
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering active intermetallic Pt–Zn sites via vapour–solid synthesis for photocatalytic hydrogen production†
Daniel
Garstenauer
 ab,
Stephen
Nagaraju Myakala
c,
Pablo
Ayala
c,
Hannah
Rabl-Wolff
c,
Ondrej
Zobač
d,
Franz
Jirsa
e,
Dominik
Eder
c,
Alexey
Cherevan
*c and
Klaus W.
Richter
*af
ab,
Stephen
Nagaraju Myakala
c,
Pablo
Ayala
c,
Hannah
Rabl-Wolff
c,
Ondrej
Zobač
d,
Franz
Jirsa
e,
Dominik
Eder
c,
Alexey
Cherevan
*c and
Klaus W.
Richter
*af
aDepartment of Functional Materials & Catalysis, University of Vienna, Josef-Holaubek-Platz 2, 1090 Vienna, Austria. E-mail: klaus.richter@univie.ac.at
bVienna Doctoral School in Chemistry, University of Vienna, Währinger Straße 42, 1090 Vienna, Austria
cInstitute of Materials Chemistry, TU Wien, Getreidemarkt 9, 1060 Vienna, Austria. E-mail: alexey.cherevan@tuwien.ac.at
dInstitute of Physics of Materials, Czech Academy of Sciences, Žižkova 22, 61600 Brno, Czech Republic
eDepartment of Inorganic Chemistry, University of Vienna, Josef-Holaubek-Platz 2, 1090 Vienna, Austria
fCore Facility Crystal Structure Analysis, University of Vienna, Währinger Straße 42, 1090 Vienna, Austria
First published on 19th May 2025
Abstract
Intermetallic compounds hold great potential owing to the possibility of fine tuning their structure- and composition-dependent catalytic properties. Herein, a series of intermetallic Pt–Zn nanoparticles decorated on a TiO2 support was designed via a novel and facile direct vapour–solid synthesis approach, and their co-catalytic performance towards the light-driven hydrogen evolution reaction (HER) was investigated. The intrinsic activity of Pt/TiO2 was almost doubled via the addition of Zn and the formation of Pt27–Zn73/TiO2, achieving a substantial increase in the apparent quantum yield (AQY) values up to 10.3%. In contrast to Pt–Zn intermetallic co-catalysts generally exhibiting higher HER rates, the interaction of Zn with surface defects of TiO2 enhanced the catalyst stability, resulting in strongly suppressed deactivation. This work introduces intermetallic cocatalysts as promising systems, highlighting the influence of composition and structure on catalyst activity and providing future research directions.
1 Introduction
Fossil fuels have long been used as primary sources of energy owing to their high energy content and facile implementation into existing infrastructures. They currently cover around 80% of global energy requirements.1 However, their excessive consumption leads to the release of greenhouse gases, particularly carbon dioxide (CO2), which is a major contributor to global warming.2 This makes the utilization of environmentally friendly, carbon-neutral, and renewable energy sources essential to replace fossil fuels in the long term.Alongside energy sources such as wind, hydropower, geothermal energy, and biomass, solar energy is considered the most abundant and sustainable alternative.3,4 Consequently, the so-called hydrogen economy is becoming increasingly important. Green hydrogen (H2) is considered a sustainable, clean and renewable energy source that is easy to store and has the potential to replace fossil fuels and tackle the associated environmental problems. H2 exhibits a high gravimetric energy density of 120 MJ kg−1 and releases water as the only by-product when burnt.5,6
There are numerous technologies for H2 production, but only a few of them are considered environmentally friendly and sustainable. Steam reforming of hydrocarbons currently dominates, requiring high temperatures and emitting large amounts of CO2.7 As a carbon-free alternative, water-based processes, such as water splitting, are promising for future energy scenarios. The possibility of carrying out water splitting using solar energy is particularly attractive.8
Photocatalytic water splitting utilizes the solar energy directly to split water into its constituent elements, hydrogen and oxygen, under mild conditions. Despite this, various technological challenges, associated with poor efficiency and scalability, hinder its wide-ranging application.9 Semiconductor materials such as TiO2, CuO, ZnO, and CdS have already been successfully tested as photocatalysts for water splitting. However, wide band gaps, low specific surface areas, and rapid carrier recombination remain as limitations for these materials.10 To overcome these challenges, different approaches such as band structure engineering,11 surface sensitisation and active site engineering,12 doping,13 coupling with co-catalysts14,15 and the formation of heterojunctions16 through the combination of multiple semiconductors have been investigated.
Therefore, latest research focusses on the development of highly active and stable catalysts to facilitate hydrogen production via photocatalytic water splitting under mild conditions. Recently, catalytic active site engineering has gained the spotlight, as it provides17–20 control over the atomic environment of catalytically active centres, their electronic structure, crystal structure and surface properties, ultimately resulting in high-performance photocatalysts.21 The material class of intermetallic compounds are of particular interest for such investigations.22 Intermetallic phases are characterised by a defined atomic arrangement of two or more metal constituents that can offer unique catalytic properties. Often, a single intermetallic system includes a variety of different phases with different compositions, electronic structures and physical properties, which define its overall catalytic performance.
