
Enabling high-performance and high-rate-capability Na4MnV(PO4)3 sodium-ion battery cathodes through tuning the NASICON framework†
Cheng-Yen
Wu
a,
Chen-En
Huang
a,
An-Chi
Luo
a,
Wen-Hsuan
Lu
a,
Shao-Chu
Huang
a,
Yu-Ming
Huang
b,
Vanessa K.
Peterson
 cd,
Hao-Yu
Liu
a,
Chi-Liang
Chen
e,
Ying-Rui
Lu
e,
Tsung-Yi
Chen
ae,
Cheng-Zhang
Lu
*f,
Wei Kong
Pang
*d and
Han-Yi
Chen
*ag
cd,
Hao-Yu
Liu
a,
Chi-Liang
Chen
e,
Ying-Rui
Lu
e,
Tsung-Yi
Chen
ae,
Cheng-Zhang
Lu
*f,
Wei Kong
Pang
*d and
Han-Yi
Chen
*ag
aDepartment of Material Science and Engineering, National Tsing Hua University, Hsinchu 300044, Taiwan. E-mail: hanyi.chen@mx.nthu.edu.tw
bMolecular Science and Digital Innovation Center, Sunrise Scientific Company Limited, 114 Taipei, Taiwan
cAustralian Centre for Neutron Scattering, Australian Nuclear Science and Technology Organisation, Sydney, NSW 2234, Australia
dInstitute for Superconducting and Electronic Materials (ISEM), Australian Institute for Innovative Materials (AIIM), Faculty of Engineering and Information Sciences, University of Wollongong, NSW 2522, Australia. E-mail: wkpang@uow.edu.au
eNational Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu 300092, Taiwan
fPatriot Green Energy Technology Co., Ltd., Hsinchu 310401, Taiwan. E-mail: czlu@ssbtek.com
gHierarchical Green-Energy Materials (Hi-GEM) Research Center, National Cheng Kung University, Tainan 70101, Taiwan
First published on 17th December 2024
Abstract
Na4MnV(PO4)3 (NMVP) has emerged as a cost-effective alternative to Na3V2(PO4)3, which is considered a promising cathode material for sodium-ion batteries. However, challenges such as low electronic conductivity, fast capacity fading resulting from the dissolution of Mn and polarization due to irreversible structural transformation impede the widespread application of NMVP. In this study, a facile sol–gel method is employed to dope NMVP with Mo, aiming to address these limitations. Synchrotron extended X-ray absorption fine structure data, neutron powder diffraction results, and density functional theory (DFT) calculations indicate a preferential occupation of the P site by Mo. Mo-doped NMVP demonstrates an outstanding discharge capacity of 97.5 mA h g−1 at 0.2C and 46.4 mA h g−1 at 20C, along with impressive long-term stability, retaining 78.8% capacity after 300 cycles at 1C. DFT calculations reveal a significant reduction in the band gap of Mo-doped NMVP, enhancing electronic conductivity and thereby improving rate capability retention. In operando X-ray absorption spectroscopy reveals changes in the valence of V, Mn, and Mo in the material during charge/discharge, confirming the complete reversibility of redox reactions. The outstanding performance of the novel Mo-doped NMVP cathode highlights its promising potential for application in large-scale energy storage systems.
Introduction
The surging demand for large-scale energy storage and electric vehicles has spurred significant research efforts into exploring promising sodium-ion batteries (NIBs), driven by the cost-effectiveness and widespread availability of sodium sources.1,2 Additionally, NIBs have analogous intercalation chemistry to Li-ion batteries (LIBs) and a redox potential of −2.71 V vs. the standard hydrogen electrode (SHE), only marginally higher than
of −2.71 V vs. the standard hydrogen electrode (SHE), only marginally higher than  3,4
3,4
Cathode materials for sodium-ion batteries (NIBs) encompass three primary categories: layered structure-type transition metal oxides,5,6 Prussian blue analogs,7 and polyanionic compounds.8–10 Polyanionic compounds with tunable crystal structures and stable frameworks, such as Na3V2(PO4)3,11–13 Na4FexP4O12+x (2 ≤ x ≤ 4),9 Na3MnTi(PO4)3,10 Na2Fe2(SO4)3,14 and Na3V2(PO4)2F3,15 offer safe operation and higher redox potentials.8 Among these, Na3V2(PO4)3 (NVP, NASICON) with a stable three-dimensional Na superionic conductor framework offers extensive interstitial sites for sodium ion diffusion.11–13 Tunnels in the NASICON structure facilitate Na ion diffusion and contribute to NVP's impressive specific capacity of 110 mA h g−1 and a flat voltage platform at 3.4 V (vs. Na+/Na), corresponding to a reversible V3+/V4+ redox reaction, providing an outstanding energy density of 400 W h kg−1.16–19 Despite these merits, the high cost and limited availability of vanadium sources hinder the widespread adoption of NVP as a cathode material for large-scale energy storage.20 Consequently, cost-effective alternatives have been sought, leading to the development of Na4MnV(PO4)3 (NMVP), in which V is partially substituted with the comparatively inexpensive transition metal element Mn.21 The NMVP cathode exhibits flat voltage plateaus at 3.3 V and 3.6 V, corresponding to the V3+/V4+ and Mn2+/Mn3+ redox reactions, respectively. NMVP demonstrates an excellent specific capacity of 101 mA h g−1 at 1C (1C = 111 mA g−1), a remarkable high-rate capability of 90 mA h g−1 at 10C, and stable long-term performance, retaining 89% capacity after 1000 cycles across various current densities.22
Despite the promising performance of the NMVP cathode, a low intrinsic electronic conductivity remains a limitation and has prompted extensive research. Various strategies to address this limitation have been explored, including embedding intertwined carbon nanotubes in the material, as well as coating the cathode with porous graphene aerogels or a reduced graphene oxide layer.23–25 Furthermore, the dissolution of Mn and undesirable side reactions in the electrolyte result in a dramatic decay of NMVP. To address this, surface coatings such as thermally stable Al(PO3)3 on NMVP can enhance the stability of the electrode/electrolyte interface and inhibit transition metal dissolution, thereby significantly improving long-cycle stability performance.26 While these approaches enhance electronic conductivity and sodium diffusion kinetics, they are not conducive to large-scale manufacture.27 Subsequently, the alternative and practical method of partial ion substitution within NMVP to enhance electrochemical performance has been explored, with several studies reporting the influence of substituent ions including Cr, Cu, Ce, Fe, Ti, Mg, Zr, Al, and F on performance.16,28–33 Carbon-coated Cu-doped Na4VMn0.9Cu0.1(PO4)3 with a cotton candy-like morphology was synthesized using modified pyrosynthesis, possessing both fast sodium ion diffusion and excellent specific capacity (117 mA h g−1 at 0.25C), with improved rate capability compared to undoped counterparts.29 Fe substituted NMVP showed enhanced electronic conductivity and specific capacity (120 mA h g−1 at 0.5C), with improved rate capability (96 mA h g−1 at 20C) compared to the pristine NMVP cathode.30 Carbon-coated and Zr-doped NMVP, Na3.95MnV0.95Zr0.05(PO4)3/C, was synthesized via the sol–gel method.34 The Zr4+ doping ion has a large number of free electrons on the d-orbital, which not only enriches the local electron density of NMVP but enhances the Na+ diffusion kinetics. As a result, Na3.95MnV0.95Zr0.05(PO4)3/C displays a high rate capability of 71.8 mA h g−1 at 30C.34 A sodium-deficient F-doped NMVP cathode, Na3.85□0.15MnV(PO3.95F0.05)3, demonstrated a high energy density of approximately 380 W h kg−1, along with improved rate capability compared to undoped NMVP and a long cycling life exceeding 2000 cycles.33 These studies underscore the efficacy of metal ion doping in augmenting NMVP's electronic conductivity, leading to improved specific capacity and rate performance.
The current study focuses on leveraging hexavalent Mo6+ substituted NMVP with a valence higher than P5+ to increase the electron concentration, thereby effectively improving electronic conductivity. Mo-doped NMVP was synthesized through a facile sol–gel approach and the structure of the as-synthesized material was investigated using X-ray diffraction (XRD), extended X-ray absorption fine structure (EXAFS) analysis, neutron powder diffraction, and density functional theory (DFT) calculations. Sodium diffusion was investigated through the potentiostatic intermittent titration technique (PITT) and galvanostatic intermittent titration technique (GITT). In operando XRD and in operando V/Mn/Mo K-edge X-ray absorption spectroscopy (XAS) were employed to study the redox mechanism and structural changes during sodium ion insertion/extraction. The Mo-doped NMVP demonstrates remarkable electrochemical performance, establishing suitability for application as a cathode material for NIBs.
