
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiquid crystal–aqueous interface renders selective detection of distinct mycobacterial cell envelope responses†
Tarang Guptaa,
Anjana P Menonb,
Shobhna Kapoor *b and
Santanu Kumar Pal*a
*b and
Santanu Kumar Pal*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Mohali, Knowledge City, Sector-81, SAS Nagar, Mohali 140306, India. E-mail: skpal@iisermohali.ac.in; santanupal.20@gmail.com
bDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: shobhnakapoor@chem.iitb.ac.in
First published on 16th June 2025
Abstract
Antibiotic-resistant latent tuberculosis mandates sensitive, label-free screening platforms that permit the detection of changes in bacterial membranes as a potential therapeutic target. Growth stage–induced alterations in the mycobacterial lipidome, which underlie drug tolerance, further intensify this goal. In this work, liquid crystal (LC)-based detection captured the selective, spatiotemporal modulation of lipid membranes induced by bacterial dormancy. The LC–aqueous interface effectively differentiated responses across the bacterial cell envelope layers. Next, subtle changes in mycobacterial membranes were detected upon interaction with specific antimicrobial peptides (AMPs) at various growth stages. This showcased the potential of the LC–aqueous interface as a label-free membrane screening diagnostic tool. Collectively, the LC–aqueous interface detected membrane order across envelope layers, dormancy-induced lipidome changes, and mycobacterial membrane disruption by AMPs. The distinct behaviour of the LC–aqueous interface in detecting alterations in lipid membranes during different infection stages could be developed in the future to diagnose latent/active pathogens in tuberculosis or other infectious diseases.
Introduction
Antibiotic-resistant Mycobacterium tuberculosis (Mtb) is the corollary of latent tuberculosis.1 The complex lipid composition of Mtb's cell envelope poses a challenge to the effective uptake of antibiotics, but it also presents a unique and tangible drug target and diagnostic avenue.2 Underlying the limited drug permeability are the constituent lipids that form the rigid cell envelope, which are unique to mycobacteria. These include long-chained (C60–C90) lipids such as mycolic acids (MAs), glycolipids such as GPLs (glycopeptidolipids), TDM (trehalose dimycolate) containing MA chains and cyclopropane rings, PDIM (phthiocerol dimycocerosate), and sulfolipids (SLs) with methyl branches and hydroxylated kinks.3 A correlation exists between the mycobacterial lipidome, infection stages, and differential sensitivity to antibiotics.4 This warrants the need for a better understanding of Mtb lipids to foster the development of membrane-centric chemotypes or the modification of existing routes for improved drug uptake.5 For the latter, antimicrobial peptides (AMPs), known for their membrane activity, are ideal candidates.6 Pertinently, the theragnostic potential of mycobacterial membranes requires the development of sensitive, label-free screening systems to detect membrane responses under various conditions.The liquid crystal (LC)–aqueous interface has emerged as a crucial platform for the investigation of diverse atmospheric and biological phenomena. LCs have garnered significant attention due to their unique characteristics, including low surface-anchoring energy, efficient communication properties, and inherent self-responsiveness.7 These features allow for the amplification of minor perturbations within thin films of LCs, which typically have a thickness of only a few micrometres. The high optical anisotropy and birefringence of LCs enable the monitoring of molecular reorientation using polarized optical microscope (POM). Consequently, this technique has been extensively utilized as an indicator of biomolecular binding, membrane transitions, and membrane–protein interactions.8 However, the potential to utilize this approach for investigating bacterial membrane modulations in diagnostic and therapeutic contexts remains uncharted.9
In this work, using an LC-based aqueous interface, we evaluated the conformational differences between the inner and outer membrane envelopes (IML and OML, respectively) of Mycobacterium smegmatis (Msm, a lab model for Mtb)6 in response to distinct growth stages. The distinct conformational attributes of constitutive lipids specific to these layers underlined their interaction with the LC–aqueous interface and, hence, the differential sensitivity. Next, we revealed distinct actions and sensitivities of two different AMPs, aurein 1.2 and maculatin 1.1 (hereafter referred to as aurein and maculatin, respectively), on the IML contingent upon the growth stage. Collectively, this work substantiates the use of LC-based optical detection to (a) correlate LC responses with mycobacterial lipid membrane organization and biophysical properties governed by lipidome differences and (b) decipher the action of AMPs, thereby serving as an efficient screening and diagnostic tool for the identification of novel membrane-centric drugs/approaches. The label-free diagnosis of changes in bacterial membranes induced by late-stage growth or infection could be further explored to detect latent Mtb in sputum samples with minimal sample processing. Lastly, this platform could also help differentiate early drug tolerance in pathogens due to resistance-induced membrane changes.
