
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene-based catalysts for electrochemical CO2 reduction reaction
Lankamullage Hasini Amanda Wijewardena
a,
Woo Seok Cheon
a,
Seol-Ha Jeong
*b,
Jungwon Park
 *b and
Ho Won Jang
*ac
*b and
Ho Won Jang
*ac
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea
bDepartment of Chemical and Biological Engineering, Seoul National University, Seoul 08826, Republic of Korea. E-mail: jsh528@snu.ac.kr; jungwonpark@snu.ac.kr
cAdvanced Institute of Convergence Technology, Seoul National University, Suwon 16229, Republic of Korea. E-mail: hwjang@snu.ac.kr
First published on 7th May 2025
Abstract
In response to environmental concerns and the energy transition, electrochemical CO2 reduction (eCO2RR) to valuable chemicals and feedstocks using renewable electricity holds significant promise. Yet the challenge remains in developing efficient, low-cost catalysts that exhibit no detrimental environmental effects. Emerging graphene-based materials have demonstrated exceptional potential for eCO2RR due to their distinctive electronic characteristics and structural advantages, providing remarkable activity, selectivity, and durability. Moreover, they are earth-abundant, non-toxic, and environmentally friendly. This review provides a comprehensive analysis of the application of graphene-based materials as a catalyst for eCO2RR, focusing on strategies to optimize catalytic performance. Key strategies, such as heteroatom doping and metal-graphene hybridization, are discussed. Furthermore, this review not only explores traditional graphene-based materials but also covers emerging graphene analogs for eCO2RR. It also highlights challenges, providing perspectives on future research directions to further improve the design and application of graphene-based catalysts for sustainable CO2 conversion.
 Lankamullage Hasini Amanda Wijewardena | Lankamullage Hasini Amanda Wijewardena is currently a master's degree candidate under the supervision of Prof. Ho Won Jang in the Department of Materials Science and Engineering at Seoul National University. She received her B.S. degree from the Department of Textile and Apparel Engineering at the University of Moratuwa, Sri Lanka, in 2021. Her research interest is focused on the design of metal-graphene-based electrocatalysts for CO2 reduction. |
 Woo Seok Cheon | Woo Seok Cheon is currently a PhD candidate under the supervision of Prof. Ho Won Jang at the Department of Materials Science and Engineering of Seoul National University. He received his B.S. degree in the Department of Materials Science and Engineering from Seoul National University in 2021. His research interest is focused on the design of nanostructured metal-based electrocatalysts for CO2 reduction. |
 Seol-Ha Jeong | Seol-Ha Jeong is a research professor at the Institute of Chemical Processes, Seoul National University, specializing in bioengineering, tissue engineering, and biomedical devices. She received a B.S. degree (2012), M.S. degree (2014), and PhD (2017) from the department of Materials Science and Engineering at Seoul National University. She has an extensive background, having completed a postdoctoral research position at Seoul National University's Department of Materials Science and Engineering (2018–2022) and worked as a research fellow at Harvard Medical School and Brigham and Women's Hospital (2022–2024). Dr Jeong also served as a senior researcher at the Seoul National University Institute of Biotechnology and the Department of Plastic Surgery at Boramae Medical Center (2024.03–2024.10). Since November 2024, she has joined Professor Jungwon Park's lab at Seoul National University, where she is using in situ liquid-phase transmission electron microscopy to analyze the dynamic structures of biomaterials. |
 Jungwon Park | Jungwon Park is currently an Associate Professor at Seoul National University, Seoul, Korea. His research field of interest is physical chemistry of nanomaterials and in situ electron microscopy. He started his professional career as an Assistant Professor in the School of Chemical and Biological Engineering at Seoul National University (2016-present), after a postdoc and a research associate position at the School of Engineering and Applied Sciences, Harvard University. He is jointly affiliated in the Center for Nanoparticle Research, Institute for Basic Science, as a Research Associate. He has received various prestigious awards such as Korea Presidential Award for Young Scientists in 2020, The 3rd Hanseong Science Award in 2020, and Wiley-KCS Young Chemist Award in 2021. He serves as an advisory board member for iScience, cell press and an early career advisory board member for ChemNanoMat (Wiley). He received a B.S. degree from the department of Chemistry at POSTECH, Korea in 2003. He then received a PhD from the department of Chemistry at the University of California, Berkeley in 2012. |
 Ho Won Jang | Ho Won Jang is a full professor at the Department of Materials Science and Engineering of Seoul National University. He received his PhD from the Department of Materials Science and Engineering at Pohang University of Science and Technology in 2004. He worked as a research associate at the University of Wisconsin–Madison from 2006 to 2009. Before he joined Seoul National University in 2012, he had worked at the Korea Institute of Science and Technology (KIST) as a senior research scientist. His research interests include materials synthesis and device fabrication for (photo)electrocatalysis, chemical sensors, memristors, micro-LEDs, and thin film transistors. |
Sustainability spotlightThe continuous rise in CO2 emissions threatens global climate stability, emphasizing the need for carbon-neutral energy solutions. Electrochemical CO2 reduction offers a sustainable pathway to convert CO2 into valuable fuels using renewable energy, relying on catalysts to improve efficiency. Conventional catalysts, however, often depend on noble metals or exhibit low efficiency, presenting sustainability challenges. In this regard, graphene-based catalysts offer a sustainable alternative due to their abundance, non-toxicity, and eco-friendliness. With its unique properties, graphene enables more efficient CO2 reduction, offering environmentally friendly and improved performance. This advancement aligns with UN SDGs 7 (Affordable and Clean Energy), 12 (Responsible Consumption and Production), and 13 (Climate Action), promoting sustainable energy conversion and reducing carbon emissions. |
1. Introduction
The sharp rise in atmospheric CO2 levels, primarily due to fossil fuel combustion and human activities, poses a significant threat to the environment and humankind. This necessitates the urgent need for sustainable solutions to achieve carbon cycle neutrality through the development of advanced carbon recycling technologies. Consequently, various strategies have emerged to reduce fossil fuel dependency and recycle excess CO2,1 with carbon capture and storage (CCS) and carbon capture and utilization (CCU) being extensively explored as promising approaches.2 While CCS is constrained by high costs and associated risks, CCU offers a more sustainable alternative by capturing CO2 at its source and converting it into fuels and value-added feedstock, thereby addressing both environmental and energy concerns.3 Among the various strategies within carbon capture and utilization (CCU), eCO2RR stands out due to its ability to operate under ambient conditions and its potential to generate carbon-based fuels and chemicals from non-fossil fuel energy sources, making it highly aligned with the goals of a sustainable energy economy.4While electrochemical CO2 reduction (ECR) presents a potential solution for fuel production and the generation of value-added chemicals, the inherent properties of CO2 pose significant challenges. CO2 is a highly stable linear molecule with a strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (750 kJ mol−1), which poses a challenge for its electrochemical conversion. This strong bond makes CO2 thermodynamically stable, resulting in a high activation barrier and sluggish conversion. Consequently, significant overpotentials are required to overcome these barriers and drive the reaction.5 Moreover, ECR becomes more challenging in an aqueous electrolyte due to the multi-proton-coupled electron transfer steps. Furthermore, the competing hydrogen evolution reaction (HER) and the complex reaction pathways leading to a broad range of products make achieving selectivity for a desired product a significant challenge.6 Therefore, to make CO2 electrochemical reduction economically viable, electrocatalysts with high selectivity, stability, and activity toward CO2 are required.7
O bond (750 kJ mol−1), which poses a challenge for its electrochemical conversion. This strong bond makes CO2 thermodynamically stable, resulting in a high activation barrier and sluggish conversion. Consequently, significant overpotentials are required to overcome these barriers and drive the reaction.5 Moreover, ECR becomes more challenging in an aqueous electrolyte due to the multi-proton-coupled electron transfer steps. Furthermore, the competing hydrogen evolution reaction (HER) and the complex reaction pathways leading to a broad range of products make achieving selectivity for a desired product a significant challenge.6 Therefore, to make CO2 electrochemical reduction economically viable, electrocatalysts with high selectivity, stability, and activity toward CO2 are required.7
The eCO2RR has been extensively studied on metal catalysts due to their high catalytic performance. Precious metal-based catalysts such as Au, Ag, and Pt have been well-established for electrochemically converting CO2 into C1 products with lower overpotentials and high current densities.8 Yet, they face several challenges, including high costs, scarcity, and negative environmental effects.9 Moreover, to date, only copper and copper oxide-derived catalysts have been demonstrated to facilitate the electrolysis of CO2 into hydrocarbons and oxygenates beyond the 2-electron reduction pathway. However, the application of these catalysts is limited by their high overpotential and stability issues,10 making them inadequate for industrial applications and global commercialization. Therefore, the development of environmentally friendly electrocatalysts with high activity and stability, capable of tuning the competitive reactivity between eCO2RR and HER, is essential for advancing electrochemical CO2 reduction.
Graphene-based materials are gaining increasing attention as promising catalysts for ECR due to their unique properties, including a large specific surface area, high electrical conductivity, and exceptional thermal stability.11 In addition, graphene is a carbon-based material that is abundant and non-toxic and can be synthesized through eco-friendly methods, reducing reliance on scarce or harmful materials.12 Thus, using graphene-based catalysts for eCO2RR approaches aims to overcome the intrinsic limitations of metal-based catalysts. Although pristine graphene is relatively inactive for ECR due to its electroneutrality, functionalization induces charge redistribution, enhancing its ability to adsorb and activate CO2, making it a promising catalyst. For example, heteroatom-doped metal-free graphene-based catalysts have shown promising results in eCO2RR, producing CO with faradaic efficiencies (FE) exceeding 90% at low overpotentials, rivaling those of silver and gold.13–17 Moreover, metal-graphene hybridized catalysts have achieved FEs for C2 products as high as 90% at overpotentials comparable to copper.18,19 Hence, advancements in graphene-based catalysts will be pivotal for driving sustainable energy conversion technologies.
This paper provides a comprehensive review of recent progress on graphene-based materials for the electrocatalytic conversion of CO2 into value-added products. We first present an overview of eCO2RR, including evaluation parameters and characterization techniques. Next, a systematic review of the role of graphene-based materials, along with their optimization strategies such as metal-free heteroatom doping and metal hybridization, is provided, along with the application of these materials for the electrochemical conversion of CO2. Following that, the advances in emerging graphene analogs as an extension of graphene-based catalysts for eCO2RR are presented. A schematic overview of the major categories of graphene-based catalysts explored for eCO2RR is illustrated in Fig. 1. Finally, an outlook on the future development of graphene-based catalysts and electrochemical systems for eCO2RR is discussed.
 | ||
| Fig. 1 Schematic representation categorizing graphene-based catalysts designed for eCO2RR. | ||
2. Key aspects of eCO2RR
2.1 Fundamentals of eCO2RR
CO2, with an oxidation state of +4, represents the highest oxidation state of carbon. Consequently, converting CO2 into a more energetic product necessitates the transfer of electrons to carbon, lowering its oxidation state.5 The conversion of CO2 into more reduced products is referred to as CO2 reduction, with eCO2RR specifically referring to the use of electrical energy to drive this transformation into various carbon-based products. Generally, this process is performed in an electrolyzer consisting of a cathode and an anode separated by an electrolyte. The electrode is solid for heterogeneous catalysts, and the electrolyte is an aqueous solution saturated with CO2. The electrolysis process typically occurs at the electrode–electrolyte interface. During the eCO2RR process, the cathode facilitates the reduction of carbon dioxide into useful hydrocarbons, aldehydes, or alcohols, while the anode undergoes the oxygen evolution reaction (OER), producing oxygen to complete the overall reaction. The reaction mechanisms are outlined below.20At the cathode
| xCO2 + nH+ + ne− → product + yH2O | (1) |
At the anode,
| 2H2O → O2 + 4H+ + 4e− | (2) |
Mechanistically, three principal steps are involved in the conversion of CO2 into carbon-based products.21 The first step involves the adsorption and activation of CO2 onto the electrode surface. This is followed by proton-coupled electron transfer (PCET) processes that generate various products through different reaction pathways involving 2 to 8 electrons. Finally, the products desorb from the catalyst surface and diffuse into the electrolyte. The final product distribution is highly dependent on the properties of the electrocatalyst and the applied electrode potential. A diverse range of products can be obtained, including C1 compounds such as carbon monoxide (CO), formic acid (HCOOH), methanol (CH3OH), formaldehyde (HCHO), and methane (CH4), as well as C2+ products like ethylene (CH2CH2), ethanol (C2H5OH), and acetate (CH3COOH). As illustrated in Fig. 2, the formation of CO and HCOOH is relatively straightforward, each involving only a single pathway.22 In contrast, methane and methanol may proceed through either the *CH2OH or *OCH3 intermediate, while the formation of C2+ products involves multiple intermediates, making product selectivity more complex. For example, acetate can be formed via hydrogenation of *CO to *CH2, which then reacts with CO to form *CH2CO, or alternatively through *CO dimerization to *COCHO followed by hydrogenation.23 Ethanol and ethylene share the intermediate *CH2CHO, with ethanol forming via further PCET steps, while ethylene results from dehydration.24 Ethane forms through sequential PCET on CO to generate CH3, which then couples to yield ethane. Ethylene glycol synthesis starts from *CHO formation, followed by CO insertion and multiple PCET steps.25 Table 1 summarizes the half-reactions and their equilibrium potentials relative to the standard hydrogen electrode (SHE) at pH 7.
 | ||
| Fig. 2 Mechanistic pathways for electrochemical CO2 reduction to C1, C2, and C3 products. Adapted with permission from ref. 22 Copyright 2023, Elsevier. | ||
| CO2 reduction half-reaction | E0 [V vs. SHE] |
|---|---|
| CO2 + 2H+ + 2e− → HCOOH + H2O | Eredox0 = −0.610 V |
| CO2 + 2H+ + 2e− → CO + H2O | Eredox0 = −0.530 V |
| 2CO2 + 2H+ + 2e− → H2C2O4 | Eredox0 = −0.913 V |
| CO2 + 4H+ + 4e− → HCHO + H2O | Eredox0 = −0.480 V |
| CO2 + 6H+ + 6e− → CH3OH + H2O | Eredox0 = −0.380 V |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | Eredox0 = −0.240 V |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | Eredox0 = −0.349 V |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O | Eredox0 = −0.329 V |
| 2CO2 + 14H+ + 14e− → C2H6 + 4H2O | Eredox0 = −0.270 V |
| 3CO2 + 18H+ + 18e− → C3H7OH + H2O | Eredox0 = −0.310 V |
| 2H+ + 2e− → H2 | Eredox0 = −0.42 V |
Steering the selectivity of catalysts towards specific products in eCO2RR is challenging due to the wide range of possible carbon-based outputs. Pioneering work by Hori et al.26 classified metal catalysts based on their FE and the types of products they generate, showing that different metals tend to produce specific products such as H2, HCOOH, CO, or more complex hydrocarbons, depending on their catalytic properties.27 According to the Sabatier principle, optimal catalytic performance requires a balanced interaction between the catalyst and reaction intermediates, neither too weak nor too strong, to ensure effective adsorption and product release, which explains the performance differences among various metal catalysts.