Experimental
Synthesis of Mo-doped Na4MnV(PO4)3
A series of Mo-doped NMVP (precursor molar ratio of Mo to NMVP = 0, 0.02, 0.03, and 0.04) composites were synthesized through a facile sol–gel method, followed by high-temperature calcination and sintering under an argon atmosphere. Initially, taking NMVP as an example, a homogeneous precursor solution was prepared by dissolving sodium acetate trihydrate (40 mmol, VETEC), manganese acetate tetrahydrate (10 mmol, ACROS), ammonium trioxovanadate (10 mmol, Alfa), ammonium dihydrogen phosphate (30 mmol, SHOWA), and citric acid (7 mmol, Sigma-Aldrich) in 80 mL of distilled water. The Mo-doped NMVP was prepared using the same method, with molybdenum(VI) oxide (Alfa) as the Mo precursor. The molar ratios of Mo to NMVP were 0, 0.02, 0.03, and 0.04. The solution was vigorously stirred, and the uniform mixture was heated to 80 °C in a water bath to form a gel. Subsequently, the gel was dried in a vacuum oven at 100 °C overnight to yield a powder. Finally, the powder was calcined at 400 °C for 4 h (the heating rate was 5 °C min−1), followed by sintering at 750 °C for 6 h (the heating rate was 1 °C min−1) under an argon atmosphere.Material characterization
The crystal structures of Mo-doped NMVP materials were characterized through XRD using a Bruker D2 Phaser with CuKα radiation (λ = 1.5417 Å). Rietveld refinement using the XRD data was performed using Topas V4.2 software35 with Na4MnV(PO4)3 as the starting structure.22 High-resolution neutron powder diffraction (NPD) data were acquired using Echidna,36 the high-resolution neutron powder diffractometer at the Australian Nuclear Science and Technology Organization (ANSTO), employing a neutron beam wavelength of 1.62270(6) Å, as determined by the La11B6 NIST standard reference material 660b. Structural refinements were carried out jointly using NPD and XRD data with GSAS-II software,37 where refined parameters include background coefficients, zero shift, lattice parameter, positional parameters, and isotropic atomic displacement parameters. The surface morphology of the synthesized samples was examined using field-emission scanning electron microscopy (SEM) on a Hitachi SU8010 at an acceleration voltage of 15 kV, complemented by spherical-aberration-corrected, high-resolution transmission electron microscopy (TEM) on a JEOL JEM-ARM200FTH at an acceleration voltage of 200 kV. Elemental composition analysis was conducted via inductively coupled plasma-mass spectroscopy (ICP-MS) and energy-dispersive X-ray spectroscopy (EDS) integrated into TEM analysis. The determination of carbon content in the synthesized powder was carried out using thermogravimetric analysis (TGA) with a Jupiter STA 449 F3. Raman spectroscopy, employing a 532 nm laser excitation source on a RAMaker PTT Micro Raman spectrometer, was utilized to characterize the degree of carbonization in the samples. High-resolution X-ray photoelectron spectroscopy (XPS) was carried out on a Versaprobe II ULVAC-PHIPHI 5000 with an Al Kα source to investigate the surface valence state of the prepared samples. In operando synchrotron XRD experiments were conducted at beamlines TPS 19A1 and TLS 01C2 at the National Synchrotron Radiation Research Center, Hsinchu, Taiwan, utilizing an X-ray energy of 20 and 12 keV, respectively. In operando Mn/V/Mo K-edge X-ray absorption near edge spectroscopy (XANES) and Mn/V/Mo/P K-edge extended X-ray absorption fine structure (EXAFS) analyses were carried out at beamlines TLS 17C1, TLS 01C1, and TPS 32A1, at the National Synchrotron Radiation Research Center, Hsinchu, Taiwan.Electrochemical measurements
The cathode was prepared by mixing 70 wt% active material, 20 wt% carbon black (Super P), and 10 wt% polyvinylidene difluoride binder in N-methyl pyrrolidinone (NMP) solvent. The slurry was evenly coated onto aluminium foil using a doctor blade and subsequently dried in an oven at 80 °C for 12 h. The mass loading of the active material reached approximately 1.3 mg cm−2. The electrochemical properties of the electrodes were systematically evaluated in CR2032 button cells manufactured within a high-purity argon-filled glovebox. For half-cell tests, the working electrode served as the cathode, with sodium metal foil acting as the counter and reference electrodes, while a porous glassy fiber (Whatman GF/C) served as the separator. The electrolyte comprised 1 M NaClO4 in propylene carbonate (PC) with 5 vol% fluoroethylene carbonate (FEC). Galvanostatic charge–discharge (GCD) investigations were conducted on a NEWARE battery test system (CT-4008-5V10mA) within an incubator at 30 °C. Furthermore, cyclic voltammetry (CV) was performed at varying scan rates of 0.1, 0.2, 0.5, 1, and 2 mV s−1 with the potential ranging from 2.5 to 3.8 V. The sodium ion diffusion coefficient (DNa+) was calculated utilizing the potentiostatic intermittent titration technique (PITT) and galvanostatic intermittent titration technique (GITT). In PITT measurements, a voltage step of 25 mV was applied with a hold time of 3600 s. Both CV and PITT analyses were executed using a BioLogic potentiostat VSP. The GITT was performed under a constant low current (0.1C), followed by an open-circuit stand for 120 min on a NEWARE battery test system (CT-4008-5V10mA). The setup for in operando XRD and Mn/V/Mo K-edge XANES measurements involved a coin cell with a hole in the bottom case, utilizing carbon paper as the substrate.Computational details
All density functional theory (DFT) calculations were executed using AMS-BAND software (BAND 2023.101).1 The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) function was employed to handle the exchange–correlation interaction.2 Intrinsic system realizations were conducted using a TZP basis set for all elements. Conversely, the single-point calculations of Mo-doped systems utilized a DZP basis for main group elements (i.e. Na, O, and P) and TZP for heavy metals (i.e. Mn, V, and Mo). Frozen core approximations were applied to optimize computational efficiency.3 Structural optimization convergence thresholds were set at 0.0005 eV for total energy and 0.05 eV Å−1 for the force acting on atoms, with a self-consistent cycle convergence criterion of 5 × 10−4 eV. Relativistic effects were addressed using the zeroth-order regular approximation formalism at the scalar relativistic level.4 For accurate electronic property calculations of the NMVP structure, the DFT + U correction was incorporated to account for the strongly correlated electronic nature of d-electrons in transition metals. The DFT + U correction was applied for the d electrons of Mn, V, and Mo atoms with the effective U values of 0.147, 0.11, and 0.26 a.u., respectively.5,6 All calculations employed dense k-point meshes of 3 × 3 × 1.In constructing the unit cell of the NMVP crystal structure, six primitive cells were utilized. To identify Mo atom sites, a total of 30 site configurations were generated. The stability of Mo-doped NMVP was assessed using the formation energy Ef, defined as follows:
| Ef = Edoped − Eintrinsic + Edopand atom − Esubstituent atom. |
Results and discussion
Material characterization of Mo-doped Na4MnV(PO4)3
Diverging from X-rays, neutrons participate in interactions with atomic nuclei, endowing neutron scattering lengths with independence from elemental atomic numbers. The amalgamation of X-ray and neutron powder diffraction analysis yields a comprehensive and accurate crystallographic elucidation. In this study, both neutron powder diffraction (NPD) and X-ray diffraction (XRD) techniques were employed to characterize the crystal structures of NASICON-typed Mo-doped NMVP (precursor molar ratio of Mo to NMVP = 0, 0.02, 0.03, 0.04, denoted as MnV, Mo0.02, Mo0.03, and Mo0.04, respectively) cathode compounds. As depicted in Fig. 1a and S1,† all samples exhibit a high-purity rhombohedral symmetry NASICON-type structured single phase with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c. The findings confirm that a low concentration of Mo6+ does not induce alterations in the crystal structure, maintaining conformity with the NMVP crystal structure.22
c. The findings confirm that a low concentration of Mo6+ does not induce alterations in the crystal structure, maintaining conformity with the NMVP crystal structure.22
 | ||
| Fig. 1 (a) XRD data of NASICON-type structured Mo-doped NMVP (precursor molar ratio of Mo to NMVP = 0, 0.02, 0.03, and 0.04). Data are shown with the offset on the y-axis for clarity; (b) Rietveld refinement profile using Mo0.03 XRD data with the calculated Na4MnV(PO4)3 XRD pattern shown below. Corresponding figures of merit include the weighted profile R-factor, Rwp = 7.95%, and the goodness of fit, GOF = 1.70; (c) Rietveld refinement profile using Mo0.03 NPD data with Rwp = 74.18% and GOF = 1.59; (d) refined structural model of Na4MnV(PO4)3; (e) lattice constants and volume of the Mo-doped NMVP (precursor molar ratio of Mo to NMVP = 0, 0.02, 0.03, and 0.04). Lines through the points are a guide to the eye. | ||
