequently, 3a reacts with 1b to form 4a, followed by cyclization to 5a and final oxidative dehydrogenation to 6a. Parallelly, 2b (derived from C–C cleavage of 1a) reacts with benzenic acid (oxidative product of 3a) to form by-product 3b.
The proposed one-pot catalytic system for flavone synthesis represents the first integration of 2-phenoxy-1-phenylethanol (1a) and aldehyde-ketone coupling within a single catalytic system, demonstrating a novel route to directly obtaining natural products from lignin β-O-4 segments. Notably, this process operates under base-free conditions, a significant advantage over most state-of-the-art systems for flavone synthesis or lignin β-O-4 segment cleavage, which require strong bases as essential additives.25–27,40 We speculate that the absence of additional alkaline additives in this one-pot transformation system is due to the inherent acidity of the ZrO2 component in Fe3@Ni1-ZrO2, which likely facilitates proton transfer during catalysis. To probe the acidic properties of Fe3@Ni1-ZrO2, pyridine-adsorbed Fourier-transform infrared spectroscopy (Py-FTIR) was employed. Characteristic peaks at 1445, 1490, 1540, and 1595 cm−1 clearly confirm the co-existence of Lewis and Brønsted acid sites (Fig. S19†).41,42 Additionally, some of the reported catalytic systems for the cleavage of lignin model compounds and for flavone synthesis are summarized in Tables S6 and S7.†
Reaction mechanism investigations
Subsequently, density functional theory (DFT) calculations were performed to provide theoretical insights into the reaction mechanism. The proposed reaction pathway was first supported by control experiments (Fig. 4a–d). In the adsorption configurations, 1a was adsorbed onto Fe3@Ni1-ZrO2 through two O atoms, one from the C–O bond and the other from the –OH group bonded to an Fe atom, with an adsorption energy of −1.29 eV. A structurally analogous adsorption mode was observed on Fe3@ZrO2 (Fig. 4a, b and S20†). In contrast, adsorption of 1a onto Ni1-ZrO2 via a single O atom of the –OH group bonded to a Zr atom (Fig. 4a and S21†). Two possible evolutionary intermediates (2a or 2b) were identified following 1a adsorption. On both Fe3@Ni1-ZrO2 and Fe3@ZrO2, the exothermic oxidation of 1a to 2a is thermodynamically favoured, with energy changes of −0.16 eV (vs. −0.05 eV for 2b) and −1.6 eV (vs. 0.17 eV for 2b), respectively. However, the conversion of 1a to 2b is more favorable than to 2a on Ni1-ZrO2, as evidenced by the lower energy barrier (1.48 eV for 2b vs. 2.55 eV for 2a) (Fig. 4a–d and S20–S22†). These findings highlight that Fe3 clusters enhance catalytic activity through two synergistic effects, i.e., strengthened substrate adsorption during the 1a to 2a (or 2b) step and preferential generation of the 2a intermediate over catalysts containing Fe3 clusters.Subsequently, the C–C bond cleavage of intermediate 2a generates the 3a intermediate, a thermodynamically favorable process with reaction energies of −0.89 eV and −0.95 eV on Fe3@Ni1-ZrO2 and Fe3@ZrO2, respectively. To assess competing pathways, we examined the alternative C–O bond cleavage route leading to acetophenone (3c). Comparative analysis revealed strong preference for C–C cleavage over C–O bond scission, as evidenced by the significantly lower reaction energies for the former pathway (−0.89 eV vs. −0.43 eV on Fe3@Ni1-ZrO2; −0.95 eV vs. 0.17 eV on Fe3@ZrO2) (Fig. 4a, c, d, S20 and S22†). Notably, Ni1-ZrO2 exhibited distinct behavior; oxidation of intermediate 2b to 3a proceeded with an exceptionally large thermodynamic driving force (−4.4 eV; Fig. 4a, d and S21†). In addition, the potential oxidation of 2b to 3a on Fe3@Ni1-ZrO2 and Fe3@ZrO2 requires an energy barrier of 0.12 eV and −1.59 eV. Collectively, these computational results demonstrate that 3a intermediate formation is energetically feasible across all catalytic systems, which aligns with experimental observations, where 3a and its derivatives were consistently detected in all three catalytic systems.