2.2 Evaluation parameters
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles), with minimal change indicating stability. The galvanostatic/potentiostatic approach monitors potential or current density over time at a constant current density (≥10 mA cm−2) for at least 10 hours, where minimal variation signifies good stability.
000 cycles), with minimal change indicating stability. The galvanostatic/potentiostatic approach monitors potential or current density over time at a constant current density (≥10 mA cm−2) for at least 10 hours, where minimal variation signifies good stability.3 Graphene-based materials as a catalyst for eCO2RR
3.1 Properties of graphene-based materials
Since its discovery by Novoselov and Geim in 2004, graphene has been the focus of research due to its unique properties, such as exceptionally high surface area, outstanding conductivity, and excellent thermal and chemical stability.28,29 This single-atom-thick carbon structure is not only easy to modify into various structures, such as graphene oxide (GO), reduced graphene oxide (rGO), and carbon nanotubes (CNTs), but also versatile in assembling into various allotropes- (0D) fullerenes, (1D) nanotubes, (2D) graphene, or (3D) stacked-graphene (Fig. 3),30–32 while retaining the two-dimensional hexagonal lattice of sp2-hybridized carbon atoms as their fundamental building block.33 This structural diversity of graphene has laid the basis for developing novel graphene-based catalysts, which have attracted increasing interest for applications in electrocatalytic energy conversion. However, pristine graphene and its derivatives exhibit limited catalytic performance for some electrochemical reactions including eCO2RR due to their highly ordered and inert carbon structure. This results in poor adsorption and activation of CO2 molecules during the eCO2RR. This provides greater flexibility for modifications to optimize the catalytic properties of graphene-based materials. The introduction of non-metal heteroatoms and metal hybrid formation with non-noble metals has been utilized to enhance the catalytic performance of graphene.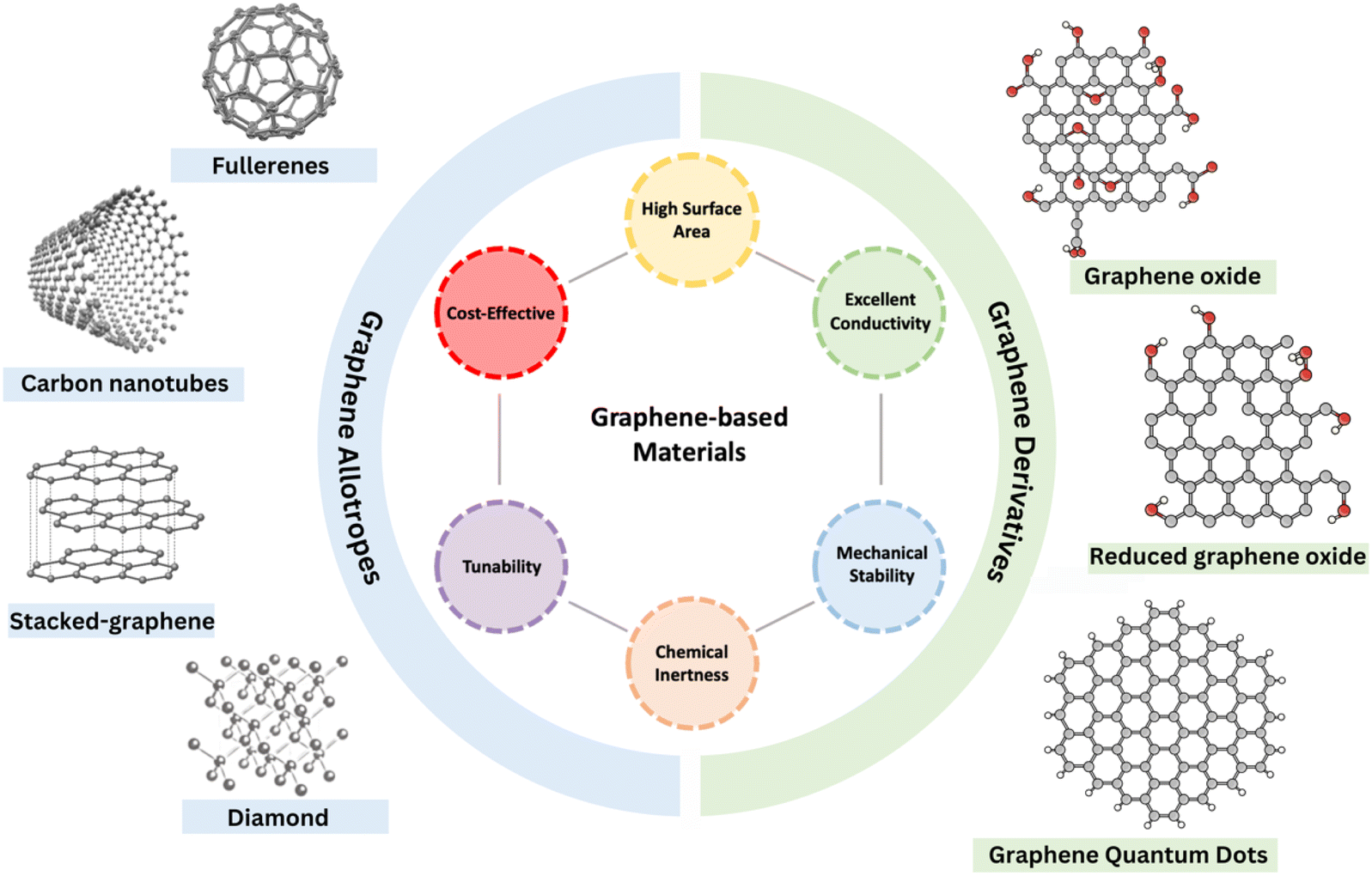 | ||
| Fig. 3 Schematic representation of graphene-based materials, including graphene allotropes and derivatives, highlighting their key properties. | ||
3.2 Graphene-based catalyst optimization strategies: a classification approach
Graphene-based catalysts can be broadly divided into three main categories based on their compositional structure, offering distinct optimization strategies for enhancing eCO2RR performance. First, metal-free heteroatom-doped graphene utilizes non-metallic dopants to introduce active sites within the graphene lattice. Second, metal–graphene hybrids combine single atoms or metal nanoparticles with graphene to utilize the synergistic effects between the metal components and the conductive carbon support. Third, graphene analogues, including materials such as bismuthene and stanene, possess graphene-like two-dimensional structures but are composed of different elements, offering unique catalytic properties.Single heteroatom doping involves the incorporation of one type of heteroatom into the graphene lattice. Among these, nitrogen doping is the most widely studied and highly effective method for enhancing catalytic properties in graphene-based materials. This is due to nitrogen's similar atomic size to carbon and its strong electron-donating ability, which facilitates seamless integration into the graphene structure and creates active sites for catalytic reactions. As presented schematically in Fig. 4a, nitrogen can exist in various N species, including pyridinic-N, pyrrolic-N, graphitic-N, and pyrazole-N.37 Pyridinic nitrogen is sp2-hybridized, substitutes a carbon atom within the hexagonal lattice, and donates a single p-electron to the π-electron system. Pyrrolic nitrogen, which is sp3-hybridized, replaces a carbon atom in a five-membered ring and provides two p-electrons to the π-electron system. Graphitic nitrogen, being sp2-hybridized, substitutes a carbon atom within the hexagonal lattice. These N-species have significantly varying impacts on the carrier concentration, leading to distinct electronic structures in the N-doped graphene.38 Recently, much work has been dedicated to unveiling the origin of activity from nitrogen doping in the eCO2RR. DFT calculations indicate that the nitrogen atom of pyridine can form a hydrogen bond with the hydrogen atom of the important intermediate *COOH, therefore decreasing the reaction's free energy and stabilizing the key intermediate. Furthermore, both graphitic and pyridinic nitrogen have been reported to initiate catalytic activity for the process. Due to the complexity of many N dopants influencing activity, the active sites for CO2 reduction on N-doped graphene-based materials are still under debate.39
 | ||
| Fig. 4 (a) Structural configurations of N-doped graphene. Adapted with permission from ref. 37 Copyright 2016, Springer Nature. (b) XPS spectra (B1s and C1s) of B-doped graphene. Adapted with permission from ref. 35 Copyright 2012, Wiley. (c) Potential structures of S-doped graphene. Adapted with permission from ref. 42 Copyright 2014, American Chemical Society. (d) Binding modes of metal single atoms on graphene. Adapted with permission from ref. 55 Copyright 2020, American Chemical Society. (e) Summary of reported active elements in SACs for eCO2RR. Adapted with permission from ref. 56 Copyright 2020, Wiley. | ||
Single heteroatom doping also includes the incorporation of other non-metallic elements such as boron, sulphur, and phosphorus into the graphene lattice. Although less studied than nitrogen, these dopants also play important roles in enhancing catalytic performance by altering the local electronic structure and inducing charge redistribution. Their different electronegativity and atomic sizes compared to carbon create unique active sites that promote CO2 adsorption and conversion through distinct mechanisms. Due to the lower electronegativity of boron compared to carbon, boron doping in graphene generates electron deficiency in the carbon atoms, resulting in a noticeable difference in electron densities across the graphene structure. As shown in Fig. 4b,35 the XPS B1s spectrum displays two peaks, indicating that boron atoms in the graphene network have two bonding types: a smaller peak at 198.5 eV for boron silane in the π-conjugated system, and a stronger peak at 200.5 eV for graphitic boron, suggesting substitutional doping of boron into the graphene lattice. Due to the relatively higher electropositivity of B compared to C atoms, B-doping generates charge imbalances in the carbon structure, which helps stabilize the negatively charged oxygen atoms of CO2, thus promoting stronger chemisorption of CO2 to the carbon surface during the eCO2RR process.40 In comparison, sulphur, with an electronegativity of 2.58, is almost identical to carbon, which has an electronegativity of 2.55, resulting in minimal charge density distribution effects in S-doped graphene. The interaction between sulphur 3p and carbon 2p orbitals, which induces sp3 hybridization, creating a tetrahedral-like distortion with neighbouring carbon atoms, leads to an uneven spin density, imparting graphene catalytic properties.41 As presented in Fig. 4c, S dopant atoms can be observed on the S-doped graphene through the adsorption of S onto the graphene surface, the replacement of C by S at the edges, the creation of S/S oxide at the edges, and an S-containing ring linking two graphene sheets.42 Due to the larger atomic radius of sulphur (1.04 Å) compared to carbon (0.77 Å), S atoms tend to incorporate at the edges of graphene.
In addition to single-atom doping, co-doping strategies involve the simultaneous incorporation of two different heteroatoms into the graphene lattice. These heteroatoms, such as nitrogen and sulphur, nitrogen and phosphorus, or boron and nitrogen, work together to combine their unique electronic and structural effects. Co-doping allows for the optimization of multiple properties at once, leading to synergistic enhancements in charge distribution, active site formation, and the adsorption of key intermediates. This often results in improved catalytic activity, increased selectivity for specific products, and enhanced stability in eCO2RR.
These metal-graphene hybrids can be classified into four key configurations based on the form and the scale of metal integration. One configuration involves isolated single metal atoms anchored onto graphene, forming single-atom graphene systems that maximize atomic utilization and provide well-defined active sites. Another configuration features paired metal atoms coordinated on the graphene lattice, creating dual-atom graphene systems that enable unique electronic interactions and synergistic effects. In contrast, fully encapsulated metal nanoparticles, covered by graphene layers, form metal-encapsulated systems, where the graphene shell provides stability while allowing selective access to the active metal core. Lastly, metal nanoparticles or clusters supported on graphene constitute metal-graphene composites, combining the advantages of nanoscale metals with graphene's conductive framework. This structural classification emphasizes how varying degrees of metal-graphene integration influence catalytic behavior, from single-site precision to multifunctional architectures.