Hitherto, active site engineering via the development of intermetallic nanoparticles itself involves considerable challenges often requiring sophisticated synthesis routes and a general lack of control over particle size, composition, and structure. Therefore, synthesis methods must be developed individually for each intermetallic compound, increasing cost and research effort.23
A promising answer to the above-mentioned synthesis challenges is the simple, direct vapour–solid (VS) synthesis method (Fig. 1), which can be used for various systems.21,24 For the preparation of intermetallic compounds using this method, stoichiometric quantities of the constituent metals are enclosed in an evacuated quartz vessel, spatially separated and equilibrated within a temperature gradient. The reactant with the higher vapour pressure condenses at the coolest point of the system. A temperature gradient between the high-temperature reaction side and the low-temperature reservoir side of the system assures that the interaction of the reactants solely occurs as a direct vapour–solid reaction. Furthermore, system pressure and, thus, the activity are controlled via the respective temperatures. The absence of any additives, the use of simple, commercially available reactants with high purity, and the good control of the composition enable the synthesis of various intermetallic systems with morphological and electronic structural tuning.
 | ||
| Fig. 1 Reaction scheme for the loading of TiO2 nano powder (P25) with Pt co-catalyst particles via in situ photoreduction (a) and the consequent vapour–solid process (b) for the formation of intermetallic Pt–Zn co-catalysts. | ||
This work devised TiO2-supported, intermetallic Pt–Zn nanoparticles as highly active and stable catalysts for the photocatalytic hydrogen evolution reaction (HER), denoted herein as Pt–Zn/TiO2. TiO2 (P25) nano-powder was loaded with a Pt co-catalyst by in situ photoreduction (Fig. 1a), followed by a further material modification via the vapour–solid reaction with Zn (Fig. 1b). These samples were then evaluated for the photocatalytic HER performance with investigations particularly focused on the effect of the composition and structure of the Pt–Zn co-catalyst, as well as the impact of Zn doping of TiO2.
2 Results and discussion
In this work, Pt/TiO2 catalysts with 1 and 5 wt% platinum loadings were prepared by in situ photodeposition. Based on these materials, a total number of seven Pt–Zn/TiO2 catalysts were prepared by the vapour–solid synthesis method (VS): three samples starting from 5 wt% Pt/TiO2 and four samples based on the 1 wt% Pt/TiO2 starting catalyst. The samples were systematically labelled as Ptx–Zny/TiO2_z, where x/y represents the respective atomic Pt/Zn ratio and z the Pt loading of the Pt/TiO2 starting catalyst. The material classification was based on the co-catalyst as either metallic (Pt) or intermetallic (Pt–Zn). Additionally, pure TiO2 (P25) was exposed to Zn vapour, with similar conditions to the Pt–Zn/TiO2 samples, to provide a reference and investigate possible interactions of neat TiO2 with Zn vapour as well as their effect on the photocatalytic performance.2.1 X-ray fluorescence spectroscopy
The elemental compositions were investigated by total X-ray fluorescence spectroscopy (TXRF) of the dissolved catalyst samples. Measured concentrations for Pt and Zn standard solutions were within a 95% confidence interval of the nominal concentration and the results for all samples were used without any further correction. The comparison between the targeted composition and the observed composition by TXRF (see Fig. S1†) shows a very good agreement. Since there is no systematic deviation from the targeted concentration, it is assumed that the slight drifts are based on balancing inaccuracies when handling samples in the milligram regime. The VS method allowed precise concentration control with a maximum deviation of −0.7 wt% Zn in the case of Pt25–Zn75/TiO2_5, which outperforms state-of-the-art synthesis methods.22 The oxidation of Zn on the reservoir side of the reaction vessel can also be excluded, since the composition deviations are not systematic towards lower zinc concentrations, nor was any residual ZnO detected on the glass vessels after reaction. Overall, TXRF hints full conversion of the reactants during the VS synthesis approach. The results for the samples are presented based on the total powder mass (Table 1).| Sample | ω TXRF (Pt) [wt%] | ω target (Pt) [wt%] | ω TXR F (Zn) [wt%] | ω target (Zn) [wt%] |
|---|---|---|---|---|
| Pt/TiO2_5 | 5.0 | 5.0 | — | — |
| Pt59–Zn41/TiO2_5 | 5.1 | 4.9 | 1.2 | 1.1 |
| Pt54–Zn46/TiO2_5 | 4.6 | 4.9 | 1.3 | 1.4 |
| Pt25–Zn75/TiO2_5 | 4.9 | 4.7 | 4.1 | 4.8 |
| Pt/TiO2_1 | 0.9 | 1.0 | — | — |
| Pt50–Zn50/TiO2_1 | 0.9 | 0.9 | 0.3 | 0.3 |
| Pt32–Zn68/TiO2_1 | 0.6 | 0.9 | 0.9 | 0.6 |
| Pt27–Zn73/TiO2_1 | 0.9 | 0.9 | 1.0 | 0.8 |
| Pt20–Zn80/TiO2_1 | 1.0 | 0.9 | 1.1 | 1.2 |
2.2 X-ray diffraction and Rietveld refinement
For a comprehensive understanding of the materials, the investigation of the crystal structure of the catalyst as well as the respective co-catalysts is indispensable. This remains particularly challenging, since typically the co-catalyst particles are rather small and only minor loading concentrations are applied. Consequently, small and broad signals of the respective co-catalyst phases are observed in the diffractogram of the X-ray diffraction analysis. It was found that the respective patterns of the intermetallic were significantly better defined in diffractograms of the 5 wt% Pt-based materials than the 1 wt% Pt-based materials, as one would expect from the corresponding loading values.Table 2 lists the relevant crystallographic information of Pt–Zn phases expected in the composition and temperature regime of interest. It must be considered that the nano-size character of the co-catalyst can lead to different stabilities of the intermetallic compared to the reported phase diagrams based on bulk studies. The phase stability in nanoparticles is greatly dependent on the morphology and size. Simpler structures are more facile to form in nanoparticles contrary to complex structures with a high unit cell volume. Furthermore, phases with a homogeneity range and their associated flexibility regarding composition and activity could benefit the stability of those phases. Complex structures also go hand in hand with lower individual peak intensities in the patterns than those of simpler structures, which lead to fewer, but more intense diffraction reflexes. Consequently, small amounts of complex patterns are more likely to be missed in the presence of dominant simple patterns, especially if the individual reflexes are extremely broadened due to their nanoparticle size.