DFT calculations further delineate distinct mechanistic pathways across catalytic systems. The oxidation of 1a to 2a emerges as the predominant pathway, though 2b forms as a minor by-product through alternative routes (Fig. 4a–d). Control experiments corroborate this mechanistic dichotomy; oxidative esterification between 3a and 2b generates by-product 3b in both Fe3-containing systems (Fe3@Ni1-ZrO2 and Fe3@ZrO2), as evidenced in Fig. 3b and g. Notably, Ni1-ZrO2 exhibits different behavior; the formation of 2a is thermodynamically unfavorable, requiring a high reaction energy of 2.55 eV (Fig. 4a–d). In contrast, 2b emerges as the dominant product on Ni1-ZrO2 with a significantly lower energy barrier of 1.48 eV. This primary intermediate (2b) then undergoes spontaneous conversion to 3a with substantial exothermicity (ΔE = −4.4 eV; Fig. 4d). These results suggest that 3a is generated later than 2b on Ni1-ZrO2, whereas the formation of 3b as a by-product is difficult in this case, consistent with experimental data (Fig. 3b and g).
The reaction pathway proceeds via aldol condensation between intermediates 3a and 1b to form 4a. This step is exothermic on Fe3@Ni1-ZrO2 (−0.39 eV), but becomes progressively less favorable on Fe3@ZrO2 (0.43 eV) and Ni1-ZrO2 (2.46 eV). This computational prediction aligns closely with experimental observations (Fig. 4a, b, S20 and S21†), where efficient 4a formation was exclusively observed in the Fe3@Ni1-ZrO2 system. These findings highlight the unique catalytic role of Fe3 clusters in facilitating the conversion of 3a to 4a. Conversely, the absence of Fe3 clusters in Ni1-ZrO2 creates an energy barrier that effectively suppresses 4a generation. The thermodynamically accessible 4a in the Fe3@Ni1-ZrO2 system serves as a key precursor, enabling subsequent transformations for 6a.
The subsequent transformation of 4a to 5a on Fe3@Ni1-ZrO2 requires overcoming an energy barrier of 2.22 eV, whereas it exhibits a reaction energy of −0.5 eV on Fe3@ZrO2. Further conversion of 5a to 6a remains energetically challenging for both Fe3-containing catalysts; Fe3@Ni1-ZrO2 demonstrates relatively low endothermicity (ΔE = 0.55 eV), while Fe3@ZrO2 encounters a substantial energy barrier (ΔE = +2.56 eV; Fig. 4a). This thermodynamic contrast manifests experimentally in the pronounced accumulation of 5a observed exclusively in Fe3@ZrO2 systems (Fig. 3b). Notably, the final desorption of 6a from Fe3@Ni1-ZrO2 requires an energy barrier of 1.66 eV, which likely contributes to the prolonged reaction duration in this system (Fig. 4a and b). For Fe3@ZrO2, the desorption of 6a requires overcoming an energy barrier as high as 3.74 eV, also consistent with the experimental observation of a low 6a product yield. For Ni1-ZrO2, initial 4a generation demands a 2.46 eV energy change, followed by a prohibitive 2.64 eV barrier for transforming 4a to 5a. This dual kinetic impediment correlates directly with experimental observations, where the Ni1-ZrO2 system showed negligible 6a production (Fig. 3b).