Graphene has been effectively utilized as a particle stabilizer for single atoms and dual atoms in eCO2RR. Its exceptional two-dimensional structure and high specific surface area help anchor these active centers, preventing agglomeration during electrochemical processes.47 Moreover, the surface of graphene can be modified with various functional groups, including those containing nitrogen and oxygen.48 These functional groups enhance graphene's ability to stabilize metal species by serving as anchoring sites for single atoms and metal nanoparticles, preventing their migration or aggregation.49 As a result, graphene serves as an ideal substrate for loading metal atoms, substantially improving their dispersion, stability, and electrocatalytic performance, ultimately contributing to enhanced reaction efficiency. Fig. 4d illustrates the various binding sites for single atoms (SAs) on graphene, including pristine graphene,50 defective graphene with vacancies,51 doped graphene,52 and edge sites.53,54 These binding modes differ in stability and efficiency, with defective and doped graphene generally enhancing the stability of metal SAs.55 The efficiency and selectivity of SAs for eCO2RR vary depending on their elemental properties and group number in the periodic table, as shown in Fig. 4e.56
Graphene can also serve as a protective cover for the active phase, such as metal catalysts, in eCO2RR. exceptional mechanical strength and impermeability prevent corrosion and oxidation of the underlying active material, ensuring long-term stability. Additionally, graphene's chemical inertness under reaction conditions preserves the catalyst's integrity, while its ability to modulate local environments, such as the concentration of reactants, further enhances the selectivity and efficiency of the reaction.
In the form of metal-graphene composite structures, graphene acts as a conductive matrix that supports and enhances the activity of metal catalysts in eCO2RR due to the excellent electrical conductivity (1 × 105 S m−1) of graphene.57,58 Therefore, graphene can serve as an ideal conductor, significantly improving the electrical conductivity of the hybrid catalyst composites. Its excellent electrical conductivity facilitates efficient electron transfer, while its mechanical strength ensures enhanced stability under reaction conditions. The synergy between the combined structures also promotes easy adsorption of CO2 on the catalyst surface.59
To outline the concepts discussed above, a schematic overview is presented in Fig. 5, illustrating the key advantages of graphene-based materials in electrochemical CO2 reduction.
 | ||
| Fig. 5 Schematic representation of the advantages of graphene-based catalysts for electrochemical CO2 Reduction. | ||
4. Characterization of graphene-based catalysts
The characterization of graphene-based catalysts for eCO2RR requires a comprehensive analysis of the intrinsic properties, such as surface morphology, defect density, and electronic structure, as well as the electrochemical reaction mechanisms, to enhance the catalytic performance. The most commonly used methods for characterizing graphene-based catalysts in electrochemical CO2 reduction can be divided into two categories: ex situ and in situ techniques. Ex situ characterization involves analyzing the catalyst before or after the electrochemical reaction, providing detailed information about its structural, morphological, and compositional properties. In situ characterization, on the other hand, focuses on real-time monitoring of the catalyst's behavior under electrochemical conditions. This approach provides crucial insights into the reaction intermediates, active sites, and dynamic changes in the catalyst's electronic structure during eCO2RR.664.1 Ex situ characterization
Methods such as scanning electron microscopy (SEM) transmission electron microscopy (TEM), and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) are commonly used to analyze the morphology and structure of graphene-based catalysts, offering detailed images of surface topography, layer thickness, defect sites, and the arrangement of metal nanoparticles or single atoms on the graphene substrate as shown in Fig. 6a and b.67 In addition, changes in catalyst morphology during electrochemical reactions, both before and after the CO2RR, can be revealed, providing insights into the structural evolution, stability, and potential aggregation of nanoparticles as presented in Fig. 6c and d.68 The crystallinity and phase composition can be characterized using X-ray diffraction (XRD) and selected area electron diffraction (SAED). As depicted in Fig. 6e, the XRD measurement confirms the successful oxidation, functionalization, and doping of graphene were achieved, as evidenced by shifts in diffraction peaks and changes in peak intensities.69 The degree of structural deformation of catalysts can be assessed using Raman spectra, as demonstrated in Fig. 6f. Raman analysis of pristine, defective, and doped graphene reveals that the pristine graphene shows strong G and 2D bands, while defective graphene, such as nitrogen-doped, exhibits an additional D band. Increased D/G intensity ratios indicate a higher density of defects due to substitutions, vacancies, and structural disruptions, thus providing insights into the defect concentration and doping level in the graphene structure.70 Additionally, as illustrated in Fig. 6g, the X-ray photoelectron spectroscopy (XPS), analysis of the N1s spectra allows for the identification of the incorporation of nitrogen into the carbon structure and the formation of various nitrogen bonding types.71 As shown in Fig. 6h and i, the synchrotron-radiation-based X-ray absorption fine structure (XAFS), including both X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), can be used to examine the coordination environment and electronic states of metal species in catalysts.72 XANES analysis offers insight into the oxidation states of metal species while the EXAFS analysis helps identify the coordination environment and differentiate between single-atom catalysts and metallic clusters. Additionally, the wavelet transform (WT) of EXAFS (Fig. 6j) signals offers a more detailed view of the coordination environment and bonding interactions in the catalyst.73 | ||
| Fig. 6 Ex situ characterization (a) TEM (b) HAADF-STEM elemental mapping images of N-doped graphene (NG-1000). Adapted with permission from ref. 67 Copyright 2021, Elsevier. (c) Unconfined and (d) confined Cu nanoparticles before and after the eCO2RR, respectively. Adapted with permission from ref. 68 Copyright 2021, Springer Nature (e) XRD patterns of graphite, GO, bare graphene, iodine-doped graphene (IG), NG, and I and N co-doped graphene (ING). Adapted with permission from ref. 69 Copyright 2015, Elsevier. (f) Raman spectra of pristine graphene, defective graphene (DG) and NG. Adapted with permission from ref. 70 Copyright 2013, Elsevier. (g) High-resolution N1s XPS spectra of carbon. Adapted with permission from ref. 71 Copyright 2023, Elsevier. (h) Structural characterization of nickel-based catalysts using Ni K-edge XANES spectra. (i) Fourier transform k3-weighted R-space χ EXAFS spectra for Ni–CN, NiSA/NCNT, SANi-Ni/NCNT, NiNx/NCNT, and reference materials (NiPc and Ni foil). (j) EXAFS fitting and wavelet transforms of Ni K-edge EXAFS signals for Ni–CN, SANi-Ni/NCNT, and NiNx/NCNT. Adapted with permission from ref. 73 Copyright 2023, Wiley. | ||
4.2 In situ characterization
The in situ/operando TEM technique enables real-time characterization of catalyst morphology changes during reactions through imaging and diffraction techniques by applying light, electrical, and thermal stimuli during TEM analysis (Fig. 7a).74 In situ spectroscopic techniques, such as UV-vis, Raman, and FTIR can facilitate real-time monitoring of catalytic processes. UV-vis absorption spectroscopy is used to study catalytic reactions in both homogeneous and heterogeneous systems. Recently, Chai et al. utilized in situ UV-visible spectroscopy to reveal that copper-doped carbon dots (Cu-CDs) retain their single-site Cu configuration, confirming their intrinsic catalytic activity without forming metallic clusters. In contrast, Cu-phthalocyanine (CuPc) transforms into Cu clusters under electrochemical conditions (Fig. 7b).75 Raman spectroscopy is widely used to analyze the evolution of catalyst surface adsorbates during ECR. As presented in Fig. 7d,76 Raman spectroscopy revealed that the Fe–N4 catalyst on an oxygenated carbon matrix forms *CO2− intermediates during ECR, while the Fe–N–C catalyst with iron nanoparticles showed no *CO2− peaks, suggesting rapid protonation to *COOH intermediates, indicating enhanced protonation on the iron surface. Compared to the above two methods, FTIR spectroscopy, with its high sensitivity and fast characterization speed, is especially effective for identifying reaction intermediates. It provides real-time, molecular-level insights into the adsorption and desorption of intermediates by analyzing the absorption of infrared light, offering insights into bond changes and molecular interactions, as shown in Fig. 7c.77 In situ X-ray absorption spectroscopy (XAS) uses synchrotron radiation to probe the chemical and structural properties of catalysts with subatomic resolution. It is divided into XANES and EXAFS. EXAFS reveals changes in coordination environments (Fig. 7e),78 while XANES helps determine the oxidation state (Fig. 7f).77 Attenuated total reflectance–surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) can be used to study how the electric double layer (EDL) structure changes under the applied potential during electrocatalytic processes, including the reduction of CO2. As shown in Fig. 7g,79 ATR-SEIRAS at −0.68 V vs. RHE reveals stable carboxyl (−COOH) groups on metal-free single-layer graphene nanodisks (GNDs) during CO2RR, with no significant change after oxygen saturation, indicating surface inertness. | ||
| Fig. 7 In situ characterization (a) In situ TEM Configuration. Adapted with permission from ref. 74 Copyright 2020, American Chemical Society. (b) In situ UV-visible spectra of CuPc and Cu-CDs. Adapted with permission from ref. 75 Copyright 2021, Springer Nature. (c) In situ FTIR spectra of CoPc/MWCNT in CO2−saturated (top) and CO-saturated (bottom) electrolytes, recorded at potentials of −0.3 V to −0.8 V vs. RHE. Adapted with permission from ref. 77 Copyright 2023 Springer Nature. (d) In situ Raman spectra of Fe-poN-C(O) and Fe-poN-C/Fe. Adapted with permission from ref. 76 Copyright 2023, Springer Nature (e) operando EXAFS spectra of p-CuNW and cAA-CuNW. Adapted with permission from ref. 78 Copyright 2024, Springer Nature. (f) Normalized Co K-edge XANES spectra for CoPc/MWCNT at open-circuit potential (OCP), E = −0.5 V, and E = −0.7 V vs. RHE in CO2 and CO-saturated electrolytes. Adapted with permission from ref. 77 Copyright 2023, Springer Nature. (g) Schematic of the ATR-SEIRAS setup, and real-time ATR-SEIRAS spectra of GNDs-160 in a CO2-saturated 0.5 M KHCO3 solution at −0.68 V vs. RHE. Adapted with permission from ref. 79 Copyright 2021, Springer Nature. | ||
5. Application of graphene-based catalysts for eCO2RR
5.1 Metal-free heteroatom doped graphene for eCO2RR
Among all nitrogen configurations in N-doped graphene catalysts, pyridinic-N, characterized by a pair of lone electrons, exhibits a strong binding affinity for CO2. This interaction facilitates the formation of the *COOH intermediate via proton-electron coupling, followed by its conversion to CO through *CO adsorption. In contrast, the electrons in graphitic-N occupy π–π antibonding orbitals, rendering them less favourable for CO2 binding. As a result, the preferential adsorption of weakly acidic CO2 molecules occurs more readily at pyridinic-N sites, promoting selective eCO2RR on these sites rather than on graphitic-N.8 Further studies showed that increasing the pyridinic-N concentration correlated with enhanced CO selectivity. Early reports on N-doped NCNTs enriched with pyridinic-N demonstrated a CO FE of ∼80% at a low overpotential of −0.18 V vs. RHE, highlighting the role of pyridinic-N as the active site for eCO2RR.80 N-doped CNTs with a high concentration of pyridinic-N sites (62.3% of all nitrogen) achieved a CO FE of 96% with a jCO of 22 mA cm−2 at −0.7 V vs. RHE, emphasizing the dominant role of pyridinic-N in promoting eCO2RR.81 Subsequent work demonstrated that local curvature modulation of CNTs could further enhance the electronic environment of pyridinic-N, boosting CO selectivity to nearly 100%.82
Similarly, Wang et al.83 designed hierarchically structured porous N-doped carbon membranes (Fig. 8a) combined with CNTs (HNCM/CNTs), achieving a formate FE of 81% at −0.9 V vs. the RHE in 0.1 M KHCO3 solution (Fig. 8b and c). The catalyst demonstrated high selectivity for formate, along with excellent long-term stability of 36 hours and a significant current density. The study attributed the pyridinic N (41.8%) as the key active sites, playing a crucial role in CO2 adsorption and activity enhancement.
 | ||
| Fig. 8 N-doped graphene for eCO2RR (a) HRTEM image of HNCM/CNT. (b) LSV curves recorded at a scan rate of 5 mV s−1 in CO2-saturated and Ar-saturated 0.1 M KHCO3 aqueous electrolyte. (c) FE for formate production as a function of applied potential at HNCM and HNCM/CNT electrodes. Adapted with permission from ref. 83 Copyright 2017, Wiley. (d) TEM image of NC-1100 and corresponding EDX maps of the NC-1100. (e) Stability test of the NC-1100 at 0.5 V versus RHE. (f) Free energy pathways for eCO2RR to CO and HER over GN-induced active sites at 0 V versus RHE. Adapted with permission from ref. 39 Copyright 2020, Elsevier. (g) TEM images of NG-1000. (h) High-resolution XPS analysis in N1s region of Pc and various NG catalysts. (i) FE for CO at −0.72 V vs. RHE for different catalysts. Adapted with permission from ref. 67 Copyright 2021, Elsevier. (j) High-magnification TEM image of NGQDs. The inset shows a single NGQD containing zigzag edges, as circled. (k) High-resolution N1s spectrum for NGQDs. (l) FEs for NGQDs at various cathodic potentials. Adapted with permission from ref. 84 Copyright 2016, Springer Nature. | ||
In contrast, Zhang et al.39 challenged conventional assumptions by demonstrating that graphitic-N, rather than pyridinic-N, promotes CO2 reduction. Using template-assisted pyrolysis, they synthesized N-doped carbon foams (Fig. 8d) with tunable N species and achieved a CO FE of 95% at −0.5 V vs. RHE in 0.5 M KHCO3 (Fig. 8e). DFT calculations revealed that the graphitic-N doped C atoms are more selective for CO2 reduction to CO over HER, whereas pyridinic-N doped C atoms tend to favor HER over CO2RR, and pyrrolic-N doping reduces activity for both reactions (Fig. 8f). In addition, compared to metal catalysts, the sp2 framework in the carbon matrix enhances the durability of heteroatom-doped graphene-based materials. The nitrogen active sites remain stable within the sp2 carbon structure, resisting reconstruction and aggregation even at low doping levels.
