
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiodegradable hyperbranched polyesters of trimethylolpropane with acrylate side chains enabling sustainable gel materials and nanomaterials for drug delivery applications†
Binglin Sui *,
Safiya Nisar and
M. D. I. H. Seneviratne
*,
Safiya Nisar and
M. D. I. H. Seneviratne
Department of Chemistry, University of North Dakota, Grand Forks, ND 58202, USA. E-mail: binglin.sui@und.edu
First published on 12th June 2025
Abstract
To date, various biodegradable polymers have been synthesized due to the merits of biodegradable polymers in biomedical applications. The current widely used biodegradable polymers generally have linear structures, such as poly(lactic-co-glycolic acid) (PLGA), which prevents polymer crosslinking and polymer modification via covalent conjugation, thus restricting their even broader applications. In this research, we report the synthesis and studies of new sustainable polymers consisting of biodegradable backbones and side chains, which endow the polymers with complete biodegradability, biocompatibility, crosslinking, and availability for covalent chemical modifications. The convenient synthesis of the polymers needs no catalyst under ambient conditions, which effectively avoids the unintended toxicity and immune response associated with the catalyst residues in the polymer materials. Therefore, these polymers are especially desired in biomedical materials and devices. Moreover, the polymers can be fabricated into gel materials and nanomaterials. Using a near-IR fluorescent probe as an indicative cargo, we have established a biodegradable and biocompatible agent-delivering nanosystem paradigm with an average nanoscale size of ∼50 nm. In the nanoarchitectures, the cargo molecules are tethered to the nanoparticulate scaffold through covalent conjugation, preventing unwanted premature release of the cargo molecules in the blood circulation and thus circumventing the related systemic toxicity and adverse effects. Further, the delivery nanosystems are available for facile decoration with targeting ligands to attain disease-targeted delivery. The new materials exhibited excellent in vivo biocompatibility, signifying the immense potential they hold for biomedical applications.
Introduction
Biodegradable polymers, referred to as polymers that can work for a limited time and then degrade into harmless species via a regulated procedure, have attracted more and more interest in recent years due to their advantageous properties of biocompatibility and biodegradability, especially in the field of medical science.1–5 The development of biodegradable polymeric materials represents a revolution bringing about significant biotechnological advancements in modern medicine as such materials can break down into nontoxic small molecules inside the body, leading to several virtues in terms of compatibility with biological systems, controlled drug release, and potential for reduced inflammation and foreign body response.6–10 At present, biodegradable polymers continue to be a focal point in biomedical research and innovation, offering solutions to address challenges associated with a wide range of biomedical materials, including temporary implants, surgical sutures and staples, tissue engineering, regenerative medicine, wound care and dressings, vascular stents, dissolvable medical electronics, and drug delivery microparticles and nanoparticles.To date, different types of biodegradable polymers have been synthesized, and some of them have been employed to construct biomedical materials and devices, such as poly(ε-caprolactone) (PCL), polycaprolactone-co-lactide (PCLA), polydioxanone (PDO), polyglycolic acid (PGA), polyhydroxyalkanoates (PHA), polylactic acid (PLA), poly(lactic-co-glycolic acid) (PLGA), poly(trimethylene carbonate) (PTMC), and poly(tetramethylene succinate) (PTMS). Among the polymers, PLGA has been broadly used in the biomedical field since it was approved for clinical applications by the U.S. Food and Drug Administration (FDA).11–14 However, all of those polymers, including PLGA, have linear polymeric structures without side chains, which prohibits polymer crosslinking and covalent agent-conjugating. Thus, their biomedical applications are limited in many aspects: (1) the synthesis of those polymers generally requires transition metal-based catalysts, the residue of which in the resulting polymer and polymer-based materials may generate severe toxicities and trigger immune responses in the body; (2) they cannot be formulated into polymeric soft gels or hard bioplastics with adjustable stiffness because of the non-crosslinkable linear architecture; (3) for agent delivery purposes, the carriers based on those polymers load cargos through physical encapsulation due to the lack of side chains, for which premature leakage of loaded contents is unavoidable; and (4) they are not eligible materials for long-term controlled drug release devices as the weak interactions between the polymer chains and drug molecules are not able to protect the drug molecules from burst release. Therefore, biodegradable branched polymers with functionalizable side chains, preferably biodegradable side chains, are highly desired in the biomedical field.
Herein, we report the synthesis of new completely biodegradable branched polymers bearing functionalizable and biocleavable side chains and the evaluation of their suitability for biomedical applications as an agent-loading platform for short- and long-term delivery. These polymers are all polyesters synthesized via catalyst-free step-growth polymerization, and their biodegradability relies on the biocleavable nature of ester bonds in their polymer backbones and side chains. The functional side chains render the polymers advantageous in carrying biomedical agents, such as diagnostic probes, therapeutic drugs, functional proteins/antibodies, and genetic DNA/RNA, through covalent chemical conjugation, providing effective protection to the cargo molecules. Moreover, the side chains can be functionalized with certain stimuli-responsiveness, depending on the agent linker employed to link the polymers and the agents. Thereby, in response to the predesigned external stimulus, controllable and even programmable transportation of loaded cargo can be accomplished by using the polymeric delivering platform. As depicted in Fig. 1(A), after covalently loading cargo molecules, the crosslinkable agent-loaded polymers can be fabricated into polymer gels and nanoparticles (NPs), both of which are biocompatible and biodegradable. Upon being administered into the body, the cargo-carried nanoparticles are available for targeted delivery and the gels can accomplish controlled and sustained release of loaded agents. More importantly, both forms of materials will ultimately break down into nontoxic small molecules after fulfilling their tasks, free of biosafety and sustainability concerns.
