Strong impacts of inter-π-chain charge transfer accelerating CO2 reduction photocatalysis of carbazole–diimine-based linear conjugated polymer/Ru complex hybrids†
Received
29th January 2025
, Accepted 1st April 2025
First published on 11th April 2025
Abstract
Conjugated polymers are promising candidates for photocatalyst materials owing to the molecular design flexibility in tuning their properties, including visible light responsiveness. The rational introduction of a molecular metal complex acting as a catalyst at a specific location is an effective approach to activate conjugated polymer photocatalysts for the selective conversion of small molecules, such as carbon dioxide. However, the photocatalytic activity of the conjugated polymer/metal complex hybrids has not been satisfactory. In particular, there is still much room for improvement in polymer structure engineering to maximise the activation of a molecular complex catalyst centre by photoexcited electrons. This work demonstrates the strong impact of side chains and ligand structures, which do not significantly affect the optical properties of the polymers, on their photocatalytic performance for CO2 reduction. The relatively rigid aromatic side chains and condensed aromatic ligand moieties enable effective inter-π-chain charge transfer to activate the isolated (i.e. low-concentration) Ru(II) complex catalyst. The manipulation of photoexcited charge transfer by structural modulation resulted in a significantly improved photocatalytic activity (quantum efficiency of 2.2% at 450 nm) compared to the counterpart photocatalysts containing the alkyl side chain and bipyridine ligand moieties.
Introduction
The catalytic conversion of CO2 into energy-added molecules is important for both decreasing the CO2 concentration in the atmosphere and generating energy and carbon resources.1–3 In particular, the utilisation of solar energy has been regarded as an attractive method for CO2 conversion. There is an urgent need to develop photocatalyst materials for harnessing the energy of visible light, which is a major component of sunlight.
Inorganic semiconductors,4–6 metal complexes,7–9 and their hybrid materials10–12 have been extensively studied as photocatalysts for visible-light-driven CO2 reduction. High selectivity for CO2 reduction, based on well-defined and tunable active sites, is one of the advantages of molecular photocatalysts, as has been demonstrated in previous works.7–9 Another important favourable property of molecular photocatalysts is the capability to design molecular orbitals to facilitate the transfer and separation of photoexcited charges, as demonstrated by highly selective and efficient photocatalysis even under visible-light illumination.13 On the other hand, semiconductor photocatalysts are advantageous with regard to formation of band structures.5,14 The band-gap excitation of semiconductors generates multiple electrons and holes in the conduction and valence bands. Therefore, simultaneous multi-electron reduction and oxidation reactions, e.g., overall water splitting15–18 and CO2 reduction using water as the electron source,6 have been achieved by a number of semiconductor photocatalysts, while they have hardly been reported with molecular photocatalysts alone.19 However, when used for CO2 reduction, semiconductor photocatalysts frequently suffer from the low selectivity of CO2 reduction due to the competition with efficient proton reduction.6 Hence, assemblies of metal complex molecules and semiconductor particles have been developed to exploit their specific advantages; however, the achievement of efficient photocatalytic CO2 reduction using these systems is still challenging.10,11
Recently, conjugated polymers have attracted significant attention as new and promising heterogeneous photocatalyst materials.20–23 Conjugated polymers exhibit visible light absorption owing to their expanded π-conjugation with flexible molecular designability for engineering the HOMO and LUMO energies. The hybridisation of molecular metal complex (photo)catalysts is an effective method for activating conjugated polymer photocatalysts for selective CO2 reduction. Although introducing an appropriate anchor unit to either conjugated polymers24 or molecular metal complex25,26 affords intermolecular hybrid for CO2 reduction, incorporating ligand units for the metal centre directly into the polymer as building blocks is a simpler and more effective method for the site-selective and robust introduction of metal complex catalysts.24,27–31
We have developed all-in-one hybrid photocatalysts consisting of linear conjugated polymers with a bipyridine unit that acts as a ligand bearing the Ru(II) carbonyl complex catalyst for photocatalytic CO2 reduction.31 The introduction of an appropriate donor–acceptor structure in the conjugated system enabled selective LUMO localisation on the Ru(II) complex catalyst moiety, leading to improved photocatalytic CO2 reduction activity. In addition, this hybrid photocatalyst exhibited selective CO2 reduction even in aqueous solution.24 However, its photocatalytic activity was not satisfactory, with a quantum efficiency of 0.4%, leaving much room for improvement.31
In this work, we demonstrate a strategy for effective inter-π-chain transfer of photoexcited electron to the Ru(II) complex catalyst through the control of the concentration of the catalyst centre and side chain modulation (Fig. 1).
 |
| | Fig. 1 (a) Synthetic procedures, (b) molecular structures, and abbreviations of conjugated polymers bearing a Ru(II) complex catalyst used in this study. | |
Results and discussion
Effects of side chains and Ru complex introduction on crystallinity, light absorption, and HOMO–LUMO potentials
Conjugated polymers [PhCz-Bpy]n, [PhCz-Phen]n, and [C8,8Cz-Bpy]n (PhCz: 9-phenyl-carbazole, C8,8Cz: 9-(9-heptadecanyl)-carbazole, Bpy: 2,2′-bipyridine, Phen: phenanthroline) were synthesised using Suzuki–Miyaura cross-coupling reactions between 5,5′-dibromo-2,2′-bipyridine or 3,8-dibromo-1,10-phenanthroline and carbazoles with diboronic ester substituents at the 2,7 positions. The newly synthesized [PhCz-Phen]n and [C8,8Cz-Bpy]n polymers were characterised using solid-state 13C cross-polarization/magic angle spinning (CP/MAS) NMR spectroscopy to confirm the existence of key building unit structures (Fig. S1†). Gel permeation chromatography (GPC) for [C8,8Cz-Bpy]n that is soluble in chloroform gave weight-average molecular weight (Mw) of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800. Scanning electron microscopy (SEM) images show aggregated structures of the polymers with sizes of several μm (Fig. S2†). A Ru(II) carbonyl complex was introduced by coordination to the diimine moieties of [PhCz-Bpy]n, [PhCz-Phen]n, and [C8,8Cz-Bpy]n to yield the corresponding hybrids, [PhCz-BpyRux]n, [PhCz-PhenRux]n, and [C8,8Cz-BpyRux]n. The polymers with the introduced Ru(II) complex exhibited characteristic CO vibrations at 1960 and 1995 cm−1 that can be assigned to the Ru(Bpy)(CO)2Cl2-type coordination environment (Fig. S3–S5†). The CO vibration absorption increased with increasing extent of the used Ru complex (x) for coordination reaction to the polymer.