| Name | Formula | Structure type | Space group | Composition [at% Zn] | Lattice parameters [nm] | Reference |
|---|---|---|---|---|---|---|
| Pt | Pt | Cu |
Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
0 | a = 0.391(5) | E. A. Owen et al.25 |
| (Pt) | (Pt, Zn) | Cu |
Fmm |
0–25 | 0.391(5) ≥ a ≥ 0.388(5) | H. Nowotny et al.26 |
| Pt3Zn | Pt3Zn | AuCu3 |
Pmm |
∼25 | a = 0.388(5) | H. Nowotny et al.26 |
| (PtZn) | PtZn | AuCu | P4/mmm | 31–48 | a = 0.405(0) | H. Nowotny et al.27 |
| c = 0.351(0) | ||||||
| (r) | PtZn1.7 | Pt7Zn12 | Pbam | 62–63 | a = 2.879(0) | W. Carl et al.28 |
| b = 0.694(0) | ||||||
| c = 0.276(0) | ||||||
| γ 1 | Pt3Zn10 | Pt3Zn10 |
F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m 3m |
73–76 | a = 1.811(2) | Johansson et al.29 |
Rietveld refinement of the commercial P25 reference, the metallic Pt/TiO2 catalyst, and the intermetallic Pt–Zn/TiO2 materials confirmed a mixture of anatase (∼87–83%) and rutile (∼13–17%) in all cases, which is in good agreement with the reported investigations.30–32 Importantly, the virtually unchanged ratio between the two phases confirms that VS synthesis did not affect the crystallinity of the supporting TiO2 particles. For the prepared materials, signals, which cannot be ascribed to anatase and rutile patterns, were attributed to the co-catalyst and refined with respective data. For the metallic catalysts, Pt/TiO2_1 and Pt/TiO2_5, the Pt co-catalyst could be observed clearly in the case of Pt/TiO2_5, while for the material with a lower Pt loading, no co-catalyst signals were detected in the diffractogram. Furthermore, for the intermetallic Pt–Zn/TiO2 products, lower Pt–Zn loadings were suitable to be detected, but as expected, the patterns were better defined for samples of higher co-catalyst loadings. Complete conversion to intermetallic phases is hinted, since no Pt patterns could anymore be detected after VS processing. From the four expected intermetallic phases (see Table 2), the two phases (Pt) and (PtZn) were successfully prepared from both 1 wt% and 5 wt%-loaded TiO2 starting catalysts. It is impossible to distinguish (Pt) form its superstructure Pt3Zn, as the additional superstructure reflexes are much too small. Therefore, we generally list (Pt) in Table 3, although we cannot rule out that the superstructure was formed in the nanoparticle. Pt3Zn10 was only identified from the starting material with a higher Pt loading, while the more complex Pt7Zn12-structure, with its rather large unit cell, was generally not observed.
| Sample | Phase | Lattice parameters [nm] |
|---|---|---|
| Pt/TiO2_5 | Pt | a = 0.391(5) |
| Pt59–Zn41/TiO2_5 | (Pt) | a = 0.391(3) |
| (PtZn) | a = 0.401(1); c = 0.354(2) | |
| Pt54–Zn46/TiO2_5 | (Pt) | a = 0.391(4) |
| (PtZn) | a = 0.401(3); c = 0.355(1) | |
| Pt25–Zn75/TiO2_5 | (PtZn) | a = 0.401(2); c = 0.338(6) |
| Pt3Zn10 | a = 1.820(3) | |
| Pt/TiO2_1 | n.a. | n.a. |
| Pt50–Zn50/TiO2_1 | (Pt) | a = 0.390(4) |
| (PtZn) | a = 0.407(6); c = 0.334(6) | |
| Pt32–Zn68/TiO2_1 | (Pt) | a = 0.390(8) |
| Pt27–Zn73/TiO2_1 | (PtZn) | a = 0.407(5); c = 0.340(6) |
| Pt20–Zn80/TiO2_1 | (PtZn) | a = 0.405(9); c = 0.341(2) |
Surprisingly, in all the samples, the refined data suggested lower Zn concentrations in the Pt–Zn co-catalyst than the targeted concentration. This is particularly interesting, since the TXRF results show good agreement of the overall composition. Hence, it is concluded that Zn did not react fully selective with the Pt co-catalyst, but resided on the surface of the TiO2 support. The direct deposition of Zn on the TiO2 particles can be ruled out by the applied temperature gradient keeping the condensed Zn on the cool side of the reaction vessel (Fig. 1). A layer formed due to the reaction of Zn with the surface must result in either fully amorphous or rather thin Zn-containing layers, since neither Zn nor ZnO was observed in the diffractograms. Nevertheless, the presence of the more complex phases Pt3Zn10 and Pt7Zn12 cannot fully be excluded. Although the smaller particle size in the lower loaded samples might benefit the simpler phases, the higher concentrated phases would most likely not be observable in PXRD since their relative intensities would be rather small compared to other present phases (‘invisible patterns’), as discussed above. Considering that Pt3Zn10 was observed in the higher loaded samples, it is most likely that this phase was also formed in the lower concentrated samples but is just not observable by PXRD in those