 | ||
| Fig. 1 (A) Schematic illustration of the fabrication of sustainable polymeric gels and nanomaterials based on biodegradable and biocompatible polymers loaded with biomedical agents. Upon being administrated into the body, the polymeric materials will degrade into nontoxic small molecules, ensuring the complete release of loaded cargo without inducing acute toxicity and chronic side effects, free of biosafety and sustainability concerns. (B) Synthesis of TMPA polymers, including TMPA-MA, TMPA-SA, TMPA-GA, TMPA-AA, TMPA-SbA, and TMPA-DTPA. | ||
Results and discussion
Polymer design and synthesis
To address the abovementioned challenges regarding advanced polymeric materials for biomedical applications, we designed a library of new biodegradable branched polymers consisting of diol and dicarboxylic acid units. These copolymers were synthesized through step-growth polymerization, a well-developed and widely used method for synthesizing polyesters under mild conditions.15–18 As shown in Fig. 1(B), the synthesis of the polymers began with two monomers: trimethylolpropane acrylate (TMPA) and a dicarboxylic acid dichloride. TMPA was synthesized with a commercially available chemical, trimethylolpropane (TMP), reacting with acryloyl chloride (Fig. S1, ESI†), and it was characterized by nuclear magnetic resonance (NMR) spectroscopy (Fig. S2 and S3, ESI†). The polymerization reactions were conducted under anhydrous conditions due to the high moisture sensitivity of the dichlorides. To establish a standard protocol for synthesizing the polymers, we optimized the reaction conditions of the polymerizations, including the ratio between the monomers, different bases for accepting the hydrochloric acid molecules generated in the polymerization, the amount of the base, and the reaction time.As hydrochloric acid is produced as a side product in the esterification reaction between an alcohol and a carboxylic acid chloride, removing the acid by a base will promote the reaction, resulting in polymer molecules with a higher degree of polymerization. First, we tested different bases, including pyridine, triethylamine (TEA), N,N-diisopropylethylamine (DIEA), N,N′-dimethylaminopyridine (DMAP), and 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), to evaluate their efficiency in facilitating the polymerization. Following the same procedure, each base was dropped into a solution of the two monomers (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by molar) in dichloromethane. The resulting polymers were analyzed by GPC to measure their molecular weights. As displayed in Fig. 2(A), the use of pyridine resulted in the highest yield (81%) and desired molecular weight (Mn: 18.0 kDa, PDI: 1.26) of the polymer TMPA-AA, exhibiting the best performance among the bases tested. DMAP was also capable of promoting the polymerization reaction, although it was not as efficient as pyridine. In contrast, stronger bases, such as TEA, DIEA, and DBU, were even less efficient in potentiating the synthesis of polymers, as indicated by the lower yields and shorter polymer chains of the resulting products. Based on these findings, further optimizations of the polymerization were carried out using pyridine as the acid acceptor.
:1 ratio by molar) in dichloromethane. The resulting polymers were analyzed by GPC to measure their molecular weights. As displayed in Fig. 2(A), the use of pyridine resulted in the highest yield (81%) and desired molecular weight (Mn: 18.0 kDa, PDI: 1.26) of the polymer TMPA-AA, exhibiting the best performance among the bases tested. DMAP was also capable of promoting the polymerization reaction, although it was not as efficient as pyridine. In contrast, stronger bases, such as TEA, DIEA, and DBU, were even less efficient in potentiating the synthesis of polymers, as indicated by the lower yields and shorter polymer chains of the resulting products. Based on these findings, further optimizations of the polymerization were carried out using pyridine as the acid acceptor.
 | ||
| Fig. 2 Summary of the yield, Mn, Mw, and PDI of polymer TMPA-AA under different conditions: (A) with different bases as the acid acceptor, (B) with varied equivalents of pyridine (*pyridine was used as the solvent for the reaction instead of dichloromethane), (C) with varying ratios of the monomers (TMPA:adipoyl chloride, by molar), and (D) with different reaction time. (E) Summary of the synthesis yield, Mn, Mw, and PDI of various TMPA polymers. The time-lapsed molecular weight changes of TMPA-AA upon incubation in (F) aqueous NaOH solutions with different concentrations (0.01, 1.0, and 100 mM) and (G) PBS solutions (pH 7.4) containing varied concentrations (0, 5, and 100 units per mL) of the PLE enzyme. Data represent the means ± SD, n = 3. (H) Modifications of polymer TMPA-AA with thiol-contained compounds. | ||
Second, we noticed that the amount of pyridine used in the reactions affected the yield and molecular weight of the polymer. To elucidate that, varied equivalents of pyridine were screened under the same conditions. As shown in Fig. 2(B), the highest yield and largest molecular weight of the polymer was obtained in the presence of 1.1 times the amount of stoichiometric pyridine (2.2 equiv. relative to the monomers). Less pyridine, e.g., 1.1 equiv., was insufficient to capture all the acid generated in the reaction, which is unfavorable for polymer propagation, resulting in a decreased degree of polymerization. Excessive pyridine (3.3 equiv. and more), however, brought negative impacts on the polymerization, too. The polymerization yield and the molecular weights of resulting polymers decreased with more pyridine presented in the reaction. Especially, when pyridine was used as the solvent for the polymerization reaction, no polymer was collected in the process. This phenomenon might be attributed to the destabilization of the ester bond by free pyridine molecules, which suppressed further propagation of polymer chains.
Third, as both the monomers are functionalized at the two ends, the ratio between the monomers plays an important role in the polymerization reaction. To figure out an optimized ratio for the reaction, the polymer TMPA-AA was synthesized with different feeding ratios of TMPA and adipoyl chloride (2:1, 1.5:1, 1:1, 1:1.5, and 1:2, by molar). The results summarized in Fig. 2(C) demonstrate that the stoichiometric ratio of the two monomers is more efficient compared to other ratios, yielding polymer with the highest yield and largest molecular weight.
Fourth, adequate reaction time is necessary for the polymer chains to grow longer. To optimize the reaction time for the polymerization, we terminated the reaction after certain time periods (24 h, 48 h, 72 h, and 96 h). As listed in Fig. 2(D), the polymer chains kept growing longer within 48 h, as indicated by the significantly larger molecular weight of the polymer generated after 48 h (15.3 kDa) than that of the polymer formed after 24 h (8.9 kDa). Thereafter, the growth of the polymer chains slowed down as the molecular weight increased by about 2.7 kDa in the following 24 h. The molecular weight of the polymer after 96 h (17.2 kDa) was approximately equal to that after 72 h (18.0 kDa), suggesting that the polymer propagation stopped after 72 h.
Combining the results of optimizing experiments, a standard polymerization protocol was established – polymerizing the monomers TMPA and adipoyl chloride at a molar ratio of 1:1 for 72 h, with 2.2 equivalents of pyridine as the acid acceptor for the reaction. Following the protocol, the dichlorides of malonic acid (MA), succinic acid (SA), glutaric acid (GA), adipic acid (AA), and suberic acid (SbA) were utilized in the polymerization reactions to synthesize polymers TMPA-MA, TMPA-SA, TMPA-GA, TMPA-AA, and TMPA-SbA, respectively. In addition to the commercially available dicarboxylic acid dichlorides, a disulfide-contained dicarboxylic acid, 3,3′-dithiodipropionic acid (DTPA), was utilized to synthesize its corresponding dichloride, 3,3′-dithiodipropionic acid chloride (DTPC, Fig. S4, ESI†), which was confirmed by NMR spectroscopy (Fig. S5 and S6, ESI†). DTPC was used in the polymerization reaction with TMPA to yield a disulfide-contained polymer TMPA-DTPA (Fig. 1(B)). All the synthesized polymers were characterized by NMR spectroscopy (Fig. S7–S12, ESI†), and their molecular weights were measured by gel permeation chromatography (GPC). Fig. 2(E) summarizes the yield, molecular weights (Mn and Mw), and polydispersity index (PDI) for the polymers, including TMPA-MA, TMPA-SA, TMPA-GA, TMPA-AA, TMPA-SbA, and TMPA-DTPA. The general yields of the polymerization reactions were around 80%, although TMPA-MA had a much lower one (42%). The low yield of TMPA-MA might be ascribed to its small molecular weight, which results in a larger loss of polymer chains in the post-reaction purification process. The molecular weights (Mn) of TMPA-MA (7.9 kDa), TMPA-SA (9.8 kDa), and TMPA-DTPA (9.9 kDa) are relatively smaller than those of TMPA-GA (15.2 kDa), TMPA-AA (18.0 kDa), and TMPA-SbA (17.7 kDa) under the same polymerization conditions, which implies the higher efficiency of the latter ones in yielding longer polymer chains. Notably, the polymers have low PDI values (1.25–1.39), indicating the narrow distributions of their polymer chains and uniform properties. It is noteworthy to point out that the incorporation of a stimuli-responsive biocleavable disulfide group in the backbone of the polymer, i.e., TMPA-DTPA, would not affect the polymerization reaction, as evidenced by the high yield and high molecular weight of the polymer, which provides a method to bring more versatile polymers for diverse biomedical purposes.