Similarly, Li et al.67 reported a 2D N-doped graphene-like carbon (Fig. 8g) achieving 95% FE for CO at −0.72 V vs. RHE (Fig. 8h), with jCO of 9.07 mA cm−2. They demonstrated that high-temperature pyrolysis increased defect density and porosity, enhancing the exposure of graphitic-N sites (Fig. 8i). DFT calculations indicated that C atoms adjacent to graphitic-N were the most catalytically active. Moreover, Zhou et al. and Liu et al. suggested that in certain nanostructured carbons (e.g., CNTs or graphene nanoribbons), both pyridinic and graphitic-N may act as indistinguishable or co-active sites, complicating the mechanistic understanding.
While N-doped graphene-based materials have been extensively studied for the electrochemical reduction of CO2 to C1 products, recent advancements have demonstrated their potential for producing economically valuable C2 products, such as ethylene and ethanol. In this context, Wu et al.84 reported the use of N-doped NGQDs (Fig. 8j and k) as effective catalysts for eCO2RR. Operating within a flow cell electrolyzer using 1 M KOH as the electrolyte, NGQDs exhibited a total CO2 reduction FE of up to 90% at −0.75 V vs. RHE. Notably, the catalyst favored C2 product formation, achieving a combined FE of 45% for ethylene and ethanol, with partial current densities of 46 mA cm−2 and 21 mA cm−2 for C2H4 and C2H5OH, respectively (Fig. 8l). Although NGQDs demonstrate the potential for selective C2 product formation, the FE for individual products remains relatively low compared to state-of-the-art Cu-based catalysts, which are currently the benchmark for multi-carbon product generation. To address this limitation, strategies such as increasing N dopant density or co-doping with complementary heteroatoms (e.g., B, S, P) may enhance both the activity and selectivity toward specific C2 products. Furthermore, the C–C bond formation mechanism over N-doped carbon catalysts remains poorly understood, posing a significant challenge for the development of next-generation metal-free graphene-based electrocatalysts tailored for efficient and selective multi-carbon product synthesis.
 | ||
| Fig. 9 Other heteroatom doped graphene for eCO2RR High-resolution XPS spectra of (a) B1s and (b) C1s (c) LSV of BG and Bi in 0.1 M KHCO3 in the presence of CO2. Adapted with permission from ref. 85 Copyright 2015, Royal Society of Chemistry. (d) SEM image of PGA-2. (e) Stability test on PGA-2. (f) FE for EtOH and yield for PGA-2 in a flow cell. Adapted with permission from ref. 19 Copyright 2022, Wiley. | ||
Phosphorus doping in graphene significantly modifies its electronic structure and active site distribution, enabling enhanced catalytic activity for ECR to C2 products. Unlike N-doping, where the adjacent carbon atoms often serve as the active sites due to electronic redistribution, P atoms themselves act as positively charged active sites due to the reversed polarity of the C–P bond, stemming from phosphorus's lower electronegativity (2.19) compared to carbon (2.55).87 Additionally, the larger atomic radius of P introduces lattice distortion and abundant edge defects, which are beneficial for C–C coupling. Motivated by these properties, Yang et al.19 synthesized phosphorus-doped graphene aerogels (PGAs) via hydrothermal reduction of GO with phosphoric acid (Fig. 9d). Using an H-type three-electrode cell with CO2-saturated 0.5 M KHCO3 electrolyte, PGA-2 achieved a FE of 48.7% for ethanol production at −0.8 V versus RHE, with an ethanol yield of 14.62 μmol h−1cm−2 and stability maintained for 70 hours (Fig. 9e and f). It was revealed that the catalyst's hierarchical porous structure enhanced CO2 adsorption and charge transfer, while DFT calculations showed that P-doping at graphene zigzag edges promoted *CO dimerization and C–C coupling. This work inspires the design of metal-free catalysts by using P-doped graphene-based materials for the conversion of CO2 into high-value C2 products with high efficiency, selectivity, and stability.
 | ||
| Fig. 10 Co-heteroatom doped graphene for eCO2RR (a) scanning TEM and N,S elemental mapping images of NS-C-900. (b) FE for CO vs. applied potential. (c) Chronoamperometric responses and CO FEs at −0.6 V on NS-C-900 for stability tests. Adapted with permission from ref. 61 Copyright 2019, Elsevier. (d) Elemental mapping of rGONB (using STEM-HAADF). (e) XPS spectra of N1s spectrum and B1s spectrum. (f) FE for CO2 reduction products and CO2 conversion rate with rGO and rGONB catalysts vs. Cu-based catalysts (Cu/rGO and Cu/rGONB) under experimental conditions. Adapted with permission from ref. 91 Copyright 2023, Elsevier. | ||
In addition, Cerrillo et al.91 demonstrated that nitrogen and boron co-doped reduced graphene oxide (rGONB) (Fig. 10d) effectively catalyzes the electrochemical reduction of CO2 in a PEM-type cell operating in gas-phase and continuous mode, and the XPS signals of N1s and B1s indicate the incorporation of boron atoms into the hexagonal graphene lattice (Fig. 10e). The catalyst achieved a CO2 conversion rate 30 times higher than undoped rGO (Fig. 10f), with formic acid and CO as the primary products. The electron-donating nitrogen species and electron-accepting oxidized boron sites synergistically enhanced catalytic activity, outperforming Cu/rGO in CO2 conversion efficiency.
5.2 Graphene-metal hybrid catalysts
Over the past decade, SACs supported on such graphene-based materials have garnered significant attention due to the synergistic benefits of atomically dispersed active sites and highly conductive and chemically tunable graphene-based frameworks.97 A notable advancement in this area is the development of M–N–C SACs (M = Fe, Co, Ni, Cu), where metal atoms are stabilized in well-defined M–Nx coordination environments. These materials feature well-defined coordination environments and exhibit excellent performance in electrocatalysis. For example, M–N4 sites on graphene support have demonstrated exceptional activity and selectivity for electrochemical CO2 reduction to CO, achieving FE > 90%.98 Despite the high per-atom activity of these catalysts, maintaining high metal loadings without aggregation remains a key challenge, driving continued research in synthesis strategies to enhance both stability and performance.
Beyond isolated M–N4 sites, emerging strategies to tailor the coordination environment of SACs have unlocked new pathways to enhance both activity and operational stability. Notably, recent advances demonstrate that certain M–Nx configurations can simultaneously achieve exceptional catalytic efficiency and durability.99 For instance, Zhang et al.100 demonstrated that single-atom Fe–N5 sites, formed when an axial ligand coordinates with Fe–N4 during prolonged thermal pyrolysis of N-doped graphene(Fig. 11a), exhibit exceptional catalytic activity and durability for the electrochemical CO2 reduction to CO in aqueous media, outperforming the Fe–N4. The FeN5 catalyst achieved a FE of 97.0% at a low overpotential of 0.35 V and demonstrated excellent durability with less than 2% current decay and stable CO selectivity (∼97%) over 24 h at −0.46 V vs. RHE, while preserving the atomic FeN5 configuration post-reaction (Fig. 11b). DFT calculations revealed that the additional axial pyrrolic nitrogen ligand in the FeN5 site causes an additional reduction in electron density of Fe 3d orbitals, which weakens the bonding interaction with CO and enables the rapid desorption of CO along with high selectivity for CO production (Fig. 11c).
 | ||
| Fig. 11 Single-atom catalysts on graphene for eCO2RR (a) magnified STEM images of uniformly distributed single Fe atoms in graphene (single Fe atoms are highlighted with dashed circles). (b) FE for CO vs. applied potential. (c) Free-energy profile with optimized intermediates for electroreduction of CO2 to CO, determined using the computational hydrogen electrode (CHE) model. Adapted with permission from ref. 100 Copyright 2019, Wiley. (d) Atomic-resolution HAADF-STEM image (Zn atoms are highlighted with yellow circles). (e) FE for methane vs. applied potential. (f) Free energy diagrams for ERC to CH4 on Zn–N4−graphene, showing the most stable structure of each step. Red and pink balls represent O and H atoms, respectively. Adapted with permission from ref. 103 Copyright 2020, American Chemical Society. (g) SEM images of Cu-SA/NPC. (h) FE for CO2 reduction products for Cu-SA/NPC. (i) Free energy diagrams calculated at a potential of −0.36 V for CO2 reduction to CH3COCH3 on Cu-pyridinic-N4 and Cu-pyrrolic-N4 sites of Cu-SA/NPC. Adapted with permission from ref. 104 Copyright 2020, Springer Nature. | ||
Although most SAs coordinated on graphene-based materials primarily produce CO as the main product,56 some catalysts have also been reported to generate hydrocarbons.101,102 The single-atom Zn catalysts supported on N-doped carbon (SA-Zn/MNC) (Fig. 11d) reported by Han et al.103 selectively reduce CO2 to methane, achieving a FE of 85% for CH4 production at −1.8 V vs. SCE (Fig. 11e) with a partial current density of −31.8 mA cm−2 and stability over 35 hours without significant performance degradation. The stable and efficient performance of the catalyst can be attributed to the graphene's high electrical conductivity, which ensures rapid charge transport. Theoretical calculations revealed that during ECR, the O atom in the *OCHO intermediate preferentially bonds with Zn instead of the C atom, thereby suppressing CO formation and favoring CH4 production (Fig. 11f).
While SACs have demonstrated excellent performance for eCO2RR to C1 products, the formation of multi-carbon products remains a significant challenge due to the absence of adjacent active sites typically required for C–C coupling. The incorporation of atomically dispersed Cu into nitrogen-doped graphene frameworks has emerged as a compelling strategy to address this limitation by enabling unconventional C–C coupling pathways within a single-atom coordination environment. Graphene provides a highly conductive, chemically tunable support, while nitrogen doping, particularly pyrrolic-N, strongly coordinates single Cu atoms and alters their electronic structure. This coordination environment allows two CO* intermediates to adsorb sequentially on the same Cu site, where intramolecular coupling occurs at a single metal center, leading to C–C bond formation. Zhao et al.104 demonstrated that Cu atoms anchored on nitrogen-doped porous carbon (Cu-SA/NPC) exhibited (Fig. 11g) a FE of 36.7% (Fig. 11h) for acetone production, with a notable rate of 336.1 μg h−1 at low overpotential. DFT studies revealed that the pyrrolic-N coordinated Cu sites lower the energy barriers for both CO2 activation and CO–CO coupling, enabling the formation of key intermediates for C3 product generation (Fig. 11i). This work highlights the ability of graphene-based SACs to host complex reaction pathways, offering new insights for the rational design of efficient and selective metal-free or atom-efficient CO2RR systems targeting C2+ products. The co-doping of multiple species of foreign atoms may generate new properties or create synergistic effects.
Li et al.105 reported a highly active N-coordinated DAC by embedding Ni–Fe sites within ZIF-derived porous carbon (Fig. 12a), aiming to overcome the limitations of traditional M–N–C SACs in eCO2RR. Among the structures studied, the 2N-bridged (Fe–Ni)N6 configuration featuring FeN4 and NiN4 sites linked by two shared nitrogen (Fig. 12b) atoms showed the most promising performance. This dual-metal synergy facilitated optimal *COOH adsorption and *CO desorption, addressing the strong CO* binding that lowers the reactivity of FeN4,106 and the weak *COOH binding of NiNx sites.107,108 DFT calculations revealed favourable charge redistribution between the Ni and Fe centres, reducing the energy barrier for *CO desorption and enhancing the onset potential for CO2RR, while suppressing HER activity (Fig. 12c). Beyond experimental advances, theoretical studies also have played a key role in guiding DACs design. For instance, Luo et al. used DFT calculations to investigate dual-metal site catalysts like Cu/Mn, Ni/Mn, and Ni/Fe, which showed enhanced activity for CO2-to-CO conversion due to the breaking of the conventional scaling relationship between *CO and *COOH adsorption, allowing more favourable reaction pathways.109
 | ||
| Fig. 12 Dual-atom catalysts on Graphene for eCO2RR (a) HAADF-STEM images illustrating metal atom dispersion on a carbon support: formation of atom pairs/clusters (cyan) versus isolated single atoms (magenta). (b) Structural model of N-coordinated dual-metal Ni–Fe sites. (c) Calculated free energy diagram for electrochemical CO2 reduction (eCO2RR) on various dual-metal Fe–Ni sites. Adapted with permission from ref. 105 and 128 Copyright 2022, Wiley. (d) Structural model of Fe2NPC dual-atom catalysts (DACs). (e) Gibbs free energy profile for electrochemical CO2 reduction on Fe2NPC. Adapted with permission from ref. 110 and 128 Copyright 2022, American Chemical Society. (f) Corresponding partial current density for CO at different applied potentials in acidic electrolyte. (g) Partial current density for CO in alkaline electrolyte. (h) Gibbs free energy diagram for CO2-to-CO conversion over NiN3–CuN3 and NiN3 with pH correction at pH = 3. (i) Gibbs free energy diagram at pH = 14. Adapted with permission from ref. 112 Copyright 2023, Wiley. | ||
Similarly, Zhao et al.110 prepared a homonuclear Fe–Fe DAC featuring Fe2N6 sites uniformly anchored on nitrogen-doped porous carbon (Fig. 12d). This Fe–Fe DAC reached a FE of 96.0% for CO production at −0.6 V vs. RHE with a low Tafel slope of 60 mVdec−1, markedly outperforming Fe SACs. DFT studies reveal that the cooperative interaction between the two Fe centres facilitates the adsorption of CO2 in a bent configuration, effectively reducing the activation barrier and promoting rapid *CO desorption (Fig. 12e). Similarly, Hao and co-workers found that Ni/Ni DMSCs could also achieve over 99% FE for CO production, indicating that even homonuclear DACs can achieve exceptional eCO2RR performance when properly configured.111
Demonstrating the potential of advanced dual-atom catalyst systems, Zhang et al.112 developed a Ni–Cu DAC embedded in hollow N-doped carbon nanocages that exhibited pH-universal activity for CO2 reduction. The catalyst demonstrated near-unity CO FE up to 99% across acidic, neutral, and alkaline electrolytes (Fig. 12f and g), with high partial current densities (up to 489 mA cm−2 in alkaline). Under acidic conditions, where HER typically dominates, the catalyst retained a CO selectivity of 98.5% and achieved a remarkable CO2 utilization efficiency of 64.3%. Operando characterizations and DFT analysis revealed that Cu shifted the Ni d-band center closer to the Fermi level, thus enhancing *COOH formation and tuning the binding energies of key intermediates (Fig. 12h and i).