Reusability and stability of Fe3@Ni1-ZrO2
The used Fe3@Ni1-ZrO2 catalyst was collected by centrifugation and demonstrated direct reusability for six runs without significant activity loss (Fig. S23†), confirming its good recyclability. Characterization results (Fig. S24–S27†) indicate that the recycled Fe3@Ni1-ZrO2 retains identical structural and compositional properties compared to the fresh one. In addition, metal leaching tests and a hot filtration experiment (Fig. S28†) confirm the robust stability of Fe3@Ni1-ZrO2 and the heterogeneous catalytic nature of this one-pot tandem reaction.Considering the practicality of the developed one-pot tandem catalytic system and the large-scale synthesis demand in the biopharmaceutical field, this one-pot tandem reaction system was evaluated in a continuous flow reactor to assess industrial scalability (Fig. 5a). The Fe3@Ni1-ZrO2 catalyst was slurry-packed with toluene and loaded into the packed column. Upon injecting a toluene solution containing 2-phenoxy-1-phenylethanol (1a) and 2′-hydroxyacetophenone (1b), the system achieved 84.3% conversion of 1a and 63.3% yield of flavone (6a) (average values). Continuous operation for 120 h showed no notable performance degradation, delivering a space-time yield (STY) of 3.3 g gcat−1 h−1 and a turnover frequency (TOF) of 139 h−1. To the best of our knowledge, this represents the first reported one-pot synthesis of flavone from lignin β-O-4 segments, highlighting the system's pioneering significance. Although a unified comparison standard is difficult to establish, we still compiled a comparison table (Table S8†) summarizing various flavonoid synthesis methodologies, in which the yields, substrates, and sustainability metrics from representative enzymatic, homogeneous catalytic, and our current work were included.