cases. Considering these deliberations, only the Pt32–Zn68/TiO2_1 catalyst sample acts as an outlier since solely the solid solution (Pt) was observed, while the more zinc rich (PtZn) phase would also be expected. This sample in TXRF had already shown a greater divergence from the expected behaviour than other samples, and according to our findings, it is most likely that a smaller co-catalyst loading with a low zinc concentration was reached than targeted for this catalyst. The observed structural characteristics are listed in Table 3, and the patterns of the samples are presented in Fig. 2a. A close-up of the refined patterns for the representative Pt54–Zn46/TiO2_5 sample is shown in Fig. 2b. The other diffractograms with their respective refined phase patterns can be found in the ESI (Fig. S2).†
 | ||
| Fig. 2 Measured PXRD patterns of all the samples (a) and close-up of Pt54–Zn46/TiO2_5 with refined phases (b). | ||
2.3 Transmission electron microscopy and energy-dispersive X-ray spectroscopy
The transmission electron microscopic (TEM) images of the Pt–Zn/TiO2 samples reveal the TiO2 support catalyst loaded with edgy, well-dispersed, and randomly oriented nanoparticles. Fig. 3 presents the brightfield images of the representative samples Pt50–Zn50/TiO2_1 and Pt25–Zn75/TiO2_5. The co-catalyst particles of the low loaded 1 wt% materials are around 2 to 15 nm in size with an average particle size of 6 nm, and no aggregation was observed. For the 5 wt% species also, bigger co-catalyst particles up to 65 nm were seen, which might be formed due to aggregation and sintering, fostered by higher particle loadings. For the graphical illustration of the particle size distribution, each of about 400 Pt–Zn particles was measured in both x and y directions. Fig. S3† shows the resulting histograms and illustrates that a narrow and sharp peak is observed for the Pt–Zn/TiO2_1 sample while Pt–Zn/TiO2_5 features a rather broad distribution, with decreasing numbers of particles towards bigger sizes, which again hints towards sintering and aggregation processes during VS synthesis. | ||
| Fig. 3 Brightfield TEM images of Pt50–Zn50/TiO2_1 (a) and Pt25–Zn75/TiO2_5 (b), along with magnified images; EDX elemental mappings of Pt25–Zn75/TiO2_5 for Pt (c, left) and Zn (c, right). | ||
Elemental mapping and line scanning via energy-dispersive X-ray spectroscopy (EDX) was applied to check on the selectivity of the Zn intake into the co-catalyst particles. Here, for the 1 wt%-loaded material, the difference between the support, the particles, and the noise was not nuanced strongly enough to prove the selective reaction of Zn and Pt. However, the higher overall concentration in the case of the 5 wt% loading makes the differentiation clearer. It appears that Zn mostly interacts with the co-catalyst particles, but a certain deposition on the TiO2 support cannot be excluded. EDX line scans of the sample Pt25–Zn75/TiO2_5 (see Fig. S4†) further reveal co-catalyst compositions, which are in good agreement with the expectations from the PXRD analysis. Based on the line profiles, it can be concluded that Zn may interact with the TiO2 support, leading to a homogeneous deposition of Zn on supporting TiO2.
2.4 X-ray photoelectron and auger electron spectroscopy
For a more profound investigation of the potential Zn deposition as well as the oxidation states of the constituting elements after VS procedure, X-ray photoelectron spectroscopy (XPS) was used. The acquired spectra show the expected C 1s, O 1s and Ti 2p signals for the measured samples (Fig. 4). The Ti 2p3/2 signal (458.3 eV) corresponds to Ti4+ in all samples, with no detectable contribution of other oxidation states. A small, negligible shift to higher binding energies was observed in the presence of Pt and/or Zn (Fig. S5†). This could indicate an interaction of the surface TiO2 layer with deposited metals. The asymmetric Pt 4f7/2 signal at 70.2 eV is attributed to purely metallic Pt0 in the metallic and intermetallic co-catalyst particles; however, the position differs slightly from 71 eV described in the literature33 (Fig. S6†). The determination of the oxidation state of the present Zn species is tricky, since the Zn 2p3/2 signals of both Zn0 and Zn2+ are hard to distinguish and the acquired spectra showed broad peaks of weak strength (Fig. S7a†). Hence, also the Auger peaks had to be investigated. Fig. S7b† shows the exemplary auger spectra for Pt59Zn41/TiO2_5 with an Auger parameter of 2009.6 eV. Those findings