Copper is widely studied for its ability to yield C2+ products such as ethylene, ethanol, and n-propanol with high selectivity. However, its structural instability under electrochemical reduction conditions is a major challenge, leading to catalyst reconstruction and reduced performance.118–123 Degradation prevention methods, such as the use of physical barriers124 on the surface and alloying with second metals,125,126 have been proposed as solutions to mitigate this issue. Among these, the physical barrier method has been the most studied, with graphene emerging as the most suitable option due to its inert nature. Kim et al.68 developed a catalyst system resistant to reconstruction by spontaneously depositing graphitic carbon, which surrounds the Cu nanoparticles in a quasi-graphitic C shell, thereby protecting them (Fig. 13a). This catalyst not only prevents Cu from reconstruction but also enables inner Cu doping to enhance activity and stability. Their copper nanoparticles encapsulated in quasi-graphitic carbon shells effectively catalyze the electrochemical conversion of CO2 to C2+ products with no reconstruction. Using a flow-cell reactor with a 1 M KOH electrolyte, boron-doped Cu catalysts achieved a FE of 68.1% for ethylene production at −0.55 V versus RHE, while nitrogen-doped Cu catalysts attained an FE of 82.3% for C2+ products with a partial current density of 329 mA cm−2 (Fig. 13b). The quasi-graphitic carbon shell stabilized the Cu nanoparticles against reconstruction and enhanced C–C coupling, as confirmed by DFT calculations (Fig. 13c).
 | ||
| Fig. 13 Graphene-shell encapsulated metal nanoparticles for eCO2RR (a) high-resolution TEM image of the confined Cu nanoparticles and quasi-graphitic C shell. (b) FE and partial current density for C2+ products at 400 mA cm−2. (c) Free energy diagram for CO dimerization on the pristine Cu, Cu (B), and Cu (N) surfaces. Adapted with permission from ref. 68 Copyright 2021, Springer Nature. (d) TEM images of Cu-600-N2. (e) LSV curves in CO2 and Ar-saturated 0.1 M KHCO3 aqueous solutions for Cu-600-N2, Cu-600-H2, and Cu-600-NH3. (f) FE and partial current density for Cu-600-N2. Adapted with permission from ref. 127 Copyright 2023, Royal Society of Chemistry. (g) FE distributions at 1.0 V vs. RHE. (h) In situ Raman spectroscopy results. Left: Potential-dependent Raman spectra of NiCu0.25, with adsorption models illustrating *CO adsorption at bridge and atop sites on the catalyst surface. Right: Raman spectra of other catalysts recorded at an applied potential of 1.0 V vs. RHE. (i) Electron donation and CO selectivity trend with increasing Cu content in NiCux. Glow intensity represents 3d electron density in molecular models. Adapted with permission from ref. 128 Copyright 2017, Royal Society of Chemistry. | ||
Similarly, Yao et al.127 revealed that copper nanoparticles encapsulated in a carbon shell (Cu-600-N2), synthesized by calcination of a copper-based metal–organic framework (Cu-MOF) in an N2 atmosphere (Fig. 13d), effectively catalyze the electrochemical reduction of CO2 to ethanol with high selectivity and stability. These Cu-MOFs were also exposed to various gas atmospheres, including H2 and NH3, to investigate their influence on the catalytic performance. The catalyst under N2 achieved a FE of 67.8% at −0.8 V vs. RHE, outperforming copper nanoparticles synthesized under H2 and NH3 atmospheres (Fig. 13e and f). The carbon shell provided structural protection, preventing Cu particle aggregation and surface reconstruction, significantly enhancing catalyst stability over 16 hours of continuous operation. Additionally, Tafel analysis revealed superior corrosion resistance for Cu-600-N2, attributed to its more positive corrosion potential than the other catalysts. This protective carbon layer and synergistic Cu–C interactions maintained high FE and current density, making Cu-600-N2 a promising electrocatalyst for ethanol production in CO2RR applications.
Moreover, Xu et al.128 demonstrated that graphene-encapsulated nickel-copper bimetallic nanoparticles (NiCu0.25) effectively catalyze the electrochemical reduction of CO2 to CO. Using a CO2-saturated 0.1 M KHCO3 electrolyte, NiCu0.25 achieved a FE of 88.5% at −1.0 V versus RHE (Fig. 13g), with enhanced stability due to the graphene encapsulation preventing particle aggregation. The study revealed that lower Cu content in NiCux alloys optimizes the electronic structure, enhancing *COOH adsorption and CO selectivity. The graphene shell stabilizes the nanoparticles, preserving structural integrity. Fig. 13h confirms *CO adsorption variations, while Fig. 13i shows that increasing Cu content shifts electron donation, reducing CO selectivity. These results demonstrate the dual role of graphene as both a physical barrier and an electronic modulator, pointing to the potential of graphene-based encapsulation strategies in designing next-generation stable and selective CO2 reduction catalysts.
 | ||
| Fig. 14 Graphene-metal composite structures for eCO2RR (a) TEM images of GO-VB6-Cu-x. (b) FEs for CO2 reduction vs. applied potential for various GO-VB6-Cu catalysts. (c) Schematic illustration of the CO2 electrochemical reduction process on the GO-VB6-Cu catalyst. Adapted with permission from ref. 129 Copyright 2019, Elsevier. (d) Schematic of magnetron sputtering delivering atoms of Cu directly onto the GNF surface. (e) FE and the current density for Cu/GNF (0.84 wt% Cu) at −0.38 V vs. RHE over time. (f) Binding energy of CO2 and H2O to Cu (111) and Cu (311) surfaces of bulk metal, Cu atom embedded in graphene vacancy defects, and pristine graphene. Adapted with permission from ref. 133 Copyright 2024, Nature. (g) TEM image of the CoPc/CNT (6%) hybrid with the chemical representation of the CoPc/CNT hybrid interface. (h) FE for CO and H2 at various applied potentials for CoPc and CoPc/CNT electrodes. (i) Corresponding partial current densities for CO and H2 for CoPc and CoPc/CNT under the same conditions. Adapted with permission from ref. 134 Copyright 2024, Nature. | ||
Moreover, despite the ability of copper-based electrocatalysts to convert CO2 into value-added multicarbon products, the traditional Cu forms, such as foils or large nanoparticles, often suffer from limited active surface area utilization,131,132 necessitating the development of nanoscale catalysts to optimize performance. Cu nanoclusters, composed of a group of atoms, have been recognized as a solution in this context. However, the effectiveness of these catalysts is greatly influenced by the properties of the support material and the particular conditions during catalyst synthesis. In this regard, Burwell et al.133 demonstrated that copper nanoparticles directly anchored onto graphitized carbon nanofibers (Cu/GNF) prepared via solvent-free atomic deposition (Fig. 14d) enhance the electrochemical reduction of CO2 with remarkable selectivity. The Cu/GNF catalyst achieved a FE of 94% for formate production at −0.38 V vs. RHE (Fig. 14e), outperforming conventional Cu-based catalysts. The Cu nanoparticles (2–5 nm) localized at graphitic step edges provided a strong metal-support interface, facilitating charge transfer and stabilizing catalytic sites (Fig. 14f). The study highlighted the critical role of the graphitic step edges in stabilizing the Cu nanoparticles, which prevented aggregation and ensured efficient CO2 conversion. These findings highlight the significance of designing well-defined nanoscale metal–graphene interfaces, which can effectively stabilize metal active sites, inhibit particle aggregation, and enhance both the activity and durability of CO2 reduction catalysts.
In addition, immobilizing M–N–C-type molecular catalysts such as metal phthalocyanine or metal porphyrins onto conductive graphene-based materials has emerged as an effective strategy to improve both catalytic activity and stability for eCO2RR. These graphene-based supports not only prevent catalyst aggregation and facilitate charge transport but also provide favorable electronic interactions that can modulate the local coordination environment of the metal center. For instance, Zhang et al.134 reported that molecularly dispersed CoPc anchored on multi-walled CNTs via π–π interactions (Fig. 14g) exhibited high CO selectivity (FE > 90%) (Fig. 14h and i) and long-term stability in aqueous KHCO3 electrolyte, with performance further enhanced by electron-withdrawing cyano substituents that promoted more favorable Co(I) formation and faster electron transfer. Beyond tuning molecular electronics, catalyst support interactions have also been shown to influence product distribution. Shen et al.135 demonstrated that cobalt protoporphyrin immobilized on pyrolytic graphite enables the formation of both CO and CH4 in acidic media, and importantly, the product selectivity was highly dependent on pH, with lower pH favoring methane and higher pH favoring CO, due to the different stabilization mechanisms of reaction intermediates. More recently, Su et al.136 highlighted the critical role of support-induced strain by showing that single-walled CNTs (SWCNTs), due to their high curvature, distort the CoPc molecular structure in a way that enhances CO binding and enables deeper reduction to methanol. This curvature-driven modulation led to a striking increase in methanol selectivity, achieving a FE of 53.4%, compared to just 13.9% on larger-diameter CNTs.
5.3 Graphene analogs
In addition to conventional graphene-based materials as electrocatalysts for CO2 reduction, emerging graphene analogs, especially 2D monoelemental materials, have gained increasing attention due to their unique properties compared to traditional CO2 electrocatalysts.137 One of the most prominent features of these 2D monoelemental materials is their atomic thickness, which offers distinct benefits over bulk materials, including a higher density of active sites and unique electronic properties due to their reduced dimensionality. This enables enhanced catalytic performance for CO2 reduction reactions, as the altered atomic arrangements in the 2D monolayer promote more efficient electron transfer and reaction kinetics.138 For instance, bulk and nanoparticle forms of metals such as Sn and Bi suffer from higher overpotentials and lower current densities for electrochemical CO2 reduction,139–141 while their 2D monoelemental structures exhibit distinct catalytic CO2RR.62Yang et al.62 demonstrated for the first time that free-standing monolayer bismuthene, synthesized through a simple and scalable wet chemical process, serves as an efficient catalyst for the electrochemical reduction of CO2 to formate with outstanding selectivity and stability. The bismuthene catalyst achieved a FE of 99% at −580 mV vs. RHE (Fig. 15a), with an onset potential of less than 90 mV and stable performance over 75 hours without degradation. DFT calculations ascribed the high performance to the structure-sensitive nature of CO2 reduction on bismuthene, where the (111) facet of atomically thin nanosheets promotes selective formate production via an OCHO*-mediated pathway and suppresses HER due to a high overpotential (Fig. 15b). In contrast, thicker bismuth nanosheets exposing the (011) facet bind intermediates too strongly, leading to surface poisoning, which explains their lower activity and stability (Fig. 15c). These findings demonstrate bismuthene's potential as a scalable, durable, and cost-effective electrocatalyst for sustainable CO2 utilization. However, one major limitation arises from the dense stacking of nanosheets, which creates a compact catalyst layer and restricts access to active sites, thereby limiting current output. This challenge was addressed by incorporating inert carbon black into the catalyst structure. The additive effectively inhibits nanosheet restacking, thereby improving mass transport and expanding the electrochemically active surface area. This strategy offers a promising pathway for developing composite catalyst architectures that enhance both scalability and performance in eCO2RR.
 | ||
| Fig. 15 Graphene analogs for eCO2RR (a) FEs for formate vs. applied potential for Bi nanosheets with different thicknesses. (b) Free energy diagrams for CO2RR and HER on (111) single-atom-thick bismuthene and (c) (011) thick Bi nanosheets at 0.0 V. HER is represented in green, CO2RR through OCHO* and COOH* in blue and orange, respectively, and the state of CO* + OH* in red. Adapted with permission from ref. 62 Copyright 2020, Springer Nature. (d) TEM images of sandwiched bismuthene nanoflakes. (e) FEs for bismuthene nanoflakes and (f) Bi powders vs. applied potential within 2 hours. Adapted with permission from ref. 142 Copyright 2023, American Chemical Society. (g) TEM image of stanine. (h) FEs for formate vs. applied potential for Sn nanosheets with different thicknesses. (i) Stability test of stanene at −0.93 V versus RHE and corresponding FEs for HCOO− and H2. Adapted with permission from ref. 143 Copyright 2024, Wiley. | ||
In addition, growing high-quality 2D bismuthene in bismuthene-based catalysts remains challenging due to its high surface energy and strong interactions with metal substrates, which often cause structural distortion or aggregation into nanospheres. To address this, Hu et al.142 developed a sandwiched epitaxy approach using a top hexagonal boron nitride layer on Cu foil to suppress Bi–Cu interactions and stabilize the 2D hexagonal bismuthene structure during growth. Their work demonstrated that single-crystalline hexagonal bismuthene (Fig. 15d), synthesized via a sandwiched epitaxy growth, exhibits excellent performance as an electrocatalyst for the selective reduction of CO2 to formate at remarkably low overpotentials. Utilizing a CO2-saturated 0.1 M KHCO3 solution, the catalyst achieved an ultrahigh FE of 96.3% for formate production at −1.0 V versus RHE. In contrast, Bi powder failed to selectively produce formate, resulting in a mixture of C1 and C2 products and H2 (Fig. 15e and f). Compared to Bi powders, bismuthene nanoflakes exhibited superior performance for formate production, achieving a higher current density and lower onset potential for the eCO2RR. The Tafel slope for bismuthene nanoflakes (352 mV dec−1) was lower than that of Bi powders (371 mV dec−1), suggesting enhanced reaction kinetics. Despite having a smaller electrochemically active surface area (ECSA) than Bi powders (0.57 mF cm−2), the bismuthene nanoflakes (0.34 mF cm−2) demonstrated enhanced selectivity, attributed to their 2D nanostructure, which contains abundant active edge sites that effectively stabilize *OCHO intermediates, thereby reducing the occurrence of competing reactions. In addition, it was found that encapsulation by the hexagonal boron nitride layer not only prevented structural degradation but also enhanced electron transfer and stabilized reaction intermediates. This study suggests extending this encapsulation approach to synthesize other 2D materials with high surface energy, although scalability remain to be explored.