800. Scanning electron microscopy (SEM) images show aggregated structures of the polymers with sizes of several μm (Fig. S2†). A Ru(II) carbonyl complex was introduced by coordination to the diimine moieties of [PhCz-Bpy]n, [PhCz-Phen]n, and [C8,8Cz-Bpy]n to yield the corresponding hybrids, [PhCz-BpyRux]n, [PhCz-PhenRux]n, and [C8,8Cz-BpyRux]n. The polymers with the introduced Ru(II) complex exhibited characteristic CO vibrations at 1960 and 1995 cm−1 that can be assigned to the Ru(Bpy)(CO)2Cl2-type coordination environment (Fig. S3–S5†). The CO vibration absorption increased with increasing extent of the used Ru complex (x) for coordination reaction to the polymer.
Fig. 2 shows powder XRD patterns of the obtained polymers. [PhCz-BpyRux]n and [PhCz-PhenRux]n exhibited typical diffraction patterns with characteristic diffraction peaks at 2θ of ∼11° and ∼25° (Fig. 2a and b), which are often observed in linear π-conjugated polymers.32–35 The diffraction peaks at higher angle correspond to the d-spacing value of ∼3.6 Å, which should be related to the π–π stacking of the conjugated chains.32,34–36 On the other hand, the d-spacing value for the diffraction at lower angle (∼8.1 Å) was similar to the distance between the conjugated backbones separated by side chains as previously assigned for linear conjugated polymers.32,34–36[PhCz-PhenRux]n exhibited sharper and stronger diffraction peaks than [PhCz-BpyRux]n, reflecting its higher crystallinity. By contrast, [C8,8Cz-BpyRux]n with long alkyl side chains showed very broad peaks with an expanded d-spacing (∼4.3 and 16°) compared with the other polymers with phenyl side chains (Fig. 2c). The lower crystallinity of the structure is likely caused by the bulky branched C8,8 alkyl chain.32,35,36 The introduction of the Ru-complex moiety decreased the sharpness of the peaks depending on the loading amounts. In the case of [C8,8Cz-BpyRux]n, low-angle shifts in the broad peak was also observed upon increasing x. This is due to the disordering of the stacking sequence by the incorporation of the relatively large