Rietveld refinement analyses of MnV and Mo0.03 samples were performed in a joint manner against XRD and NPD simultaneously. The refinement profiles using data for Mo0.03 are presented in Fig. 1b and c, with that for MnV shown in Fig. S1a and d.† Crystallographic details of Mo0.02 and Mo0.04 were obtained using XRD data and the refinement profiles presented in Fig. S1b and c.† The details of the refined structure are tabulated in Tables S1–S6† and the refined structure of NMVP is illustrated in Fig. 1d, showing a three-dimensional framework formed by corner-sharing Mn/VO6 octahedra and PO4 tetrahedra, facilitating rapid sodium ion diffusion.38 Sodium ions occupy two independent sites (Na(1) at 6b sites and Na(2) at 18e sites) with different oxygen coordination.20,39,40 Na(1) has six-fold coordination and the remaining three sodium ions at Na(2) sites have ten-fold coordination. Further, the lower energy of the Na(2)–O bond leads to the redox reaction involving sodium insertion and extraction at Na(2) sites.24,30 The variation of lattice constants a and c and crystal volume with Mo concentration is presented in Fig. 1e, where lattice constant a remains relatively unchanged and lattice constant c contracts, leading to volume reduction from 1501.885(5) Å3 to 1494.552(8) Å3 with increasing Mo content in the formula from x = 0 to 0.03, demonstrating the successful incorporation of Mo into the NMVP structure.16,41
Owing to the limited resolution of NPD and the low concentration of Mo, the crystal structure location of Mo could not be determined from diffraction data alone and was probed using V/Mn/Mo/P K-edge EXAFS of the Mo0.03 material, yielding short-range structural information. The derived lengths from the spectra are 0.2–0.5 Å, shorter than real bond lengths due to phaser factors and energy dependence.42Fig. 2a displays characteristic peaks in V K-edge EXAFS spectra, corresponding to the V–O bond of VO6 units and V–P distances. The peaks in Mn K-edge EXAFS spectra are consistent with the Mn–O bond length in MnO6 units and Mn–P distances. V/Mn K-edge EXAFS spectra confirm that Mo0.03 possesses a NASICON-type structure, where V/Mn bonds with O, forming VO6/MnO6 octahedra. Model fits to EXAFS data, from which V–O and V–P bond lengths of 2.06(2) and 3.61(1) Å, respectively, are derived, are shown in Fig. S2 and Table S7.† Mn–O and Mn–P distances of 2.15(7) and 3.57(9) Å, respectively, are also derived. Fig. 2b shows that the characteristic peaks in P and Mo K-edge EXAFS spectra are similar and correspond to P–O and Mo–O bonds, implying that Mo is located at P sites. The degree of carbonization of MnV, Mo0.02, Mo0.03, and Mo0.04 was investigated using Raman spectroscopy (Fig. S3†). Raman spectra reveal that all samples exhibit two characteristic carbon peaks, the D-band peak at 1350 cm−1 and the G-band peak at 1590 cm−1. The ID/IG ratio (sp3 to sp2 carbon ratio) characterizing the degree of carbonization of MnV, Mo0.02, Mo0.03, and Mo0.04 is 0.975, 0.985, 0.975, and 0.986, respectively. The similar and less than 1 ID/IG ratios for all samples demonstrate that Mo concentration does not significantly impact the degree of carbonization, which is generally high, leading to enhanced electronic conductivity.29,30,43 To determine the carbon content of Mo0.03, TGA was performed and the results are shown in Fig. S4.† The calculated carbon ratio44 is approximately 1.6%, with a clear weight increase after 400 °C due to the oxidation of low valence metal ions (Mn2+ and V3+).16
 | ||
| Fig. 2 (a) V/Mn K-edge and (b) P/Mo K-edge EXAFS spectra of Mo0.03. | ||
SEM, TEM, EDS, and ICP-MS were conducted to investigate particle morphology, size, and elemental composition. Fig. 3a and S5† present the particle morphology and size of MnV, Mo0.02, Mo0.03, and Mo0.04 powders. The particle size of all as-prepared powders is approximately 1–2 µm, with microparticles aggregating into irregularly shaped particles. A typical TEM image of Mo0.03 is shown in Fig. 3b, demonstrating a particle size of approximately 1 µm, consistent with SEM results. EDS of Mo0.03 was conducted and the results are shown in Fig. 3c and S6,† revealing a homogeneous distribution of Na, V, Mn, Mo, P, and O, with a Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V of 0.024(1), an average of 3 selected areas, which is close to the theoretical ratio (0.03). The TEM images and corresponding EDS mapping of MnV, Mo0.02, and Mo0.04 are shown in Fig. S7–S9,† which also exhibit a similar particle size of approximately 1 µm and a homogeneous distribution of Na, V, Mn, Mo, P, and O. ICP-MS indicates the elemental compositions of MnV, Mo0.02, Mo0.03, and Mo0.04 (Table S8†), which are in excellent agreement with nominal compositions. Taken together, these results confirm the successful synthesis of Mo-substituted NMVP using a straightforward sol–gel method. Fig. 3d and e present a typical TEM image and corresponding fast Fourier transform of Mo0.03 powder, respectively. Atomic layer distances of 0.37 and 0.44 nm are clearly identified and correspond to the (1 1 3) and (2
:V of 0.024(1), an average of 3 selected areas, which is close to the theoretical ratio (0.03). The TEM images and corresponding EDS mapping of MnV, Mo0.02, and Mo0.04 are shown in Fig. S7–S9,† which also exhibit a similar particle size of approximately 1 µm and a homogeneous distribution of Na, V, Mn, Mo, P, and O. ICP-MS indicates the elemental compositions of MnV, Mo0.02, Mo0.03, and Mo0.04 (Table S8†), which are in excellent agreement with nominal compositions. Taken together, these results confirm the successful synthesis of Mo-substituted NMVP using a straightforward sol–gel method. Fig. 3d and e present a typical TEM image and corresponding fast Fourier transform of Mo0.03 powder, respectively. Atomic layer distances of 0.37 and 0.44 nm are clearly identified and correspond to the (1 1 3) and (2 ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) planes of the Mo0.03 crystal structure.16 A few-nm carbon layer could be seen, which is known to enhance electronic conductivity.29,30,45 Further analysis of the data identifies interatomic layer distances also corresponding to the (0 0 3) plane, consistent with the determined crystal structure.
0) planes of the Mo0.03 crystal structure.16 A few-nm carbon layer could be seen, which is known to enhance electronic conductivity.29,30,45 Further analysis of the data identifies interatomic layer distances also corresponding to the (0 0 3) plane, consistent with the determined crystal structure.
 | ||
| Fig. 3 Typical (a) SEM image, (b) TEM image with the corresponding (c) elemental mapping obtained using EDS, and (d) TEM image with the corresponding (e) fast Fourier transform of Mo0.03. | ||
To elucidate the valence of Mn, V, and Mo at the surface of Mo0.03, XPS was conducted. Mn 2p XPS spectra, shown in Fig. 4a, reveal two distinct peaks centered at 652.5 and 645.5 eV, along with a broad satellite peak at 643.0 eV, corresponding to Mn2+ 2p1/2, Mn2+ 2p3/2, and the Mn satellite peak, respectively.46 Similarly, V 2p XPS data, shown in Fig. 4b, exhibit two characteristic peaks at 522.6 and 515.7 eV, along with a broad satellite peak at 518.9 eV, corresponding to V3+ 2p1/2, V3+ 2p3/2, and the V satellite peak, respectively.30 Analysis of Mn and V 2p XPS data indicates the existence of bivalent Mn and trivalent V in Mo0.03. Notably, the broad satellite peaks in Mn and V XPS data correspond to paramagnetic metal states.24,47 These are characteristic of transition metals (Cu, Mn, Ni, Co, Fe, and V) and arise from strong electron configuration reactions such as ligand–metal charge transfer in the initial state, producing a final state with an extra electron in the 3d or 4f orbital compared to the initial state.48,49Fig. 4c presents Mo 3d XPS data of the Mo0.03 powder sample, revealing two distinct peaks at 235.0 and 231.9 eV, corresponding to Mo6+ 2d3/2 and Mo6+ 2d5/2, respectively.50,51 These results unequivocally demonstrate the successful introduction of Mo6+ into NMVP via a facile sol–gel method. Additionally, Mn and V 2p XPS data of the MnV powder are shown in Fig. S10.† The extracted binding energy of Mn2+ 2p1/2, Mn2+ 2p3/2, V3+ 2p1/2, and V3+ 2p3/2 in MnV closely aligns with that for Mo0.03, indicating little influence of Mo on the structure. These results conclusively establish that Mo does not substitute for Mn in NMVP, consistent with the findings from EXAFS.