Degradation of polymers
Complete degradation in physiological environments is one of the primary merits expected for the polymers, owing to their polyester backbone and ester bond-contained side chains. As known, the ester bond can be hydrolyzed under basic conditions through a saponification reaction. To study the degradability of TMPA-AA, the polymer was incubated in mixed media of DMSO and aqueous NaOH (0.01, 1.0, and 100 mM) and sampled at predetermined time points (0.5 h, 1 h, 2 h, 3 h, 5 h, 8 h, 12 h, and 24 h) for GPC analysis. As shown in Fig. 2(F), the molecular weight of TMPA-AA decreased at varied rates upon incubation in basic media, depending on the concentration of the base in the media. In the presence of 100 mM NaOH, the polymer decomposed drastically, and no considerable amount of polymer was detected within 5 h. The degradation of polymers slowed down in basic solutions with lower concentrations of NaOH, as evidenced by the prolonged decomposition process. These results confirmed the anticipated degradability of the polymers.Since the hydrolysis of ester bonds can be catalyzed by enzymes, we further examined the polymer in phosphate-buffered solutions (PBS, pH 7.4) with and without an esterase enzyme porcine liver esterase (PLE), mimicking different biological environments, to verify its biodegradability. The polymer-contained PBS samples were analyzed by GPC at certain time points (0.5 h, 1 h, 2 h, 3 h, 5 h, 8 h, 12 h, and 24 h) upon incubation at 37 °C. As shown in Fig. 2(G), the molecular weight of TMPA-AA remained nearly unchanged in PLE-free buffer media over 24 h, demonstrating the high stability of the polymer. However, in the presence of PLE (5 units per mL), TMPA-AA was gradually decomposed, as indicated by the decreasing molecular weight. A higher level of PLE (100 units per mL) led to a more rapid degradation of the polymer. After 24 h, no detectable amount of polymer was found in the buffer solutions, which suggests that the polymer completely degraded into small molecules, signifying the desired biodegradability of the polymers for biomedical utility. In addition, NMR analysis of the products of degradation confirmed that the polymer was degraded into TMP, adipic acid, and acrylic acid (Fig. S13, ESI†), which are biocompatible molecules.19–21
Post-synthesis modifications of polymers
Apart from being biodegradable via hydrolysis, the side chains of TMPA-AA are reactive toward thiol groups due to the activated double bond of the acrylate group. We hypothesized that small thiol-contained molecules could be smoothly conjugated to the polymer through thiol–ene reactions. To verify that, various compounds containing thiol groups were used to react with TMPA-AA under the same conditions. As shown in Fig. 2(H), thiol-contained chemicals 1-dodecanethiol (DDT), 2-mercaptoethanol (BME), 3-mercaptopropionic acid (MPA), 3-mercaptopropanyl-N-hydroxysuccinimide ester (MPA-NHS), 1H,1H,2H,2H-perfluorodecanethiol (PFDT), and thiol-modified polyethylene glycol (PEG-SH) were attached to polymer TMPA-AA to yield new polymers TMPA-AA-DDT, TMPA-AA-BME, TMPA-AA-MPA, TMPA-AA-MPA-NHS, TMPA-AA-PFDT, and TMPA-AA-PEG, respectively. MPA-NHS was synthesized following a literature procedure,22 and its chemical structure was confirmed by NMR spectroscopy (Fig. S14 and S15, ESI†). The successful conjugation of those molecules was verified by the NMR characterization of the resulting polymers (Fig. S16–S21, ESI†). More interestingly, simultaneous conjugation of multiple thiol-contained compounds to the polymer through a one-pot reaction can be achieved efficaciously. For example, by feeding the reaction with a mixture of PEG-SH and MPA-NHS, both thiol-contained compounds were conjugated to the polymer TMPA-AA, and a new polymer TMPA-AA-PEG-NHS was obtained. Its chemical composition was confirmed by NMR spectroscopy (Fig. S22, ESI†). These results demonstrated the high efficiency of the polymer in loading diverse agent molecules, which signifies the vast potential to carry various biomolecular cargos such as DNAs/RNAs, proteins, polypeptides, and organic drugs for biomedical agent delivery applications.Gelation of polymers