Ru-complex core (∼1 nm). However, the characteristic diffraction patterns of [PhCz-BpyRux]n and [PhCz-PhenRux]n were retained even when the Ru complex was incorporated. Hence, the integrated structure of conjugated chains was distinctly different for each side chain (phenyl vs. alkyl), regardless of the incorporation of the Ru complex.
 |
| | Fig. 2 XRD patterns of (a) [PhCz-BpyRux]n, (b) [PhCz-PhenRux]n, and (c) [C8,8Cz-BpyRux]n. | |
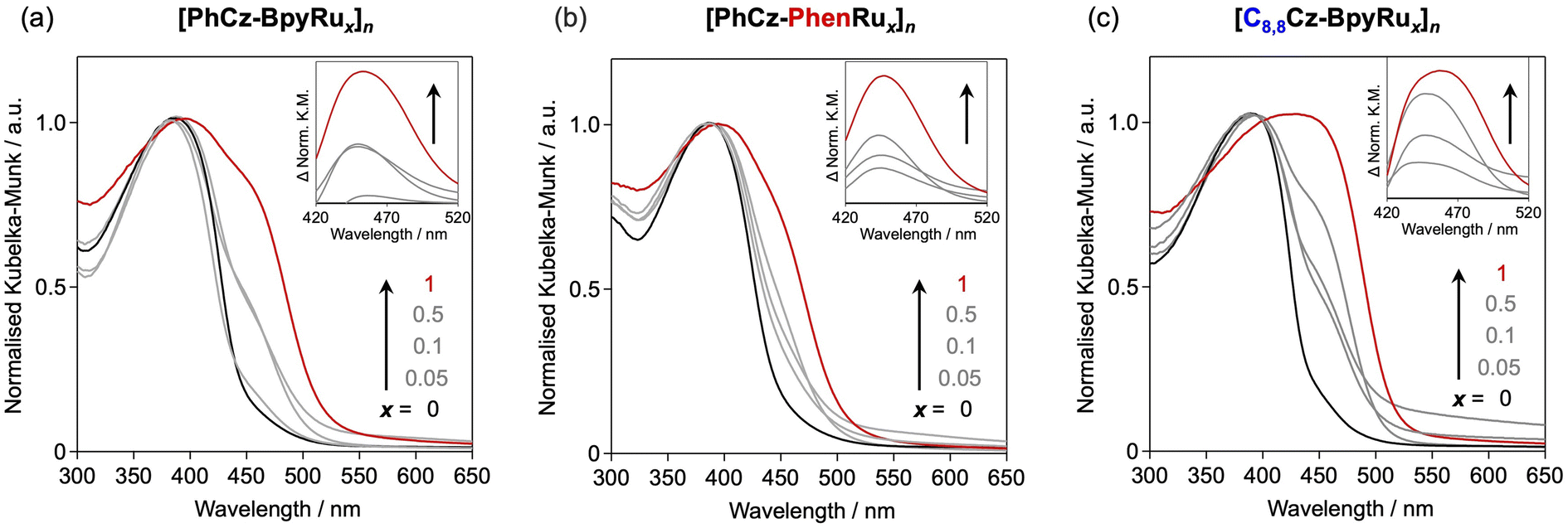
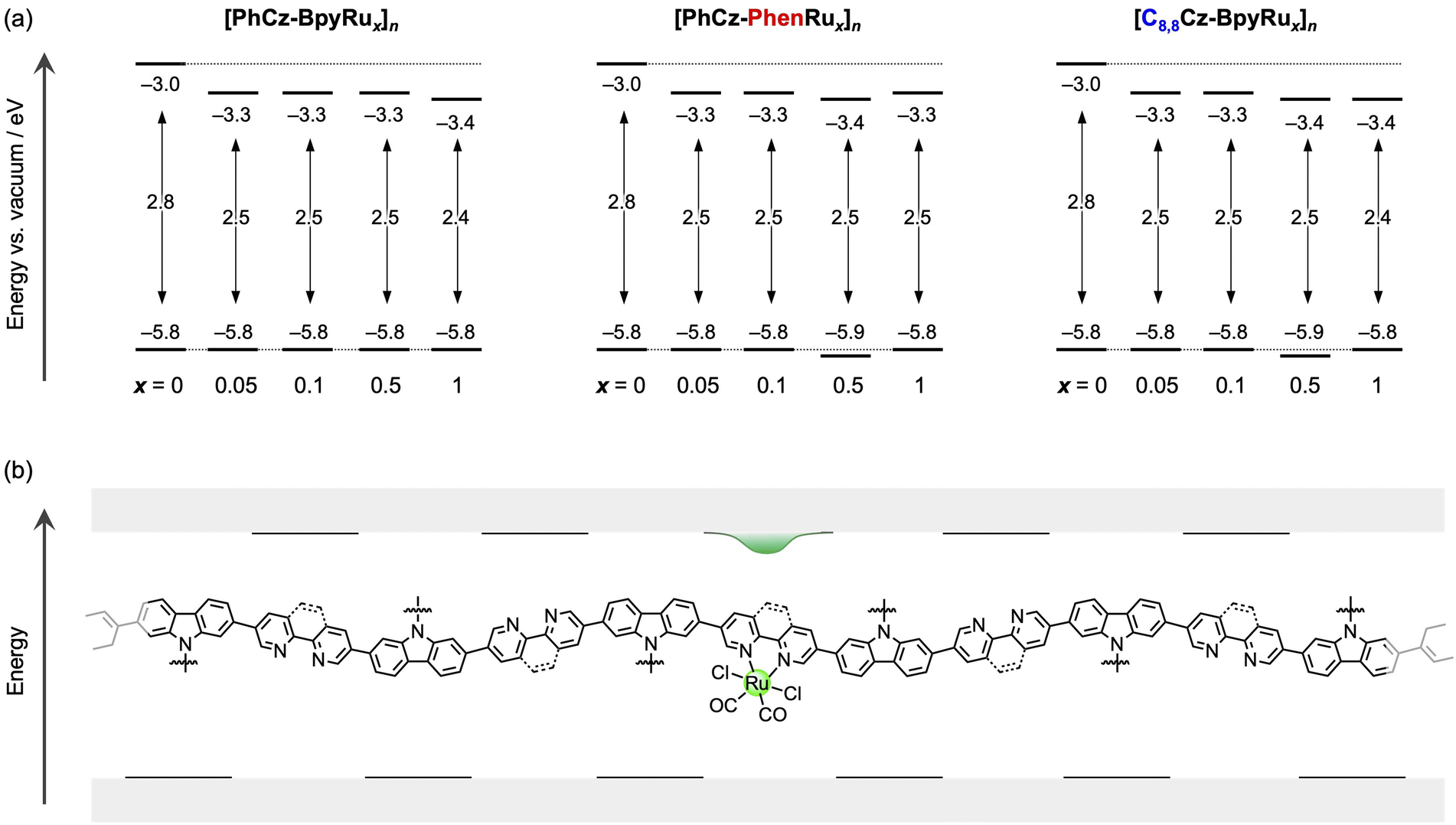
Polymerisation of the colourless monomers afforded yellow powders with visible light absorption. The absorption wavelengths were negligibly affected by the side chain structures and diimine moieties of [PhCz-Bpy]n, [PhCz-Phen]n, and [C8,8Cz-Bpy]n (Fig. 3). Coordination of the diimine moiety to the Ru complex generated a new absorption peak at approximately 450 nm (Fig. 3, inset). The HOMO and LUMO levels derived from the HOMO–LUMO energy gaps (Eg) and the ionisation energies measured by photoelectron yield spectroscopy (PYS; Fig. S6–S8†), were negligibly affected by changing the side chains and the diimine moiety (Fig. 4a). By contrast, the coordination to the Ru complex induced selective shifts of the LUMO to a lower energy by ∼0.3 eV, while maintaining the HOMO energies for all polymers. This LUMO shift due to the Ru complex introduction is consistent with our previous report on [PhCz-BpyRux]n (x = 0 and 1).31 The theoretical calculation for the corresponding monomers suggests that the HOMO and LUMO are respectively localized on carbazole and diimine, and the LUMO energy was stabilized because of the electron-donating coordination of diimine to Ru (Fig. S9†). Importantly, the LUMO energies of the Ru-complex-bearing polymers were almost constant, regardless of the amount of the Ru complex introduced (Fig. 4a). Therefore, we can conclude that the Ru complex modification produced isolated LUMO distributed on diimines coordinated to the Ru centre, which has ca. 0.3–0.4 eV lower energy than that of the free diimine moiety (Fig. 4b).