Results and discussion
The liquid crystal–aqueous interface distinctly detects the alignment of the inner and outer cell membrane (IML and OML) of Mycobacteria as a function of the growth stage
Using the nematic thermotropic liquid crystal, 5CB (4′-pentyl-4-biphenylcarbonitrile), we first probed the interaction of Msm's inner and outer membrane lipids (IMLs and OMLs) with the 5CB–aqueous interface. A thin 5CB film was developed, supported by a gold grid on a substrate coated with dimethyloctadecyl[3-(trimethoxysilyl)propyl]-ammonium chloride (DMOAP) and immersed in a tris buffer solution. As expected, a bright optical appearance of 5CB was observed due to its hybrid orientations at the aqueous interface.7 Then, increasing concentrations of OML and IML liposomes, reconstituted with lipids extracted from Msm grown at two different optical densities (OD600 of 0.8 and 3.0), reflective of early and late growth stages,4 respectively, were introduced. At 0.08 mg mL−1 of IML 3.0, a dark appearance of the 5CB–aqueous interface was observed within 5 min (Fig. 1(a)). In contrast, a completely bright optical appearance of the interface was observed in the presence of OML 3.0 (up to 4 mg mL−1) (Fig. 1(b)). This suggests a differential alignment of IML and OML (extracted at the same OD600 from intact bacteria) at the interface, which could potentially be guided by variations in their lipid composition and conformations.10 | ||
| Fig. 1 Polarized optical images (a)–(d) of the 5CB–aqueous interface captured 10 min after the addition of (a) 0.08 mg mL−1 IML 3.0, (b) 4 mg mL−1 OML 3.0, (c) 2.3 mg mL−1 IML 0.8, and (d) 4 mg mL−1 OML 0.8. Scale bar = 200 μm. The graph in (e) represents the surface pressure (π)–area density Langmuir monolayer isotherms of spatiotemporally controlled extracted lipids from Msm. The graph in (f) indicates the abundance of saturated lipids in the different extracted lipids from Msm (statistically significant with *p = 0.033, **p = 0.002, and ***p < 0.001). | ||
Next, as the growth stage has been shown to remodel the bacterial lipidome in a cell envelope layer-dependent fashion,4b we probed whether the 5CB–aqueous interface could detect such variations. The responses of the LC to OML and IML extracted from two growth stages (early: 0.8 OD600 and late: 3.0 OD600) were investigated. Increasing amounts of IML 0.8 induced a consistent dark optical appearance of 5CB–aqueous interface at 2.3 mg mL−1 (Fig. 1(c)). In contrast, the bright appearance of the 5CB–aqueous interface remained unaltered for OML 0.8, even at increasing concentrations (Fig. 1(d)). IML showed higher sensitivity to alternations in 5CB alignment, even at low concentrations, when extracted from bacteria at the late growth stage. We validated these findings using atomic force microscopy (AFM) imaging, where OML failed to generate a stable solid-supported bilayer, in contrast to IML (Fig. S1, ESI†).
To elucidate the correlation between the molecular configurations of layer-selective lipids and their behaviour at aqueous–5CB interfaces, we fabricated Langmuir monolayers and evaluated their characteristics at air–water interfaces (Fig. 1(e)). We derived four key insights: (i) OML exhibited a lower collapse pressure (CP) compared to IML; (ii) lipids with a lower OD600 displayed a lower CP; (iii) IML experienced a sharp increase in surface pressure at a smaller molecular area compared to OML; and (iv) IML attained a smaller molecular area at the CP compared to OML (Table S1, ESI†). The sharp rise in CP, reduced molecular area at collapse, and higher CP observed for IML, particularly at higher OD600, indicated a consistent perpendicular alignment, densely packed, and well-organized assemblies within the compressed monolayer interface. In summary, a stable arrangement of lipids in the order of IML 3.0 > IML 0.8 > OML 3.0 > OML 0.8 was observed. We then correlated the distinct 5CB responsiveness of Msm membrane layers with lipidome profiling. IML 3.0 exhibited the highest abundance of saturated bond content of fatty acyl moieties (Fig. 1(f)), rendering stronger van der Waals and hydrophobic interactions, thus leading to a densely packed bilayer assembly on the 5CB–aqueous interface. However, despite very long acyl chains (Fig. 1(f)), the higher unsaturation in OML lipids at both growth stages led to hydrophobic mismatch, which mitigated a compact/defined lipid layer alignment on the 5CB–aqueous interface (Table S2, ESI†).
Furthermore, the IML assembly remained stable at the 5CB–aqueous interface for up to 2 h and endured washing, indicating irreversible binding at the interface (Fig. S2, ESI†). These findings reveal that variations in lipid composition with growth and associated conformational properties dictate the interfacial arrangement of lipids in mycobacterial membranes and are acutely discerned via the 5CB–aqueous interface. Most importantly, the bacterial lipidome changed during early and late infection stages (latent TB),4 which underlines the increased resistance to antibiotics in the latent stage, and are effectively sensed by the LC platform. This highlights the practical usage of the LC–aqueous interface for screening drug-resistant bacterial strains in a time- and cost-effective manner. It further showcases, for the first time, the scope of LC-based sensors as a label-free diagnostic tool for latent TB.