strongly suggest the presence of Zn2+ in the form of a thin ZnO layer on top of the TiO2 catalyst particles which are likely formed due to the interaction between Zn and surface defects. It is assumed that the Zn vapour locally reacts with these defects (e.g. oxygen vacancies), forming colour centres, and in turn, is oxidized during the process. Since clear signals for titanium were still obtained despite the low XPS penetration depths, it can be assumed that the thickness of the ZnO layer must be in the range of nanometres. Considering its invisibility during our TEM investigations, we further suggest it to be as thin as a few monolayers. Besides the XPS observations, the colour change due to thin ZnO layer formation was observed in our reference experiments during the reaction of neat P25 with Zn under the same reaction conditions as the VS preparation for Pt–Zn/TiO2 catalysts (see Fig. S8†), which complements the above-mentioned conclusions.34 Furthermore, the metallic Zn0 component observed in XPS is attributed to the intermetallic Pt–Zn phases of the co-catalyst particles expected from intermetallic composition.35 | ||
| Fig. 4 XPS survey spectra of the pristine TiO2 and intermetallic composites. | ||
2.5 Photocatalytic performance
In order to understand the photocatalytic performance of the novel intermetallic Pt–Zn/TiO2 materials for the photocatalytic hydrogen evolution reaction (HER), it is necessary to recognise the various influences of phase formation, composition, and zinc vapor treatment as well as their combined properties. The investigation of not only the intermetallic catalysts, but also the metallic Pt/TiO2 samples, as well as the TiO2 educt materials, with and without exposure to Zn vapour, enables the attribution of various impacts on the activity and stability of the studied photosystems. Since the intermetallic catalysts of high loading showed particle sintering and weaker photocatalytic performance, we decided to base the following discussions on the Pt–Zn/TiO2_1 species and present data of the Pt–Zn/TiO2_5 samples in the supplementary.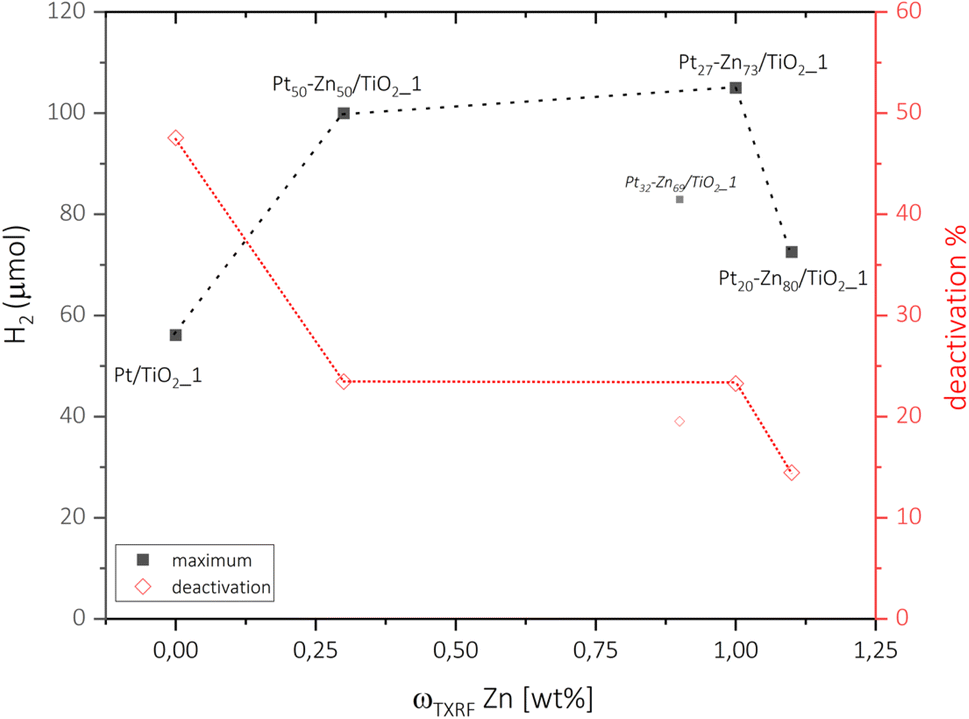 | ||
| Fig. 5 Maximum (■) hydrogen release and the respective catalyst deactivation (◊) after 2 h of steady operation of Pt/TiO2_1 and Pt–Zn/TiO2_1 catalysts. As discussed above, Pt32–Zn68/TiO2_1 is assumed to be an outlier according to characterization methods. | ||
3 Concluding remarks
In summary, TiO2-supported Pt–Zn nanoparticles of different crystal structures and compositions have been prepared by a novel, facile vapour–solid synthesis route and studied for their photocatalytic performance towards hydrogen evolution. The uniform reaction conditions allowed direct comparison of the different intermetallic phases of the Pt–Zn system. Investigations particularly focused on the relationship between the crystal structure, composition and activity of the co-catalysts. In our operation setup, the formation of intermetallic Pt–Zn could increase the intrinsic activity of neat Pt co-catalysts and almost double the H2 generation rate from 56.1 μmol for Pt/TiO2 to 105.0 μmol for Pt27–Zn73/TiO2, corresponding to an increase in apparent quantum yield from 5.4% up to 10.3%. The performance trend of Pt–Zn phases for photocatalytic hydrogen evolution was (PtZn) > (Pt) > Pt and agrees well with expectations from previously published (de)hydrogenation studies. The boosted performance of (PtZn) was assigned to the optimized availability and electron density of the Pt sites. In addition, the interaction of Zn vapour with the TiO2 support was found to be beneficial for the stability and durability of the photocatalysts. This resulted in lowered HER deactivation, which we assigned to reduced encapsulation by the support and higher stability of Pt–Zn surfaces. This work demonstrates the potential of Pt–Zn intermetallic compounds as co-catalysts for H2 evolution, presents an innovative perspective for their vapor–solid synthesis and gives directions to follow-up studies.4 Experimental
4.1 Chemicals
Zinc, >99.999% (Alfa Aesar, United States of America); nitric acid, 69% HNO3 suprapur (Merck, Germany); Inoxline H5, 95 vol% Ar/5 vol% H2 (Messer, Austria); methanol, HPLC-grade (VWR, Germany); chloroplatinic acid, H2PtCl6 8 wt% in water (Sigma-Aldrich, Germany); titanium(IV) oxide, P25 (Sigma-Aldrich, Germany) were used in this experiment.All chemicals were used as purchased. Water was purified and deionized (18.2 MΩ cm) in-house using a Milli-Q® (Merck, Germany) system.