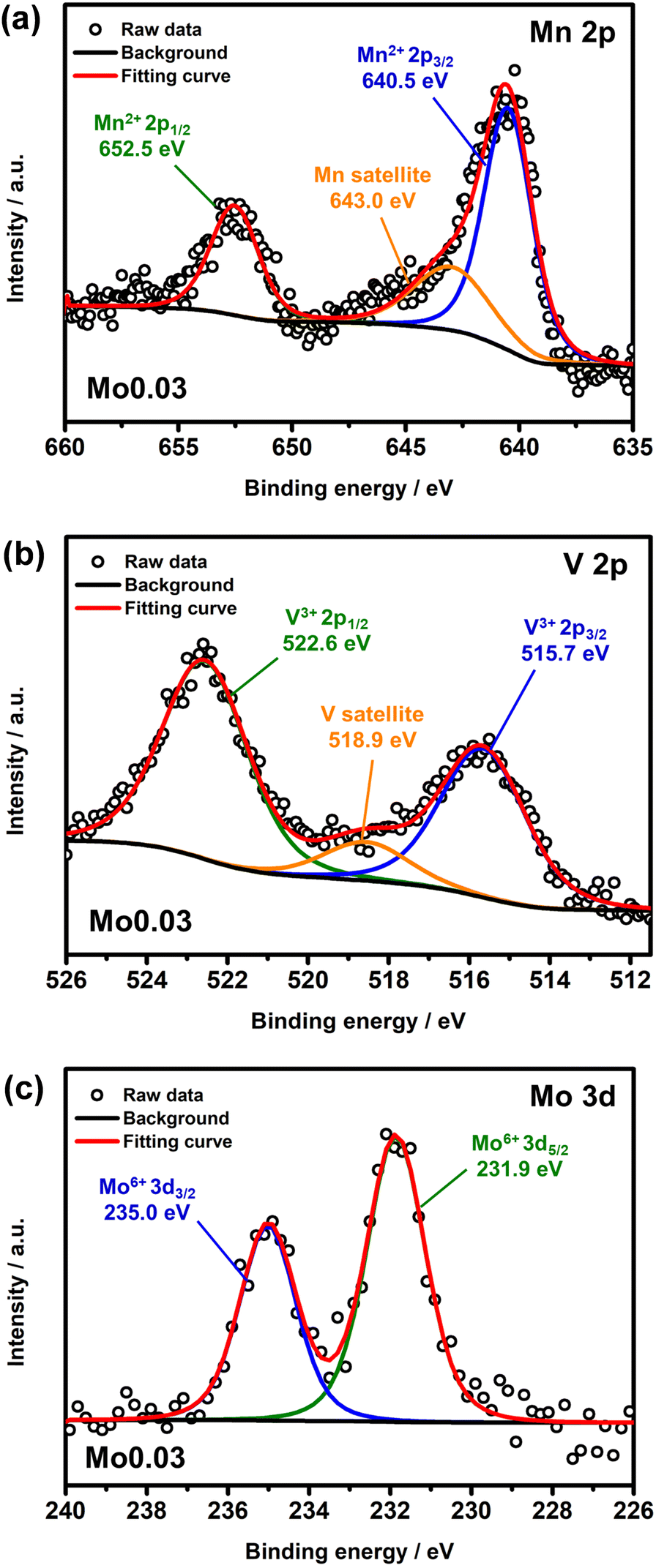 | ||
| Fig. 4 (a) Mn 2p XPS data; (b) V 2p XPS data; (c) Mo 3d XPS data of Mo0.03. Intensity is shown in arbitrary units (a.u.). | ||
DFT calculations
To elucidate the impact of Mo on the electronic properties of NMVP, DFT calculations were conducted. The calculated and experimentally determined lattice constants of pristine Na4MnV(PO4)3 (tabulated in Table S9†) differ by approximately 0.07%, underscoring the reliability of our DFT calculations.7 The electronic structure of the NMVP was investigated using the DFT + U method, with the total and partial density of states (DOS) shown in Fig. S11.† Our calculations suggest that unoccupied states near the Fermi level are predominantly composed of Mn 3d-states, while V 3d-states dominate the conduction band. The resulting band gap of 0.54 eV indicates semi-conductive behavior, consistent with previous reports.6Subsequent analysis focused on evaluating the stability of Mo-containing structures by examining formation energies (Table 1), with the lowest of 1.37 eV corresponding to Mo substituting for P. Conversely, the substitution of Mo for Mn is energetically unfavorable, with a significantly higher formation energy of 4.82 eV. This observation aligns with the energetically unfavorable substitution of Mo for Mn observed in our EXAFS results.
| Formation energy (eV) | Electronic band gap (eV) | |
|---|---|---|
| Na4MnV(PO4)3 | — | 0.54 |
| Mo at P sites | 1.37 | 0.24 |
| Mo at Mn sites | 4.82 | 0.0 |
| Mo at V sites | 2.28 | 0.34 |
To assess the impact of Mo inclusion in NMVP on conductivity, we calculated the total (Fig. 5a) and partial DOS (Fig. 5b and 4c) of the Mo containing NMVP structures. A slight reduction in the band gap was found for all structures with Mo, with gaps of 0.24 and 0.34 eV calculated for Mo at P and V sites, respectively. Remarkably, the substitution of Mo for Mn resulted in conductive behavior, despite the high formation energy suggesting an unfavorable Mo substitution at this site. Further analysis reveals that the unoccupied states of the Mo 3d orbital primarily contribute above 1 eV (Fig. 5b), while the partially occupied states are situated between −5 and 0 eV within the valence band. This suggests that conductivity is not predominantly influenced by Mo 3d states. In contrast, the appearance of new V 3d states near the Fermi level after the incorporation of Mo (Fig. 5c) implies that V 3d states play a crucial role in reducing the band gap when Mo is incorporated into NMVP. More specifically, V 3d states intrude upon the Fermi level in the cases of Mo at P and Mn sites, decreasing the band gap, particularly when Mo is included at P sites. This result is consistent with the EXAFS findings. In summary, our results underscore the interplay between Mo and the electronic structure of NMVP, shedding light on the mechanisms governing conductivity.
 | ||
| Fig. 5 (a) The total DOS for Na4MnV(PO4)3 with different arrangements of Mo. The partial DOS arising from (b) Mo 3d-states and (c) V 3d-states. The Fermi level is set to zero as displayed by the dashed line. | ||
Electrochemical performance of Na4MoxMn1−xV(PO4)3
To explore the impact of Mo inclusion on electrochemical performance, the electrochemical properties of MnV, Mo0.02, Mo0.03, and Mo0.04 cathodes were evaluated in CR2032 coin cells using 1 M NaClO4 in PC with 5 vol% FEC as the electrolyte. The galvanostatic charge/discharge profiles of all samples within the working potential range of 2.5–3.8 V at a current density of 0.2C are shown in Fig. 6a. Notably, all electrodes exhibit two discernible plateaus at 3.4 V and 3.6 V, corresponding to the reversible redox reactions of V3+/V4+ and Mn2+/Mn3+,22,44 respectively. Remarkably, Mo0.03 demonstrates the highest discharge capacity, reaching 94.9 mA h g−1, attributed to enhanced electron conductivity. Furthermore, Mo0.03 exhibits the lowest electronic polarization, indicative of improved electronic conductivity and superior electrochemical performance.29Fig. 6b shows cyclic voltammetry (CV) curves for the Mo0.03 electrode at various scan rates between 2.5 and 3.8 V. The Mo0.03 cathode undergoes two-step reactions during charge/discharge, with two redox pairs, one at 3.32 V corresponding to V3+/V4+ and one at 3.58 V corresponding to Mn2+/Mn3+ with two Na+ insertions/extractions. Oxidation peaks at 3.50 V and 3.67 V correspond to the oxidation of V3+ to V4+ and Mn2+ to Mn3+, respectively, each involving one Na+ extraction. In contrast, reduction peaks at 3.49 V and 3.14 V correspond to the reduction of Mn3+ to Mn2+ and V4+ to V3+, respectively, each involving one Na+ insertion. Furthermore, the current in the CV curve is influenced by both capacitive-limited (k1v) and diffusion-limited (k2v0.5) processes, the quantitative analysis of which is expressed by:29,52,53| i = k1v + k2v1/2 | (1) |
 | ||
| Fig. 6 (a) Galvanostatic charge/discharge profiles of MnV, Mo0.02, Mo0.03, and Mo0.04 cathodes at a current density of 0.2C (1C = 110 mA g−1); (b) cyclic voltammetry of Mo0.03 at different scan rates; (c) rate retention of MnV, Mo0.02, Mo0.03, and Mo0.04; (d) cycle stability of Mo0.03 at a current density of 1C. | ||
Fig. 6c presents the capacities of all samples at various C rates. The initial discharge capacities (and corresponding coulombic efficiencies) of MnV, Mo0.02, Mo0.03, and Mo0.04 at 0.2C are 93 mA h g−1 (91%), 95 mA h g−1 (88%), 98 mA h g−1 (91%), and 84 mA h g−1 (90%), respectively. The relatively low initial coulombic efficiencies can be attributed to side reactions involving the carbon composite, electrolyte decomposition, and the formation of a solid/electrolyte interface film during the first cycle.54 However, in the subsequent cycles, the coulombic efficiencies for all samples stabilize at >99%, indicating excellent cycling stability. The rate retention from 0.2C to 20C for MnV, Mo0.02, Mo0.03, and Mo0.04 is 27.3%, 35.8%, 47.6%, and 51.2%, respectively. The poor rate capability of the NMVP cathode is attributed to its low electronic conductivity. However, the inclusion of even a small amount of Mo6+ significantly enhances electronic conductivity, thereby substantially improving rate retention.50 This result is further supported by electrochemical impedance spectroscopy (EIS), as shown in Fig. S13,† along with the equivalent circuit model.