Antimicrobial peptide-induced membrane modulation is sensed by the LC–aqueous interface
Given the sensitivity of the 5CB–aqueous interface to detect changes in the Msm membranes, which belong to compositionally and spatially distinct layers of the mycobacterial envelope and vary with growth stage, we aimed to detect antimicrobial peptide–induced membrane modulation, if any, in the Msm IML system. Aurein and maculatin, derived from Australian frog skin (Fig. S3a and b, ESI†), are recognized for their interactions with other bacterial membranes and were investigated on the Msm IML-laden 5CB–aqueous interface.11 Bright optical domains were observed with 2–8 μM aurein over time (Fig. 2(a)), appearing at specific time points and persisting for up to 20 min. Subsequently, upon increasing the aurein concentration, we observed initial modest domain formation immediately after addition. However, at 5 min, a distinct secondary response characterized by completely bright optical features was observed. Remarkably, at 10 min, we noticed that the appearance returned to the dark state due to the homeotropic alignment of 5CB. This transition occurred more rapidly at high aurein concentrations, i.e., 20 μM aurein. Furthermore, this homeotropic alignment appeared considerably darker than the previous alignment observed with lipid anchoring alone. We hypothesize that the binding of aurein to the IML lipid-laden 5CB–aqueous results in tilting and removal of the upper lipid-laden 5CB layer. The tilting and exfoliation of the top layer would expose the 5CB to the solution, causing a bright appearance. However, this brightness soon reverted due to lipid rearrangement and the influence of DMOAP on the thinned 5CB films.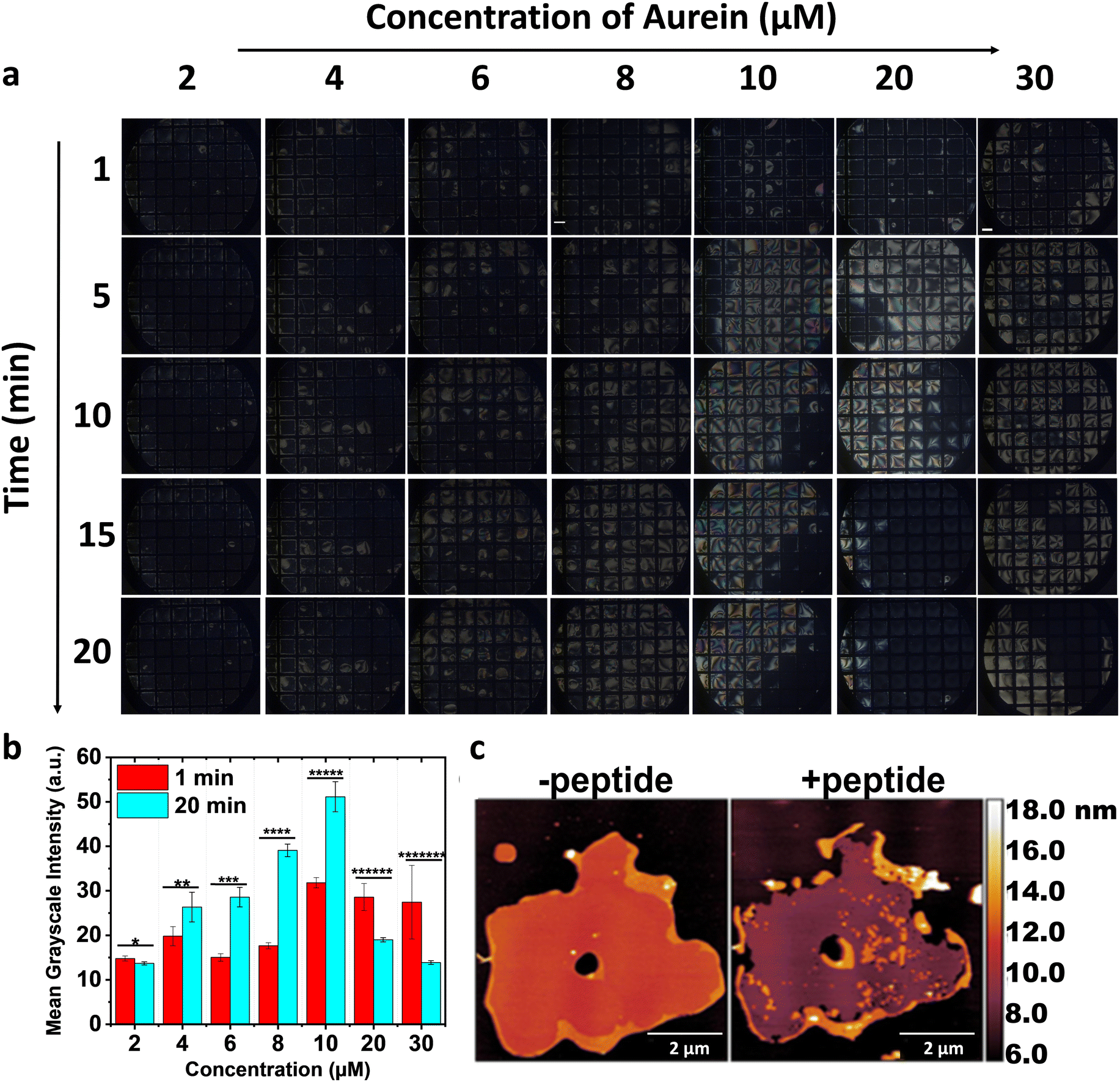 | ||
| Fig. 2 Polarized optical images in (a) depict the responses of IML 3.0-laden 5CB–aqueous interface in the presence of 2–30 μM aurein. The images illustrate the domain formation up to 6 μM aurein. In contrast, a uniform brightening of the optical images is noticed at high concentrations (10–30 μM aurein) at 5 min, which reverts to dark at 10 and 20 μM aurein. The removal of 5CB is observed at 30 μM aurein. Scale bar = 200 μm. The bar graph in (b) depicts the mean grayscale intensities (four grid squares) at 1 and 20 min after adding aurein. Statistically significant with *p = 0.02, **p = 0.02, ***p = 2 × 10−5, ****p = 1 × 10−7, *****p = 3 × 10−5, ******p = 8 × 10−4, and *******p = 0.002. AFM images in (c) represent the solid-supported bilayer of inner membrane lipids (IML 3.0) on activated mica before and after adding aurein. The image demonstrates the removal of IML 3.0 in the presence of aurein. Scale bar = 2 μm. | ||
To confirm this, we increased the aurein concentration to 30 μM and observed a noticeable removal of 5CB from the grid squares. To quantify these 5CB–aqueous interface responses, we calculated the average mean grayscale intensity after 1- and 20-min incubations with aurein (Fig. 2(b)). Increased intensities were observed for IML 3.0-laden 5CB–aqueous interface in the presence of 4–10 μM aurein up to 20 min, in contrast to high concentrations, i.e., 20–30 μM (Fig. 2(b)). These findings indicate that aurein interacted with IML 3.0, and at high peptide–lipid ratios, the interaction induced a tilt in the membrane structure, reorienting 5CB and subsequently causing lipid removal from the interface. This was orthogonally validated using AFM images of IML 3.0 solid-supported bilayers (Fig. 2(c)), wherein aurein caused lipid membrane removal. Surprisingly, no such response was observed with IML 3.0 and 5CB in the presence of maculatin (Fig. 3(a)). However, upon increasing the AMP concentration, faint, short-lived bright domains originating from the grid walls were observed, and these persisted for ∼3–4 min (Fig. S4, ESI†). Upon further increasing the maculatin's concentration, a secondary response was observed, characterized by the emergence of small dots that gradually evolved into branched domains (Fig. 3(a) and (b)), which intensified with time. An increase in average mean grayscale intensities of the IML 3.0-laden 5CB–aqueous interfaces from 1 to 20 min, upon increasing the concentration of maculatin, is shown in Fig. 3(c). Notably, this effect with maculatin occurred at much higher concentrations than with aurein. This implies that aurein is significantly more effective at infiltrating the lipid domains in the bacterial membrane, underlying its enhanced penetration and interaction. The 5CB–aqueous interface responses observed at variable concentrations of AMPs indicate that a specific threshold concentration is essential to achieve disruption of the bacterial membrane; otherwise, recovery occurs.