In addition to loading functional agent molecules, we also found that TMPA-AA can be crosslinked by dithiol-containing molecules. We used 1,2-ethanedithiol (EDT) as a prototypical crosslinker to react with the polymer chains. As shown in Fig. 3(A), the liquid polymer is converted into an easy-to-manipulate gel upon crosslinking, with triethylamine (TEA) as a catalyst to initiate the thiol–ene addition between TMPA-AA and EDT at room temperature. It is worthwhile to point out that, different from transition metal-based catalysts, TEA has low toxicity and high biocompatibility, and more importantly, it can be easily washed away from the polymer gel due to its miscibility with water. In the presence of TEA, thiols are deprotonated into nucleophilic thiolates, which react with the acrylate groups of the polymer through the Michael addition (Fig. S23, ESI†), resulting in a polymeric gel (TMPA-AA-EDT). Its chemical composition was confirmed by NMR spectroscopy (Fig. S24, ESI†). Comparing the NMR spectra of TMPA-AA and TMPA-AA-EDT, we found that the three characteristic peaks between 6.50 ppm and 5.50 ppm, which correspond to the double bond of the polymer side chain, disappeared after the crosslinking (Fig. 3(B)), indicating the consumption of acrylate double bonds in the crosslinking process. Meanwhile, the emergence of NMR signals at 3.00–2.63 ppm further confirmed the occurrence of thiol–ene additions. Therefore, the crosslinking of highly branched polymer chains led to the formation of a polymeric gel.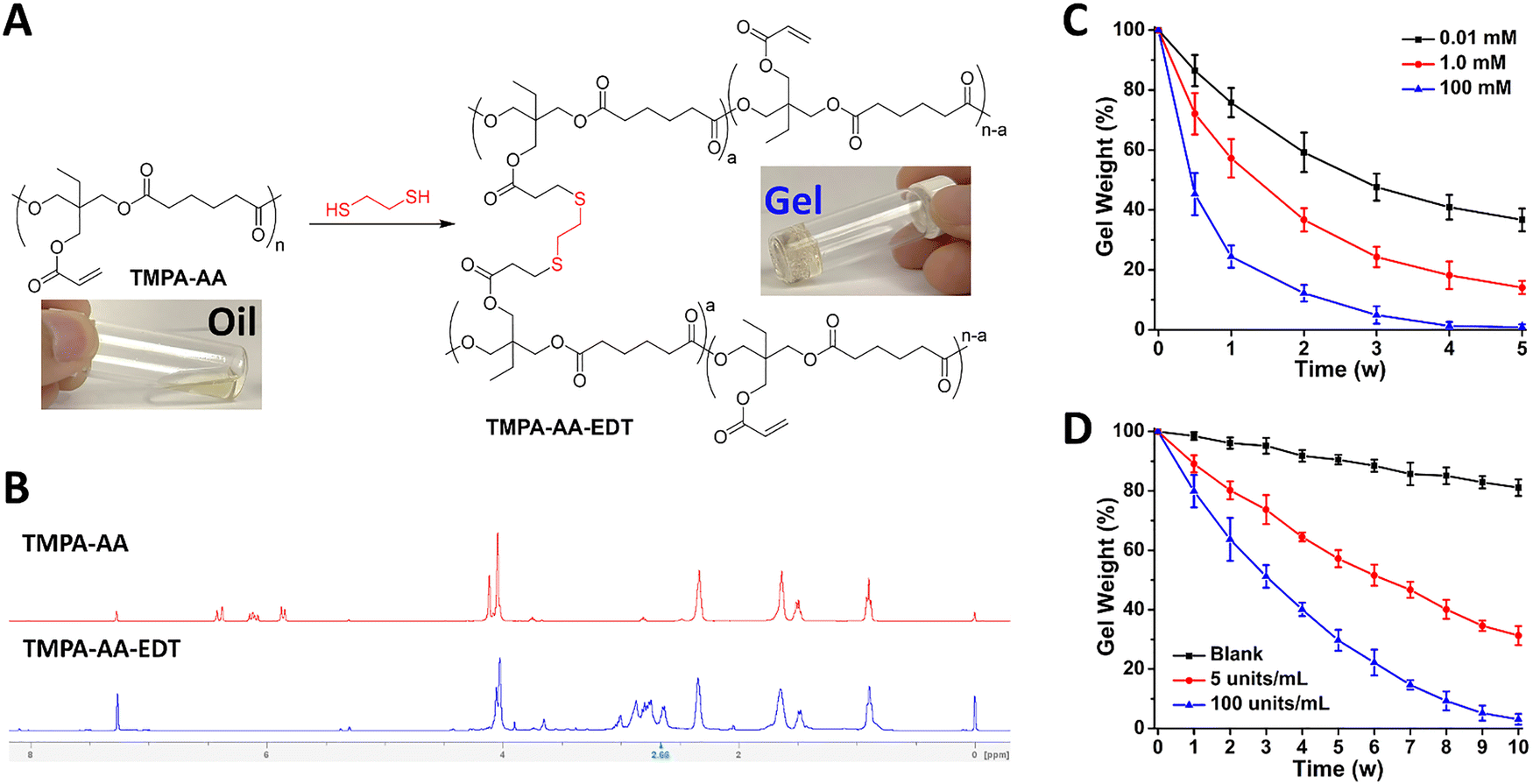 | ||
| Fig. 3 (A) The crosslinking of polymer TMPA-AA by the crosslinker EDT and the resulting polymeric gel. (B) 1H NMR spectra of TMPA-AA and the crosslinked TMPA-AA-EDT. The time-lapsed weight changes of gels upon incubation in (C) aqueous NaOH solutions with different concentrations (0.01, 1.0, and 100 mM) and (D) PBS solutions (pH 7.4) containing varied concentrations (0, 5, and 100 units per mL) of the PLE enzyme. Data represent the means ± SD, n = 3. | ||
Degradation of gels
Based on the complete biodegradability of the polymers, we hypothesized that the resulting polymeric gels can be degraded as well. To verify that, we examined the degradation of gels under varied chemical and biological conditions. First, the gels were incubated in aqueous NaOH solutions (0.01, 1.0, and 100 mM). After certain time intervals (0.5, 1, 2, 3, 4, and 5 weeks), the gel residues were collected and weighed for quantitative analysis. As shown in Fig. 3(C), the weight of gels decreased gradually at varied rates, depending on the concentration of NaOH. Faster degradation was found in solutions with higher concentrations of the base. In the presence of 100 mM NaOH, no significant amount of gels was detected within 4 weeks, indicating the complete degradation of gels.The degradability of gels was further determined in PBS buffer solutions (pH 7.4) with and without PLE, mimicking different biological environments. After incubation at 37 °C for different time intervals (1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 weeks), the gel residues were collected for weight analysis. As shown in Fig. 3(D), in the PBS solutions without PLE, the weight of gels was reduced by >15% over 10 weeks, which demonstrated the slow degradation of gels under biological conditions. In contrast, the addition of PLE at the concentration of 5 units per mL substantially accelerated the degradation rate, resulting in a ∼70% weight loss of gels during the same time period. Furthermore, in the presence of higher-level PLE (100 units per mL), the gels decomposed nearly completely by the end of 10 weeks, signifying the excellent biodegradability of the gels.