 |
| | Fig. 3 UV-visible diffuse reflectance spectra of (a) [PhCz-BpyRux]n, (b) [PhCz-PhenRux]n and (c) [C8,8Cz-BpyRux]n. | |
 |
| | Fig. 4 (a) HOMO and LUMO energies of [PhCz-BpyRux]n, [PhCz-PhenRux]n, and [C8,8Cz-BpyRux]n. (b) Assumed energy structure of the linear polymer/Ru complex hybrid. | |
Effects of concentration of Ru complex hybridised on the photocatalytic CO2 reduction activity
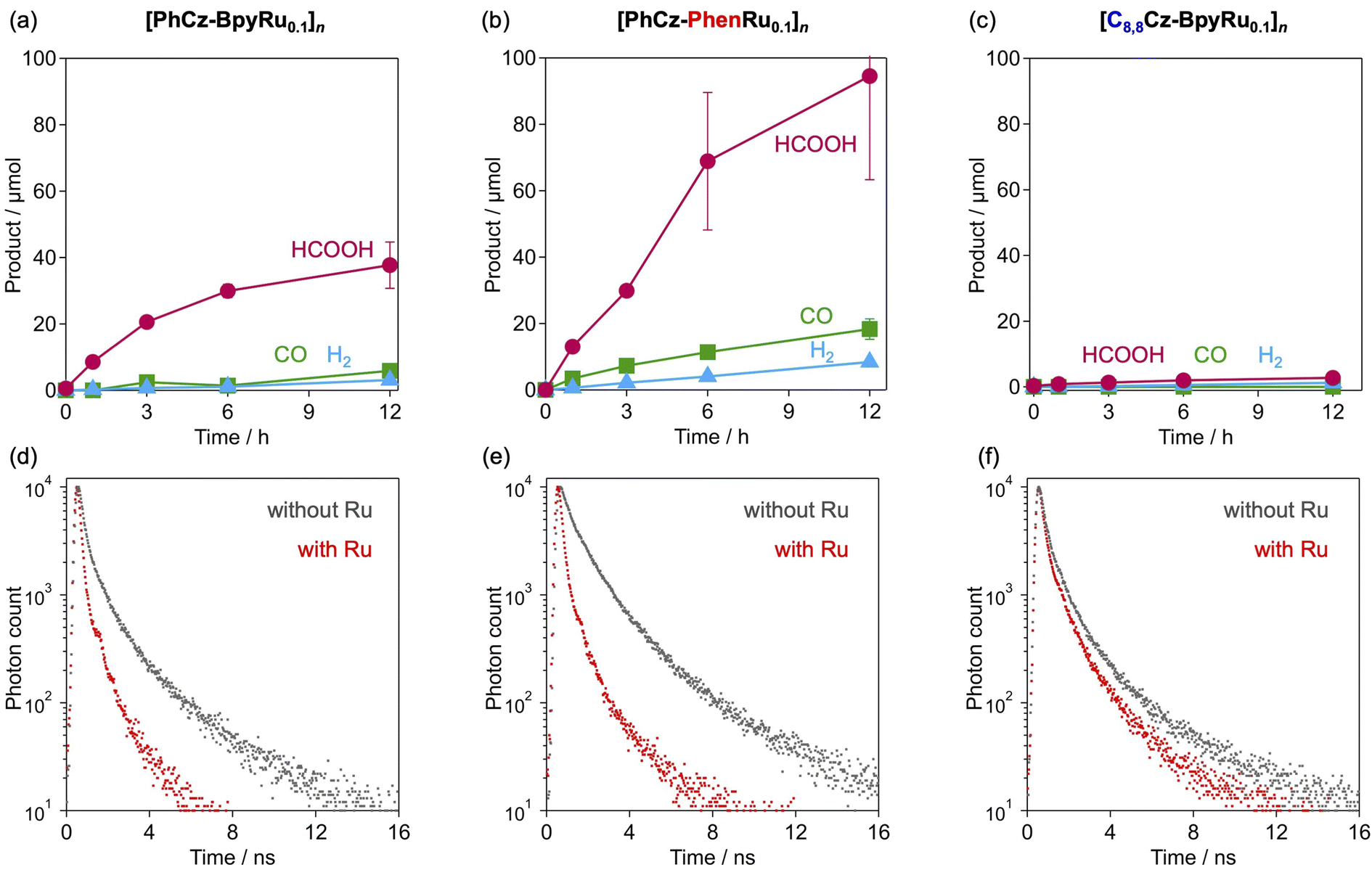
Photocatalytic CO2 reduction was performed in a mixed solution of acetonitrile (MeCN) and triethanolamine (TEOA) (4:1), where hybrid polymers were dispersed, under a CO2 atmosphere with a light (λ > 380 nm) irradiation (Fig. 5). As indicated in our previous work,31[PhCz-BpyRu1]n generated formate as the main product of CO2 reduction using TEOA as an electron donor. By contrast, [PhCz-Bpy]n without the Ru complex (x = 0) did not give formate or CO, clearly indicating that the Ru complex moiety acted as a selective CO2 reduction catalyst. The best photocatalytic performance was obtained at x = 0.1. Surprisingly, the amount of generated formate increased with decreasing Ru-complex incorporation, from x = 1 to 0.1 (Fig. 5). This trend is unusual and differs from the trends observed for the conventional semiconductor/molecular hybrid photocatalysts; in most cases, photocatalytic activity increases with increasing number of molecular catalyst units because electron transport from the semiconductor to the molecular catalyst is the limiting process.37,38 By contrast, the unusual trend found in this study, i.e., enhanced activity with decreasing Ru-complex hybridisation, implies that the conjugated polymer enables more effective activation of isolated Ru-complex catalysts by their photoexcited electrons. Consequently, the turnover number for formate formation increased drastically with decreasing Ru-complex catalyst loading (Fig. 5).
 |
| | Fig. 5 Amounts of formate, CO, and H2 formation generated over [PhCz-BpyRux]n (2 mg) with different loading amounts of Ru complex moiety (x = 0, 0.05, 0.1, 0.3, 0.5, and 1) in MeCN–TEOA (4:1, v/v, 2 mL) under irradiation of λ > 380 nm light for 6 h. | |
Because CO2 reduction into formate is a two-electron reduction process, frequent transfer of photoexcited electrons to one catalyst molecule would enhance the photocatalytic activity if the chemical reaction were the rate-determining process. To gain further insight into the photoinduced charge-transfer behaviour, time-resolved microwave conductivity measurements, which provide a powerful tool for the visualization of the photoconductivity of a material, were performed. The photoconductivity ϕΣμ, where ϕ is the quantum efficiency of charge carrier generation and Σμ is the sum of the photogenerated carrier mobilities, first increased within the instrumental time resolution (∼10−7 s) and then gradually decreased due to charge recombination and/or trapping. Compared to [PhCz-Bpy]n, the maximum photoconductivity ϕΣμmax decreased upon loading the Ru complex in [PhCz-BpyRux]n (Fig. 6a). Interestingly, the introduction of only a small amount of the Ru complex (x = 0.05) significantly decreased the photoconductivity, although it did not affect their crystallinity. According to our previous report,31 the decreased photoconductivity can be attributed to the trapping of photoexcited electrons by the Ru-complex moiety, which results in an increased electronic density of the Ru centre, as indicated by transient IR measurements. Importantly, the decreased photoconductivity was saturated at x = 0.1 and became constant at 0.1 ≦ x ≦ 1 (Fig. 6b). This indicated that the Ru complex coordinated to only 10% of the bipyridine unit in [PhCz-BpyRu0.1]n is enough for accepting the photogenerated electrons in the conjugated skeleton. In other words, one Ru-complex catalyst can more frequently accept photoexcited electrons in the case of lower loading amount; therefore, [PhCz-BpyRu0.1]n showed a higher rate of photocatalytic CO2 reduction.