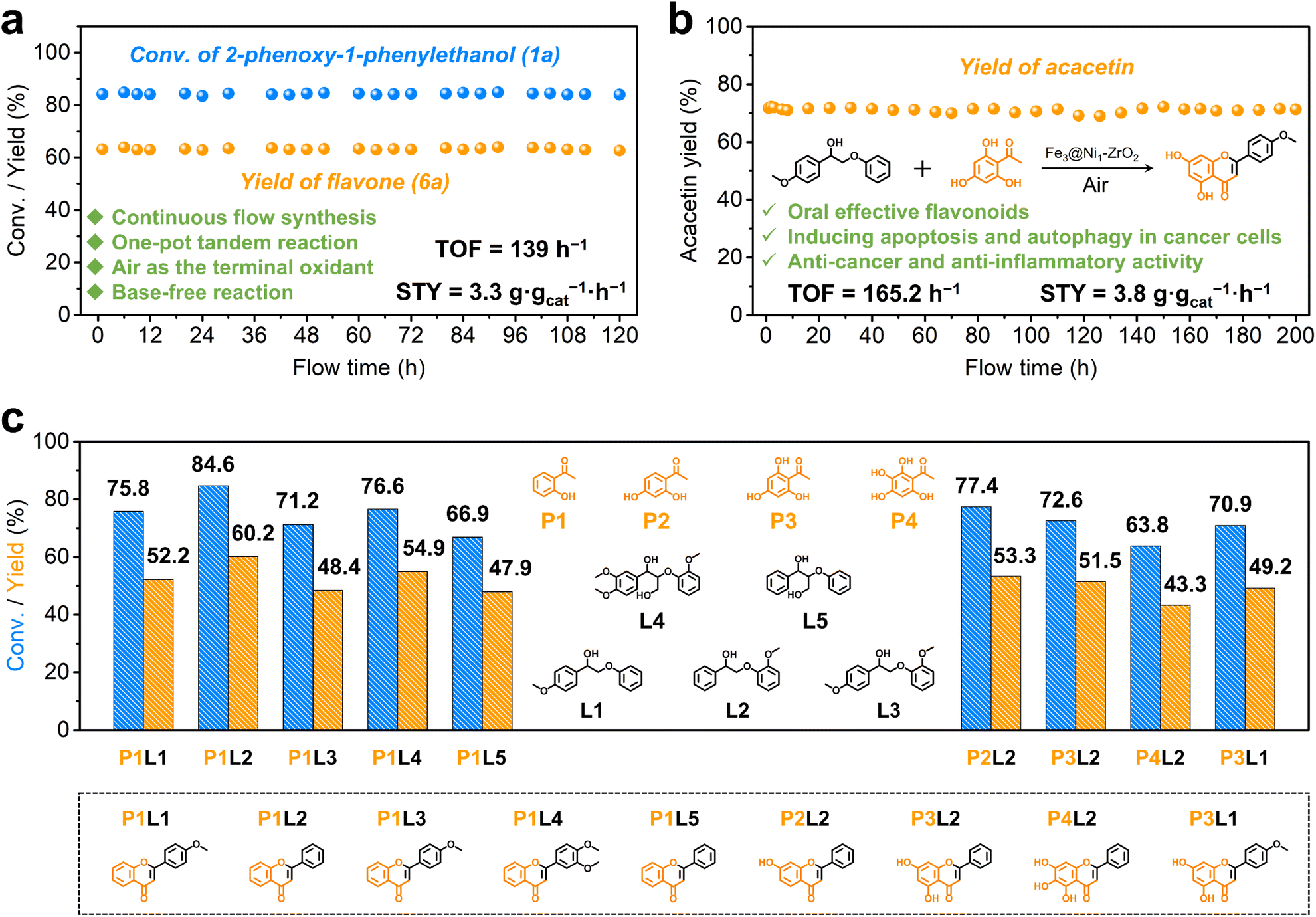 | ||
| Fig. 5 (a) Catalytic system (for flavone synthesis) proceeds in a continuous flow reactor over Fe3@Ni1-ZrO2. (b) The continuous flow reaction for acacetin synthesis over Fe3@Ni1-ZrO2. (c) Scope of the one-pot tandem cleavage-coupling reaction for flavone synthesis over Fe3@Ni1-ZrO2. Conversion and yield were determined by GC and GC-MS based on L1–L4. | ||
Catalytic system flexibility and product practicality
In order to further demonstrate the progressiveness of the developed one-pot tandem lignin-upgrading system, we evaluated the catalytic performance using additional lignin β-O-4 dimers (L1–L5) as substrates. Experimental results demonstrate substrate conversions of 71.2–84.6% with the corresponding flavonoid yields of 48.4–60.2% (Fig. 5c, left columns). To assess broader applicability, the system was extended to synthesize flavonoids with fantastic biological activities from substrates with different substituted groups (P1–P4), achieving yields of 43.3–53.3% (Fig. 5c, right columns).For comprehensive assessment of the application prospective, a scale-up production of acacetin from L1 and P3 was conducted in a continuous flow reactor (Fig. 5b). The system maintained stable operation for >200 h without significant performance decay, delivering a STY up to 3.8 g gcat−1 h−1 and a TOF of 165.2 h−1. Acacetin was easily isolated from the crude reaction mixture by simple centrifugation and purification treatments, with structural confirmation by 1H-NMR and 13C-NMR spectroscopy (Fig. S29 and S30†). These results demonstrate the great application potential of this catalysis system for sustainable and large-scale flavonoid synthesis.