In the equivalent circuit, Rs represents the bulk resistance of the cell; Rinter and CPE1 correspond to the resistance and capacitance of the interface layer; Rct and CPE2 represent the charge-transfer resistance and double-layer capacitance, respectively. The ion diffusion in the electrode materials is related to the Warburg impedance (Wo), derived from the slope of the diagonal line. The charge-transfer resistances for MnV, Mo0.02, Mo0.03, and Mo0.04 are 4548, 1884, 1304, and 817 Ω, respectively. These results demonstrate that Mo6+ doping effectively reduces charge-transfer resistance, thereby enhancing sodium-ion diffusion kinetics. Consequently, the rate performance of Mo-doped electrodes is significantly better than that of the undoped MnV electrode.
At a low current density (e.g., 0.1C), sodium ions have sufficient time to insert into and extract from the structure. However, excessive Mo6+ ions in the NMVP cathode induce slight lattice distortion, resulting in the MnV electrode exhibiting a higher specific capacity than Mo0.04 under these conditions. Conversely, Mo6+ ion doping improves sodium-ion kinetics, allowing the Mo0.04 electrode to achieve superior rate capabilities at very high current densities (e.g., 10C and 20C). Nevertheless, the performance of Mo0.04 is slightly inferior to that of Mo0.03, possibly due to the rapid decrease in crystal volume (Fig. 1e), which adversely affects sodium-ion diffusion.
Long-term cycling of Mo0.03 (Fig. 6d) reveals that the 93.4 and 91.7 mA h g−1 initial charge and discharge capacities, respectively, have a coulombic efficiency of 98.3% and a capacity retention of 78.8% in the first 300 cycles. The high reversibility of the Na+ insertion/extraction process is attributed to the stability of the structural framework. Compared with other similar studies in the literature,29,32,55 as shown in Table S10,† Mo0.03 exhibits relatively good capacity, rate retention, and cycling stability, demonstrating the practical viability of the proposed strategy for high-performance sodium-ion batteries.
In-depth insights into the valence changes during the charge/discharge process of Mo0.03 electrodes were obtained through in operando V/Mn/Mo K-edge XANES. Fig. 7a presents the normalized V K-edge XANES data of Mo0.03 during the first cycle at a current density of 0.1C, revealing a reversible energy shift. Fig. 7b displays the normalized V K-edge XANES data at different potentials. At the open circuit voltage (OCV), the spectra resemble that of V2O3, indicating that V is close to V3+. At 3.61 V, a clear shift to higher energy occurs, signifying the oxidation from V3+ to V4+. Near the fully charged potential (3.75 V), the spectra resemble that of VO2, indicating V4+. Conversely, during discharge (at 3.32 V), the spectra return to lower energy, confirming the reduction from V4+ to V3+. At the fully discharged potential (2.5 V), the spectra match those at the OCV, indicating the stability of the Mo0.03 electrode structure. The valence variation of V during charge/discharge and the corresponding charge–discharge profile are illustrated in Fig. 7c, demonstrating oxidation of V from V3+ to V4+ at the 3.4 V plateau during charge and a near V3.9+ valence at the 3.6 V plateau. V completely oxidizes to V4+ at the fully charged state (3.8 V), confirming the change during charge from V3+ to V4+. Furthermore, the V remains nearly V4+ until the end of the 3.6 V plateau in the discharge process, indicating that the plateau at 3.6 V does not contribute to the V redox reaction. During the 3.4 V plateau, the V valence in Mo0.03 undergoes significant reduction and reduces to V3+ at a fully discharged state (2.5 V), highlighting a change from V4+ to V3+ during discharge, with the 3.4 V plateau originating from the redox reaction of V. Fig. 7d displays the normalized Mn K-edge XANES data of Mo0.03 in the initial cycle at a current density of 0.1C. The variation in the absorption edge and apparent peak shift suggests a valence change of Mn during the charge/discharge process. Normalized Mn K-edge XANES data at different potentials are presented in Fig. 7e. At OCV, the spectrum resembles that of MnO, indicating Mn2+. During charge (at approximately 3.69 V), there is a significant shift to higher energy and a reduction of the absorption edge, revealing the oxidation of Mn from Mn2+ to Mn3+. At the fully charged state (3.8 V), the spectrum resembles that for Mn2O3, suggesting Mn in Mo0.03 is Mn3+. During discharge (at 3.49 V), the spectrum returns to lower energy and the absorption edge intensity increases, suggesting the reduction of Mn3+ to Mn2+. Finally, at the fully discharged state (at 2.5 V), the spectrum matches that at OCV, indicating the return to Mn2+ and demonstrating complete reversibility of the process. The Mn valence variation and corresponding charge–discharge profile are displayed in Fig. 7f. These results reveal that Mn in Mo0.03 remains approximately in the Mn2.1+ state from OCV to the 3.4 V plateau and significantly oxidizes to Mn2.9+ during the plateau at 3.6 V during charge. The Mn completely oxidizes to Mn3+ at the fully charged potential of 3.8 V, confirming the activity of the Mn2+/Mn3+ redox couple on charge. During discharge, the Mn valency reduces from 3.8 V to the end of the plateau at 3.5 V, indicating that the plateau at 3.5–3.6 V arises from Mn redox activity. The Mn valence remains stable at approximately 2.2+ during the plateau at 3.4 V, ruling out contribution from Mn to the plateau at 3.4 V. The Mn valence reduces to 2+ at the fully discharged state of 2.5 V, as Mn3+ reduces to Mn2+. Our in operando V and Mn K-edge XANES data of the Mo0.03 electrode support the attribution of the redox activity of V to the 3.4 V plateau and the redox activity of Mn to the 3.6 V plateau. To assess whether the Mo dopant in the Mo0.03 electrode remains inactive or undergoes redox reactions during charge/discharge, normalized Mo K-edge XANES was performed during the first cycle at a current density of 0.1C. Fig. 7g reveals a similar absorption edge intensity for both charge and discharge spectra, indicating minimal variation in Mo valence. Further exploration of Mo valence variation during charge/discharge is presented in Fig. 7h. The edge position of the Mo K-edge XANES spectrum of the Mo0.03 electrode at OCV is between those in the spectrum of MoO2 and MoO3, suggesting a mixture of Mo4+ and Mo6+ at OCV. During charge (approximately 3.56 V), a slight shift to higher energy and a significant decrease in the absorption edge intensity indicate Mo oxidation. At the fully charged potential (3.8 V), the spectrum closely resembles that of MoO3, confirming the presence of Mo6+. Conversely, during discharge (at 3.50 V), the edge position in the spectrum returns to lower energy, signifying a reduction of Mo. At the fully discharged potential (2.5 V), the spectrum exhibits an increased absorption edge intensity, resembling the OCV spectrum, indicating the return to a mixture of Mo4+ and Mo6+ and highlighting the reversibility of the Mo redox activity in the Mo0.03 electrode. The variation of Mo valence during charge/discharge and the corresponding GCD profile are illustrated in Fig. 7i. Mo in the Mo0.03 electrode is Mo5.2+, which oxidizes to Mo5.6+ at 3.4 V and remains stable during the first and second plateaus. Complete oxidation to Mo6+ occurs at the fully charged potential (3.8 V). Mo6+ converts to Mo5.8+ just prior to the 3.4 V plateau and remains stable at approximately Mo5.7+ until the end of the plateau, before returning to Mo5.2+ at the fully discharged OCV potential of 2.5 V.