Polymeric nanoparticles
We found that the PEG-modified polymer TMPA-AA-PEG could be formulated into nanostructures by crosslinking and subsequent dispersion into aqueous media, which should be attributed to its amphiphilic nature. The amphiphilicity of TMPA-AA-PEG results from the hydrophobicity of the polymer backbone and the hydrophilicity of attached PEG chains. As shown in Fig. 4(A), the polymer dissolved in DMSO was crosslinked by the crosslinker EDT. When the DMSO solution was dropped into deionized water or PBS buffer solutions under vigorous stirring, the crosslinked polymer self-assembled into nanoscale particles, with the nanoparticulate scaffold formed with the hydrophobic polymer backbone and coated with a hydrophilic PEG shell. The following dialysis in a dialysis tube (MWCO: 6–8 kDa) against deionized water or PBS buffer completed the fabrication of TMPA-AA-PEG nanoparticles (TMPA-AA-PEG NPs). Similar to the polymer gel, the NPs exhibited desired biodegradability in the presence of esterase enzymes as well (Fig. S25, ESI†). | ||
| Fig. 4 (A) Construction of polymeric nanoparticles. A polymer or polymer mixture is crosslinked by the crosslinker (e.g., EDT) and then assembled into polymeric nanostructures upon dispersion in aqueous media. (B) Size distribution and (C) TEM image of TMPA-AA-PEG NPs. (D) Size changes of TMPA-AA-PEG NPs in PBS (pH 7.4) solutions with and without 10% FBS. Data represent the means ± SD, n = 3. | ||
The resulting TMPA-AA-PEG NPs were characterized by dynamic light scattering (DLS) and transmission electron microscope (TEM). DLS analysis showed that TMPA-AA-PEG NPs had an average hydrodynamic diameter of 81.4 nm with a narrow size distribution as indicated by a polydispersity index (PDI) of 0.13 (Fig. 4(B)), and a negative surface charge with a zeta potential at −31.8 mV. TEM imaging observed a spherical morphology of the nanostructures with a size of ∼67 nm in diameter at the dry state (Fig. 4(C)). In addition, to examine the dynamic stability of TMPA-AA-PEG NPs, we monitored their hydrodynamic size in deionized water for a week. No significant change was detected in the hydrodynamic diameter (Fig. S26, ESI†), denoting the high colloidal stability of the nanostructures. Further, they were incubated in PBS buffter solutions (pH 7.4) with and without fetal bovine serum (FBS), mimicking different biological conditions, to assess their suitability for applications in physiological enviroments. As shown in Fig. 4(D), TMPA-AA-PEG NPs maintained high stability in the PBS buffer, as evidenced by their unaltered hydrodynamic size. In PBS buffer solutions containing 10% FBS (by volume), the hydrodynamic size of TMPA-AA-PEG NPs was enlarged at the beginning of the incubation, which should be ascribed to the protein adsorption on the surface of the NPs.23–26 Afterwards, the average hydrodynamic diameter remained constant. These observation reflected the desirable serum stability of TMPA-AA-PEG NPs.
NIR fluorophore labeling of polymers
Owing to the virtues of fluorescent dyes and advanced fluorescence microscopy, fluorescence labeling has been regarded as a highly effective tool for studying the properties and behaviors of biological molecules. Compared to dyes fluorescing in the visible light region, fluorophores emitting near-infrared (NIR) fluorescence have additional advantages, such as higher signal-to-noise ratios, deeper tissue penetration, lower interrupting autofluorescence from tissues, reduced light scattering, superior imaging resolution, and diminished phototoxicities, for biomedical imaging purposes.27–30 Amongst various NIR fluorophores, cyanine 7 (Cy7) has been widely employed in NIR fluorescent biosensors and probes due to its high biocompatibility and photostability. Hence, to further reveal the eligibility of the new polymers for in vitro and in vivo biomedical applications, we labeled polymer TMPA-AA-PEG with the NIR fluorophore Cy7.Given the proven efficiency of the polymer side chains in reacting with thiol-contained agents, we synthesized a thiol-modified derivative of Cy7 using Cy7-Cl as the starting material (Fig. S27, ESI†). Cy7-Cl was prepared according to a reported procedure.31 Upon reacting with 1,4-dithiothreitol (DTT) under ambient conditions, the meso-Cl atom of Cy7-Cl was replaced by a thiol group of DTT due to its high reactivity toward thiol groups, producing a DTT-substituted Cy7 fluorophore (Cy7-DTT). This new fluorescent dye was characterized by NMR spectroscopy (Fig. S28 and S29, ESI†). Its photophysical properties were measured by UV-vis spectroscopy and fluorescence spectroscopy. An absorption peak at 778 nm and an emission maximum at 801 nm in the NIR wavelength range were recorded in its UV-vis absorption and fluorescence emission spectra (Fig. S30, ESI†). The fluorescence emission quantum yield of Cy7-DTT was determined to be 0.12 using indocyanine green (ICG) as the reference. NIR fluorescent probes with such a high fluorescence emission quantum yield are advantageous for biomedical imaging in vivo. Further, we studied the stability of Cy7-DTT to assess its qualification for applications in biological environments. The fluorescence emission of Cy7-DTT was examined under varied conditions with different temperatures and pH values. Constant fluorescence intensities were recorded at physiological temperatures (Fig. S31, ESI†) and over a broad range of pH values (Fig. S32, ESI†). A high photostability of Cy7-DTT was also observed, as its fluorescence emission stayed nearly unchanged upon consecutive repeated excitations (Fig. S33, ESI†). In addition, we studied the cellular uptake of Cy7-DTT using confocal laser scanning microscopy (CLSM). CLSM images of A549 cells showed that cells emitted striking fluorescence after incubation with 5 μM Cy7-DTT for 3 h, which validated the efficient cellular uptake of Cy7-DTT (Fig. S34, ESI†). The desirable photophysical properties of Cy7-DTT render it an excellent NIR fluorescent probe for cellular bioimaging.
Taking advantage of the free thiol group in the Cy7-DTT molecule, we conjugated Cy7-DTT to polymer TMPA-AA-PEG via thiol–ene reactions to synthesize a Cy7-labeled polymer TMPA-AA-PEG-Cy7 (Fig. S35, ESI†). Its chemical composition was confirmed by NMR spectroscopy (Fig. S36, ESI†). UV-vis absorption and fluorescence emission spectra displayed that the polymer absorbs NIR light at 780 nm and emits fluorescence at 804 nm due to the covalently attached Cy7-DTT pendants (Fig. S37, ESI†). The NIR fluorescence labeling endows the polymer availability for in vitro and in vivo bioimaging studies. To verify that the new polymers could transport loaded cargo molecules into cells, the Cy7-labeled polymer TMPA-AA-PEG-Cy7 was examined by CLSM cell imaging. Upon incubation with a fresh cell culture medium containing polymer TMPA-AA-PEG-Cy7 for 3 h, cells emitted strong NIR fluorescence (Fig. S38, ESI†), suggesting the effective uptake of TMPA-AA polymers by human cells.