 | ||
| Fig. 3 Polarized optical images in (a) depict the responses of IML 3.0-laden 5CB–aqueous interface in the presence of 2–200 μM maculatin. Scale bar = 200 μm. The magnified optical images in (b) represent the responses of the IML 3.0-laden 5CB–aqueous interface in the presence of maculatin under crossed polars and without the analyzer. Scale bar = 100 μm. The double-headed arrows at the top-right corner of each image in (b) show the relative orientation of the polarizers. The graph in (c) illustrates the mean grayscale intensities (four grid squares) at 1- and 20-min incubation of maculatin with the IML 3.0-laden 5CB–aqueous interfaces. | ||
Next, the impact of AMPs on early growth IML systems was studied. With IML 0.8, the responses of 5CB–aqueous interface were similar to those observed with IML 3.0 (Fig. S5 and S6, ESI†), but less pronounced and required higher concentrations of AMPs. Thus, the interaction of AMPs with mycobacterial membranes is growth stage–specific. As a control, we saw no changes in the 5CB–aqueous interface when 500 μM AMPs were added to 5CB–aqueous interface without lipids (Fig. S7, ESI†). This fortifies the idea that the observed response of 5CB–aqueous interface arises from interactions between AMPs and the lipids at the interface.
Next, to compare the action of AMPs, the equilibrium responses of the IML-laden 5CB–aqueous interface at high and equal concentrations (i.e., 300 μM of AMPs) were studied. Aurein with IML 3.0 showed a vivid optical appearance, but in most grid squares, 5CB was still concealed (Fig. S8a, ESI†).
In contrast, with maculatin, a consistently bright optical appearance was observed (Fig. S8b, ESI†). Moreover, the observed texture of 5CB in both cases was distinct. These intriguing observations prompted the question: why do such distinct 5CB–aqueous interface responses emerge? This curiosity led us to explore whether these differences are also reflected in the interactions of AMPs with IML 0.8. Upon exposing AMPs to IML 0.8-laden 5CB–aqueous interface, the ensuing responses resembled those observed with IML 3.0 (Fig. S8c and d, ESI†) but were less pronounced than those observed with IML 3.0-laden 5CB–aqueous interface, as noted earlier. We attribute these differences to lipidome variations and abundances that govern the differential interaction of AMPs (Fig. S9, ESI†) with the IML layers. For example, a 2-fold increase in cardiolipin levels (net charge: −2), a prominent constituent of the IML, was observed during the late growth stage. This underlines the effective electrostatic interactions between peptides and IML 3.0.
LC–aqueous interface subtly captures the AMP–membrane interaction mechanisms
To evaluate changes in AMP conformation upon interaction with IML, circular dichroism (CD) studies were conducted. AMPs exhibited different conformations in solution (Fig. S10, ESI†). While aurein displayed a maximal negative band at ∼208 nm, maculatin exhibited a peak at ∼225 nm at the IML 3.0/0.8 vesicles (Fig. 4(a) and (b)). Secondary structure analysis (Fig. 4(c)) revealed a predominance of α-helical structure in aurein and β-sheet conformation in maculatin, with substantial modulation of these structures upon interaction with IML 0.8 and 3.0. This acquisition of different conformations of AMPs with IML is attributed to lipidome differences that guide lipid–peptide interactions. Subsequently, we investigated the impact of electrostatics on the interplay between AMPs and IML (Fig. 4(d)). The significantly higher zeta potential of IML 3.0-laden 5CB droplets, when exposed to cationic AMPs, validates higher IML 3.0-laden 5CB–aqueous interface responses. This is attributed to the increased abundance of cardiolipin in IML (Fig. S9, ESI†). Collectively, the 5CB–aqueous interfaces enhanced the varied responses of AMPs, contingent upon their conformations and secondary structure composition, when exposed to IML lipids from Mycobacteria. | ||
| Fig. 4 Circular dichroism (CD) spectra in (a) and (b) illustrate the conformational changes of aurein and maculatin upon interaction with IML 3.0 and IML 0.8, respectively. The histogram in (c) represents the percentage (%) of secondary structure contents of AMPs upon exposure to IML 3.0 and 0.8. The bar graph in (d) displays the variations in the zeta potential of the lipids in the presence of AMPs. | ||
Next, the lateral distribution of IML and AMPs at the aqueous–5CB interfaces was explored by doping FITC-labelled aurein and rhodamine DHPE into the IML 3.0-laden 5CB–aqueous interface. Upon introducing FITC-labelled aurein, the initially uniform homeotropic alignment of the 5CB–IML interface was disrupted, forming a distinct bright domain in the polarized field (Fig. S11a, ESI†), which exhibited green fluorescence (Fig. S11b–d, ESI†), indicative of AMP-rich regions. To investigate the potential aggregation of AMPs at the interface, we subjected the IML 3.0-laden 5CB–aqueous interface to a drying process. The dried interface revealed a uniform dark optical appearance of 5CB in the presence of aurein (Fig. S12a and c, ESI†). Conversely, bright domains were observed with maculatin, indicating potential aggregation (Fig. S12b and d, ESI†). The aggregation of maculatin was confirmed via AFM (Fig. S12e and f, ESI†), wherein lipid membrane disruption due to aggregation was observed.