 |
| | Fig. 6 (a) Transient photoconductivities upon laser pulse excitation at 355 nm of [PhCz-BpyRux]n with different loading amounts of Ru complex moiety. (b) Dependence of maximum transient conductivities ϕΣμmax and amounts of formate production on the amount of Ru complex hybridised. | |
Manipulation of interchain charge transfer for improving the photocatalytic CO2 reduction activity
Fig. 7a–c compare the photocatalytic CO2 reduction activities of [PhCz-BpyRu0.1]n, [PhCz-PhenRu0.1]n, and [C8,8Cz-BpyRu0.1]n. Despite the lack of a significant difference in the light absorption properties (Fig. 3), the choice of the side chain and diimine moiety strongly affected the photocatalytic activity. Compared to [PhCz-BpyRu0.1]n (Fig. 7a), substitution of the bipyridine moiety with phenanthroline ([PhCz-PhenRu0.1]n) improved the rate of formate formation (Fig. 7b). IR spectra at 400–1700 cm−1 that corresponds to the organic backbone in [PhCz-PhenRu0.1]n was maintained after photocatalysis (Fig. S10†). On the other hand, characteristic CO vibration peaks at 2060 and 1995 cm−1 assignable to Ru(diimine)(CO)2Cl2-type coordination were shifted to 2036 and 1956 cm−1 just after photoirradiation and the structure was maintained after the long-term photocatalysis (Fig. S10†). Although the obtained structure could not be precisely characterized, ligand-substituted complex, e.g., Ru(diimine)(CO)LCl2,39,40 may form as the real catalyst species. By contrast, the photocatalytic activity was almost completely diminished by the introduction of the C8,8 branched long alkyl side chains on the carbazole moiety (Fig. 7c). The initial rates of formate formation on [PhCz-PhenRu0.1]n were 2.3 and 37 times higher than those on [PhCz-BpyRu0.1]n and [C8,8Cz-BpyRu0.1]n, respectively. The difference in their π-chain orientation is likely a key factor for their distinct photocatalytic activities because [PhCz-PhenRu0.1]n has a more oriented inter-π-chain structure than [PhCz-BpyRu0.1]n while the bulky C8,8 side chain in [C8,8Cz-BpyRu0.1]n induces a disordered structure, as shown in Fig. 2.
 |
| | Fig. 7 Time courses of formate, CO, and H2 formation over (a) [PhCz-BpyRu0.1]n, (b) [PhCz-PhenRu0.1]n and (c) [C8,8Cz-BpyRu0.1]n in MeCN–TEOA (4:1, v/v, 2 mL) under irradiation of λ > 380 nm light. Emission decays from (d) [PhCz-BpyRux]n, (e) [PhCz-PhenRux]n and (f) [C8,8Cz-BpyRux]n (x = 0 or 0.1) dispersed in MeCN with the excitation wavelength of λ = 440 nm. | |
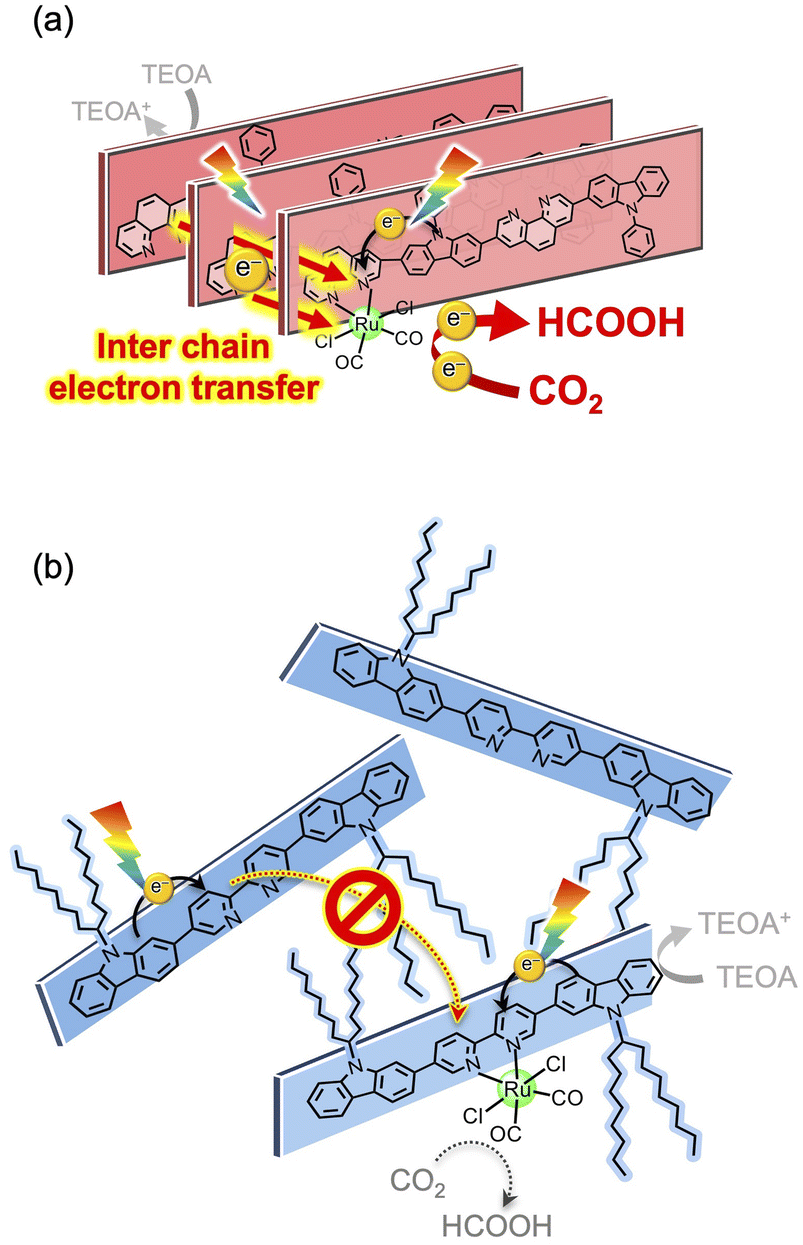
The behaviour of the photoexcited state was compared using emission spectroscopy (Fig. 7d–f and S11, S12†). The MeCN dispersions of the polymers without the Ru complex (x = 0) showed emission maxima at approximately 500–550 nm upon photoexcitation at 440 nm (Fig. S11†). Introduction of the Ru-complex moiety resulted in decreased emission intensities (Fig. S11†) with shortened emission lifetimes (Fig. 7d–f) to different extents, depending on the conjugated skeleton. Importantly, decreased emission, which was exhibited in the MeCN dispersion, was not observed in dichloromethane solutions of [C8,8Cz-BpyRux]n (compare x = 0 and 0.1 in Fig. S12a†). By contrast, [PhCz-BpyRux]n, which is insoluble in dichloromethane, exhibited emission deactivation upon the introduction of the Ru complex unit in its dispersion form (Fig. S12b†), similar to that in MeCN (Fig. 7a). These observations imply that the non-radiative deactivation process originates from the inter-π-chain charge-transfer states which can be promoted in the solid-state