 | ||
| Fig. 7 In operando XANES data of the Mo0.03 electrode in the first cycle at a current density of 0.1C (blue: charge; red: discharge) at the (a) V, (d) Mn, and (g) Mo K-edges. Normalized in operando XANES data of the Mo0.03 electrode at different potentials compared to the spectra of V2O3 and VO2 as identified in the charge/discharge profile shown in the inset at the (b) V, (e) Mn, and (h) Mo K-edges. Valence variation as a function of time shown alongside the corresponding charge/discharge profile for (c) V, (f) Mn, and (i) Mo in the Mo0.03 electrode. | ||
In operando synchrotron XRD investigation of the MnV and Mo0.03 electrodes was performed during charge to 3.8 V and discharge to 2.5 V at a current density of 0.1C. As depicted in Fig. 8a and S14,† the 1 1 3, 2 1 1, 1 1 6, and 0 3 0 reflections of the NASICON structure shift to higher angles during charge, signifying contraction of the unit cell due to Na+ extraction. Importantly, the absence of additional reflections before reaching 3.6 V suggests a single-phase solid solution reaction mechanism involving the V3+/V4+ redox couple. Upon further charging the cell from 3.6 V to 3.8 V, the (113) and (116) reflections disappear, and new peaks emerge at slightly higher angles, indicating a two-phase Mn2+/Mn3+ redox transition within this potential range.16,29,55 At the end of 3.8 V, the Na-poor phase, Na2MnV(PO4)3, is formed.56,57 During discharge, the (113) and (116) peaks reappear, signifying the occurrence of the Mn3+/Mn2+ redox reaction, accompanied by the two-phase transition. The return of reflections to lower angles indicates the expansion of the unit cell during Na+ insertion. The fully discharged XRD pattern matches that at the OCV, confirming the reversibility of the redox reactions in the MnV and Mo0.03 electrodes. Sequential two-phase reactions involving two sites for reversible Na+ insertion/extraction were observed, suggesting that the inclusion of minor amounts of Mo6+ preserves the structural mechanism and enhances conductivity, improving electrochemical performance.58
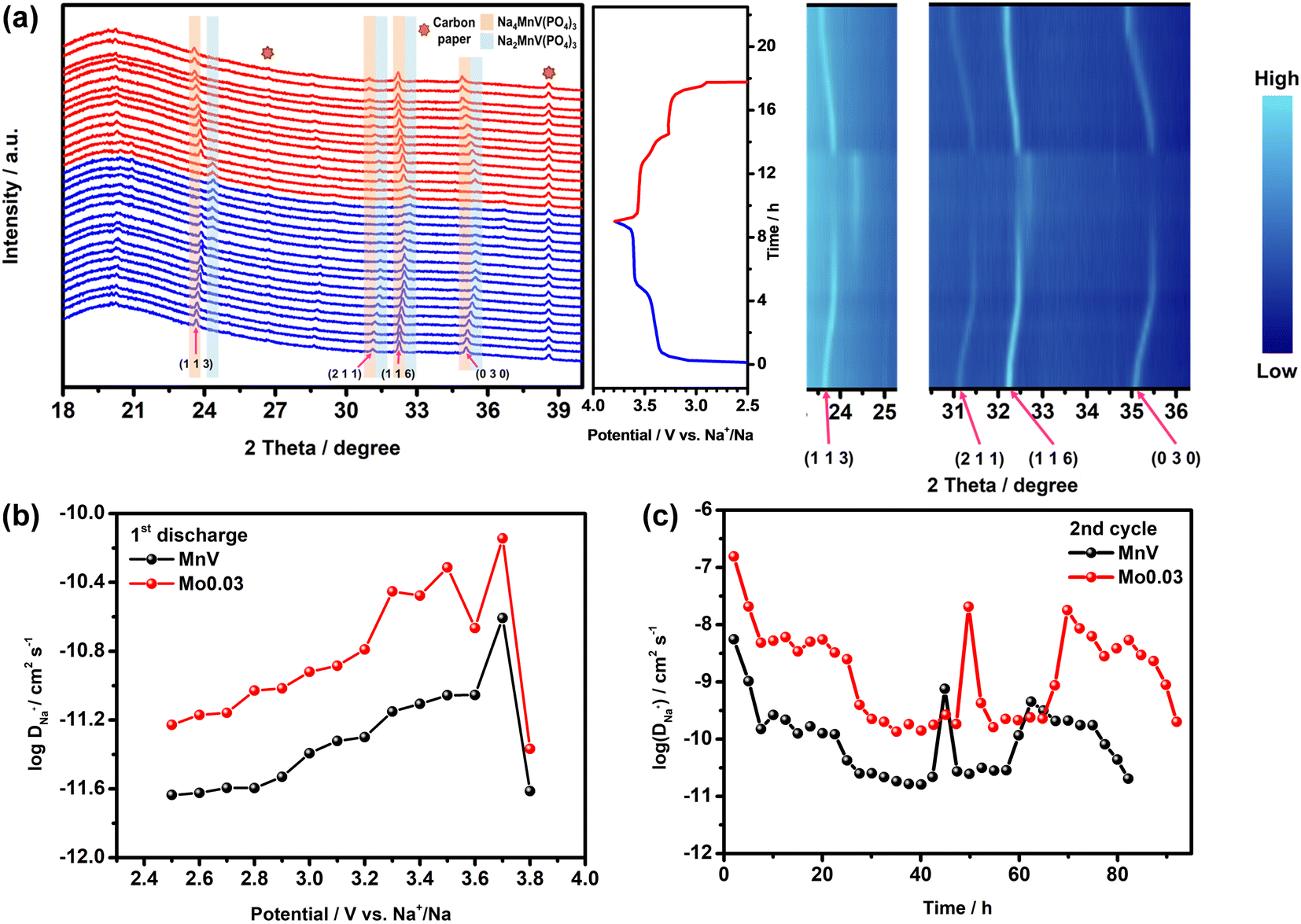 | ||
| Fig. 8 (a) Operando synchrotron XRD pattern and the contour map of the Mo0.03 electrode; (b) PITT results of the 1st discharge for MnV and Mo0.03 electrodes; (c) GITT results of the 2nd cycle for MnV and Mo0.03 cathodes at a current density of 0.1C. | ||
Potentiostatic intermittent titration technique (PITT) and galvanostatic intermittent titration technique (GITT) measurements were used to calculate the sodium ion diffusion coefficient (DNa+). PITT investigations, as shown in Fig. S15a and S16a,† involved a potential step of 25 mV with a hold time of one hour at each step. Plots of ln(I)–t at different potentials for the MnV and Mo0.03 electrodes are presented in Fig. S15b and S16b,† where I is the current and t is the time in seconds. The sodium ion diffusion coefficient (DNa+) is evaluated using:59

 is the slope of the ln(I)–t plot, L is the diffusion length (L = VMnM/S), where VM is the molar volume of the MnV and Mo0.03 cathodes, nM is the molar number of MnV and Mo0.03 composites in the electrode, and S is the area of the electrode. Fig. 8b shows the relationship between log(DNa+) and potential for the MnV and Mo0.03 electrodes obtained from PITT measurements. The diffusion coefficient (DNa+) for the MnV cathode ranges from 2.32 × 10−12 cm2 s−1 to 8.84 × 10−12 cm2 s−1, while for the Mo0.03 cathode, it is estimated to range from 1.63 × 10−11 cm2 s−1 to 7.18 × 10−11 cm2 s−1. This significantly higher DNa+ upon inclusion of Mo6+ in the cathode demonstrates substantial improvement in intrinsic electronic conductivity.
is the slope of the ln(I)–t plot, L is the diffusion length (L = VMnM/S), where VM is the molar volume of the MnV and Mo0.03 cathodes, nM is the molar number of MnV and Mo0.03 composites in the electrode, and S is the area of the electrode. Fig. 8b shows the relationship between log(DNa+) and potential for the MnV and Mo0.03 electrodes obtained from PITT measurements. The diffusion coefficient (DNa+) for the MnV cathode ranges from 2.32 × 10−12 cm2 s−1 to 8.84 × 10−12 cm2 s−1, while for the Mo0.03 cathode, it is estimated to range from 1.63 × 10−11 cm2 s−1 to 7.18 × 10−11 cm2 s−1. This significantly higher DNa+ upon inclusion of Mo6+ in the cathode demonstrates substantial improvement in intrinsic electronic conductivity.
GITT measurements involving a charge time of 30 min and an open-circuit rest of 120 min were performed. Fig. S17a and b† illustrate the second-cycle GITT charge/discharge profile and the corresponding log(DNa+) plots for MnV and Mo0.03 electrodes. DNa+ is calculated using:60

To assess the practical viability of the Mo0.03 cathode in energy storage systems, a comprehensive investigation of full cells was conducted. The full cell configuration comprised a Mo0.03 cathode and a NaMo0.05Ti1.95(PO4)3 anode. Our prior work demonstrated that the inclusion of Mo6+ in NaTi2(PO4)3 enhanced electronic conductivity and improved sodium ion diffusion.62 In this previous work, DNa+ in NaMo0.05Ti1.95(PO4)3 reached 1.43 × 10−7 cm2 s−1, surpassing that of unmodified NaTi2(PO4)3 (5.31 × 10−8 cm2 s−1) in an aqueous sodium ion battery. This enhanced sodium ion diffusion translated to superior rate retention, particularly at high current density, where in an aqueous electrolyte sodium ion battery, NaMo0.05Ti1.95(PO4)3 exhibited a specific capacity of 71.9 mA h g−1 at 10 A g−1, significantly outperforming NaTi2(PO4)3 (51.5 mA h g−1). Leveraging its potential in non-aqueous electrolyte systems, NaMo0.05Ti1.95(PO4)3 was chosen here as the anode material. The rate performance of the NaMo0.05Ti1.95(PO4)3 anode and Mo0.03 cathode (Fig. S18†) at 1C demonstrated a comparable capacity of 82.7 and 85.4 mA h g−1, respectively. Consequently, the mass ratio of the cathode to the anode in all full cells was fixed at ∼0.97. To establish the functional voltage window of the full cell, CV investigations (Fig. S19†) on the NaMo0.05Ti1.95(PO4)3 anode were conducted under the same electrolyte conditions as the Mo0.03 cathode half-test. The stable voltage window of the Mo0.03 cathode//NaMo0.05Ti1.95(PO4)3 anode full cell was determined to be 0.4–2 V. CV data of the Mo0.03 cathode and NaMo0.05Ti1.95(PO4)3 anode full cell, presented in Fig. 9a, exhibited two redox peaks centered at 1.29 and 1.46 V, corresponding to those exhibited for the Mo0.03 cathode and NaMo0.05Ti1.95(PO4)3 anode, respectively. The charge/discharge profiles in Fig. 9b further corroborate these findings, with two clear plateaus at low current density (0.2C), the first arising from V3+/V4+ and the second from Mn2+/Mn3+ and Ti4+/Ti3+ reactions. The full cell exhibited a charge capacity of 89.6 mA h g−1 and a discharge capacity of 85.6 mA h g−1, yielding a coulombic efficiency of approximately 95.5%. At higher current density (5C), the coulombic efficiency remained close to 100%; however, voltage hysteresis increased and plateaus vanished. Rate retention, shown in Fig. 9c, revealed a discharge capacity of 85.6, 77.4, 55.9, and 39.8 mA h g−1 at 0.2C, 1C, 5C, and 10C, respectively. The discharge capacity recovered to 76.1 mA h g−1 when the cell was cycled at a low current density of 1C, demonstrating excellent structural reversibility and rate capability. The Ragone plot in Fig. 9d illustrates promising energy density, with the full cell delivering 72.9 W h kg−1 at a power density of 18.4 W kg−1, 24.3 W h kg−1 at 678.0 W kg−1, and 12.4 W h kg−1 at 2027.6 W kg−1 (based on the total active mass of cathode and anode materials). These results indicate a high energy output even at elevated power density, positioning the Mo0.03 cathode and NaMo0.05Ti1.95(PO4)3 anode full cell as a promising candidate for energy storage applications. The energy density performance is comparable with that of other full cells containing NASICON-type structured electrodes, as shown in Fig. 9d (Na3V2(PO4)3/rGO//NaTi2(PO4)3/rGO;63 Na3V2(PO4)3/C//NaTi2(PO4)3@rGO64). Notably, the Mo0.03 cathode and NaMo0.05Ti1.95(PO4)3 anode are cost-effective, being synthesized through a facile sol–gel method without the need for graphene oxide or other expensive carbon layers, underscoring their potential for deployment in batteries for large-scale energy storage and electric vehicles.