Conclusions
In summary, we have developed a robust Fe3@Ni1-ZrO2 catalyst through atomic-level engineering of ZrO2 nanosheets with dual metal species, which features subnanometric Fe–O clusters anchored on atomically dispersed Ni-doped ZrO2 mesoporous nanosheets. This synergistic architecture enables concerted catalysis of selective C–C bond oxidative cleavage in lignin β-O-4 segments and in situ transformation of the cleavage intermediate into flavone, establishing a highly coupled tandem process for one-pot β-O-4 model upgrading. Our findings propose a versatile strategy for integrating atomic-scale metal species within mesoporous metal oxide catalysts. This approach opens avenues for continuous synthesis of value-added bio-products by overcoming challenges in multistep reaction cascades, thereby advancing sustainable utilization of biomass-derived feedstocks.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. L. and X. Z. developed the idea and designed experiments. X. Z. and J. W. performed the catalyst synthesis and most of the characterization studies. X. Z. and J. W. performed the electron microscopy characterization. J. W. and R. F. performed most of the catalytic reactions. Q. Q. and D. T. synthesized substrates for the catalytic performance evaluation and performed some reaction tests. X. Z., R. F., and F. W. conducted and discussed the theoretical calculations. X. Z., C. L., R. F., and Y. L. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the State Key Laboratory of Pulp and Paper Engineering (2024ZD09), the National Natural Science Foundation of China (22138003, 21825802, 22408109, and 22478135), the China National Postdoctoral Program for Innovative Talents (BX20230131), the China Postdoctoral Science Foundation (2023M741209), the Natural Science Foundation of Guangdong Province (2023B1515040005), the Guangdong Basic and Applied Basic Research Foundation (2023B1515020033 and 2025A1515011981), the Young Elite Scientists Sponsorship Program by CAST (2023QNRC001), the Fundamental Research Funds for the Central Universities (2024ZYGXZR071, 2024ZYGXZR099), and the Science and Technology Program of Guangzhou City (2023A04J0673).References
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Y. H. Liao, S.-F. Koelewijn, G. V. Bossche, J. V. Aelst, S. V. Bosch, T. Renders, K. Navare, T. Nicolaï, K. V. Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. V. Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- G. Meng, W. Lan, L. Zhang, S. Wang, T. Zhang, S. Zhang, M. Xu, Y. Wang, J. Zhang, F. Yue, Y. Wu and D. Wang, J. Am. Chem. Soc., 2023, 145, 12884–12893 CrossRef CAS PubMed.
- W. Lan, M. T. Amiri, C. M. Hunston and J. S. Luterbacher, Angew. Chem., Int. Ed., 2018, 57, 1356–1360 CrossRef CAS PubMed.
- Y. X. Cao, G. Zhu, Y. Li, N. L. Breton, C. Gourlaouen, S. Choua, J. Boixel, H.-P. J. Rouville and J.-F. Soulé, J. Am. Chem. Soc., 2022, 144, 5902–5909 CrossRef CAS PubMed.
- J. Yan, Q. Meng, X. Shen, B. Chen, Y. Sun, J. Xiang, H. Liu and B. Han, Sci. Adv., 2020, 6, eabd1951 CrossRef CAS PubMed.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- E. Subbotina, T. Rukkijakan, M. D. Marquez-Medina, X. Yu, M. Johnsson and J. S. M. Samec, Nat. Chem., 2021, 13, 1118–1125 CrossRef CAS PubMed.
- P. Sudarsanam, R. Y. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- L. Li, L. Dong, D. Li, Y. Guo, X. Liu and Y. Wang, ACS Catal., 2020, 10, 15197–15206 CrossRef CAS.
- Q. Wang, L.-P. Xiao, Y.-H. Lv, W.-Z. Yin, C.-J. Hou and R.-C. Sun, ACS Catal., 2022, 12, 11899–11909 CrossRef CAS.
- C. Zhang, H. Li, J. Lu, X. Zhang, K. E. MacArthur, M. Heggen and F. Wang, ACS Catal., 2017, 7, 3419–3429 CrossRef CAS.
- M. Wang, M. Liu, H. Li, Z. Zhao, X. Zhang and F. Wang, ACS Catal., 2018, 8, 6837–6843 CrossRef CAS.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994 RSC.
- H. Li and G. Song, ACS Catal., 2019, 9, 4054–4064 CrossRef CAS.
- T. Cui, L. Ma, S. Wang, C. Ye, X. Liang, Z. Zhang, G. Meng, L. Zheng, H.-S. Hu, J. Zhang, H. Duan, D. Wang and Y. Li, J. Am. Chem. Soc., 2021, 143, 9429–9439 CrossRef CAS PubMed.
- H. Luo, E. P. Weeda, M. Alherech, C. W. Anson, S. D. Karlen, Y. Cui, C. E. Foster and S. S. Stahl, J. Am. Chem. Soc., 2021, 143, 15462–15470 CrossRef CAS PubMed.
- S. Z. Wang, K. L. Zhang, H. L. Li, L. P. Xiao and G. Y. Song, Nat. Commun., 2021, 12, 416 CrossRef CAS PubMed.