with integration of the conjugated chains. Taking this into account, the order of magnitude of the pronounced emission decays induced the by introduction of the Ru complex ([PhCz-PhenRu0.1]n > [PhCz-BpyRu0.1]n ≫ [C8,8Cz-BpyRu0.1]n) shown in Fig. 7d–f likely reflects the degree of occurrence of inter-π-chain charge transfer depending on their inter-π-chain distance and orientation. Namely, the relatively more-oriented structure with shorter inter-π-chain distances in [PhCz-PhenRu0.1]n (and [PhCz-BpyRu0.1]n) possibly enables more extended charge separation between the π-chains (Fig. 8a), and thus, more efficient photocatalysis than [C8,8Cz-BpyRu0.1]n with a disordered structure (Fig. 8b). The fact that the photocatalytic activities of the conjugated polymer/Ru complex hybrids were highly sensitive to inter-π-chain charge transfer (Fig. 7) indicates the importance of inter-chain structural engineering.
 |
| | Fig. 8 Proposed effects of interchain electron transfer on the photocatalytic activities of (a) [PhCz-PhenRu0.1]n and [PhCz-BpyRu0.1]n, and (b) [C8,8Cz-BpyRu0.1]n. | |
As shown in Fig. 9, the maximum external quantum efficiency for formate formation (EQEFormate) is 2.2% at 450 nm by using [PhCz-PhenRu0.1]n. This value is approximately six times higher than that of our previously reported [PhCz-BpyRu1]n (ref. 31) because of the improved transport of photoexcited electrons to the isolated Ru complex catalyst. Interestingly, the wavelength dependence of EQEFormate did not simply follow the absorption spectra for [PhCz-PhenRu0.1]n (Fig. 9a), but was similar to the newly generated absorption peak obtained by introducing the Ru complex (Fig. 9b). Because the peak absorption can be assigned to charge transfer from carbazole to the diimine-Ru moiety (vide supra), direct charge transfer to the Ru-complex catalyst is still more effective than higher excited states (such as carbazole to free diimine units) for CO2 reduction. This result implies that there is further room for improvement in the remote transport of photogenerated electrons by higher excited states (generated spatially far from the centre of the Ru complex).
 |
| | Fig. 9 Wavelength dependence of external quantum efficiency for formate formation (EQEFormate) using [PhCz-PhenRu0.1]n along with (a) diffuse reflectance spectra of [PhCz-PhenRu0.1]n and (b) differential diffuse reflectance spectra between [PhCz-PhenRu0.1]n and [PhCz-Phen]n. | |
Conclusions
In this study, we demonstrated polymer structure modulation to manipulate photoinduced charge transfer and improve the photocatalytic performance of conjugated polymer/metal complex hybrids for CO2 reduction. First, the isolation (i.e. decreasing concentration) of the molecular Ru-complex catalyst enabled much more frequent activation of the Ru-complex catalyst by photoexcited electrons in the conjugated polymer [PhCz-BpyRux]n. Second, the introduction of a rigid aromatic side chain on the carbazole moiety and condensed phenanthroline ligand enabled a further improvement in the inter-π-chain charge transfer owing to their more ordered stacking structure. Owing to structural modulation, the optimised hybrid [PhCz-PhenRu0.1]n exhibited ca. 6 times higher photocatalytic activity for CO2 reduction with an external quantum efficiency of 2.2% at 450 nm compared with its previously reported [PhCz-BpyRu1]n analogue (0.4%).31 This study demonstrates the impact of rational polymer engineering on inter-π-chain charge transfer for effective activation of a molecular catalytic site.