Overall, the interaction of the two AMPs with IML-laden 5CB–aqueous interface revealed several significant findings. Aurein induced a bright optical appearance without discernible domains, eventually expelling IML lipids from the interface. This suggests a detergent-like mechanism akin to the carpet model of AMP action.12 In contrast, maculatin induced the emergence of branched dendritic domains, indicating a β-sheet-rich conformation in contact with IML at the interface.13 The observation of aggregation at the interface suggests the involvement of β-sheet strands and multimeric structures in membrane disintegration, resembling the barrel-stave model.12 The final noteworthy observation is the diminished responses of 5CB–aqueous interface with IML 0.8 compared to IML 3.0 in the presence of AMPs. This may be explained by lipid density fluctuations and reduced electrostatic charges, accounting for the attenuated influence of AMPs during the initial phase of Mycobacterium growth.
Methods
Piranha cleaning and coating of glass slides
Following established protocols, the glass slides were meticulously cleaned using piranha solution (comprising H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2 in a volumetric ratio of 70:30).14 Briefly, it is mentioned below for reader's convenience. Freshly prepared piranha solution was applied to the glass slides within a glass container, which were then immersed in a water bath set at 80 °C for 1 h. After this treatment, the slides were subjected to sequential rinses with ethanol and deionized water. A thorough drying process was then carried out using nitrogen gas purging, followed by an incubation period within an oven maintained at 100 °C for a minimum of 3 h.
:H2O2 in a volumetric ratio of 70:30).14 Briefly, it is mentioned below for reader's convenience. Freshly prepared piranha solution was applied to the glass slides within a glass container, which were then immersed in a water bath set at 80 °C for 1 h. After this treatment, the slides were subjected to sequential rinses with ethanol and deionized water. A thorough drying process was then carried out using nitrogen gas purging, followed by an incubation period within an oven maintained at 100 °C for a minimum of 3 h.
To impart hydrophobic properties to the glass slides, they were immersed in a 0.1% (v/v) aqueous solution of DMOAP at room temperature for 30 min as reported earlier.8i Subsequently, a meticulous washing procedure was employed to remove excess DMOAP before subjecting the slides to nitrogen drying. Furthermore, these slides underwent an additional heating process in the oven for 3 to 6 h to facilitate cross-linking and the formation of siloxane bonds.
The chemical modification of the glass slide surface with DMOAP resulted in the establishment of hydrophobic interactions between the octadecyl chains of DMOAP and the alkyl tails of 5CB molecules. This modification encouraged the perpendicular alignment of 5CB molecules with respect to the substrate surface.
Culturing of Mycobacterium smegmatis
Mycobacterium smegmatis MC2155 (Msm) was cultured following established protocols,4b,c with a brief description provided for convenience. Msm was cultured at 37 °C with agitation (120 rpm) in Middlebrook 7H9 broth supplemented with 10% (v/v) in-house prepared ADC (albumin-dextrose-sodium chloride-catalase), 0.5% glycerol, and 0.1% tyloxapol. Log-phase cells (OD600 ≈ 0.8) were harvested under the above conditions.15 For the early stationary phase, cells were grown without tyloxapol to ∼OD600 3.0, representing nutrient-deprived, aggregated populations.17 All harvested cells were washed twice with phosphate buffered saline (PBS; pH 7.4) before lipid extraction.Extraction of lipids
Lipid extraction was performed as previously reported,18 with a summary provided. Outer leaflet lipids were isolated by treating cells with 10 mM AOT (sulfosuccinic acid 1,4-bis (2-ethylhexyl) ester sodium salt) in n-heptane (1 mL per 10 mg dry weight). RMS-treated cells were washed with water, and inner membrane lipids were extracted using chloroform:methanol:water (2:1:0.1, v/v/v; 3 mL per 10 mg biomass) under monophasic conditions: an overnight extraction followed by three 30 min extractions. Extracts were pooled, dried under reduced pressure, and lyophilized.