4.2 Preparation of Pt/TiO2
The pristine Pt/TiO2_1 sample was prepared by a photodeposition method using a commercially available aqueous solution of H2PtCl6 (Sigma-Aldrich, 8 wt% in water). Briefly, 1.3 mL of the solution was diluted in 65 mL of DI H2O resulting in a 4.1 mM Pt stock solution. In a large round-bottom flask, 5 g of TiO2 (P25, Sigma-Aldrich) was dispersed via ultrasonication in a mixture of 250 mL CH3OH (HPLC) and 200 mL of H2O (DI) for 10 min. Following this, 62.5 mL of Pt-stock solution was added to the flask and the solution was purged with argon gas (Ar 5.0) for 10 min at 10 mL min−1 to remove any dissolved oxygen. The reactor was then illuminated using two 365 nm LED lamps (SOLIS LED, Thorlabs) for 16 h to ensure complete photoreduction of Pt onto TiO2. Finally, the powder was washed with 100 mL CH3OH and 100 mL H2O via vacuum filtration. A similar method was used to prepare 1 g of pristine Pt/TiO2_5, using appropriate amounts of Pt stock solution, resulting in 5 wt% on 1 g of P25/TiO2.4.3 Preparation of Pt–Zn/TiO2via the vapour–solid route
The formation of the intermetallic species by the direct vapour–solid method was carried out similarly as described in our previous works. For pre-treatment, the Pt/TiO2 powders were reduced for two hours in an Inoxline (Ar/H2 = 95/5 = v/v) gas stream of about 30 mLN min−1 at 350 °C. The gases were removed in a follow-up step at 350 °C for two hours under reduced pressure.Zinc was freed from potential surface oxides by melting the zinc granules and filtering through chemically pure quartz wool under an argon atmosphere. The respective stoichiometric amount of purified zinc was weighed to an accuracy of ±0.1 mg and condensed on one side of a custom-built quartz glass tube under a static, reduced pressure of <2 × 10−2 mbar by heating with an H2/O2-torch.
Consequently, the respective amount of as-prepared Pt/TiO2 was transferred into a reaction tube, without direct contact to the zinc. The reaction tube, loaded with the Pt/TiO2 powder on the bottom and the zinc reservoir on top, was subsequently sealed under a reduced pressure of <2 × 10−2 mbar. The evacuated reaction vessel was then transferred into a two-zone furnace (HTM Reetz, Germany) and aligned in a temperature gradient of 422 °C at the zinc side and 448 °C at the Pt/TiO2 side of the final experimental setup. The reaction progress could be tracked by the disappearance of zinc from the quartz wall during two days of reaction. An isothermal post-reaction step of five days at 400 °C in a muffle furnace (Nabertherm, Germany) was applied to improve homogeneity of the samples. The reaction vessels were opened in the glovebox (MBraun, Germany) and the produced Pt–Zn/TiO2 catalysts were stored under argon.
4.4 Materials characterisation
The sample compositions were determined using a Picofox S2 (Bruker, Germany) total X-ray fluorescence spectrometer. Therefore, about 30 mg of the powders, respectively, were dispersed in 20 mL of 69% HNO3 and kept under constant stirring at 105 °C for one hour. The dispersions were filtered and the solutions each diluted up to 100 mL with ultra-pure water. Then 500 μL of the respective solutions were mixed with 500 μL of 10 mg L−1 chromium standard solution and 100 μL of 0.3 g L−1 polyvinyl alcohol solution. For analysis, 5 μL of the as-prepared sample solutions were transferred onto quartz plates and dried under infra-red irradiation and reduced pressure for 45 minutes. Measurement times of 400 seconds per plate were used with the Picofox S2 apparatus. Gain correction was carried out with an arsenic standard mounted to a quartz plate, similar to the samples.Powder X-ray diffraction was carried out using a D8 Advance diffractometer (Bruker, Germany) to investigate the phase composition and structural information of the prepared catalysts. A thin layer of grease was used to fix the powders on zero-background silicon single-crystal plates. The diffractometer was operated in Bragg–Brentano pseudo focusing mode with theta/theta geometry. A one-dimensional silicon strip detector, Lynxeye (Bruker, Germany) was used to record the diffractograms. The patterns were determined in the relevant 30° to 90° 2-theta range over three hours at an accelerating voltage of 40 kV and a beam current of 40 mA. Rietveld refinements were performed using the Topas 7.13 software (Bruker, Germany).
Transmission electron microscopy (TEM) and electron-dispersive X-ray spectroscopy (EDS) were performed using a Talos F200i (Thermo Scientific™, Germany) electron microscope operating at a voltage of 200 kV, equipped with a field electron gun and a 4k × 4k Ceta 16M camera. Data evaluation was done using the Velox software (Thermo Scientific™, Germany).
X-ray photoelectron spectroscopy (XPS) investigations were carried out using a custom-built SPECS XPS-spectrometer equipped with a monochromatic Al Kα X-ray source (μFocus 350) along with a hemispherical WAL 150 analyser at an acceptance angle of 60°. Powder samples were mounted on the sample holder plate using double-sided carbon tape.
Pass energies of 100 eV and 30 eV with energy resolutions of 1 eV and 100 meV, respectively, were used while acquiring the surveys and detailed spectra of each element, respectively. The excitation energy of 1486.6 eV was used along with a beam energy and a spot size of 70 onto 400 μm, respectively, at an angle of 51° to sample surface normal. The base pressure achieved was 5 × 10−10 mbar with a pressure during measurements of 2 × 10−9 mbar.