Polymeric nanosystems for agent delivery
Aside from fabricating biosafe and eco-friendly polymer gels, another important biomedical application of biodegradable and biocompatible polymers is to construct nontoxic and sustainable polymeric nanomaterials for the purposes of delivering diagnostic, therapeutic, and genetic agents. Thanks to the crosslinkable nature of the TMPA-AA polymers, either a single polymer, such as TMPA-AA-PEG, or a mixture of different polymers can be formulated into polymeric nanomaterials, providing a flexible and convenient platform for constructing multi-agent-loaded nanosystems. To demonstrate the strategy, a polymer mixture of TMPA-AA-PEG-Cy7 and TMPA-AA-PEG-NHS was crosslinked by the crosslinker EDT in DMSO and then dispersed into deionized water and PBS buffer solutions under robust stirring. The crosslinked polymers were spontaneously assembled into nanostructures. The assembly of nanostructures was driven by the amphiphilicity of the crosslinked polymer, with the hydrophobic Cy7 fluorophore molecules enclosed by a hydrophilic shell made of PEG chains. The resulting nanoassemblies were purified by dialysis in dialysis tubes (MWCO: 6–8 kDa) to produce Cy7-labeled nanoparticles (nano-Cy7 NPs). The average hydrodynamic diameter of the nano-Cy7 NPs was measured to be 64.1 nm with a PDI of 0.21 (Fig. 5(A)). Spherical morphology and a diameter of ∼54 nm at the dry state were observed by TEM microscopy (Fig. 5(B)). The NPs also exhibited a negatively charged surface, and the zeta potential was −26.5 mV (Fig. 5(C)). The hydrodynamic size of nano-Cy7 NPs remained constant in deionized water over a week, indicating their high colloidal stability (Fig. S39, ESI†). Such a desirable stability of NPs was observed in PBS buffer solutions (pH 7.4) with and without FBS as well (Fig. 5(D)), which suggested that the hydrodynamic stability of the NPs would not be affected by blood serum. | ||
| Fig. 5 (A) Size distribution and (B) TEM image of nano-Cy7 NPs. (C) Zeta potential of nano-Cy7 NPs and nano-Cy7-RGD NPs. (D) Size changes of nano-Cy7 NPs in PBS (pH 7.4) solutions with and without 10% FBS. (E) Size distribution and (F) TEM image of nano-Cy7-RGD NPs. (G) Size changes of nano-Cy7-RGD NPs in PBS (pH 7.4) solutions with and without 10% FBS. Cell viability of (H) HUVEC cells, (I) A549 cells, and (J) BxPC-3 cells after incubation with nano-Cy7 NPs and nano-Cy7-RGD NPs for 24 h. Data represent the means ± SD, n = 3. (K) CLSM images of A549 cells after incubation with nano-Cy7 NPs and nano-Cy7-RGD NPs for 3 h. | ||
To showcase the availability of the resulting NPs for post-formulation modifications, we further decorated nano-Cy7 NPs with a tumor-homing cyclic peptide sequence RGD (cyclo-Arg-Gly-Asp-D-Phe-Lys), which interacts specifically with the αvβ3 and αvβ5 integrin receptors overexpressed on the surface of various types of cancer cells.32–35 The RGD ligand was attached to the NPs through reactions between the free amino groups of lysine units of the peptide and the NHS groups of the NPs under mild conditions, followed by dialysis in dialysis tubes (MWCO: 6–8 kDa) to generate RGD-decorated nano-Cy7 NPs (nano-Cy7-RGD NPs). TEM images of nano-Cy7-RGD NPs showed a spherical morphology and a dry-state diameter of ∼52 nm (Fig. 5(E)). It is noteworthy to point out that nanoparticles of such dimensions are highly desired for the purpose of intracellular drug delivery,36 as nanoparticles of diameters 5–50 nm are more efficient in penetrating cancer cells, whereas those with a size of 50–200 nm tend to circulate longer in blood circulation and accumulate more in tumor tissues with a longer retention time.37–40 DLS analysis found that nano-Cy7-RGD NPs had an average hydrodynamic diameter of 61.3 nm with a PDI of 0.23 (Fig. 5(F)), and a negative surface charge with a zeta potential at −9.4 mV (Fig. 5(C)). The reduced negative charge of nano-Cy7-RGD NPs compared to that of nano-Cy7 NPs is attributed to the RGD decoration. Analogous to nano-Cy7 NPs, nano-Cy7-RGD NPs had excellent dynamic stability in deionized water (Fig. S40, ESI†). Also, they demonstrated high serum stability in FBS-contained PBS buffer solutions (Fig. 5(G)), comparable to that of nano-Cy7 NPs.
In vitro studies of nanomaterials
To serve as an applicable agent delivery vehicle, the nanosystems are expected to possess high biocompatibility and cell-entering capacity. The cytotoxicity of nano-Cy7 NPs and nano-Cy7-RGD NPs were determined in human umbilical vein endothelial cells (HUVEC), human lung cancer A549 cells, and human pancreatic cancer BxPC-3 cells by the 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The viability of those cells was scarcely affected after incubation with both NPs at varied concentrations (0–200 μg mL−1) for 24 h (Fig. 5(H)–(J)), reflecting the low cytotoxicity of the NPs. Next, to certify that the NPs are able to enter cells, A549 cells were incubated with the NPs at a concentration of 1 μM equivalent to Cy7 for 3 h and then imaged by fluorescence microscopy. As shown in Fig. 5(K), the observed NIR fluorescence signals proved that both NPs were taken up by the cells. Furthermore, the fluorescence intensity of cells incubated with nano-Cy7-RGD NPs was apparently stronger than that of cells treated with nano-Cy7 NPs. The enhanced cellular uptake of nano-Cy7-RGD NPs should be ascribed to the RGD ligands attached to the NPs, which further consolidated the effective post-formulation decoration of the polymeric nanosystems.In vivo tumor-targeted delivery
Encouraged by the excellent in vitro performances of the NPs, we further investigated their efficiencies for tumor-targeted delivery in mice. A xenograft tumor model of A549 cancer was established in nude mice. The in vivo targeted delivery efficiency of the NPs was demonstrated by their distributions in tumor-bearing mice. After intravenous injection of the NPs at a dose of 1 mg kg−1 equivalent to Cy7 for 6 h, mice were imaged under anesthesia by a noninvasive Lago X in vivo imaging system. As shown in Fig. 6(A), an intense NIR fluorescence signal was observed in the tumor area of mice administrated with nano-Cy7 NPs, which suggested that the NPs accumulated in the tumor tissues. This phenomenon resulted from the enhanced permeability and retention (EPR) effect that the NPs inherently possess.41,42 More importantly, tumor tissues of mice injected with nano-Cy7-RGD NPs emitted significantly stronger fluorescence than those with nano-Cy7 NPs, which is attributed to the elevated tumor-targeting efficacy of the RGD-decorated NPs. The visual observation was further supported by the quantitative analysis of the fluorescence intensity (Fig. 6(B)). Afterward, the mice were sacrificed, and their tumor tissues and main organs, including liver, spleen, kidney, heart, and lung, were excised to study the biodistribution of the NPs. As displayed in Fig. 6(C), the ex vivo imaging of those organs and tissues showcased that the NPs were largely distributed in the livers, kidneys, and tumors of mice. In addition, more nano-Cy7-RGD NPs were detected in the tumor tissues than nano-Cy7 NPs, indicated by the higher fluorescence emission intensity (Fig. 6(D)), which was in good agreement with the outcomes of in vivo bioimaging. These findings corroborated the potency of the functionalizable polymer-based nanomaterials for biomedical agent transportation and targeted delivery. | ||