 | ||
| Fig. 9 (a) CV data of the Mo0.03 cathode//NaMo0.05Ti1.95(PO4)3 anode full cell at a scan rate of 0.1 mV s−1; (b) GCD profiles of the Mo0.03 cathode//NaMo0.05Ti1.95(PO4) anode full cell at different current densities (1C = 110 mA g−1); (c) rate retention of the Mo0.03 cathode//NaMo0.05Ti1.95(PO4) anode full cell; (d) Ragone plot of the Mo0.03 cathode//NaMo0.05Ti1.95(PO4) anode full cell and other full cells. Lines through points are a guide to the eye. | ||
Conclusions
In summary, this study demonstrates the significant enhancement of rate retention in cells containing a Na4MnV(PO4)3 cathode by the inclusion of Mo in the crystal structure using a facile sol–gel synthesis method. DFT calculations, supported by experimental EXAFS, reveal the location of Mo at P sites, leading to a reduced band gap and improved electronic conductivity in Mo-doped NMVP structures. The optimized Mo-doped NMVP composite (Mo0.03) displays excellent specific capacities of 97.5 mA h g−1 at 0.2C and 46.4 mA h g−1 at 20C, surpassing those of its pristine counterparts. Mo0.03 possesses remarkable cycle stability, retaining 78.8% capacity after 300 cycles at 1C and excellent rate retention arising from enhanced sodium ion diffusion. In operando XANES at the V/Mn/Mo K-edge and in operando XRD provide evidence of reversible V, Mn, and Mo valence changes, with fully reversible Mo valence being observed for an electrode for the first time. Full cells containing a Mo0.03 cathode and NaMo0.05Ti1.95(PO4)3 anode exhibited an excellent energy density of 72.9 W h kg−1 at 18.4 W kg−1 and 24.3 W h kg−1 at 678.0 W kg−1. This study introduces a promising Mo-substituted cathode material with exceptionally high rate performance and cycle stability, positioning it as a potential candidate for application in large-scale energy storage and electric vehicles.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Cheng-Yen Wu: conceptualization, formal analysis, investigation, validation, and writing – original draft. Chen-En Huang: formal analysis and validation. An-Chi Luo: formal analysis and validation. Wen-Hsuan Lu: formal analysis. Shao-Chu Huang: formal analysis and writing – review & editing. Yu-Ming Huang: formal analysis, software, and writing – original draft. Vanessa K. Peterson: resources and writing – review & editing. Hao-Yu Liu: formal analysis. Chi-Liang Chen: formal analysis. Ying-Rui Lu: formal analysis. Tsung-Yi Chen: formal analysis. Cheng-Zhang Lu: funding acquisition, resources, supervision, and writing – review & editing. Wei Kong Pang: formal analysis, resources, supervision, and writing – review & editing. Han-Yi Chen: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the 2030 Cross-Generation Young Scholars Program of the National Science and Technology Council of Taiwan under Grant No. NSTC 112-2628-E-007-014-MY4. It was also financially supported by the Hierarchical Green-Energy Materials (Hi-GEM) Research Center, from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. The authors thank the Instrumentation Center at the National Tsing Hua University for aiding in XPS, TEM, and XRD analysis. The authors thank Dr Jeng-Lung Chen, Dr Ting-Shan Chan, Dr Yu-Chun Chuang, and Dr Bo-Hao Chen for their help with in operando synchrotron XAS and TXM (beamlines TLS 17C1, TLS 01C1 and TPS 19A1) at the NSRRC, Hsinchu, Taiwan. W. K. P. acknowledges support from the Australian Research Council (ARC) for DP230100198. Part of the work was carried out at the Echidna instrument (beamtime: MI17241) at the Australian Centre for Neutron Scattering.References
- P.-H. Chiang, S.-F. Liu, Y.-H. Hung, H. Tseng, C.-H. Guo and H.-Y. Chen, Energy Fuels, 2020, 34, 7666–7675 CrossRef CAS.
- C.-C. Lin, W.-H. Lin, S.-C. Huang, C.-W. Hu, T.-Y. Chen, C.-T. Hsu, H. Yang, A. Haider, Z. Lin, U. Kortz, U. Stimming and H.-Y. Chen, Adv. Mater. Interfaces, 2018, 5, 1800491 CrossRef.
- Y. M. Chang, H. W. Lin, L. J. Li and H. Y. Chen, Mater. Today Adv., 2020, 6, 100054 CrossRef.
- H.-Y. Chen, N. Bucher, S. Hartung, L. Li, J. Friedl, H.-P. Liou, C.-L. Sun, U. Stimming and M. Srinivasan, Adv. Mater. Interfaces, 2016, 3, 1600357 CrossRef.
- C.-H. Yeh, J.-W. Kang, Y.-L. Chen, H.-J. Chen, H.-H. Chang, W.-H. Lu, S.-Y. Chen, H.-L. Chen, C.-W. Hu, L.-Y. Chueh, Y.-T. F. Pan and H.-Y. Chen, ACS Sustain. Chem. Eng., 2024, 12, 12795–12807 CrossRef CAS.
- C.-C. Lin, H.-Y. Liu, J.-W. Kang, C.-C. Yang, C.-H. Li, H.-Y. T. Chen, S.-C. Huang, C.-S. Ni, Y.-C. Chuang, B.-H. Chen, C.-K. Chang and H.-Y. Chen, Energy Storage Mater., 2022, 51, 159–171 CrossRef.
- T.-Y. Pan, Ruqia, C.-Y. Wu, C.-S. Ni, S. Gull, A. Haider and H.-Y. Chen, Electrochim. Acta, 2022, 427, 140778 CrossRef CAS.
- L. Xiao, F. Ji, J. Zhang, X. Chen and Y. Fang, Small, 2023, 19, 2205732 CrossRef CAS.
- A. Zhao, C. Liu, F. Ji, S. Zhang, H. Fan, W. Ni, Y. Fang, X. Ai, H. Yang and Y. Cao, ACS Energy Lett., 2023, 8, 753–761 CrossRef CAS.
- F. Ji, K. Chen, F. Li, A. Zhao, Y. Fang and Y. Cao, Energy Storage Mater., 2024, 68, 103371 CrossRef.
- Z. Jian, W. Han, X. Lu, H. Yang, Y. S. Hu, J. Zhou, Z. Zhou, J. Li, W. Chen and D. Chen, Adv. Energy Mater., 2013, 3, 156–160 CrossRef CAS.
- K. Saravanan, C. W. Mason, A. Rudola, K. H. Wong and P. Balaya, Adv. Energy Mater., 2013, 3, 444–450 CrossRef CAS.
- Y. Liu, C. Wang, S. Zhao, L. Zhang, K. Zhang, F. Li and J. Chen, Chem. Sci., 2021, 12, 1062–1067 RSC.
- G. Yao, X. Zhang, Y. Yan, J. Zhang, K. Song, J. Shi, L. Mi, J. Zheng, X. Feng and W. Chen, J. Energy Chem., 2020, 50, 387–394 CrossRef.
- S. T. Dacek, W. D. Richards, D. A. Kitchaev and G. Ceder, Chem. Mater., 2016, 28, 5450–5460 CrossRef CAS.
- K. Wang, X. Huang, T. Zhou, D. Sun, H. Wang and Z. Zhang, J. Mater. Chem. A, 2022, 10, 10625–10637 RSC.