Data analysis was carried out using the CASA XPS software, employing transmission corrections (as per the instrument vendor's specifications), Shirley/Tougaard backgrounds41,42 and Scofield sensitivity factors.43 Charge correction was done, so that the adventitious carbon peak (C–C peak) was shifted to 284.8 eV binding energy (BE).
4.5 Photocatalytic experiments
Photocatalytic hydrogen evolution tests were carried out using a custom-built slurry type reactor with a total volume of 100 mL, equipped with a water-cooling jacket for keeping a constant reaction temperature of 15 °C. Briefly, 10 mg of the catalyst material was homogenously dispersed in a beaker containing an aqueous mixture of 20 mL CH3OH (HPLC) and 20 mL H2O (DI) via ultrasonication for 180 s. The reaction mixture was then transferred to the reactor vessel and purged for 10 min using argon gas (Ar5.0) to remove any dissolved oxygen in the solution. Next, the reactor was sealed with a constant flow of argon set to 30 mL min−1 carrying any reaction products to the detector system. The produced H2 gas was detected using a thermal conductivity detector (TCD, X-stream by Emerson Process Management) recording a datapoint every 10 s. First, a baseline zero was obtained for 30 min in the absence of light, followed by an illumination of 2 h. The measured H2 values in ppm were converted to the H2 evolution rate (μmol h−1) using the ideal gas equation and the reaction flow rate (30 mL min−1) as described below:In general, 1 ppm is equal to 1/100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 volume of gas flowing through the detector. In our case, the flow rate is 30 mL min−1, which can be converted from volume to μmoles using the ideal gas equation PV = nRT. Therefore, the final activity can be written as follows:
000 volume of gas flowing through the detector. In our case, the flow rate is 30 mL min−1, which can be converted from volume to μmoles using the ideal gas equation PV = nRT. Therefore, the final activity can be written as follows:

Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
Daniel Garstenauer: writing – original draft, methodology, investigation, formal analysis, data curation, conceptualization, visualization. Stephen Nagaraju Myakala: writing – review & editing, methodology, investigation, formal analysis. Pablo Ayala: writing – review & editing, methodology, investigation, formal analysis. Hannah Rabl-Wolff: writing – review & editing, methodology, investigation, formal analysis. Ondřej Zobač: writing – review & editing, methodology, investigation, formal analysis, resources. Franz Jirsa: writing – review & editing, formal analysis, resources. Dominik Eder: writing – review & editing, methodology, validation, resources. Alexey Cherevan: writing – review & editing, methodology, formal analysis, conceptualization, resources, project administration, validation, supervision. Klaus W. Richter: writing – review & editing, methodology, formal analysis, conceptualization, resources, project administration, validation, supervision.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Dr Ivo Kuběna, Institute of Physics of Materials, Czech Academy of Sciences, deserves our thanks for the advice on the interpretation of the microscopy results. The authors acknowledge the funding support from the University of Vienna, Austria. This research was further supported by Austria's Agency for Education and Internationalisation (OEAD) within the scientific & technological cooperation programme (CZ 06/2023). This research was funded in part by the Austrian Science Fund (FWF) [10.55776/COE5] (Cluster of Excellence MECS). H. R-W. would like to acknowledge funding from the Austrian Science Fund (FWF) [10.55776/DOC142] (Doctoral college TU-Dx). The authors acknowledge the analytical instrumentation center (AIC) at TU Wien for facilitating XPS measurements.References
- IEA, World Energy Outlook, 2024, https://www.iea.org/.
- E. A. Rosa and T. Dietz, Nat. Clim. Change, 2012, 2, 581–586 CrossRef CAS.
- A. Hussain, S. M. Arif and M. Aslam, Renewable Sustainable Energy Rev., 2017, 71, 12–28 CrossRef.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Genovese, A. Schlüter, E. Scionti, F. Piraino, O. Corigliano and P. Fragiacomo, Int. J. Hydrogen Energy, 2023, 48, 16545–16568 CrossRef CAS.
- Y. Gong, J. Yao, P. Wang, Z. Li, H. Zhou and C. Xu, Chin. J. Chem. Eng., 2022, 43, 282–296 CrossRef CAS.
- IEA, Global Hydrogen Review, 2024, https://www.iea.org/.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- P. C. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS.
- A. B, J. A, A. S. Rao, S. S. Nagarkar, A. Dutta, S. P. Duttagupta, S. S. Prabhu and R. Pinto, Int. J. Hydrogen Energy, 2024, 81, 1442–1466 CrossRef CAS.
- M. Tahir, S. Tasleem and B. Tahir, Int. J. Hydrogen Energy, 2020, 45, 15985–16038 CrossRef CAS.
- J. Huo, Y. Dou, C. Wu, H. Liu, S. Dou and D. Yuan, Adv. Mater., 2024, 2416483–2416525 Search PubMed.
- Y. He, T. Lv, T. Zhou, B. Liu, B. Xiao, H. Zheng, B. Zi, J. Zhang, Y. Zhang, G. Zhang and Q. Liu, Int. J. Hydrogen Energy, 2024, 57, 491–499 CrossRef CAS.
- J. S. Schubert, L. Kalantari, A. Lechner, A. Giesriegl, S. P. Nandan, P. Alaya, S. Kashiwaya, M. Sauer, A. Foelske, J. Rosen, P. Blaha, A. Cherevan and D. Eder, J. Mater. Chem. A, 2021, 9, 21958–21971 RSC.