Experimental section
General procedure
Solid-state 13C CP/MAS NMR spectra were recorded using a wide-bore 14.1 T magnet and Bruker AVANCE NEO 600 NMR spectrometer equipped with a 1H–13C double resonance 3.2 mm MAS probe. UV-visible diffuse reflectance spectra were recorded at room temperature using a Shimadzu UV-2600i spectrophotometer equipped with an integral sphere (Shimadzu ISR-2600Plus). Attenuated total reflection FT-IR (ATR-FT-IR) spectra were recorded using a JASCO FT/IR-4200 spectrometer equipped with a diamond prism (JASCO ATR PRO ONE). Powder XRD patterns were recorded on MiniFlex600-C (Rigaku, X-ray source: Cu Kα). The emission spectra were measured under an Ar atmosphere at room temperature using a JASCO FP-8550 spectrometer. To determine the emission quantum yield, an integrated sphere (JASCO ILF-135) was equipped with the spectrometer. Emission decay data were collected by the single-photon counting method using a HORIBA DeltaFlex and a Nano-LED (HORIBA NanoLED-440L, λ = 440 nm) as the excitation light source. Scanning electron microscopy (SEM) images were obtained using an NVision 40 instrument (Carl ZeissSII Nano Technology). Photoelectron yield spectra (PYS) were measured using a Bunko Keiki BIP-KV100K instrument. PYS measurements provide the lowest ionisation energies corresponding to the valence band maximum potentials.41,42
Material
Acetonitrile (MeCN; dehydrated, >99.5%), 1,2-dichlorobenzene (>98.0%), methanol (MeOH, >99.8%), CH2Cl2 (>99.5%), ruthenium(III) chloride hydrate (RuCl3·nH2O; 36–44% as Ru), potassium bicarbonate (K2CO3; >99.5%), tetrakis(triphenylphosphine)palladium (Pd(PPh3)4; >95.0%), ethylenediamine tetra acetic acid (>99.0%), Bis-Tris (>99.0%) and potassium bromide (KBr, >99.0%) were purchased from Wako Pure Chemical Industries Ltd., 5,5-dibromo-2,2′-bipyridine (>98.0%), 3,8-dibromo-1,10-phenanthroline (>96.0%), 9-phenyl-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole (>96.0%), 9-(9-heptadecanyl)-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)carbazole (>97.0%) and p-toluenesulfonic acid (>99.0%) were purchased from Tokyo Chemical Industry Co., Ltd. Triethanolamine (TEOA; >98.0%) was purchased from Sigma-Aldrich Co., LLC. Barium sulfate (BaSO4, >99.0%) was purchased from Nacalai Tesque, Inc. The materials were used as received without further purification. [Ru(CO)2Cl2]n was prepared according to a procedure reported in the literature.43[PhCz-Bpy]n was synthesised according to our previous work.31
Synthesis
[PhCz-Phen]n
.
A 1,2-dichlorobenzene–H2O (5:1, v/v; 12 mL) solution containing 3,8-dibromo-1,10-phenanthroline (169 mg, 0.50 mmol), 9-phenyl-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole (247 mg, 0.50 mmol), K2CO3 (524 mg, 3.75 mmol), and Pd(PPh3)4 (11.7 mg, 0.01 mmol) was stirred under reflux for 2 days under a N2 atmosphere. After refluxing, MeOH (20 mL) was added to the reaction mixture, which was then filtered and washed with Milli-Q water (60 mL) and MeOH (20 mL). Yield: 194 mg (93% based on ideal formula of [PhCz-Phen]n:[C30H17N3]n).
[C
8,8
Cz-Bpy]
n
.
A 1,2-dichlorobenzene–H2O (5:1, v/v; 12 mL) solution containing 5,5′-dibromo-2,2-bipyridine (157 mg, 0.50 mmol), 9-(9-heptadecanyl)-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole (329 mg, 0.50 mmol), K2CO3 (525 mg, 3.75 mmol), and Pd(PPh3)4 (11.6 mg, 0.01 mmol) was stirred under reflux for 2 days under N2 atmosphere. After refluxing, MeOH (20 mL) was added to the reaction mixture, which was then filtered and washed with Milli-Q water (60 mL) and MeOH (20 mL). Yield: 250 mg (88% based on ideal formula of [C8,8Cz-Bpy]n:[C39H47N3]n).
[PhCz-PhenRu1]n
.
A MeOH dispersion (6 mL) containing 1 equiv. [Ru(CO)2Cl2]n (11.4 mg, 0.05 mmol) and [PhCz-Phen]n (20.5 mg, 0.05 mmol) was refluxed through stirring for 17 h under a N2 atmosphere. After refluxing, the obtained precipitate was filtered and washed with MeOH (20 mL). Yield: 17.9 mg (55% based on ideal formula of [PhCz-PhenRu]n:[C30H17N3Ru]n). FT-IR (ATR): νCO = 2060, 1995 cm−1.
The same protocol was applied to the synthesis of [PhCz-BpyRux]n, [PhCz-PhenRux]n, and [C8,8Cz-BpyRux]n by changing the mother polymer and the equivalents (x) of reacting [Ru(CO)2Cl2]n to the mother polymer.
Time-resolved microwave conductivity measurement
Time-resolved microwave conductivity experiments44 were performed using a third harmonic generator (THG; 355 nm) of an Nd:YAG laser (Continuum Inc., Surelite II, 5–8 ns pulse duration, 10 Hz) as the excitation source (9.1 × 1015 photons per cm2 per pulse) and X-band microwave (∼9.1 GHz) as the probe. The photoconductivity, Δσ, was obtained using eqn (1):where ΔPr, A, and Pr are the transient power changes in the reflected microwave power, the sensitivity factor, and the reflected microwave power, respectively. The transient photoconductivity Δσ was converted to the product of the quantum yield (ϕ) and sum of the charge carrier mobilities Σμ using eqn (2) and (3), respectively:where e and Flight are the unit charge and the correction (or filling) factor, respectively. The experiments were performed in air at room temperature (298 K).
Photocatalytic reactions
Photocatalytic CO2 reduction reactions were conducted using a merry-go-round system with a batch test tube for each sampling. A MeCN–TEOA (4:1) suspension (2 mL) of the photocatalyst (2 μmol) was placed in a Pyrex test tube (inner diameter: 10 mm; volume: 8.4 mL). The reaction suspension was purged by CO2 bubbling (20 min) and sealed using a rubber septum prior to photoirradiation. The sample tube was placed in an LED merry-go-round reactor (Iris-MG-S, Cell System Inc.) and irradiated with UV-visible light (380 < λ < 700 nm) while stirring. The CO and H2 gaseous products were analysed using a GL Science GC 3210 gas chromatograph (MS-5A column, Ar carrier) equipped with a TCD detector. The formate produced in the liquid phase was analysed using a Shimadzu LC-20AT HPLC system equipped with two Shimadzu Shim-pack FAST-OA columns (100 × 7.8 mm) and a Shimadzu CDD-10A conductivity detector. An aqueous solution containing p-toluenesulfonic acid (0.95 g L−1) was used as the eluent at a flow rate of 0.8 mL min−1 (column temperature: 313 K). After column separation, the eluent was mixed with an aqueous solution containing p-toluenesulfonic acid (0.95 g L−1), ethylenediaminetetraacetic acid (0.03 g L−1), and Bis–Tris (4.18 g L−1). The turnover number for formate formation was estimated using eqn (4):| | | Turnover number = amount of formate produced (mol)/[amount of polymer (mol) × introduction ratio of Ru complex (x)] | (4) |
For wavelength dependence measurements, an LED lamp with an independent wavelength centred at 365, 405, 430, 450, 470, 505, or 525 nm (CL-H1 series, Asahi Spectra Co.) was used at a fixed photon flux of 9.1 × 10−8 einstein s−1, instead of the aforementioned LED merry-go-round system. The external quantum efficiencies of photocatalytic formate formation (EQEformate) were determined using eqn (5):
| | | EQEformate = n × amount of formate produced (mol)/input photon (einstein) | (5) |
where
n is the number of electrons required to generate one molecule of the product (
i.e. two for formate formation by CO
2 reduction).