Residual AOT was removed from outer leaflet extracts via alumina column chromatography (chloroform:methanol gradient, up to 8%), and lipid purity was confirmed by TLC with 1% anthrone spray.19
Preparation of vesicles of inner and outer membrane lipid
The preparation of vesicles was conducted using a previously established method.16 It is mentioned below for reader's convenience. In brief, a predetermined quantity of lipid was measured and introduced into a round-bottom flask, suspended in chloroform. Subsequently, the required quantity of lipid was subjected to a drying process for 3 h at room temperature under vacuum conditions. Following this, the dried lipid was rehydrated with Tris buffer containing 5 mM of magnesium chloride (MgCl2) for 3 h period. Freeze–thaw cycles were then implemented to facilitate the formation of lipid vesicles. Small unilamellar vesicles were generated through probe sonication of the aqueous lipid dispersion, which lasted for 30 min. Notably, all prepared lipid solutions were utilized within 24 h.Preparation of LC–aqueous interface
The LC–aqueous interface was established following protocols previously detailed in the literature.17 It is mentioned below for reader's convenience. In brief, glass slides coated with DMOAP were fragmented into small fragments and subsequently overlaid with gold grids. Subsequently, approximately 0.2 μL of 5CB was dispensed onto the grids, and any excess 5CB was removed via aspiration, ensuring a uniform LC film was formed. This assembled system was then positioned within an optical well, wherein a 10 mM buffer solution was introduced to generate the LC–aqueous interface.Preparation and optical characterization of lipid-laden-LC-filled specimen grid
The LC–aqueous interface was enveloped within lipid layers comprised of synthesised lipid vesicles and subsequently allowed to incubate.8i Following the lipid encapsulation, the buffer solution was substituted with various concentrations of the AMPs of interest. Subsequently, the optical response of the LC system was examined using a POM to gain insights into the interactions between the protein and the lipid-rich LC–aqueous interface. To maintain the alignment of the lipid layer and avoid turbulence, the AMPs were gently introduced from the walls of the optical well. As documented in earlier publications,18 optical characterization procedures have been outlined. In summary, optical characterization involved the use of a Zeiss Scope.A1 polarizing optical microscope in transmission mode to observe the optical behaviour of the LC system. Each image was meticulously focused and acquired using a Q-imaging digital camera, which was integrated with the POM and set with an exposure time of 80 milliseconds.18The LC sample, enclosed within an optical well, was positioned on a rotating platform. The orientation of the LC material was assessed by ensuring the orthogonal alignment of the polarizer and analyzer. During each experimental trial, images were acquired while maintaining a source intensity level approximately 40% of the maximum brightness setting.
Circular dichroism measurements
Far-UV circular dichroism (CD) studies were conducted using a Chirascan spectrophotometer manufactured by Applied Photophysics in the United Kingdom. The instrument possessed a scan range spanning from 190 to 280 nm, with a step size of 1 nm. The solution under investigation was placed within a quartz cell featuring a 1 mm path length quartz cell. Freshly prepared aqueous solutions containing 10 μM concentrations of aurein 1.2 and maculatin 1.1 were employed for the experiments. These solutions were prepared in a 10 mM Tris buffer at a pH of 7.4. To investigate conformational alterations in AMPs during their interaction with IML, a solution containing 10 μM of AMPs was subjected to a 1 h incubation period with lipids at a concentration of 1 mg mL−1.Data acquisition and analysis were performed using the ProData software provided with the CD instrument. Each spectrum was generated by averaging the results of 5 scans and subsequently adjusted against the buffer signal for accurate analysis. The data pertaining to the percentage secondary structure content of AMPs was processed using the BestSel software.
LC droplet preparation and zeta potential measurements
To investigate variations in zeta potential at the surface of the LC, we prepared LC droplets18 through the addition of 1 μl of 5CB to either 100 μl of DI water or a lipid solution at a concentration of 1 mg mL−1. The resulting dispersions were prepared via vortex mixing for 1 min at a rate of 3000 revolutions per minute (rpm), followed by 1-min emulsification using a homogenizer. In the context of the AMP studies, we introduced 10 μL of the AMP stock solution into a mixture containing 40 μL of the lipid–5CB droplet emulsion, thus achieving a final AMP concentration of 10 μM. After a 15-min incubation period, the solution was diluted to a final volume of 800 μL with DI water. The measurement of zeta potential under these experimental conditions was performed using the Zetasizer Nano ZS90 instrument manufactured by Malvern Instruments Inc., at room temperature, applying a cell-driven voltage of 30 V.FITC-labelling of Aurein 1.2 AMP
To fluorescently label Aurein 1.2 with FITC, a solution of 1 mg mL−1 FITC was prepared by dissolving FITC in dimethyl sulfoxide (DMSO). Subsequently, 100 μL of this FITC solution was mixed with 5 mg mL−1 of Aurein 1.2. The resulting mixture was then placed in a dark environment at a temperature of 4 °C for 5 to 6 h. Following this incubation period, the ongoing reaction of labeling was halted by adding 50 mM of ammonium chloride, and the mixture was allowed to react for an additional 2 h. Subsequently, the reaction mixture was passed through a Sephadex G-25 column (gel filtration), and the formed conjugate was purified by elution through the column using a 10 mM Tris buffer (pH = 7.4). The eluted samples were collected in separate vials for further analysis. UV absorption spectra were recorded for the collected samples, revealing absorption maxima at 280 nm for the FITC–Aurein conjugate and 495 nm for FITC alone. The concentration of the protein was determined based on the absorbance value at 280 nm.Confocal microscope imaging