| Fig. 6 (A) In vivo whole-body fluorescence images of mice intravenously injected with nano-Cy7 and nano-Cy7-RGD NPs. (B) The average intensity of the fluorescence emitted from tumor tissues of mice injected with nano-Cy7 and nano-Cy7-RGD NPs. (C) Ex vivo fluorescence images and (D) the average fluorescence intensity of main organs (liver, spleen, kidney, heart, and lung) harvested from sacrificed mice intravenously injected with nano-Cy7 and nano-Cy7-RGD NPs. (E) Body weight changes of mice during the administration with polymer TMPA-AA-PEG (100 mg kg−1 and 300 mg kg−1 body weight). (F) H&E staining of organ tissues (liver, spleen, kidney, heart, and lung) collected from mice in different treatment groups. Data represent the means ± SD, n = 5. | ||
In vivo biocompatibility studies
High in vivo biocompatibility is necessary for materials that are expected to be extensively used in biomedical applications. To evaluate the qualification of the new polymers, we studied their biocompatibility in BALB/c mice. Three randomly assigned groups of mice (1 control group and 2 experimental groups) with 5 mice per group were separately treated with saline in the control group and saline solutions of polymer TMPA-AA-PEG at the contractions of 100 mg kg−1 and 300 mg kg−1 body weight in the experimental groups. The treatments were intravenously administered to mice through the tail vein twice per week for 4 weeks. Following the first administration, the body weight and health conditions of mice were monitored every day. During the observation period, no deterioration in health was witnessed in those mice, and their overall behavior was no different from that observed in untreated mice. Also, no considerable body weight loss was shown in the three groups of mice after the four-week treatments (Fig. 6(E)). One week after the final injection, all treated mice were sacrificed, and their major organs (liver, spleen, kidney, heart, and lung) were harvested for histological analysis. As shown in Fig. 6(F), hematoxylin and eosin (H&E) staining of the collected organ tissues demonstrated that the main organs of mice maintained their normal architectures, and no obvious histological damage was observed in the organ tissues. The combined results of histological analysis and body weight monitoring validated the excellent biocompatibility and suitability of the new polymers for biomedical purposes.Conclusions
We have developed completely biodegradable, biocompatible, crosslinkable, hyperbranched polymers consisting of a biodegradable polyester backbone and crosslinkable and functionalizable branches. The polymers are synthesized under ambient conditions conveniently with no need of any catalyst, which is highly desired for developing biomedical materials because the catalyst remnants in the polymers, especially those from transition metal-based catalysts, may induce systemic toxicities and provoke immune responses in vivo. The optimized synthetic methodologies of the polymers were established by screening the polymerization reaction conditions. Comprehensive evaluations of the polymers under varied chemical and biological conditions revealed that polymer TMPA-AA and its derivative gels and nanomaterials are promising materials for diverse biomedical applications.A compelling advantage of the polymers is their hyperbranched structure with biodegradable side chains. Such a structure allows for crosslinking of polymer chains and carrying functional molecules through covalent conjugation, which can be manipulated to fabricate polymeric gels and nanomaterials with tunable properties to meet the extensive need for biosafe and sustainable materials in biomedical devices and applications. Given the excellent biodegradability of the polymers, the polymer-derived materials break down into nontoxic molecules upon fulfilling their predesigned tasks in living systems, free of systemic toxicities and immune reactions. For instance, the polymeric nanomaterials successfully delivered fluorescent probe molecules into cancerous tissues and cells, based on which the in vivo biodistribution of the delivering nanosystems was investigated. Thanks to the covalent conjugation and nanoscale structures, the NPs exhibited high accumulation and retention at tumor sites in vivo. The facile post-formulation decoration with tumor-homing ligands further elevated the targeted delivery of the nanosystems to tumor tissues. In vivo experiments confirmed the low toxicity and high biocompatibility of the polymers in the body. In a word, our work has resulted in advanced sustainable polymers and related polymeric gels and nanomaterials that hold vast potential for versatile applications in the biomedical field.
Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
We acknowledge the support from the National Science Foundation (CHE-2213445 and 2117699) and the University of North Dakota (the Early Career Scholars Award). We also acknowledge the UND Behavioral Research Core Facility for animal studies.References
- R. A. Gross and B. Kalra, Biodegradable Polymers for the Environment, Science, 2002, 297(5582), 803–807 CrossRef CAS PubMed.
- M. S. Kim, H. Chang, L. Zheng, Q. Yan, B. F. Pfleger, J. Klier, K. Nelson, E. L. W. Majumder and G. W. Huber, A Review of Biodegradable Plastics: Chemistry, Applications, Properties, and Future Research Needs, Chem. Rev., 2023, 123(16), 9915–9939 CrossRef CAS PubMed.
- A. Samir, F. H. Ashour, A. A. A. Hakim and M. Bassyouni, Recent advances in biodegradable polymers for sustainable applications, npj Mater. Degrad., 2022, 6(1), 68 CrossRef CAS.
- I. Vroman and L. Tighzert, Biodegradable Polymers, Materials, 2009, 2(2), 307–344 CrossRef CAS.
- S. Doppalapudi, A. Jain, W. Khan and A. J. Domb, Biodegradable polymers—an overview, Polym. Adv. Technol., 2014, 25(5), 427–435 CrossRef CAS.
- R. Haag and F. Kratz, Polymer Therapeutics: Concepts and Applications, Angew. Chem., Int. Ed., 2006, 45(8), 1198–1215 CrossRef CAS PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release, Chem. Rev., 2016, 116(4), 2602–2663 CrossRef CAS PubMed.
- B. D. Ulery, L. S. Nair and C. T. Laurencin, Biomedical applications of biodegradable polymers, J. Polym. Sci., Part B: Polym. Phys., 2011, 49(12), 832–864 CrossRef CAS PubMed.
- I. Manavitehrani, A. Fathi, H. Badr, S. Daly, A. Negahi Shirazi and F. Dehghani, Biomedical Applications of Biodegradable Polyesters, Polymers, 2016, 8(1), 20 CrossRef PubMed.
- G. E. Luckachan and C. K. S. Pillai, Biodegradable Polymers- A Review on Recent Trends and Emerging Perspectives, J. Polym. Environ., 2011, 19(3), 637–676 CrossRef CAS.
- H. K. Makadia and S. J. Siegel, Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier, Polymers, 2011, 3(3), 1377–1397 CrossRef CAS PubMed.
- J.-S. Choi, K. Seo and J.-W. Yoo, Recent Advances in PLGA Particulate Systems for Drug Delivery, J. Pharm. Investig., 2012, 42(3), 155–163 CrossRef CAS.
- S. Sharma, A. Parmar, S. Kori and R. Sandhir, PLGA-Based Nanoparticles: a New Paradigm in Biomedical Applications, TrAC, Trends Anal. Chem., 2016, 80, 30–40 CrossRef CAS.
- S. Rezvantalab, N. I. Drude, M. K. Moraveji, N. Güvener, E. K. Koons, Y. Shi, T. Lammers and F. Kiessling, PLGA-Based Nanoparticles in Cancer Treatment, Front. Pharmacol., 2018, 9, 1260 CrossRef CAS PubMed.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y. X. Chen, Recyclable and (Bio)degradable Polyesters in a Circular Plastics Economy, Chem. Rev., 2024, 124(7), 4393–4478 CrossRef CAS PubMed.