- Z. H. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- H. Li, A. Bunrit, J. Lu, Z. Gao, N. Luo, H. Liu and F. Wang, ACS Catal., 2019, 9, 8843–8851 CrossRef CAS.
- Y. Ding, T. Guo, Z. Li, B. Zhang, F. E. Kühn, C. Liu, J. Zhang, D. Xu, M. Lei, T. Zhang and C. Li, Angew. Chem., Int. Ed., 2022, 61, e202206284 CrossRef CAS PubMed.
- Y. Liu, Q. Luo, Q. Qiang, H. Wang, Y. Ding, C. Wang, J. Xiao, C. Li and T. Zhang, ChemSusChem, 2022, 15, e202201401 CrossRef CAS PubMed.
- B. Zhang, T. L. Guo, Z. W. Li, F. E. Kuhn, M. Lei, Z. B. K. Zhao, J. L. Xiao, J. Zhang, D. Z. Xu, T. Zhang and C. Z. Li, Nat. Commun., 2022, 13, 3365 CrossRef CAS PubMed.
- H. Xiong, H. Zhou, G. Sun, Z. Liu, L. Zhang, L. Zhang, F. Du, Z.-A. Qiao and S. Dai, Angew. Chem., Int. Ed., 2020, 59, 11053–11060 CrossRef CAS PubMed.
- Y. Wang, H. Arandiyan, J. Scott, A. Bagheri, H. Dai and R. Amal, J. Mater. Chem. A, 2017, 5, 8825–8846 RSC.
- M. Chen, Y. Zhang, L. Xing, Y. Liao, Y. Qiu, S. Yang and W. Li, Adv. Mater., 2017, 29, 1607015 CrossRef PubMed.
- K. Liu, H. Jin, L. Huang, Y. Luo, Z. Zhu, S. Dai, X. Zhuang, Z. Wang, L. Huang and J. Zhou, Sci. Adv., 2022, 8, eabn2030 CrossRef CAS PubMed.
- J. Ma, Y. Ren, X. Zhou, L. Liu, Y. Zhu, X. Cheng, P. Xu, X. Li, Y. Deng and D. Zhao, Adv. Funct. Mater., 2018, 28, 1705268 CrossRef.
- Y. Huang, S. L. Zhang, X. F. Lu, Z.-P. Wu, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 11841–11846 CrossRef CAS PubMed.
- Q. Wang, T. Liu, K. Chen, D. Wu, C. Chen, M. Chen, X. Ma, J. Xu, T. Yao, Y. Li, H. Zhou and Y. Wu, Small, 2022, 2204015 CrossRef CAS PubMed.
- X. F. Lu, Y. Chen, S. Wang, S. Gao and X. W. Lou, Adv. Mater., 2019, 31, 1902339 CrossRef PubMed.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- J. Fu, F. M. Hassan, C. Zhong, J. Lu, H. Liu, A. Yu and Z. Chen, Adv. Mater., 2017, 29, 1702526 CrossRef PubMed.
- W. Lyu, Y. Liu, J. Zhou, D. Chen, X. Zhao, R. Fang, F. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202310733 CrossRef CAS PubMed.
- W. Zhao, Y. Shi, Y. Jiang, X. Zhang, C. Long, P. An, Y. Zhu, S. Shao, Z. Yan, G. Li and Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 5811–5815 CrossRef CAS PubMed.
- X. Zhao, R. Fang, F. Wang, X. Kong and Y. Li, Nat. Commun., 2022, 13, 7873 CrossRef CAS PubMed.
- W. Yao, J. Chen, Y. Wang, R. Fang, Z. Qin, X. Yang, L. Chen and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 23729–23734 CrossRef CAS PubMed.
- C. Dai, A. Zhang, M. Liu, X. Guo and C. Song, Adv. Funct. Mater., 2015, 25, 7479–7487 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02215k |
| This journal is © The Royal Society of Chemistry 2025 |