Moreover, free-standing stanine (Fig. 15g), synthesized by Mei et al.143 using a bottom-up approach through a simple wet chemical method with scalable production potential, was shown to effectively catalyze the electrochemical reduction of CO2 to formate in an aqueous solution. Using a CO2-saturated 0.5 M KHCO3 electrolyte (Fig. 15h), the stanene catalyst achieved a high FE of 93% at −0.93 V versus RHE with excellent stability over 60 hours (Fig. 15i). The superior performance was attributed to the high density of active edge sites on the Sn (100) step, as confirmed by DFT calculations and in situ Mössbauer spectroscopy. These results highlight stanene as a promising, scalable, and efficient electrocatalyst for sustainable CO2 utilization.
6 Summary and outlook
The eCO2RR is a valuable process for advancing sustainable development. Over the past few years, materials for ECR have undergone significant advancements, largely driven by the limitations of traditional metal-based catalysts. Graphene-based materials have emerged as a promising class of electrocatalysts for CO2 reduction as an alternative to commercial metal catalysts. This is due to their unique structural versatility and exceptional properties, which enable superior catalytic performance. In this review, we highlight graphene-based electrocatalysts as sustainable alternatives for ECR. Graphene-based materials serve diverse roles in eCO2RR, functioning as metal-free catalysts through heteroatom doping, as well as in combination with metals ranging from single atoms to nanoparticles and clusters. Furthermore, we provide an overview of emerging catalysts, including advanced graphene analog materials. Table 2 illustrates the recent advancements in the development of graphene-based electrocatalysts for CO2 reduction. Despite the advantages of carbon-based catalysts and the notable progress made over the past decade, several constraints still hinder their industrial applicability.| Catalyst | Main product (FE) | Potential (V vs. RHE) | Current density (mA cm−2) | Stability | Electrolyte | Cell type | Ref. |
|---|---|---|---|---|---|---|---|
| N-doped graphene | |||||||
| N-CNF | CO (98%) | −0.57 vs. SHE | 1.3 | 9 h | EMIMBF4 | H-cell | 144 |
| N-graphene foam | CO (85%) | −0.47 | 1.8 | 5 h | 0.1 M KHCO3 | H-cell | 145 |
| N-CNTs | CO (85%) | −0.9 | 5.8 | 60 h | 0.5 M NaHCO3 | H-cell | 146 |
| g-C3N4-MWCNTs | CO (60%) | −0.75 | −1.02 | 50 h | 0.1 M KHCO3 | H-cell | 147 |
| N-graphene sheet | HCOO− (73%) | −0.84 | 7.5 | 12 h | 0.5 M NaHCO3 | H-cell | 148 |
| N-graphene like carbon | CH4 (93.5%) | −1.40 | 1.42 | 5 h | [Bmim]BF4 | H-cell | 149 |
| Reduced oxidized-GQDs | CH4 (70%) | −0.9 | 200 | 10 h | 1.0M KOH |
Flow cell | 150 |
| N-diamond | CH3COO− (91.8%) | −1.0 | — | 3 h | 0.5 M NaHCO3 | H-cell | 18 |
| N-functionalized GO | C2H5OH (37%) | −0.4 | 0.745 | — | 0.1 M KHCO3 | H-cell | 151 |
| N-mesoporous carbon | C2H5OH (77%) | −0.56 | — | 6 h | 0.1 M KHCO3 | H-cell | 87 |
|
|||||||
| Other metal free heteroatom doped graphene | |||||||
| P-OLC | CO (81%) | −0.9 | 4.9 | 27 h | 0.1 M NaHCO3 | H-cell | 152 |
| B-diamond | HCHO (74%) | −1.7 Ag/Ag+ | — | 20 h | 0.1 M CH3OH (TBAP) | H-cell | 153 |
| B-diamond | HCOO-(94.7%) | — | 2 | 24 h | 0.5 M KCl | Flow cell | 86 |
|
|||||||
| Co-doped graphene | |||||||
| N,S-NCF | CO (94%) | −0.7 | 103 | 36 h | 0.1 M KHCO3 | H-cell | 13 |
| N,S-carbon Nanoweb | CO (93.4%) | −0.6 | 5.93 | 20 h | 0.1 M KHCO3 | H-cell | 14 |
| N,P-mesoporous carbon | CO (100%) | −0.65 | 8 | 7 h | 0.5 M NaHCO3 | H-cell | 17 |
| BN─C | CH4 (68%) | −0.5 | 1 | 12 h | 0.1 M KHCO3 | H-cell | 154 |
| B,N-nanodiamond | C2H5OH (93.2%) | −1.00 | — | 3 h | 0.1 M KHCO3 | H-cell | 155 |
|
|||||||
| Single atom supported on graphene | |||||||
| Ni-NrGO | CO (97%) | −0.8 | 42 | — | 0.5M KHCO3 |
H-cell | 156 |
| Zn-NG | CO (91%) | −0.8 | 11.2 | 15 h | 0.5M KHCO3 |
H-cell | 157 |
| Ni-NG | CO (95%) | −0.5 | — | 20 h | 0.1 M KHCO3 | H-cell | 158 |
| Fe-NG | CO (80%) | −0.60 | — | 10 h | 0.1 M KHCO3 | H-cell | 159 |
| Cu–N4−NG | CO (80.6%) | −1.0 | — | — | 0.1 M KHCO3 | H-cell | 160 |
| Ni–N-MEGO | CO (92.1%) | −0.7 | 26.8 | 21 h | 0.1 M KHCO3 | H-cell | 161 |
| Sb-graphene | HCOOH | −1.06 | — | 12 h | 0.1 M NaHCO3 | H-cell | 162 |
| Bi−rGO | HCOOH | −0.57 | — | 30 h | 0.5M KHCO3 |
H-cell | 163 |
| Cu–CD | CH4 (78%) | −1.14 | 40 | 6 h | 0.5M KHCO3 |
H-cell | 75 |
| Dual Cu SAC | C2+ (91%) | −1.66 | 90 | — | 0.1 M KHCO3 | H-cell | 164 |
| Cu–N–C-800 | C2H4 (24.8%) | −1.4 | 6.84 | 10 h | 0.1 M KHCO3 | H-cell | 165 |
| Cu0·5NC | Ethanol (55%) | −1.2 | 16.2 | — | 0.1 M CsHCO3 | H-cell | 102 |
| Cu–SA/NPC | Acetone (36.7%) | −0.36 | — | 5 cycles | 0.1 M KHCO3 | H-cell | 104 |
|
|||||||
| Graphene-metal composite | |||||||
| Bi-rGO | HCOOH (92.1%) | −0.97 | 28.1 | 30 h | 0.5 M KHCO3 | H-cell | 163 |
| Sb NS-G | HCOO− (88.5%) | −1.0 | 7.5 | 12 h | 0.5 M NaHCO3 | H-cell | 162 |
| Cu Pc_CNT | CH4 (66%) | −1.06 | 13 | — | 0.5 M KHCO3 | H-cell | 166 |
| Cu2O NCS | C2H4 (24.7%) | −1.3 | — | 4 h | 0.1M KHCO3 |
H-cell | 167 |
| Cu NPs-N,B-graphene | C2H5OH (58%) | −1.0 | 20.4 | 24 h | 0.5 M KHCO3 | H-cell | 168 |
| Co3O4 nanocrystals-rGO | C2H5OH (45.9%) C2H4 (28.8%) | −0.4 | 3.2 | 5 h | 0.5 M KHCO3 | H-cell | 169 |
| Cu–Cu2O-CeOx-rgo | C2+ products (74.5%) | −0.9 | 230 | 5 h | 2 M KOH | Flow cell | 170 |
|
|||||||
| Graphene encapsulated metals | |||||||
| In2O3 ⊃ NC@GO | HCOO− (91.2) | −0.8 | 40.4 | 10 h | 0.5 M KHCO3 | H-cell | 171 |
| Sn quantum Sheets@Graphene | HCOOH (85%) | −1.8 V s SCE | 21.1 | 50 h | 0.5 M NaHCO3 | H-cell | 172 |
| Cu2O/Cu@C/NG | HCOOH (82.1 ± 1.2%) | −0.78 | — | 30 h | 0.1 M KHCO3 | H-cell | |
|
|||||||
| Graphene analogs | |||||||
| Bismuthene with Bi–O bond | HCOOH (84.5%) | −0.9 | 41.5 | 12 h | 0.5 M KHCO3 | H-cell | 173 |
| Few layer bismuthene | HCOOH (96.9%) | −0.88 | 419.4 | 45 h | 1 M KOH | Flow cell | 174 |
(1) The controlled synthesis of graphene-based materials with a desired structure is essential for efficient eCO2RR. Even minor defects, such as metal impurities or vacancies introduced during the graphene manufacturing process, can significantly influence catalytic performance. Additionally, achieving reproducible control over doping and obtaining a specific composition of heteroatom doping remains a significant challenge. For instance, in N-doped carbon materials used as catalysts for eCO2RR, the catalytic activity is primarily attributed to active sites originating from pyridinic nitrogen, which tends to localize at the edges of carbon structures. However, precise manipulation of the types and distribution of heteroatom functionalities that enhance catalytic performance continues to be a complex task.
(2) While graphene-based catalysts exhibit promising performance for C1 products, often rivaling or surpassing noble metals, their efficiency in producing C2+ products remains significantly lower than that of copper-based catalysts. Although some carbon-based materials have shown the ability to produce C2+ products, their partial current densities typically fall below practical thresholds resulting in limited production rates. This highlights an ongoing challenge in improving the catalytic performance of graphene-based materials for multi-carbon product synthesis.
(3) Understanding the origin of active sites and the reaction mechanism in graphene-based catalysts remains a significant challenge due to their structural complexity, the presence of multiple reaction pathways, and the rapid kinetics associated with eCO2RR. Thus, efforts should focus on combining theoretical simulations with advanced in situ/operando techniques to elucidate the reaction mechanisms at the molecular level, providing deeper insights into the nature of active sites and the overall catalytic process. Such an understanding is vital for enhancing the performance of graphene-based catalysts.
(4) For practical implementation of eCO2RR, it is essential to improve the stability of graphene-based catalysts for over 2000 hours at high current densities. Currently, most existing graphene-based catalysts only exhibit stability of less than 50 h, which falls significantly short of the durability required for industrial applications, making long-term stable operation a critical challenge for advancing the industrial application of eCO2RR.
(5) The discovery of catalysis for electrochemical processes has greatly benefited from the integration of machine learning (ML) techniques. ML algorithms enable the analysis of vast datasets to identify trends and predict optimal doping configurations and composite designs. High-throughput computational screening, driven by ML, can significantly accelerate the material discovery and optimization process. This approach helps design graphene catalysts with properties tailored to specific reactions.
In summary, recent research highlights a growing interest in the development of graphene-based electrocatalysts for eCO2RR. The advancements, emerging challenges, and future perspectives on graphene-based materials, including heteroatom-doped graphene and metal-graphene composites, underline their great potential for eCO2RR applications. Ongoing research continues to pave the way for the successful application of graphene-based catalysts as sustainable and efficient solutions in CO2 reduction.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Author contributions
Wijewardena Lankamullage Hasini Amanda: conceptualization, investigation, visualization, writing (original draft, review and editing). Woo Seok Cheon: supervision, writing (review and editing). Jungwon Park: supervision. Seol-Ha Jeong: supervision. Ho Won Jang: conceptualization, writing (review and editing), supervision, funding.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT), South Korea (RS-2024-00421181, and RS-2024-00405016). The Inter-University Semiconductor Research Center, Institute of Engineering Research, and SOFT Foundry at Seoul National University provided research facilities for this work. This work was also supported by the KRISS (Korea Research Institute of Standards and Science) MPI Lab. Program.References
- S. A. Bandh and F. A. Malla, Biofuels in Circular Economy, 2023 Search PubMed.
- A. Parekh, G. Chaturvedi and A. Dutta, Sustain. Energy Technol. Assessments, 2023, 55, 102942 CrossRef.
- N. Bahman, M. Al-Khalifa, S. Al Baharna, Z. Abdulmohsen and E. Khan, Rev. Environ. Sci. Biotechnol., 2023, 22, 451–470 CrossRef CAS.
- A. M. Abdellah, F. Ismail, O. W. Siig, J. Yang, C. M. Andrei, L. A. DiCecco, A. Rakhsha, K. E. Salem, K. Grandfield, N. Bassim, R. Black, G. Kastlunger, L. Soleymani and D. Higgins, Nat. Commun., 2024, 15, 1–15 CrossRef PubMed.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, 1–17 Search PubMed.
- B. Chang, H. Pang, F. Raziq, S. Wang, K. W. Huang, J. Ye and H. Zhang, Energy Environ. Sci., 2023, 16, 4714–4758 RSC.
- J. Han, X. Bai, X. Xu, X. Bai, A. Husile, S. Zhang, L. Qi and J. Guan, Chem. Sci., 2024, 15, 7870–7907 RSC.