Confocal microscopic imaging was employed to visualize the localization of lipid-doped components, specifically utilizing a 2.5% concentration of rhodamine DHPE fluorescent dye. To prepare the samples, a mixture of lipids consisting of IML 3.0 and the doped DHPE fluorescent dye was created. This lipid mixture was dissolved in high-performance liquid chromatography (HPLC) chloroform to generate vesicles. These vesicles were subsequently combined with the liquid crystal (LC)–aqueous interface. For imaging purposes, a specialized setup was employed, which included a 63× long-distance water-immersed objective equipped with hybrid detectors. The image acquisition settings were configured to capture images at a resolution of 512 × 512 pixels, with a scanning rate of 400 Hz. A diode-pumped solid-state (DPSS) laser source emitting light at a wavelength of 561 nm was utilized to excite the rhodamine DHPE fluorescent dye. Confocal image stacks were acquired using a photomultiplier tube (PMT) detector within an SP8 upright confocal microscope system. This approach allowed for the precise visualization and examination of the lipid-doped localization within the samples.Liquid chromatography and mass spectrometry
Lipid profiling was performed using an Agilent 1260 High-Performance Liquid Chromatography (HPLC) system (Agilent Technologies, Palo Alto, CA) coupled to an Agilent AdvanceBio 6545XT Q-TOF mass spectrometer equipped with an electrospray ionization (ESI)/APCI multimode ion source. Chromatographic separation was achieved using a 2.1 mm × 150 mm, 3.5 μm XBridge C18 column (Waters Corp., Milford, MA). HPLC and MS parameters were maintained as previously described.4b,19 Feature lists were exported as analysis reports and subsequently analyzed to determine mole fractions for quantitative lipid profiling.Statistics
All results are expressed as the mean ± standard error of the mean (SEM) for the indicated number of experiments (n = 3). Statistical analyses were performed using GraphPad Prism 10.2.0 (GraphPad Software Inc.) and are presented in the figure legends. Differences were considered significant if p < 0.05.Conclusions
In summary, our research highlights the LC-based detection of Mycobacteria's ability to dynamically alter their lipidome, resulting in variable membrane composition and charge distributions across different growth stages. These lipid adaptations are crucial for their resilience in adverse conditions and drug resistance. Notably, the OML consistently displayed lower organization than the IML across all growth stages. Furthermore, this work provides insights into how LC-based detection offers an exploration of AMP's distinct mechanisms in disrupting bacterial membranes. Interestingly, variations in lipid composition and charge modulation during early bacterial growth mitigate the impact of AMPs, as ascertained by the LC responses. Lastly, the findings suggest that combinatorial therapy with aurein or maculatin could synergistically prove more efficient in dismantling bacterial envelopes. This work lays the foundation for developing a robust label-free platform to investigate the specific alterations and deficiencies induced in lipid membranes by various conditions, including exposure to antibiotics or membrane-active agents. The LC–aqueous interface could foster screening for drug-resistant bacterial strains in a time- and cost-effective manner, especially in hospital-acquired infections, and serve as a label-free diagnostic tool for latent TB. All of these factors can advance our understanding of bacterial membrane-based therapeutic approaches in a broader context and target other infectious diseases. Taken together, we envision this strategy as a valuable tool for gaining molecular insights into bacterial envelopes and antibiotic potentiators, with potential applications in point-of-care diagnostics and therapeutic interventions.Author contributions
SKP and SK collaborated to conceptualize and devise the framework for the project. APM purified the lipids from Msm, performed lipidomics and AFM experiments, and conducted data analysis. TG executed the remaining experimental procedures and thoroughly analysed the resulting data. All authors contributed to the writing of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. K. P. acknowledges MOE/STARS/284 and SERB-TARE grant (TAR/2021/000146). T. G. thanks CSIR (09/947/(0223)/2019EMR-1) for the fellowship. We have used the PyMOL Molecular Graphics System, Chimera, Protein Data Bank (PDB), and Power Point 3D models for the TOC graphic. This work is part of the PhD thesis of one of the co-authors, T. G., at IISER Mohali (PhD thesis defended: Year 2024).19Notes and references
- R. Singh, S. P. Dwivedi, U. S. Gaharwar, R. Meena, P. Rajamani and T. Prasad, J. Appl. Microbiol., 2020, 128, 1547–1567 CrossRef CAS PubMed.
- (a) D. J. Hawkes and J. Mak, Curr. Drug Targets, 2006, 7, 1615–1621 CrossRef CAS PubMed; (b) M. P. Mingeot-Leclercq and J. L. Décout, Med. Chem. Commun., 2016, 7, 586–611 RSC; (c) Y. P. Subedi, M. N. AlFindee, J. Y. Takemoto and C. T. Chang, Med. Chem. Commun., 2018, 9, 909–919 RSC.
- M. Jackson, G. Stadthagen and B. Gicquel, Tuberculosis, 2007, 87, 78–86 CrossRef CAS PubMed.
- (a) P. J. Brennan and H. Nikaido, Annu. Rev. Biochem., 1995, 64, 29–63 CrossRef CAS PubMed; (b) A. P. Menon, T. H. Lee, M. I. Aguilar and S. Kapoor, Chem. Sci., 2024, 15(45), 19084–19093 RSC; (c) A. P. Menon, W. Dong, T. H. Lee, M. I. Aguilar, M. Duan and S. Kapoor, ACS bio & med Chem Au., 2022, 2(4), 395–408 Search PubMed.
- R. Pal, S. Hameed, P. Kumar, S. Singh and Z. Fatima, 3 Biotechnol., 2017, 7, 1–10 CAS.
- L. R. Pizzolato-Cezar, N. M. Okuda-Shinagawa and M. T. Machini, Front. Microbiol., 2019, 10, 1703 CrossRef PubMed.