- A. Inoishi, Y. Yoshioka, L. Zhao, A. Kitajou and S. Okada, ChemElectroChem, 2017, 4, 2755–2759 CrossRef CAS.
- Y. Chen, Y. Xu, S. He, B. Zhang and L. Guo, Ceram. Int., 2021, 47, 26579–26583 CrossRef CAS.
- J. Lee, S. Park, Y. Park, J. Song, B. Sambandam, V. Mathew, J.-Y. Hwang and J. Kim, Chem. Eng. J., 2021, 422, 130052 CrossRef CAS.
- V. Soundharrajan, S. Nithiananth, K. Sakthiabirami, J. H. Kim, C.-Y. Su and J.-K. Chang, J. Mater. Chem. A, 2022, 10, 1022–1046 RSC.
- D. V. Anishchenko, M. V. Zakharkin, V. A. Nikitina, K. J. Stevenson and E. V. Antipov, Electrochim. Acta, 2020, 354, 136761 CrossRef CAS.
- W. Zhou, L. Xue, X. Lü, H. Gao, Y. Li, S. Xin, G. Fu, Z. Cui, Y. Zhu and J. B. Goodenough, Nano Lett., 2016, 16, 7836–7841 CrossRef CAS PubMed.
- W. Zhang, Z. Zhang, H. Li, D. Wang, T. Wang, X. Sun, J. Zheng and Y. Lai, ACS Appl. Mater. Interfaces, 2019, 11, 35746–35754 CrossRef CAS.
- H. Li, T. Jin, X. Chen, Y. Lai, Z. Zhang, W. Bao and L. Jiao, Adv. Energy Mater., 2018, 8, 1801418 CrossRef.
- P. R. Kumar, A. Kheireddine, U. Nisar, R. Shakoor, R. Essehli, R. Amin and I. Belharouak, J. Power Sources, 2019, 429, 149–155 CrossRef.
- K. Wang, X. Huang, C. Luo, Y. Shen, H. Wang and T. Zhou, J. Colloid Interface Sci., 2023, 642, 705–713 CrossRef CAS PubMed.
- N. Voronina, J. H. Jo, J. U. Choi, C.-H. Jo, J. Kim and S.-T. Myung, J. Mater. Chem. A, 2019, 7, 5748–5759 RSC.
- P. Lavela, R. Klee and J. Tirado, J. Power Sources, 2021, 495, 229811 CrossRef CAS.
- V. Soundharrajan, M. H. Alfaruqi, S. Lee, B. Sambandam, S. Kim, S. Kim, V. Mathew, D. T. Pham, J.-Y. Hwang and Y.-K. Sun, J. Mater. Chem. A, 2020, 8, 12055–12068 RSC.
- C. Xu, J. Zhao, E. Wang, X. Liu, X. Shen, X. Rong, Q. Zheng, G. Ren, N. Zhang and X. Liu, Adv. Energy Mater., 2021, 11, 2100729 CrossRef CAS.
- Q. Shi, R. Qi, X. Feng, J. Wang, Y. Li, Z. Yao, X. Wang, Q. Li, X. Lu, J. Zhang and Y. Zhao, Nat. Commun., 2022, 13, 3205 CrossRef.
- S. Ghosh, N. Barman and P. Senguttuvan, Small, 2020, 16, 2003973 CrossRef CAS.
- J. Hou, M. Hadouchi, L. Sui, J. Liu, M. Tang, W. H. Kan, M. Avdeev, G. Zhong, Y.-K. Liao, Y.-H. Lai, Y.-H. Chu, H.-J. Lin, C.-T. Chen, Z. Hu, Y. Huang and J. Ma, Energy Storage Mater., 2021, 42, 307–316 CrossRef.
- L. Wang, J. Wang, L. Wang, H. Dong, C. Yang, H. Yan, Y. Xiao, Y. Wang, S. Chou and S. Chen, ACS Nano, 2024, 18, 10863–10873 CrossRef CAS.
- B. A. Topas, User's Manual, Bruker AXS, Karlsruhe, Germany, 2008 Search PubMed.
- K.-D. Liss, B. Hunter, M. Hagen, T. Noakes and S. Kennedy, Phys. B, 2006, 385–386, 1010–1012 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- Y. Niu, M. Xu, C. Guo and C. M. Li, J. Colloid Interface Sci., 2016, 474, 88–92 CrossRef CAS PubMed.
- X. Rui, W. Sun, C. Wu, Y. Yu and Q. Yan, Adv. Mater., 2015, 27, 6670–6676 CrossRef CAS.
- Y. Jiang, X. Zhou, D. Li, X. Cheng, F. Liu and Y. Yu, Adv. Energy Mater., 2018, 8, 1800068 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- D. Koningsberger, B. Mojet, G. Van Dorssen and D. Ramaker, Top. Catal., 2000, 10, 143–155 CrossRef CAS.
- X. Cao, A. Pan, B. Yin, G. Fang, Y. Wang, X. Kong, T. Zhu, J. Zhou, G. Cao and S. Liang, Nano Energy, 2019, 60, 312–323 CrossRef CAS.
- J. Peng, Y. Gao, H. Zhang, Z. Liu, W. Zhang, L. Li, Y. Qiao, W. Yang, J. Wang and S. Dou, Angew. Chem., Int. Ed., 2022, 61, e202205867 CrossRef CAS PubMed.
- H. Li, M. Xu, H. Long, J. Zheng, L. Zhang, S. Li, C. Guan, Y. Lai and Z. Zhang, Adv. Sci., 2022, 9, 2202082 CrossRef CAS.
- Y. Zhou, S. Guo, S. Li, Y. Ling, W. Ren, H. Zhou, L. Wang, X. Cao and S. Liang, J. Power Sources, 2022, 521, 230974 CrossRef CAS.
- S. Y. Kim, D. Kundu and L. F. Nazar, Adv. Energy Mater., 2018, 8, 1701729 CrossRef.
- M. C. Biesinger, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- X. Li, Y. Huang, J. Wang, L. Miao, Y. Li, Y. Liu, Y. Qiu, C. Fang, J. Han and Y. Huang, J. Mater. Chem. A, 2018, 6, 1390–1396 RSC.
- L. Liang, X. Li, F. Zhao, J. Zhang, Y. Liu, L. Hou and C. Yuan, Adv. Energy Mater., 2021, 11, 2100287 CrossRef CAS.
- Z. Peng, Q. Wei, S. Tan, P. He, W. Luo, Q. An and L. Mai, Chem. Commun., 2018, 54, 4041–4044 RSC.
- B. Sambandam, V. Soundharrajan, S. Kim, M. H. Alfaruqi, J. Jo, S. Kim, V. Mathew, Y.-k. Sun and J. Kim, J. Mater. Chem. A, 2018, 6, 15530–15539 RSC.
- H. He, D. Sun, Y. Tang, H. Wang and M. Shao, Energy Storage Mater., 2019, 23, 233–251 CrossRef.
- Z. Wang, G. Cui, Q. Zheng, X. Ren, Q. Yang, S. Yuan, X. Bao, C. Shu, Y. Zhang and L. Li, Small, 2023, 2206987 CrossRef CAS.
- M. V. Zakharkin, O. A. Drozhzhin, I. V. Tereshchenko, D. Chernyshov, A. M. Abakumov, E. V. Antipov and K. J. Stevenson, ACS Appl. Energy Mater., 2018, 1, 5842–5846 CrossRef CAS.
- M. V. Zakharkin, O. A. Drozhzhin, I. V. Tereshchenko, A. M. Abakumov, E. V. Antipov and K. J. Stevenson, High Voltage Activation of the Nasicon-Type Na4mnv(PO4)3 Cathode Studied By Operando X-Ray Diffraction, ECS Meeting Abstracts, 2019, vol. MA2019-04(2), p. 89 Search PubMed.
- U. Nisar, R. Shakoor, R. Essehli, R. Amin, B. Orayech, Z. Ahmad, P. R. Kumar, R. Kahraman, S. Al-Qaradawi and A. Soliman, Electrochim. Acta, 2018, 292, 98–106 CrossRef CAS.
- C. J. Wen, B. Boukamp, R. A. Huggins and W. Weppner, J. Electrochem. Soc., 1979, 126, 2258 CrossRef CAS.
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569 CrossRef CAS.
- M. Wang, K. Wang, X. Huang, T. Zhou, H. Xie and Y. Ren, Ceram. Int., 2020, 46, 28490–28498 CrossRef CAS.
- C.-Y. Wu, S.-C. Huang, J. Patra, C.-C. Lin, C.-S. Ni, J.-K. Chang, H.-Y. Chen and C.-Z. Lu, MRS Energy Sustain., 2022, 9, 350–359 CrossRef.
- X. Jiang, Z. Zeng, L. Xiao, X. Ai, H. Yang and Y. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 43733–43738 CrossRef CAS.
- Y. Fang, L. Xiao, J. Qian, Y. Cao, X. Ai, Y. Huang and H. Yang, Adv. Energy Mater., 2016, 6, 1502197 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06069e |
| This journal is © The Royal Society of Chemistry 2025 |