Computational details
Geometry optimisations were performed using density functional theory (DFT) calculations with the ωB97X-D exchange–correlation functional.45 For the core electrons of Ru ([Ar]3d10), Stuttgart/Dresden pseudopotentials46 at the multi Dirac–Fock level were used. Corresponding double-ζ basis sets were adopted for the Ru valence orbitals. For other elements, namely, C, H, Cl, N, O and S, correlation-consistent basis sets at the double-ζ level (cc-pVDZ)47,48 were employed. Harmonic vibrational frequencies were also analytically calculated to confirm that the optimised structure converged to an equilibrium geometry point. All calculations were performed using the Gaussian 16 program.49
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JST PRESTO grant JPMJPR20T5 (Controlled Reaction), JSPS Grant-in-Aid for Transformative Research Areas (B) “Concerto Photocatalysis” (Grant Numbers JP23H03832 and JP23H03830) and Areas (A) “Dynamic Exciton” (JP20H05836, JP20H05837, and JP20H05840), as well as JSPS KAKENHI Grants JP24K01603 and JP24H00484.
References
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol. C, 2015, 25, 106–137 CrossRef CAS.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2016, 7, 70–88 CrossRef.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, S. Isabell and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- A. Nakada, H. Kumagai, M. Robert, O. Ishitani and K. Maeda, Acc. Mater. Res., 2021, 2, 458–470 CrossRef CAS.
- T. Morikawa, S. Sato, K. Sekizawa, T. M. Suzuki and T. Arai, Acc. Chem. Res., 2022, 55, 933–943 CrossRef CAS PubMed.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- K. Maeda, Phys. Chem. Chem. Phys., 2013, 15, 10537–10548 RSC.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- Y. H. Hong, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2022, 144, 695–700 CrossRef CAS PubMed.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- J. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2020, 6, 168–190 CrossRef.
- K. Ishihara, A. Nakada, H. Suzuki, O. Tomita, S. Nozawa, A. Saeki and R. Abe, J. Mater. Chem. A, 2024, 12, 30279–30288 RSC.
- R. Kuriki and K. Maeda, Phys. Chem. Chem. Phys., 2017, 19, 4938–4950 RSC.
- E. McQueen, N. Sakakibara, K. Kamogawa, M. A. Zwijnenburg, Y. Tamaki, O. Ishitani and R. S. Sprick, Chem. Sci., 2024, 15, 18146–18160 RSC.
- H.-P. Liang, A. Acharjya, D. A. Anito, S. Vogl, T.-X. Wang, A. Thomas and B.-H. Han, ACS Catal., 2019, 9, 3959–3968 CrossRef CAS.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- X. Y. Dong, Y. N. Si, Q. Y. Wang, S. Wang and S. Q. Zang, Adv. Mater., 2021, 33, 2101568 CrossRef CAS PubMed.
- J. Li, K. Ma, Y. He, S. Ren, C. Li, X.-B. Chen, Z. Shi and S. Feng, Catal. Sci. Technol., 2021, 11, 7300–7306 RSC.
- A. Nakada, R. Miyakawa, R. Itagaki, K. Kato, C. Takashima, A. Saeki, A. Yamakata, R. Abe, H. Nakai and H.-C. Chang, J. Mater. Chem. A, 2022, 10, 19821–19828 RSC.
- T. Yamamoto, M. Arai, H. Kokubo and S. Sasaki, Macromolecules, 2003, 36, 7986–7993 CrossRef CAS.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- B. S. Ong, Y. Wu, P. Liu and S. Gardner, J. Am. Chem. Soc., 2004, 126, 3378–3379 CrossRef CAS PubMed.
- H.-P. Fang, J.-W. Lin, I.-H. Chiang, C.-W. Chu, K.-H. Wei and H.-C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5011–5022 CrossRef CAS.
- I. Osaka, T. Abe, S. Shinamura, E. Miyazaki and K. Takimiya, J. Am. Chem. Soc., 2010, 132, 5000–5001 CrossRef CAS PubMed.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- A. Gabrielsson, S. Záliš, P. Matousek, M. Towrie and A. Vlček, Inorg. Chem., 2004, 43, 7380–7388 CrossRef CAS PubMed.
- E. Eskelinen, M. Haukka, T. Venäläinen, T. A. Pakkanen, M. Wasberg, S. Chardon-Noblat and A. Deronzier, Organometallics, 2000, 19, 163–169 CrossRef CAS.
- Y. Nakayama, S. Machida, D. Tsunami, Y. Kimura, M. Niwano, Y. Noguchi and H. Ishii, Appl. Phys. Lett., 2008, 92, 153306 CrossRef.
- A. Nakada, M. Higashi, T. Kimura, H. Suzuki, D. Kato, H. Okajima, T. Yamamoto, A. Saeki, H. Kageyama and R. Abe, Chem. Mater., 2019, 31, 3419–3429 CrossRef CAS.
- P. A. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse, N. C. Thomas, J. A. Treadway and A. H. White, Inorg. Chem., 1995, 34, 6145–6157 CrossRef CAS.
- A. Saeki, S. Yoshikawa, M. Tsuji, Y. Koizumi, M. Ide, C. Vijayakumar and S. Seki, J. Am. Chem. Soc., 2012, 134, 19035–19042 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101–124112 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Electron microscope images, spectroscopic data, and calculated molecular orbitals. See DOI: https://doi.org/10.1039/d5se00142k |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Shunsuke
Asai
a,
Chen
Zhang
a,
Kotaro
Ishihara
a,
Hajime
Suzuki
*ab,
Shunsuke
Asai
a,
Chen
Zhang
a,
Kotaro
Ishihara
a,
Hajime
Suzuki