- (a) V. K. Gupta, J. J. Skaife, T. B. Dubrovsky and N. L. Abbott, Science, 1998, 279, 2077–2080 CrossRef CAS PubMed; (b) A. M. Lowe and N. L. Abbott, Chem. Mater., 2012, 24, 746–758 CrossRef CAS PubMed; (c) I. H. Lin, D. S. Miller, P. J. Bertics, C. J. Murphy, J. J. de Pablo and N. L. Abbott, Science, 2011, 332, 1297–1300 CrossRef CAS PubMed; (d) P. M. Naveenkumar, S. Mann and K. P. Sharma, Adv. Mater. Interfaces, 2019, 6, 1801593 CrossRef; (e) S. Aery, A. Parry, A. A. Calahorra, S. D. Evans, H. F. Gleeson, A. Dan and A. Sarkar, J. Mater. Chem. C, 2023, 11, 5831–5845 RSC; (f) X. Yang, Y. Tian, F. Li, Q. Yu, S. F. Tan, Y. Chen and Z. Yang, Langmuir, 2019, 35, 2490–2497 CrossRef CAS PubMed; (g) X. Yang, X. Zhao, F. Liu, H. Li, C. X. Zhang and Z. Yang, Soft Matter, 2021, 17, 4842–4847 RSC; (h) X. Yang and Z. Yang, Langmuir, 2022, 38, 282–288 CrossRef CAS PubMed.
- (a) J. S. Park and N. L. Abbott, Adv. Mater., 2008, 20, 1185–1190 CrossRef CAS; (b) K. Perera, T. M. Dassanayake, M. Jeewanthi, N. P. Haputhanthrige, S. D. Huang, E. Kooijman, E. Mann and A. Jákli, Adv. Mater. Interfaces, 2022, 9, 2200891 CrossRef CAS; (c) P. S. Noonan, P. Mohan, A. P. Goodwin and D. K. Schwartz, Adv. Funct. Mater., 2014, 24, 3206–3212 CrossRef CAS PubMed; (d) X. Yang, H. Li, X. Zhao, W. Liao, C. X. Zhang and Z. Yang, Chem. Commun., 2020, 56, 5441–5444 RSC; (e) A. Borbora and U. Manna, Langmuir, 2022, 38, 9221–9228 CrossRef CAS PubMed; (f) X. Yang, X. Zhao, H. Zhao, F. Liu, S. Zhang, C. X. Zhang and Z. Yang, Chem. – Asian J., 2022, 17, e202101251 CrossRef CAS PubMed; (g) U. Manna, Y. M. Zayas-Gonzalez, R. J. Carlton, F. Caruso, N. L. Abbott and D. M. Lynn, Angew. Chem., Int. Ed., 2013, 52, 14011–14015 CrossRef CAS PubMed; (h) L. Yang, M. Khan and S. Y. Park, RSC Adv., 2015, 5, 97264–97271 RSC; (i) T. Gupta, A. K. Mondal, I. Pani, K. Chattopadhyay and S. K. Pal, Soft Matter, 2022, 18, 5293–5301 RSC.
- (a) Q. Z. Hu and C. H. Jang, Analyst, 2012, 137, 567–570 RSC; (b) P. Bao, D. A. Paterson, P. L. Harrison, K. Miller, S. Peyman, J. C. Jones, J. Sandoe, S. D. Evans, R. J. Bushby and H. F. Gleeson, Lab Chip, 2019, 19(6), 1082–1089 RSC; (c) D. Das and S. K. Pal, ChemistrySelect, 2017, 2(17), 4779–4786 CrossRef CAS.
- P. Adhyapak, A. T. Srivatsav, M. Mishra, A. Singh, R. Narayan and S. Kapoor, Biophys. J., 2020, 118, 1279–1291 CrossRef CAS PubMed.
- A. H. Benfield and S. T. Henriques, Front. Med. Technol., 2020, 2, 610997 CrossRef PubMed.
- Y. Huan, Q. Kong, H. Mou and H. Yi, Front. Microbiol., 2020, 11, 582779 CrossRef PubMed.
- (a) X. Wang, P. Yang, F. Mondiot, Y. Li, D. S. Miller, Z. Chen and N. L. Abbott, Chem. Commun., 2015, 51, 16844–16847 RSC; (b) M. Sadati, A. I. Apik, J. C. Armas-Perez, J. Martinez-Gonzalez, J. P. Hernandez-Ortiz, N. L. Abbott and J. J. de Pablo, Adv. Funct. Mater., 2015, 25, 6050–6060 CrossRef CAS.
- J. M. Brake and N. L. Abbott, Langmuir, 2002, 18, 6101–6109 CrossRef CAS.
- M. J. Sartain, D. L. Dick, C. D. Rithner, D. C. Crick and J. T. Belisle, J. Lipid Res., 2011, 52, 861–872 CrossRef CAS PubMed.
- T. Gupta, L. Arora, S. Mukhopadhyay and S. K. Pal, J. Phys. Chem. Lett., 2024, 15, 2117–2122 CrossRef CAS PubMed.
- M. V. Meli, I. H. Lin and N. L. Abbott, J. Am. Chem. Soc., 2008, 130, 4326–4333 CrossRef CAS PubMed.
- I. Verma, S. Sidiq and S. K. Pal, ACS Omega, 2017, 2, 7936–7945 CrossRef CAS PubMed.
- T. Gupta, Toxin-Induced Ordering Transitions of Liquid Crystals at Biomolecular Interfaces, PhD thesis, IISER Mohali, 2024 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and supplementary figures. See DOI: https://doi.org/10.1039/d4tb02602k |
| This journal is © The Royal Society of Chemistry 2025 |