- Y. Zhao, J. Raj, X. Xu, J. Jiang, J. Wu and M. Fan, Small, 2024, 1–28 Search PubMed.
- R. Paul, L. Zhu, H. Chen, J. Qu and L. Dai, Adv. Mater., 2019, 31, 1–24 Search PubMed.
- F. Yu, K. Deng, M. Du, W. Wang, F. Liu and D. Liang, Carbon Capture Sci. Technol., 2023, 6, 100081 CrossRef CAS.
- M. S. K. Chowdury, Y. J. Park, S. B. Park and Y. il Park, Sustain. Mater. Technol., 2024, 42, e01124 CAS.
- M. A. Zafar, Y. Liu, S. Allende and M. V. Jacob, Nano-Struct. Nano-Objects, 2024, 38, 101129 CrossRef CAS.
- H. Yang, Y. Wu, Q. Lin, L. Fan, X. Chai, Q. Zhang, J. Liu, C. He and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 15476–15480 CrossRef CAS PubMed.
- H. Han, S. Park, D. Jang, S. Lee and W. B. Kim, ChemSusChem, 2020, 13, 539–547 CrossRef CAS PubMed.
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Angew. Chem., Int. Ed., 2020, 59, 11123–11129 CrossRef CAS PubMed.
- F. Yang, H. Yu, X. Mao, Q. Meng, S. Chen, Q. Deng, Z. Zeng, J. Wang and S. Deng, Chem. Eng. J., 2021, 425, 131661 CrossRef CAS.
- B. Pan, X. Zhu, Y. Wu, T. Liu, X. Bi, K. Feng, N. Han, J. Zhong, J. Lu, Y. Li and Y. Li, Adv. Sci., 2020, 7, 2–7 Search PubMed.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- F. Yang, C. Liang, H. Yu, Z. Zeng, Y. M. Lam, S. Deng and J. Wang, Adv. Sci., 2022, 9, 1–8 Search PubMed.
- S. M. Lee, W. S. Cheon, M. G. Lee and H. W. Jang, Small Struct., 2023, 4(6), 2200236 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- J. Zhang, J. Ding, Y. Liu, C. Su, H. Yang, Y. Huang and B. Liu, Joule, 2023, 7(8), 1700–1744 CrossRef CAS.
- Y. Ma, J. Yu, M. Sun, B. Chen, X. Zhou, C. Ye, Z. Guan, W. Guo, G. Wang, S. Lu, D. Xia, Y. Wang, Z. He, L. Zheng, Q. Yun, L. Wang, J. Zhou, P. Lu, J. Yin, Y. Zhao, Z. Luo, L. Zhai, L. Liao, Z. Zhu, R. Ye, Y. Chen, Y. Lu, S. Xi, B. Huang, C. S. Lee and Z. Fan, Adv. Mater., 2022, 34, 1–9 Search PubMed.
- X. G. Zhang, S. Feng, C. Zhan, D. Y. Wu, Y. Zhao and Z. Q. Tian, J. Phys. Chem. Lett., 2020, 11, 6593–6599 CrossRef CAS PubMed.
- H. Q. Liang, T. Beweries, R. Francke and M. Beller, Angew. Chem., Int. Ed., 2022, 61(19), e202200723 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- Y. Sun, D. Song, J. Yu, J. Zhu, J. Liu, R. Chen, Q. Liu, P. Liu and J. Wang, Surf. Sci. Technol., 2023, 1, 1–14 CrossRef.
- A. E. Adetayo, T. N. Ahmed, A. Zakhidov and G. W. Beall, Adv. Opt. Mater., 2021, 9, 1–23 Search PubMed.
- L. Wang and J. Choi, Micro Nano Syst. Lett., 2022, 10, 17 CrossRef.
- M. K. Fathy, A. H. Zaki, H. A. Shawkey and H. R. Tantawy, Trans. Electr. Electron. Mater., 2024, 732–744 CrossRef.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- R. Kumar, S. Sahoo, E. Joanni, R. K. Singh, K. Maegawa, W. K. Tan, G. Kawamura, K. K. Kar and A. Matsuda, Mater. Today, 2020, 39, 47–65 CrossRef CAS.
- X. Li, L. Fan, Z. Li, K. Wang, M. Zhong, J. Wei, D. Wu and H. Zhu, Adv. Energy Mater., 2012, 2, 425–429 CrossRef CAS.
- Y. Du, X. Meng, Z. Wang, X. Zhao and J. Qiu, Acta Phys.-Chim. Sin., 2021, 38, 1–17 Search PubMed.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- X. F. Li, K. Y. Lian, L. Liu, Y. Wu, Q. Qiu, J. Jiang, M. Deng and Y. Luo, Sci. Rep., 2016, 6, 1–10 CrossRef CAS PubMed.
- Z. Zhang, L. Yu, Y. Tu, R. Chen, L. Wu, J. Zhu and D. Deng, Cell Reports Phys. Sci., 2020, 1, 100145 CrossRef CAS.
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1–20 Search PubMed.
- T. Ma, Q. Fan, X. Li, J. Qiu, T. Wu and Z. Sun, J. CO2 Util., 2019, 30, 168–182 CrossRef CAS.
- L. Zhang, J. Niu, M. Li and Z. Xia, J. Phys. Chem. C, 2014, 118, 3545–3553 CrossRef CAS.
- A. Kumar, M. Ubaidullah, P. V. Pham and R. K. Gupta, Chem. Eng. J., 2024, 499, 156664 CrossRef CAS.
- M. B. Burkholder, F. B. A. Rahman, E. H. Chandler, J. R. Regalbuto, B. F. Gupton and J. M. M. Tengco, Carbon Trends, 2022, 9, 100196 CrossRef CAS.
- K. S. M. Manimehalai, S. B. Venkatesh and T. T. Subash, Ionics, 2025, 1467–1481 Search PubMed.
- J. Yan, F. Wang, S. Yin, J. Zhang and W. Jiang, Rare Met., 2025, 44, 2239–2267 CrossRef CAS.
- K. M. Yam, N. Guo, Z. Jiang, S. Li and C. Zhang, Catalysts, 2020, 10(1), 53 CrossRef CAS.
- X. Tong, M. Cherif, G. Zhang, X. Zhan, J. Ma, A. Almesrati, F. Vidal, Y. Song, J. P. Claverie and S. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 30512–30523 CrossRef CAS PubMed.
- H. Song, M. Wu, Z. Tang, J. S. Tse, B. Yang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 7234–7244 CrossRef CAS PubMed.
- X. Liu, C. Z. Wang, M. Hupalo, W. C. Lu, M. C. Tringides, Y. X. Yao and K. M. Ho, Phys. Chem. Chem. Phys., 2012, 14, 9157–9166 RSC.
- Y. Tang, Z. Yang and X. Dai, J. Chem. Phys., 2011, 135(22), 224704 CrossRef PubMed.
- Z. Ning, Z. Chen, X. Du, R. Ran, W. Dong and C. Chen, J. Supercond. Nov. Magn., 2014, 27, 115–120 CrossRef CAS.
- Y. Zhou, G. Gao, Y. Li, W. Chu and L. W. Wang, Phys. Chem. Chem. Phys., 2019, 21, 3024–3032 RSC.
- Y. Cheng, S. Zhao, H. Li, S. He, J. P. Veder, B. Johannessen, J. Xiao, S. Lu, J. Pan, M. F. Chisholm, S. Z. Yang, C. Liu, J. G. Chen and S. P. Jiang, Appl. Catal., B, 2019, 243, 294–303 CrossRef CAS.
- H. Y. Zhuo, X. Zhang, J. X. Liang, Q. Yu, H. Xiao and J. Li, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS PubMed.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 1–24 Search PubMed.
- A. K. Singh and S. P. Singh, Micro Nano Syst. Lett., 2023, 11, 20 CrossRef.
- Y. S. Ye, M. Y. Cheng, X. L. Xie, J. Rick, Y. J. Huang, F. C. Chang and B. J. Hwang, J. Power Sources, 2013, 239, 424–432 CrossRef CAS.
- P. Yang, X. Yang, W. Liu, R. Guo and Z. Yao, Green Energy Environ., 2023, 8, 1265–1278 CrossRef CAS.
- W. Tao, N. Kong, X. Ji, Y. Zhang, A. Sharma, J. Ouyang, B. Qi, J. Wang, N. Xie, C. Kang, H. Zhang, O. C. Farokhzad and J. S. Kim, Chem. Soc. Rev., 2019, 48, 2891–2912 RSC.
- F. Pan, B. Li, W. Deng, Z. Du, Y. Gang, G. Wang and Y. Li, Appl. Catal., B, 2019, 252, 240–249 CrossRef CAS.
- F. Yang, A. O. Elnabawy, R. Schimmenti, P. Song, J. Wang, Z. Peng, S. Yao, R. Deng, S. Song, Y. Lin, M. Mavrikakis and W. Xu, Nat. Commun., 2020, 11, 1088 CrossRef CAS PubMed.
- F. Liu and Z. Fan, Chem. Soc. Rev., 2023, 52, 1723–1772 RSC.
- S. Abdullah, S. Hussain and M. Liaquat, Dialogue Soc. Sci. Rev., 2025, 3(2), 167–185 Search PubMed.
- X. Du Liang, N. Tian, S. N. Hu, Z. Y. Zhou and S. G. Sun, Mater. Reports Energy, 2023, 3, 100191 CrossRef.
- L. Chen, X. Ding, Z. Wang, S. Xu, Q. Jiang, C. Dun and J. J. Urban, Surf. Sci. Technol., 2024, 2, 9 CrossRef.
- J. Li, W. Y. Zan, H. Kang, Z. Dong, X. Zhang, Y. Lin, Y. W. Mu, F. Zhang, X. M. Zhang and J. Gu, Appl. Catal., B, 2021, 298, 120510 CrossRef CAS.
- J. Y. Kim, D. Hong, J. C. Lee, H. G. Kim, S. Lee, S. Shin, B. Kim, H. Lee, M. Kim, J. Oh, G. Do Lee, D. H. Nam and Y. C. Joo, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- Y. Zhan, J. Huang, Z. Lin, X. Yu, D. Zeng, X. Zhang, F. Xie, W. Zhang, J. Chen and H. Meng, Carbon NY, 2015, 95, 930–939 CrossRef CAS.
- Z. Zafar, Z. H. Ni, X. Wu, Z. X. Shi, H. Y. Nan, J. Bai and L. T. Sun, Carbon NY, 2013, 61, 57–62 CrossRef CAS.
- G. D. Sun, Y. N. Cao, M. Z. Hu, X. H. Liang, Z. Wang, Z. J. Cai, F. Y. Shen, H. He, Z. X. Wang and K. Bin Zhou, Carbon NY, 2023, 214, 118320 CrossRef CAS.
- B. Sheng, D. Cao, S. Chen, N. Zhang, J. Wang and L. Song, Commun. Mater., 2025, 6, 64 CrossRef CAS.
- R. Zhao, Y. Wang, G. Ji, J. Zhong, F. Zhang, M. Chen, S. Tong, P. Wang, Z. Wu, B. Han and Z. Liu, Adv. Mater., 2023, 35, 1–11 Search PubMed.
- X. Li, S. Wang, L. Li, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2020, 142, 9567–9581 CAS.
- Y. Cai, J. Fu, Y. Zhou, Y. C. Chang, Q. Min, J. J. Zhu, Y. Lin and W. Zhu, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- C. Wang, X. Wang, H. Ren, Y. Zhang, X. Zhou, J. Wang, Q. Guan, Y. Liu and W. Li, Nat. Commun., 2023, 14, 5108 CrossRef CAS PubMed.
- X. Ren, J. Zhao, X. Li, J. Shao, B. Pan, A. Salamé, E. Boutin, T. Groizard, S. Wang, J. Ding, X. Zhang, W. Y. Huang, W. J. Zeng, C. Liu, Y. Li, S. F. Hung, Y. Huang, M. Robert and B. Liu, Nat. Commun., 2023, 3401 CrossRef CAS PubMed.
- J. Kim, T. Lee, H. D. Jung, M. Kim, J. Eo, B. Kang, H. Jung, J. Park, D. Bae, Y. Lee, S. Park, W. Kim, S. Back, Y. Lee and D. H. Nam, Nat. Commun., 2024, 192 CrossRef PubMed.
- F. Yang, X. Ma, W. Bin Cai, P. Song and W. Xu, J. Am. Chem. Soc., 2019, 141, 20451–20459 CrossRef CAS PubMed.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano., 2015, 9(5), 5364–5371 CrossRef CAS PubMed.
- C. Ma, P. Hou, X. Wang, Z. Wang, W. Li and P. Kang, Appl. Catal., B, 2019, 250, 347–354 CrossRef CAS.
- Y. Zhao, Q. Yuan, K. Sun, A. Wang, R. Xu, J. Xu, Y. Wang, M. Fan and J. Jiang, ACS Appl. Mater. Interfaces, 2023, 15, 37593–37601 CrossRef CAS PubMed.
- H. Wang, J. Jia, P. Song, Q. Wang, D. Li, S. Min, C. Qian, L. Wang, Y. F. Li, C. Ma, T. Wu, J. Yuan, M. Antonietti and G. A. Ozin, Angew. Chem., Int. Ed., 2017, 56, 7847–7852 CrossRef CAS PubMed.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- N. Sreekanth, M. A. Nazrulla, T. V. Vineesh, K. Sailaja and K. L. Phani, Chem. Commun., 2015, 51, 16061–16064 RSC.
- K. Natsui, H. Iwakawa, N. Ikemiya, K. Nakata and Y. Einaga, Angew. Chem., Int. Ed., 2018, 57, 2639–2643 CrossRef CAS PubMed.