- C. V. Aarsen, A. Liguori, R. Mattsson, M. H. Sipponen and M. Hakkarainen, Designed to Degrade: Tailoring Polyesters for Circularity, Chem. Rev., 2024, 124(13), 8473–8515 CrossRef CAS PubMed.
- Y. Yan and D. J. Siegwart, Scalable synthesis and derivation of functional polyesters bearing ene and epoxide side chains, Polym. Chem., 2014, 5(4), 1362–1371 RSC.
- S. S. Panchal and D. V. Vasava, Biodegradable Polymeric Materials: Synthetic Approach, ACS Omega, 2020, 5(9), 4370–4379 CrossRef CAS PubMed.
- K. Fukushima, Poly(trimethylene carbonate)-based polymers engineered for biodegradable functional biomaterials, Biomater. Sci., 2016, 4(1), 9–24 RSC.
- L. Navarro, N. Ceaglio and I. Rintoul, Structure and properties of biocompatible poly(glycerol adipate) elastomers modified with ethylene glycol, Polym. J., 2017, 49(8), 625–632 CrossRef CAS.
- H. Eslami, M. Ansari, A. Darvishi, H. R. Pisheh, M. Shami and F. Kazemi, Polyacrylic Acid: A Biocompatible and Biodegradable Polymer for Controlled Drug Delivery, Polym. Sci., Ser. A, 2023, 65(6), 702–713 CrossRef CAS.
- S. Connolly, S. N. Rao and D. Fitzmaurice, Characterization of Protein Aggregated Gold Nanocrystals, J. Phys. Chem. B, 2000, 104(19), 4765–4776 CrossRef CAS.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, Time Evolution of the Nanoparticle Protein Corona, ACS Nano, 2010, 4(7), 3623–3632 CrossRef CAS PubMed.
- D. Docter, D. Westmeier, M. Markiewicz, S. Stolte, S. K. Knauer and R. H. Stauber, The nanoparticle biomolecule corona: lessons learned – challenge accepted?, Chem. Soc. Rev., 2015, 44(17), 6094–6121 RSC.
- M. Mahmoudi, M. P. Landry, A. Moore and R. Coreas, The protein corona from nanomedicine to environmental science, Nat. Rev. Mater., 2023, 8(7), 422–438 CrossRef PubMed.
- M. J. Hajipour, R. Safavi-Sohi, S. Sharifi, N. Mahmoud, A. A. Ashkarran, E. Voke, V. Serpooshan, M. Ramezankhani, A. S. Milani, M. P. Landry and M. Mahmoudi, An Overview of Nanoparticle Protein Corona Literature, Small, 2023, 19(36), 2301838 CrossRef CAS PubMed.
- Z. Guo, S. Park, J. Yoon and I. Shin, Recent progress in the development of near-infrared fluorescent probes for bioimaging applications, Chem. Soc. Rev., 2014, 43(1), 16–29 RSC.
- J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, NIR dyes for bioimaging applications, Curr. Opin. Chem. Biol., 2010, 14(1), 64–70 CrossRef CAS PubMed.
- C. Sun, W. Du, B. Wang, B. Dong and B. Wang, Research progress of near-infrared fluorescence probes based on indole heptamethine cyanine dyes in vivo and in vitro, BMC Chem., 2020, 14(1), 21 CrossRef CAS PubMed.
- J. Zhao, T. Ma, B. Chang and J. Fang, Recent Progress on NIR Fluorescent Probes for Enzymes, Molecules, 2022, 27(18), 5922 CrossRef CAS PubMed.
- S. Nisar and B. Sui, A nitroreductase-sensitive near-IR fluorescent biosensor for detecting tumor hypoxia in vivo, Sens. Diagn., 2024, 3(9), 1505–1512 RSC.
- K. Ley, J. Rivera-Nieves, W. J. Sandborn and S. Shattil, Integrin-based therapeutics: biological basis, clinical use and new drugs, Nat. Rev. Drug Discovery, 2016, 15(3), 173–183 CrossRef CAS PubMed.
- M. Alipour, M. Baneshi, S. Hosseinkhani, R. Mahmoudi, A. Jabari Arabzadeh, M. Akrami, J. Mehrzad and H. Bardania, Recent progress in biomedical applications of RGD-based ligand: From precise cancer theranostics to biomaterial engineering: A systematic review, J. Biomed. Mater. Res., Part A, 2020, 108(4), 839–850 CrossRef CAS PubMed.
- E. F. Plow, T. A. Haas, L. Zhang, J. Loftus and J. W. Smith, Ligand Binding to Integrins, J. Biol. Chem., 2000, 275(29), 21785–21788 CrossRef CAS PubMed.
- Y. Sun, C. Kang, F. Liu, Y. Zhou, L. Luo and H. Qiao, RGD Peptide-Based Target Drug Delivery of Doxorubicin Nanomedicine, Drug Dev. Res., 2017, 78(6), 283–291 CrossRef CAS PubMed.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon, J. A. Hartman, I. T. Dobrucki, L. W. Dobrucki, L. B. Borst, S. Lezmi, W. G. Helferich, A. L. Ferguson, T. M. Fan and J. Cheng, Investigating the optimal size of anticancer nanomedicine, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(43), 15344–15349 CrossRef CAS PubMed.
- X. Li, E. C. Montague, A. Pollinzi, A. Lofts and T. Hoare, Design of Smart Size-, Surface-, and Shape-Switching Nanoparticles to Improve Therapeutic Efficacy, Small, 2022, 18(6), 2104632 CrossRef CAS PubMed.
- J. Xu, M. Song, Z. Fang, L. Zheng, X. Huang and K. Liu, Applications and challenges of ultra-small particle size nanoparticles in tumor therapy, J. Controlled Release, 2023, 353, 699–712 CrossRef CAS PubMed.
- J. Dolai, K. Mandal and N. R. Jana, Nanoparticle Size Effects in Biomedical Applications, ACS Appl. Nano Mater., 2021, 4(7), 6471–6496 CrossRef CAS.
- Y. Niu, J. Zhu, Y. Li, H. Shi, Y. Gong, R. Li, Q. Huo, T. Ma and Y. Liu, Size shrinkable drug delivery nanosystems and priming the tumor microenvironment for deep intratumoral penetration of nanoparticles, J. Controlled Release, 2018, 277, 35–47 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, The EPR Effect: Unique Features of Tumor Blood Vessels for Drug Delivery, Factors Involved, and Limitations and Augmentation of the Effect, Adv. Drug Delivery Rev., 2011, 63(3), 136–151 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, The EPR Effect for Macromolecular Drug Delivery to Solid Tumors: Improvement of Tumor Uptake, Lowering of Systemic Toxicity, and Distinct Tumor Imaging In Vivo, Adv. Drug Delivery Rev, 2013, 65(1), 71–79 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental conditions and methods; Fig. S1–S40 (PDF). See DOI: https://doi.org/10.1039/d5tb00806a |
| This journal is © The Royal Society of Chemistry 2025 |