- J. S. Schubert, J. Popovic, G. M. Haselmann, S. P. Nandan, J. Wang, A. Giesriegl, A. S. Cherevan and D. Eder, J. Mater. Chem. A, 2019, 7, 18568–18579 RSC.
- M. Faraji, M. Yousefi, S. Yousefzadeh, M. Zirak, N. Naseri, T. H. Jeon, W. Choi and A. Z. Moshfegh, Energy Environ. Sci., 2019, 12, 59–95 RSC.
- S. Mitchell, R. Qin, N. Zheng and J. Perez-Ramirez, Nat. Nanotechnol., 2021, 16, 129–139 CrossRef CAS.
- Y. Chen, L. Soler, C. Cazorla, J. Oliveras, N. G. Bastus, V. F. Puntes and J. Llorca, Nat. Commun., 2023, 14, 6165–6175 CrossRef CAS.
- W. Yang, Z. Wang, W. Zhang and S. Guo, Trends Chem., 2019, 1, 259–271 CrossRef CAS.
- S. Batool, S. P. Nandan, S. N. Myakala, A. Rajagopal, J. S. Schubert, P. Ayala, S. Naghdi, H. Saito, J. Bernardi, C. Streb, A. Cherevan and D. Eder, ACS Catal., 2022, 12, 6641–6650 CrossRef CAS PubMed.
- D. Garstenauer, P. Guggenberger, O. Zobac, F. Jirsa and K. W. Richter, Nanoscale, 2024, 16, 20168–20181 RSC.
- Y. Yang and M. Wei, J. Mater. Chem. A, 2020, 8, 2207–2221 RSC.
- B. P. Williams, Z. Qi, W. Huang and C. K. Tsung, Nanoscale, 2020, 12, 18545–18562 RSC.
- P. Wibner, R. Kriegel, D. Garstenauer, O. Zobač, I. Kuběna, N. Rösch, T. Seyller, M. Armbrüster and K. W. Richter, J. Phys. Chem. C, 2024, 128, 6906–6916 CrossRef CAS.
- E. A. Owen and E. L. Yates, London, Edinburgh Dublin Phil. Mag. J. Sci., 2009, 15, 472–488 CrossRef.
- H. Nowotny, E. Bauer, A. Stempfl and H. Bittner, Monatsh. Chem. Verw. Teile Anderer Wiss., 1952, 83, 221–236 CrossRef CAS.
- H. Nowotny, E. Bauer, A. Stempfl and H. Bittner, Monatsh. Chem. Verw. Teile Anderer Wiss., 1950, 81, 1164–1180 CrossRef CAS.
- W. Carl and K. Schubert, J. Less-Common Met., 1962, 19, 279–283 CrossRef.
- A. Johansson and S. Westman, Acta Chem. Scand., 1970, 24, 3471–3479 CrossRef CAS.
- N. R. C. Fernandes Machado and V. S. Santana, Catal. Today, 2005, 107–108, 595–601 CrossRef CAS.
- G. M. Haselmann and D. Eder, ACS Catal., 2017, 7, 4668–4675 CrossRef CAS.
- X. Jiang, M. Manawan, T. Feng, R. Qian, T. Zhao, G. Zhou, F. Kong, Q. Wang, S. Dai and J. H. Pan, Catal. Today, 2018, 300, 12–17 CrossRef CAS.
- A. P. Tsai, S. Kameoka, K. Nozawa, M. Shimoda and Y. Ishii, Acc. Chem. Res., 2017, 50, 2879–2885 CrossRef CAS.
- D. M. King, X. Liang, C. S. Carney, L. F. Hakim, P. Li and A. W. Weimer, Adv. Funct. Mater., 2008, 18, 607–615 CrossRef CAS.
- L. S. Dake, D. R. Baer and J. M. Zachara, Surf. Interface Anal., 2004, 14, 71–75 CrossRef.
- B. Zhang, G. Li, Z. Zhai, D. Chen, Y. Tian, R. Yang, L. Wang, X. Zhang and G. Liu, AIChE J., 2021, 67, 1–12 CrossRef.
- A. Han, J. Zhang, W. Sun, W. Chen, S. Zhang, Y. Han, Q. Feng, L. Zheng, L. Gu, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2019, 10, 3787 CrossRef.
- C. Rodaum, P. Chaipornchalerm, W. Nunthakitgoson, A. Thivasasith, T. Maihom, T. Atithep, P. Kidkhunthod, C. Uthayopas, S. Nutanong, S. Thongratkaew, K. Faungnawakij and C. Wattanakit, Fuel, 2022, 325, 124833 CrossRef CAS.
- V. J. Cybulskis, B. C. Bukowski, H.-T. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro, J. Greeley and J. T. Miller, ACS Catal., 2017, 7, 4173–4181 CrossRef CAS.
- M. Wang, Z. Shi, W. Shi, J. Jiang, J. Lan, R. Li, Y. Yan, Z. Liu, L. Fu, X. Liu, S. Sang, Y. Hu and J. Zhou, Adv. Mater., 2025, 37, e2409575 CrossRef PubMed.
- D. A. Shirley, Phys. Rev. B, 1972, 5, 4709–4714 CrossRef.
- S. Tougaard, Surf. Interface Anal., 1997, 25, 137–154 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- M. Qureshi and K. Takanabe, Chem. Mater., 2016, 29, 158–167 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5se00487j |
| This journal is © The Royal Society of Chemistry 2025 |