- Y. Song, W. Chen, C. Zhao, S. Li, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 10840–10844 CrossRef CAS PubMed.
- D. Davis and Y. Sajeev, Phys. Chem. Chem. Phys., 2014, 16, 17408–17411 RSC.
- Q. Yun, Q. Lu, X. Zhang, C. Tan and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 626–646 CrossRef CAS PubMed.
- K. Qu, Y. Zheng, Y. Jiao, X. Zhang, S. Dai and S. Z. Qiao, Adv. Energy Mater., 2017, 1602068 CrossRef.
- M. I. Cerrillo, C. Jiménez, M. Á. Ortiz, R. Camarillo, J. Rincón and F. Martínez, J. Ind. Eng. Chem., 2023, 127, 101–109 CrossRef CAS.
- B. Qiao, J. X. Liang, A. Wang, C. Q. Xu, J. Li, T. Zhang and J. J. Liu, Nano Res., 2015, 8, 2913–2924 CrossRef CAS.
- B. Qiao, J. Liu, Y. G. Wang, Q. Lin, X. Liu, A. Wang, J. Li, T. Zhang and J. Liu, ACS Catal., 2015, 5, 6249–6254 CrossRef CAS.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- H. Zhou, X. Yang, L. Li, X. Liu, Y. Huang, X. Pan, A. Wang, J. Li and T. Zhang, ACS Catal., 2016, 6, 1054–1061 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- C. Jia, Q. Sun, R. Liu, G. Mao, T. Maschmeyer, J. J. Gooding, T. Zhang, L. Dai and C. Zhao, Adv. Mater., 2024, 2404659 CrossRef CAS PubMed.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo, H. L. Xin, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- Y. Li, W. Shan, M. J. Zachman, M. Wang, S. Hwang, H. Tabassum, J. Yang, X. Yang, S. Karakalos, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2022, e202205632 CAS.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed.
- H. Bin Yang, S. F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H. Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef.
- X. Wang, Y. Wang, X. Sang, W. Zheng, S. Zhang, L. Shuai, B. Yang, Z. Li, J. Chen, L. Lei, N. M. Adli, M. K. H. Leung, M. Qiu, G. Wu and Y. Hou, Angew. Chem., 2021, 133, 4238–4244 CrossRef.
- G. Luo, Y. Jing and Y. Li, J. Mater. Chem. A, 2020, 8, 15809–15815 RSC.
- X. Zhao, K. Zhao, Y. Liu, Y. Su, S. Chen, H. Yu and X. Quan, ACS Catal., 2022, 12, 11412–11420 CrossRef CAS.
- Q. Hao, H. xia Zhong, J. zhi Wang, K. hua Liu, J. min Yan, Z. hong Ren, N. Zhou, X. Zhao, H. Zhang, D. xue Liu, X. Liu, L. wei Chen, J. Luo and X. bo Zhang, Nat. Synth., 2022, 1, 719–728 CrossRef CAS.
- L. Zhang, J. Feng, S. Liu, X. Tan, L. Wu, S. Jia, L. Xu, X. Ma, X. Song, J. Ma, X. Sun and B. Han, Adv. Mater., 2023, 35, 1–10 Search PubMed.
- M. Karuppannan, Y. Kim, S. Gok, E. Lee, J. Y. Hwang, J. H. Jang, Y. H. Cho, T. Lim, Y. E. Sung and O. J. Kwon, Energy Environ. Sci., 2019, 12, 2820–2829 RSC.
- Y. Zhang, P. He, D. Zhuo, J. Zhang, N. Zhang, X. Wang, G. Lin and Z. Kong, Electron. Mater. Lett., 2024, 20, 95–101 CrossRef CAS.
- V. Kumar, R. K. Mishra, L. G. Trung, P. Kumar, S. M. Mane, J. C. Shin and J. S. Gwag, Electron. Mater. Lett., 2024, 20, 414–424 CrossRef CAS.
- H. Y. Kim, J. Y. Kim and S. H. Joo, Bull. Korean Chem. Soc., 2021, 42, 724–736 CrossRef CAS.
- M. Sharma, J. H. Jang, D. Y. Shin, J. A. Kwon, D. H. Lim, D. Choi, H. Sung, J. Jang, S. Y. Lee, K. Y. Lee, H. Y. Park, N. Jung and S. J. Yoo, Energy Environ. Sci., 2019, 12, 2200–2211 RSC.
- S. Popović, M. Smiljanić, P. Jovanovič, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef PubMed.
- W. T. Osowiecki, J. J. Nussbaum, G. A. Kamat, G. Katsoukis, M. Ledendecker, H. Frei, A. T. Bell and A. P. Alivisatos, ACS Appl. Energy Mater., 2019, 2, 7744–7749 CrossRef CAS.
- Y. G. Kim, J. H. Baricuatro, A. Javier, J. M. Gregoire and M. P. Soriaga, Langmuir, 2014, 30, 15053–15056 CrossRef CAS PubMed.
- J. Vavra, T. H. Shen, D. Stoian, V. Tileli and R. Buonsanti, Angew. Chem., Int. Ed., 2021, 60, 1347–1354 CrossRef CAS PubMed.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., 2018, 130, 6300–6305 CrossRef.
- W. Drisdell, S. H. Lee, D. Lee, D. Larson, H. Li, J. Chen, S. Blair, A. Gallo, H. Zheng, C. Tassone and T. Jaramillo, Preprint, 2023, 1–21 Search PubMed.
- J. M. Yoo, H. Shin, D. Y. Chung and Y. E. Sung, Acc. Chem. Res., 2022, 55, 1278–1289 CrossRef CAS PubMed.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, J. Am. Chem. Soc., 2023, 145, 5370–5383 CrossRef CAS PubMed.
- K. Maekawa, K. Mori, N. Suzumura, K. Honda, Y. Hirose, K. Asai, A. Uedono and M. Kojima, Microelectron. Eng., 2008, 85, 2137–2141 CrossRef CAS.
- T. Yao, W. Xia, S. Han, S. Jia, X. Dong, M. Wang, J. Jiao, D. Zhou, J. Yang, X. Xing, C. Chen, M. He, H. Wu and B. Han, Chem. Sci., 2023, 14, 14308–14315 RSC.
- C. Xu, A. Vasileff, B. Jin, D. Wang, H. Xu, Y. Zheng and S. Z. Qiao, Chem. Commun., 2020, 56, 11275–11278 RSC.
- J. Yuan, M. P. Yang, W. Y. Zhi, H. Wang, H. Wang and J. X. Lu, J. CO2 Util., 2019, 33, 452–460 CrossRef CAS.
- D. C. B. Alves, R. Silva, D. Voiry, T. Asefa and M. Chhowalla, Mater. Renew. Sustain. Energy, 2015, 4, 2 CrossRef.
- S. Asperti, R. Hendrikx, Y. Gonzalez-Garcia and R. Kortlever, ChemCatChem, 2022, 14, 1–8 Search PubMed.
- J. Darayen, O. Chailapakul, P. Praserthdam, J. Panpranot, D. N. Tungasmita and Y. Boonyongmaneerat, Appl. Sci., 2022, 11104 Search PubMed.
- T. Burwell, M. Thangamuthu, G. N. Aliev, S. Ghaderzadeh, E. C. Kohlrausch, Y. Chen, W. Theis, L. T. Norman, J. A. Fernandes, E. Besley, P. Licence and A. N. Khlobystov, Commun. Chem., 2024, 7, 1–10 CrossRef PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 1–8 CrossRef.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 8177 CrossRef PubMed.
- J. Su, C. B. Musgrave, Y. Song, L. Huang, Y. Liu, G. Li, Y. Xin, P. Xiong, M. M. J. Li, H. Wu, M. Zhu, H. M. Chen, J. Zhang, H. Shen, B. Z. Tang, M. Robert, W. A. Goddard and R. Ye, Nat. Catal., 2023, 6, 818–828 CrossRef CAS.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- W. Yang, X. Zhang and Y. Xie, Nano Today, 2016, 11, 793–816 CrossRef CAS.
- X. Yu, Y. Fautrelle, Z. Ren and X. Li, Mater. Lett., 2015, 161, 144–148 CrossRef CAS.
- B. Ávila-Bolívar, L. García-Cruz, V. Montiel and J. Solla-Gullón, Molecules, 2019, 24, 1–15 CrossRef PubMed.
- X. Tan, S. Jia, X. Song, X. Ma, J. Feng, L. Zhang, L. Wu, J. Du, A. Chen, Q. Zhu, X. Sun and B. Han, Chem. Sci., 2023, 14, 8214–8221 RSC.
- Y. Hu, J. Liang, Y. Gu, S. Yang, W. Zhang, Z. Tie, J. Ma and Z. Jin, Nano Lett., 2023, 23, 10512–10521 CrossRef CAS PubMed.
- X. Mei, C. Liu, D. Zhang, J. Cao, R. Ge, J. Wang and W. Xu, Adv. Energy Mater., 2024, 14, 1–7 Search PubMed.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X. D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2016, 16, 466–470 CrossRef CAS PubMed.
- J. Xu, Y. Kan, R. Huang, B. Zhang, B. Wang, K. H. Wu, Y. Lin, X. Sun, Q. Li, G. Centi and D. Su, ChemSusChem, 2016, 9, 1085–1089 CrossRef CAS PubMed.
- X. Lu, T. H. Tan, Y. H. Ng and R. Amal, Chem.–A Eur. J., 2016, 22, 11991–11996 CrossRef CAS PubMed.
- H. Wang, Y. Chen, X. Hou, C. Ma and T. Tan, Green Chem., 2016, 18, 3250–3256 RSC.
- X. Sun, X. Kang, Q. Zhu, J. Ma, G. Yang, Z. Liu and B. Han, Chem. Sci., 2016, 7, 2883–2887 RSC.
- T. Zhang, W. Li, K. Huang, H. Guo, Z. Li, Y. Fang, R. M. Yadav, V. Shanov, P. M. Ajayan, L. Wang, C. Lian and J. Wu, Nat. Commun., 2021, 12, 1–9 Search PubMed.
- J. Yuan, W. Y. Zhi, L. Liu, M. P. Yang, H. Wang and J. X. Lu, Electrochim. Acta, 2018, 282, 694–701 CrossRef CAS.
- T. Liu, S. Ali, Z. Lian, C. Si, D. S. Su and B. Li, J. Mater. Chem. A, 2018, 6, 19998–20004 RSC.
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., Int. Ed., 2014, 53, 871–874 CrossRef CAS PubMed.
- B. Yuying Zhaoa, b, Q. Yuanb, M. Fana, A. O. Wanga, K. Suna, Z. Wangc and J. Jianga, Chinese Chem. Lett., 2023, 34, 108120 CrossRef.
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang, H. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607–15611 CrossRef CAS PubMed.
- H. Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, J. Jo, H. Baik, V. Subramanian, M. Kim, U. Sim and K. T. Nam, Chem.–A Eur. J., 2018, 24, 18444–18454 CrossRef CAS PubMed.
- Z. Chen, K. Mou, S. Yao and L. Liu, ChemSusChem, 2018, 11, 2944–2952 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1–9 Search PubMed.
- C. Xu, X. Zhi, A. Vasileff, D. Wang, B. Jin, Y. Jiao, Y. Zheng and S. Z. Qiao, Small Struct., 2021, 2, 1–7 Search PubMed.
- Y. Cheng, S. Zhao, H. Li, S. He, J. P. Veder, B. Johannessen, J. Xiao, S. Lu, J. Pan, M. F. Chisholm, S. Z. Yang, C. Liu, J. G. Chen and S. P. Jiang, Appl. Catal., B, 2019, 243, 294–303 CrossRef CAS.
- F. Li, M. Xue, J. Li, X. Ma, L. Chen, X. Zhang, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14718–14722 CrossRef CAS PubMed.
- D. Wu, W. Chen, X. Wang, X. Z. Fu and J. L. Luo, J. CO2 Util., 2020, 37, 353–359 CrossRef CAS.
- S. Li, A. Guan, C. Yang, C. Peng, X. Lv, Y. Ji, Y. Quan, Q. Wang, L. Zhang and G. Zheng, ACS Mater. Lett., 2021, 3, 1729–1737 CrossRef CAS.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T. K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- H. Ning, X. Wang, W. Wang, Q. Mao, Z. Yang, Q. Zhao, Y. Song and M. Wu, Carbon NY, 2019, 146, 218–223 CrossRef CAS.
- Y. Zhao, Q. Yuan, R. Xu, C. Zhang, K. Sun, A. Wang, A. Zhang, Z. Wang, J. Jiang and M. Fan, Appl. Catal., B, 2024, 355, 124168 CrossRef CAS.
- S. Saha, S. Maitra, M. Chattopadhyaya, A. Sarkar, S. Haque, S. Roy and K. Kargupta, ACS Appl. Nano Mater., 2022, 5, 10369–10382 CrossRef CAS.
- Z. Zhang, X. Wang, H. Tian, H. Jiao, N. Tian, L. Bian, Y. Liu and Z. L. Wang, J. Colloid Interface Sci., 2024, 661, 966–976 CrossRef CAS PubMed.
- Y. Wang, J. Ding, J. Zhao, J. Wang, X. Han, Y. Deng and W. Hu, J. Mater. Sci. Technol., 2022, 121, 220–226 CrossRef CAS.
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Nat. Commun., 2016, 7, 1–8 CrossRef.
- B. Ning, Q. Xu, M. Liu, H. Jiang, Y. Hu and C. Li, Chem. Eng. Sci., 2022, 251, 117409 CrossRef CAS.
- X. Wang, D. Song, C. Wang, Y. Wang, J. H. Jiang, J. Huo and Z. Li, ACS Appl. Nano Mater., 2023, 6, 17357–17363 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |