
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of heme peroxo nucleophilicities with axial ligands reveal key insights into the mechanistic landscape of nitric oxide synthase†
Shanuk
Rajapakse
 a,
Yuri
Lee
b,
Samith B.
Jayawardana
a,
Joshua
Helms
a,
Pritam
Mondal
ac,
Akhil
Singh
a,
Brad S.
Pierce
*a,
Hannah S.
Shafaat
*b and
Gayan B.
Wijeratne
*a
a,
Yuri
Lee
b,
Samith B.
Jayawardana
a,
Joshua
Helms
a,
Pritam
Mondal
ac,
Akhil
Singh
a,
Brad S.
Pierce
*a,
Hannah S.
Shafaat
*b and
Gayan B.
Wijeratne
*a
aDepartment of Chemistry and Biochemistry, University of Alabama, Tuscaloosa, AL 35401, USA. E-mail: gwijeratne@ua.edu
bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, USA. E-mail: shafaat@ucla.edu
cDepartment of Chemical Sciences, Indian Institute of Science Education and Research, Mohali, Punjab 140306, India
First published on 28th April 2025
Abstract
Mid-valent heme–oxygen intermediates are central to a medley of pivotal physiological transformations in humans, and such systems are increasingly becoming more relevant therapeutic targets for challenging disease conditions. Nonetheless, precise mechanistic details pertaining to mid-valent heme intermediates as well as key structure–activity relationships remain enigmatic. To this end, this study strives to describe the influence of heme proximal ligation on the nucleophilic reactivity patterns of heme peroxo intermediates. A functional model system in which organic oxime substrates are used as N-hydroxy-L-arginine mimics reproduces the second mechanistic step of nitric oxide synthase. Our findings reveal that axial ligation of heme peroxo adducts escalates the rates of nucleophilic reactivity, wherein the anionic ligands exhibited the most pronounced “push effect”. Coordination of these axial ligands are accompanied by distinct geometric and electronic perturbations, which are supported by complementary theoretical studies. Kinetic interrogations reveal that the axially ligated heme peroxo adducts presumably mediate oxime oxidation via the same mechanism as the parent (i.e., with only solvent ligation) heme peroxo adduct, where the initial nucleophilic attack from the peroxo moiety on the oxime substrate is rate-limiting. All reaction products, including the final ketone as well as NO−, have been characterized in detail.
Introduction
Heme enzymes are the cornerstones for a broad diversity of biological functionalities, and, hence, are ubiquitous within various aerobic and anaerobic organisms.1–3 One of the key parameters that dictate the functional divergence of heme enzymes is the choice of the proximally ligating amino acid residue. Nature has meticulously designated these proximal amino acids to tailor the electronic properties of heme centers toward precise biological roles.4,5 In dioxygen-activating heme enzymes, the proximal ligands are directly involved in orchestrating the extent of dioxygen reduction, as well as in governing the attributes of heme oxygen intermediates that modulate their reactivities. For example, in heme enzymes where dioxygen activation leads to O–O bond cleavage giving high-valent intermediates such as Compound-I (i.e., (P˙+)FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (Cmpd-I); formed via heterolytic O–O bond scission (P = porphyrin supporting ligand)) or Compound-II (i.e., (P)FeIVO (Cmpd-II); formed via homolytic O–O bond scission or reduction of Cmpd-I), strongly donating anionic ligands typically occupy the heme proximal site (e.g., cysteinate, tyrosinate, or histidinate/imidazolate (Fig. 1A, C and D)).1,6,7 In contrast, in oxygen carrier proteins (e.g., hemoglobin or myoglobin) or heme dioxygenase enzymes (e.g., indoleamine/tryptophan 2,3-dioxygenases), where O–O bond cleavage is initially unwarranted, a neutral ligand (e.g., histidine (Fig. 1B)) resides at the proximal site.1–3,8–11 Moreover, non-covalent interactions with these proximally ligated amino acids further modulate their electronic properties, which Nature has evolutionarily optimized to fine-tune the chemistry carried out at a specific heme center (see Fig. 1A–D dotted lines).12–16 Correspondingly, intrinsic dissimilarities in the kinetics and thermodynamics of formation as well as fundamental structural, spectroscopic, and reactivity properties exist across various heme–oxygen intermediates, often paralleling the donor ability of the proximally ligating amino acid side chain.17–23 In light of these trends, the influence of various axial ligands on the geometric and electronic properties of high-valent heme intermediates, such as Cmpd-I or Cmpd-II, has been well-established utilizing both enzymatic systems as well as their synthetic models.24–34 Nonetheless, how those same parameters influence the reactivities of mid-valent heme oxygen intermediates (i.e., Fe(III) containing adducts such as heme superoxo, peroxo, hydroperoxo, or alkylperoxo) are far less understood.35–41 This lack of systematic understanding of structure–function relationships is notable, particularly in light of the growing body of evidence on heme systems where heme peroxo species are either active oxidants or key intermediates being pivotal to challenging disease situations in humans (e.g., COVID-19, autoimmune and/or neurodegenerative conditions, and cancer).42–46 These include nitric oxide synthase (NOS),47 aromatase (CYP19A1),48 sterol 14α-demethylase (CYP51),49 cytochrome P450 17α-hydroxylase-17,20-lyase (CYP17A1),50 and non-canonical heme oxygenase (IsdG-type),51 which have emerged as multifaceted therapeutic targets in recent years; consequently, identifying potential inhibitory pathways of these systems are of prime interest for rational drug design.
O (Cmpd-I); formed via heterolytic O–O bond scission (P = porphyrin supporting ligand)) or Compound-II (i.e., (P)FeIVO (Cmpd-II); formed via homolytic O–O bond scission or reduction of Cmpd-I), strongly donating anionic ligands typically occupy the heme proximal site (e.g., cysteinate, tyrosinate, or histidinate/imidazolate (Fig. 1A, C and D)).1,6,7 In contrast, in oxygen carrier proteins (e.g., hemoglobin or myoglobin) or heme dioxygenase enzymes (e.g., indoleamine/tryptophan 2,3-dioxygenases), where O–O bond cleavage is initially unwarranted, a neutral ligand (e.g., histidine (Fig. 1B)) resides at the proximal site.1–3,8–11 Moreover, non-covalent interactions with these proximally ligated amino acids further modulate their electronic properties, which Nature has evolutionarily optimized to fine-tune the chemistry carried out at a specific heme center (see Fig. 1A–D dotted lines).12–16 Correspondingly, intrinsic dissimilarities in the kinetics and thermodynamics of formation as well as fundamental structural, spectroscopic, and reactivity properties exist across various heme–oxygen intermediates, often paralleling the donor ability of the proximally ligating amino acid side chain.17–23 In light of these trends, the influence of various axial ligands on the geometric and electronic properties of high-valent heme intermediates, such as Cmpd-I or Cmpd-II, has been well-established utilizing both enzymatic systems as well as their synthetic models.24–34 Nonetheless, how those same parameters influence the reactivities of mid-valent heme oxygen intermediates (i.e., Fe(III) containing adducts such as heme superoxo, peroxo, hydroperoxo, or alkylperoxo) are far less understood.35–41 This lack of systematic understanding of structure–function relationships is notable, particularly in light of the growing body of evidence on heme systems where heme peroxo species are either active oxidants or key intermediates being pivotal to challenging disease situations in humans (e.g., COVID-19, autoimmune and/or neurodegenerative conditions, and cancer).42–46 These include nitric oxide synthase (NOS),47 aromatase (CYP19A1),48 sterol 14α-demethylase (CYP51),49 cytochrome P450 17α-hydroxylase-17,20-lyase (CYP17A1),50 and non-canonical heme oxygenase (IsdG-type),51 which have emerged as multifaceted therapeutic targets in recent years; consequently, identifying potential inhibitory pathways of these systems are of prime interest for rational drug design.
 | ||
| Fig. 1 Active site structures of (A) nitric oxide synthase (PDB: 1NSI52), (B) deoxy-hemoglobin (PDB: 4HHB53), (C) catalase (PDB: 2IQF54), and (D) peroxidase (PDB: 1ATJ55) which encompass cysteinate, histidine, tyrosinate, and histidinate/imidazolate axially ligated amino acid residues, respectively. (E) Sequential reactions proposed to be catalyzed by nitric oxide synthase, transforming L-arginine into L-citrulline and ˙NO. | ||
In particular, NOS has been implicated in a wide variety of pathogenic situations, from neurodegenerative disorders (e.g., Alzheimer's,56 Parkinson's,57 Huntington's58,59 diseases, amyotrophic lateral sclerosis60) to a variety of cancers (e.g., breast,61,62 prostate,63 colorectal,64 cervical,65 lung,66 brain,67,68 colon,69 ovarian70), due to both its proposed direct involvement in prognosis and/or the detrimental effects imparted by its main reaction product, nitric oxide (˙NO).71,72 Indeed, NOS is central to the primary pathway that generates ˙NO in humans via arginine degradation under aerobic conditions.73 ˙NO serves as a crucial signaling agent within the physiological space, with paramount roles in blood pressure regulation and cell signaling, among others.74 Nonetheless, elevated ˙NO concentrations can impart a multitude of harmful effects via the elevation of nitrative and nitrosative stress, leading to undesirable protein nitration and disruption of key cell signaling pathways, often resulting in apoptosis.75 Accordingly, NOS inhibitors have acquired significant momentum in recent years as highly potent therapeutic agents.76–78 However, their rational design is impeded by the lack of a precise understanding into the NOS mechanism.79 Some proposals exist wherein NOS has been proposed to operate via a two-step mechanism; the first involves the monooxygenation of L-arginine to N-hydroxy-L-arginine (NHA), followed by further oxidation of NHA to L-citrulline in the second step, with the concomitant production of ˙NO (Fig. 1E).47,74,80–83 Exact details pertaining to the latter step have been under debate, where several mechanistic propositions have been put forward, implicating largely different heme-based active oxidants. These involve both high-valent and mid-valent oxidants, although heme ferric peroxo or heme superoxo (i.e., mid-valent) based proposals have been heavily reproduced in the literature, while also being corroborated by distinct experimental observations.1,84–91 Interestingly, when heme peroxo is the active oxidant, the final NOx product is nitroxide (NO−). We have recently utilized synthetic model systems of both heme peroxo (heme–PO) and superoxo intermediates to shine light on the relevant mechanistic ambiguities, where we demonstrated that heme–PO adducts can indeed oxidize oxime substrates (i.e., as NHA mimics) in a bioinspired fashion, producing the corresponding ketone and nitroxide anions (NO−).92 Moreover, the rate-limiting step of this reaction landscape was found to be the initial nucleophilic attack mediated by the heme–PO adduct on the oxime substrate. Nonetheless, key unknowns still exist with respect to the exact structure–activity relationships of both the NOS heme center as well as the substrate that precisely choreograph the feasibility and efficiency of this unique mechanistic step.
Synthetic model complexes can be powerful tools in addressing such mechanistic uncertainties, mainly owing to a medley of unique advantages (e.g., the feasibility of (i) precise structural alterations and (ii) detailed mechanistic investigations under cryogenic conditions, etc.) inherent to such systems compared to their biological/enzymatic counterparts.93 In this study, we evaluate the effect of proximal ligands on the reactivity patterns of heme–PO adducts, especially highlighting the details that pertain to the second mechanistic step of NOS. In that, we utilize a series of axially ligated (either neutral or anionic ligands) synthetic heme–PO oxidants, wherein the donor groups are biologically relevant. Therein, we utilize acetophenone oxime as the NHA mimic substrate (Scheme 1). Intriguingly, the nucleophilic reactivity between heme–PO adducts and oxime substrate was observed to be tightly regulated by the identity of the proximal ligand on heme, where increased donor abilities (i.e., the “push effect”) led to faster kinetic rates. Furthermore, we herein demonstrate for the first time how key thermodynamic and kinetic parameters of heme–PO driven reactivities are affected by various axial ligands, in turn resulting in measurable differences in substrate reaction rates. Systematic studies that evaluate the effect of axial ligands on bio-relevant reactivity landscapes of heme–PO intermediates (in either heme or non-heme biomimetic platforms) are sorely lacking in the contemporary literature, which underscores the importance of this current study.
 | ||
| Scheme 1 Generalized oxime oxidation reaction scheme facilitated by heme peroxo intermediates and the divergent exogenously added axial ligands evaluated in this study. | ||
Results and discussion
Formation of axially ligated heme peroxo adducts
In order to survey the effects of various axial ligands on heme–PO driven nucleophilic reactivities, herein we utilize the ferric peroxo complex, [(THF)(TPP)FeIII(O22−)]−, where TPP = 5,10,15,20-tetraphenylporphyrin.92 The preparation and spectroscopic characterization of the side-on ferric peroxo species, [(THF)(TPP)FeIII(O22−)]− were carried out as previously described,10,92 and the spectroscopic characterization details are in excellent agreement with those reports. In particular, the prominent electronic absorption features of the starting ferrous complex, [(THF)2(TPP)FeII], centered at 426 (Soret; ε = 3.4 × 105 M−1 cm−1), 538 (ε = 1.08 × 104 M−1 cm−1) and 558 (ε = 1.1 × 104 M−1 cm−1) nm shifted to 416 (Soret; ε = 2.0 × 105 M−1 cm−1) and 540 (ε = 1.58 × 104 M−1 cm−1) nm upon its dioxygenation, converting into at −40 °C in 9
at −40 °C in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DCM:THF solvent mixture (Fig. 2A). Its subsequent reduction with 1 equiv. of cobaltocene led to the formation of the corresponding side-on ferric peroxo species, [(THF)(TPP)FeIII(O22−)]−, with electronic absorption features at 436 (Soret; ε = 2.7 × 105 M−1 cm−1) and 564 (ε = 2.1 × 104 M−1 cm−1) nm. The neutral or anionic axial ligands (e.g., 4-methylimidazole (4-MeIm), 1,5-dicyclohexylimidazole (DCHIm), 4-dimethylaminopyridine (4-DMAP), thiophenolate (ArS−), and 3,5-dimethoxyphenolate (ArO−); see Scheme 1) were then introduced into [(THF)(TPP)FeIII(O22−)]− in order to prepare the axially ligated heme–PO adducts.94 In that, the addition of 2 equiv. of 4-MeIm into a solution of [(THF)(TPP)FeIII(O22−)]− in 9:1 DCM:THF at −40 °C led to the formation of the corresponding axially ligated ferric peroxo species, [(4-MeIm)(TPP)FeIII(O22−)]−, which was accompanied by only minor electronic absorption perturbations with main features observed at 436 (Soret; ε = 2.5 × 105 M−1 cm−1) and 565 (ε = 2.0 × 104 M−1 cm−1) nm. EPR spectroscopic analysis of [(4-MeIm)(TPP)FeIII(O22−)]− resulted in a prominent feature at g = 4.2, which again resembles that of the parent heme–PO species, [(THF)(TPP)FeIII(O22−)]− (Fig. S2†); this is indicative of a high-spin rhombic heme Fe(III) center in [(4-MeIm)(TPP)FeIII(O22−)]−. Nonetheless, the isotope-sensitive ν(Fe–O) and ν(O–O) resonance Raman features of [(THF)(TPP)FeIII(O22−)]− shifted from 472 (Δ18O2 = −21) cm−1 and 807 (Δ18O2 = −44) cm−1 (Fig. S3A†) to 479 (Δ18O2 = −23) cm−1 and 803 (Δ18O2 = −47) cm−1, respectively (Fig. 2B), upon the axial ligation of 4-MeIm, implicating an effective strengthening of the Fe–O bond of heme–PO, and a corresponding slight weakening of the O–O bond. This change in bonding character reflects an inflow of electron density into the π*O–O(peroxo) manifold (i.e., backbonding from the heme Fe center) upon axial ligation.95 We argue that despite these subtle electronic perturbations, the heme–PO unit still remains side-on bound to the heme center, particularly due to the lack of any isotope-sensitive Fe–O stretching frequencies in the 550–600 cm−1 region,96–98 which would be indicative of an end-on configuration. Our spectroscopic findings align closely with the previous heme–PO model complex, [(TMPIm)FeIII(O22−)]− (TMPIm = 3-imidazole-1-ylmethyl-N-{2-[10,15,20-tris-(2,4,6-trimethyl-phenyl)-porphyrin-5-yl]-phenyl}-benzamidel),95 which encompassed a covalently linked imidazole axial ligand, and was described as a seven-coordinate side-on heme–PO species with a high-spin (S = 5/2) ferric center (Table S1†).95
:1 DCM:THF solvent mixture (Fig. 2A). Its subsequent reduction with 1 equiv. of cobaltocene led to the formation of the corresponding side-on ferric peroxo species, [(THF)(TPP)FeIII(O22−)]−, with electronic absorption features at 436 (Soret; ε = 2.7 × 105 M−1 cm−1) and 564 (ε = 2.1 × 104 M−1 cm−1) nm. The neutral or anionic axial ligands (e.g., 4-methylimidazole (4-MeIm), 1,5-dicyclohexylimidazole (DCHIm), 4-dimethylaminopyridine (4-DMAP), thiophenolate (ArS−), and 3,5-dimethoxyphenolate (ArO−); see Scheme 1) were then introduced into [(THF)(TPP)FeIII(O22−)]− in order to prepare the axially ligated heme–PO adducts.94 In that, the addition of 2 equiv. of 4-MeIm into a solution of [(THF)(TPP)FeIII(O22−)]− in 9:1 DCM:THF at −40 °C led to the formation of the corresponding axially ligated ferric peroxo species, [(4-MeIm)(TPP)FeIII(O22−)]−, which was accompanied by only minor electronic absorption perturbations with main features observed at 436 (Soret; ε = 2.5 × 105 M−1 cm−1) and 565 (ε = 2.0 × 104 M−1 cm−1) nm. EPR spectroscopic analysis of [(4-MeIm)(TPP)FeIII(O22−)]− resulted in a prominent feature at g = 4.2, which again resembles that of the parent heme–PO species, [(THF)(TPP)FeIII(O22−)]− (Fig. S2†); this is indicative of a high-spin rhombic heme Fe(III) center in [(4-MeIm)(TPP)FeIII(O22−)]−. Nonetheless, the isotope-sensitive ν(Fe–O) and ν(O–O) resonance Raman features of [(THF)(TPP)FeIII(O22−)]− shifted from 472 (Δ18O2 = −21) cm−1 and 807 (Δ18O2 = −44) cm−1 (Fig. S3A†) to 479 (Δ18O2 = −23) cm−1 and 803 (Δ18O2 = −47) cm−1, respectively (Fig. 2B), upon the axial ligation of 4-MeIm, implicating an effective strengthening of the Fe–O bond of heme–PO, and a corresponding slight weakening of the O–O bond. This change in bonding character reflects an inflow of electron density into the π*O–O(peroxo) manifold (i.e., backbonding from the heme Fe center) upon axial ligation.95 We argue that despite these subtle electronic perturbations, the heme–PO unit still remains side-on bound to the heme center, particularly due to the lack of any isotope-sensitive Fe–O stretching frequencies in the 550–600 cm−1 region,96–98 which would be indicative of an end-on configuration. Our spectroscopic findings align closely with the previous heme–PO model complex, [(TMPIm)FeIII(O22−)]− (TMPIm = 3-imidazole-1-ylmethyl-N-{2-[10,15,20-tris-(2,4,6-trimethyl-phenyl)-porphyrin-5-yl]-phenyl}-benzamidel),95 which encompassed a covalently linked imidazole axial ligand, and was described as a seven-coordinate side-on heme–PO species with a high-spin (S = 5/2) ferric center (Table S1†).95
 | ||
Fig. 2 (A) UV-vis spectra (in 9:1 DCM:THF at −40 °C) for 50 μM solutions of [(THF)2(TPP)FeII] (black), 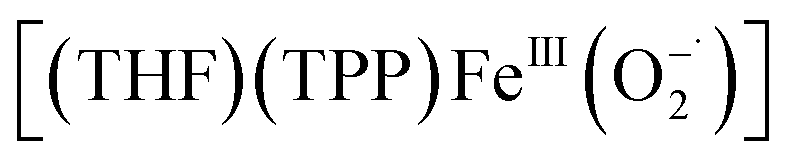 (red), [(THF)(TPP)FeIII(O22−)]− (pink), and [(4-MeIm)(TPP)FeIII(O22−)]− (green). Inset shows the expanded Q-band region. (B) Resonance Raman spectra (λex = 457 nm) collected for a 2 mM 9:1 DCM:THF frozen solution sample of [(4-MeIm)(TPP)FeIII(O22−)]− prepared with 16O2(g) (light green) and 18O2(g) (dark green); the difference spectrum is shown in black. (C) 2H NMR spectra (in 9:1 DCM:THF at −40 °C) for [(THF)(TPP-d8)FeIII(O22−)]− (pink), and [(4-MeIm)(TPP-d8)FeIII(O22−)]− (green); *peaks correspond to the solvent. (red), [(THF)(TPP)FeIII(O22−)]− (pink), and [(4-MeIm)(TPP)FeIII(O22−)]− (green). Inset shows the expanded Q-band region. (B) Resonance Raman spectra (λex = 457 nm) collected for a 2 mM 9:1 DCM:THF frozen solution sample of [(4-MeIm)(TPP)FeIII(O22−)]− prepared with 16O2(g) (light green) and 18O2(g) (dark green); the difference spectrum is shown in black. (C) 2H NMR spectra (in 9:1 DCM:THF at −40 °C) for [(THF)(TPP-d8)FeIII(O22−)]− (pink), and [(4-MeIm)(TPP-d8)FeIII(O22−)]− (green); *peaks correspond to the solvent. | ||
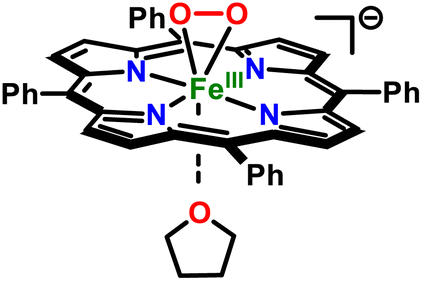
The high-spin nature of the axially ligated heme–PO adduct, [(4-MeIm)(TPP)FeIII(O22−)]−, contrasts with heme/copper peroxo adducts of cytochrome c oxidase model compounds, where the addition of axial ligands (e.g., DCHIm) flips the spin state of the heme iron center from high-spin to low-spin, with the concomitant interconversion of side-on bound peroxo moiety (with respect to heme) to end-on.99 Nonetheless, these differences should be viewed within the caveat that the copper(II) center in those model systems presumably imparts a significant “pull effect”, which is absent in heme-only systems. This phenomenon has also been discussed by Naruta and co-workers,98 where both heme (1) secondary sphere interactions (i.e., “pull effect”), as well as (2) axial ligation/donation (“push effect”) were deemed to play a key role in the formation of truly end-on low-spin heme–PO adducts (Table S1†). This finding nicely complements the analogous biological systems, where the intricate interplay between the primary and secondary coordination sphere fine-tunes the salient geometric and electronic parameters vital for their functionality.1 Moreover, in previous heme–PO models with tethered axial ligands (i.e., TMPIm vs. F8TPPIm (or PIm)), the spin state of the iron center was observed to be influenced by the electronic properties of the meso-substituents, wherein electron-donating groups gave rise to high-spin peroxo complexes,95 while electron-deficient meso-substituents have predominantly led to low-spin systems (Table S1†).97 Despite the lack of large perturbations in the peroxo binding mode of [(4-MeIm)(TPP)FeIII(O22−)]−, sizable electronic modifications within the heme peroxo moiety were clearly evident in NMR spectroscopy. The pyrrole-position deuterated TPP-d8 porphyrinate system was utilized to evaluate these heme–PO systems by 2H NMR spectroscopy, where the parent heme–PO adduct, [(THF)(TPP-d8)FeIII(O22−)]−, exhibited a single 2H NMR feature at δpyrrole = 38.5 ppm. This feature completely shifted upfield to δpyrrole = 9.1 ppm (Fig. 2C) upon axial ligation of 4-MeIm. Similar changes in NMR were also observed upon 4-DMAP and DCHIm ligation, where the final 2H NMR features were observed at δpyrrole = 12.5 ppm and 9.0 ppm, respectively (Fig. S4†). Given the relevance of proximal thiolate ligation in NOS, we also utilized strongly-donating thiophenolate as an axial ligand (Scheme 1), where the electronic absorption features of [(ArS−)(TPP)FeIII(O22−)]2− were centered at 436 nm (Soret; ε = 2.5 × 105 M−1 cm−1) and 564 nm (ε = 1.6 × 104 M−1 cm−1; Fig. S5A†). Intriguingly, attributes of [(ArS−)(TPP)FeIII(O22−)]2− was observed to be somewhat unique, exhibiting EPR spectroscopic features consistent with a mixture of high- and low-spin ferric sites. As shown in Fig. S5B,† two high spin ferric sites (S = 5/2) can be identified at low field. The sharp feature at g ∼6 (■) represents the g┴ -region of a near axial (E/D = 0.005) heme site. This feature is most prominent at low temperature as this resonance originates from the ground doublet of a S = 5/2 manifold. The second transition observed at g ∼4.2 (□) is attributed to a highly rhombic (E/D = 0.27) high-spin ferric site. This signal originates from a transition within the middle doublet of the S = 5/2 spin system. Consequently, the decrease in its signal intensity with increasing temperature is much less pronounced relative to the axial site. The temperature dependent signal intensity is diagnostic of the Boltzmann population distribution for each doublet. Therefore, the magnitude of the axial zero field splitting parameter (D) was determined for both species by matching the simulated and experimental signal intensity observed across multiple temperatures between 4 and 20 K (Fig. S5B†). Among the other spectroscopic parameters provided in Table S2,† the D-values for the axial (■) and rhombic (□) high-spin ferric sites were determined to be 8.0 ± 1.0 and 0.9 ± 0.1 cm−1, respectively. Notably, the high-spin axial EPR signature at g ∼6 (■) parallels previous reports describing heme ferric hydroxide species, and the rhombic g ∼4.2 (□) is the starting heme peroxo complex. Taking this into consideration, we can state that the parent heme peroxo complex accounts for over 30% of the observed iron whereas the ferric hydroxide species accounts for less than 4%. The two rhombic signals near g ∼2 are consistent with low-spin (S = 1/2) ferric sites (Fig. S5B†), which originate from the low-spin heme peroxo complex (●; g = 2.28, 2.13, and 1.97; Table S1† and vide infra) and its decay product (○; g = 2.46, 2.03, 1.91),95 where the former accounts for 40% of the observable iron in the sample. This speciation observed in EPR of [(ArS−)(TPP)FeIII(O22−)]2− reflects the high reactivity of this complex. Thus, we infer that the donor ability of axial ligands is key, wherein the superior donor in the series, ArS−, results in the strongest ligand-field at the heme iron center, accordingly resulting in an equilibrium mixture of high-spin and low-spin heme–PO species.
Resonance Raman characterization of [(ArS−)(TPP)FeIII(O22−)]2− is consistent this finding, where the isotope-sensitive ν(Fe–O) feature was observed at 484 cm−1 (Δ18O2 = −21 cm−1) (Fig. S3B†), indicative of greater Fe–O interaction when compared to the parent THF-bound species. In further support of relative increases in Fe–O bond strength, the ν(Fe–O) features of [(ArO−)(TPP)FeIII(O22−)]2− and [(ArO−)(F20TPP)FeIII(O22−)]2− were observed at 474 (Δ18O2 = −23) cm−1 and 492 (Δ18O2 = −18) cm−1, respectively. Unfortunately for spectroscopic characterization purposes, the addition of anionic axial ligands destabilizes the peroxo adducts, preventing the precise assignment of their isotope-sensitive ν(O–O) bands (Fig. S3B–D†). This trend in axial ligand donor abilities clearly dictates the observed second-order reaction rates of corresponding heme–PO adducts (vide-infra), where stronger axial donors lead to more nucleophilic peroxo units, and thereby resulting in faster reaction rates.
Oxime oxidation reactivities of axially ligated heme peroxo adducts
Our recent work has demonstrated that heme–PO intermediates can oxidize oxime substrates leading to the corresponding ketone with the simultaneous production of nitroxide anion (i.e., NO−), which marks the only functional heme model for the second mechanistic step of NOS.92 In this work, we set out to interrogate how this unique reactivity between heme–PO adducts and the oxime is modulated by the ligation of aforementioned axial ligands, and thereby define how key reactivity parameters are altered accordingly. Thus, we carried out rigorous reactivity studies into the nucleophilic reactivities of a series of [(B)(TPP)FeIII(O22−)]− adducts (where B = axially coordinated ligands shown in Scheme 1), with acetophenone oxime as the NHA mimic substrate. When 200 equiv. of acetophenone oxime was introduced into a 50 μM solution of [(4-MeIm)(TPP)FeIII(O22−)]− in 9:1 DCM:THF at −40 °C, prominent changes in electronic absorption were observed from 436 to 421 nm (Soret; ε = 1.4 × 105 M−1 cm−1) and 565 to 550 nm (ε = 2.5 × 103 M−1 cm−1; Fig. 3A). The final heme product of this reaction was characterized to be the corresponding six-coordinate heme ferric hydroxo adduct, [(4-MeIm)(TPP)FeIII(OH)] (Fig. 3A). When other axial ligands were employed, the final heme species was observed to be the five-coordinate heme ferric hydroxo adduct, [(TPP)FeIII(OH)] (Fig. 3B & S6†). Spectroscopic features of the five-coordinate [(TPP)FeIII(OH)] final species are in close agreement with those of an authentic standard reported previously (Fig. S7†).92 EPR spectroscopy confirms the low-spin rhombic ferric center of [(4-MeIm)(TPP)FeIII(OH)] with features centered at g = 2.38, 2.17 and 1.90 (Fig. S8A†). 2H NMR analysis of this [(4-MeIm)(TPP-d8)FeIII(OH)] final product resulted in a single resonance at δpyrrole = −18 ppm (Fig. S8B†). These characterization details are in excellent agreement with those of an authentically prepared standard of [(4-MeIm)(TPP-d8)FeIII(OH)] (Fig. S9†), further solidifying its identity. The final organic product, acetophenone, was characterized via1H NMR and GC-MS (Fig. S10 and S11A†), revealing a yield of ∼34%. [(4-DMAP)(TPP)FeIII(O22−)]− and [(ArO−)(TPP)FeIII(O22−)]2− resulted in similar acetophenone yields of 34% and 37%, respectively (Fig. S11B†). Notably, these yields of acetophenone are comparable to those observed for the parent heme–PO complex under identical reaction conditions.92 Finally, the formation of nitroxide (NO−) was determined by carrying out oxime oxidation in the presence of an excess of PPh3,88,92 where LC-MS analysis confirmed the formation of both triphenylphosphine oxide (O=PPh3; ∼20% yield (Fig. S12B and C†)) and triphenylphosphine aza-ylide (HN = PPh3; Fig. S12A†).
 | ||
| Fig. 3 (A) Electronic absorption spectral changes observed (in 9:1 DCM: THF at −40 °C) during the reaction of a 50 μM solution of (A) [(4-MeIm)(TPP)FeIII(O22−)]− and (B) [(4-DMAP)(TPP)FeIII(O22−)]− with 200 equiv. of acetophenone oxime (green: initial six coordinate heme–PO complex; blue: final ferric product). Insets show the expanded Q-band regions, and arrows indicate the direction of peak transition. | ||
In light of our previous findings describing an initial nucleophilic attack mediated by the heme–PO adduct on oxime substrate being rate-limiting, one would expect axially ligated heme–PO adducts to mediate oxime oxidation reactions much faster compared to their parent peroxo compound (i.e., with only weak solvent ligation).35 To test this hypothesis, we carried out kinetic analysis into acetophenone oxime oxidation reactions facilitated by the series of [(B)(TPP)FeIII(O22−)]− adducts. In that, the pseudo-first-order kinetic rates were computed upon the addition of 200–500 equiv. of the substrate in DCM:THF 9:1 at −40 °C. In all cases, the reaction rates (kobs) were observed to increase linearly as a function of substrate concentration (Fig. S13†), indicating the rate-limiting nature of initial heme–PO attack on the oxime substrate. Hence, the reaction mechanism presumably remains unaltered as compared with the parent heme–PO adduct. Accordingly, the second-order kinetic rates (k2) resulting from these studies illustrated an unequivocal relationship with the axial ligand donor abilities, wherein the stronger anionic donor ligands led to faster oxime oxidation rates (Table 1). Particularly, the neutral axial ligands, 4-MeIm, DCHIm, and 4-DMAP (Scheme 1), wherein the donor abilities are known to increase in the same order, exhibited second-order oxime oxidation reaction rates of 0.79 ± 0.06, 0.87 ± 0.02, and 1.11 ± 0.08 M−1 s−1, respectively (Fig. S13†). Interestingly, all of these rates are faster than that of the parent heme–PO adduct reported under identical conditions (0.26 ± 0.01 M−1 s−1). When anionic axial ligands, ArO− and ArS− (Scheme 1), were utilized, these rates escalated even further, up to 1.45 ± 0.07 and 1.53 ± 0.04 M−1 s−1, respectively (Fig. S13†). Thus, the strongest axial donor within the series, ArS− (soft base), most enhanced the nucleophilicity of the heme–PO moiety, resulting in the fastest oxime oxidation reaction within the series.
This “push effect” of the axial ligand can be further clarified in terms of the orbital overlap between the donor and the acceptor (i.e., axial ligand and Fe center, respectively).106 Metal–ligand interactions can be either σ, π, or a combination of both in nature, and the pKa of the conjugate acid provides a reasonable handle for surveying the extent of σ-donation resulting from a given ligand. The observed increase in oxime oxidation rates in this case show a nice correlation with the corresponding pKa values of the axially ligating neutral (i.e., only σ-donor) ligands (Fig. 4 and Table 1). Moreover, the anionic axial ligands have π-donor properties as well, where the lone pairs on the donor atom (i.e., O and S in ArO− and ArS−, respectively) can be donated to the metal center via a px–dπ type overlap (Fig. 4; S13† and Table 1).107 Our theoretical analysis lends credence to such a π-donation from the thiophenolate axial ligand, where it employs an orientation (with respect to the heme ring) that maximizes a px–dπ type overlap, while minimizing steric interactions with the meso-phenyl substituents on heme (Fig. 7; vide infra). Variable temperature kinetic (Eyring) analysis for acetophenone oxime oxidation by the parent heme–PO, [(THF)(TPP)FeIII(O22−)]−, as well as the axially ligated heme–PO adducts, [(4-MeIm)(TPP)FeIII(O22−)]− and [(ArO−)(TPP)FeIII(O22−)]2−, was then conducted in a 9:1 DCM:THF solvent mixture (Fig. S14†). Evidently, both enthalpy and entropy of activation (i.e., ΔH≠ and ΔS≠) increase upon axial ligation of 4-MeIm and ArO− to [(TPP)FeIII(O22−)]− (Fig. 5 and S15†). A similar study was recently conducted for a Cmpd-I analog, where axial ligation of an anionic cyanide ligand led to more positive ΔS≠ and lowered ΔH≠ values, ultimately resulting in a diminished activation barrier when compared to its five-coordinate analog toward oxygen atom transfer substrates.108
 | ||
| Fig. 4 The correlation between logk2 values for [(B)(TPP)FeIII(O22−)]− mediated acetophenone oxime oxidation reactions (in 9:1 DCM:THF at −40 °C) and the pKa values of conjugate acids of the axially ligating B ligands, where B = THF (magenta), 4-MeIm (green), DCHIm (purple), 4-DMAP (orange), ArO− (blue) and ArS− (brick red). | ||
 | ||
| Fig. 5 Eyring analysis data showing the dependence of ln(k/T) on 1/T for the reaction between a 50 μM solution of [(ArO−)(TPP)FeIII(O22−)]2− and acetophenone oxime at −40, −50, −60 and −70 °C. Solvent = 9:1 DCM:THF. | ||
Electronic structure characterization of axially ligated heme–PO adducts
Given the significant reactivity perturbations observed in heme–PO adducts upon axial ligation, we investigated whether such modifications could be utilized to ‘turn-on’ the nucleophilic reactivity of an otherwise inert,35,92 electron-deficient heme–PO adduct, [(THF)(F20TPP)FeIII(O22−)]− (where F20TPP = 5,10,15,20-tetra(pentafluorophenyl)porphyrin). To this end, we prepared various axially ligated [(B)(F20TPP)FeIII(O22−)]− complexes, where B = 4-MeIm, DCHIm, 4-DMAP, ArS− (Fig. S16†). Interestingly, EPR investigations into this series revealed that the [(4-MeIm)(F20TPP)FeIII(O22−)]− adduct maintains its rhombic high-spin iron center even after the axial ligation of 4-MeIm (Fig. S17A†). In contrast, the neutral yet stronger donor ligands, 4-DMAP and DCHIm, as well as the anionic ArS− all led to a mixture of two distinct high-spin and low-spin species as observed by EPR (Fig. S17B, C,† and 6A). Both low field transitions observed in Fig. 6A (i.e., for [(ArS−)(F20TPP)FeIII(O22−)]2−) can be attributed to the high-spin side-on Fe(III)-η2-peroxo species. Accordingly, the weak resonance observed at g ∼9.2 and the intense signal at 4.2 are assigned to transitions within the ground and middle doublet of a S = 5/2 spin manifold, respectively. In the absence of inter-doublet mixing, the observed position of these g-values is consistent with a highly rhombic zero-field splitting (E/D ∼0.24) term. As the side-on (η2) coordination of the peroxo ligand is expected to have considerable overlap with Fe(III) dp-orbitals (dxz and dyz), the high rhombicity of [(ArS−)(F20TPP)FeIII(O22−)]2− is not unexpected.109–112 Indeed, similar rhombicity and high-spin electronic configuration appears to be a shared characteristic among crystallographically and spectroscopically characterized Fe(III)- η2-peroxo complexes.95,113 Similar to the aforementioned EPR characterization of [(ArS−)(TPP)FeIII(O22−)]2−, the axial zero-field splitting term for the high-spin fraction of [(ArS−)(F20TPP)FeIII(O22−)]2− (D = 0.6 ± 0.1 cm−1) was obtained by simultaneous simulation of EPR spectra collected at temperatures ranging from 4 to 20 K (Fig. S18†). Within this temperature regime, EPR simulations faithfully reproduce the relative intensity of transitions within the ground and first excited state. The quantitative simulations (Fig. 6A, Sim) indicate that the high-spin ferric site accounts for 82% of the observable spin concentration (i.e., ∼1.6 mM). The low-spin (S = 1/2) [(ArS−)(F20TPP)FeIII(O22−)]2− complex has nearly axial symmetry, with observed g-values of 2.31, 2.29, and 1.92. Analytical simulations of the low-spin Fe(III)-site account for ∼18% of the observed spin concentration (0.4 mM). Relative to the rhombic g-values observed for the low-spin side-on peroxo complexes produced with TPP (2.28, 2.13, and 1.97), the axial g-values observed for [(ArS−)(F20TPP)FeIII(O22−)]2− suggest a change in the F20TPP ligand field strength, thereby altering the relative energies of Fe(III) t2g d-orbitals.114–116 This is consistent with the differing D-values measured for the high-spin Fe(III)-η2-peroxo species for TPP and F20-TPP. The 2H NMR spectrum of [(THF)(F20TPP-d8)FeIII(O22−)]− in DCM:THF 9:1 at −40 °C exhibited a single feature at δpyrrole = 38.7 ppm, which shifted upfield to δpyrrole = 8.8 ppm upon ligation of 4-MeIm (Fig. S19†). On the contrary, [(ArS−)(F20TPP-d8)FeIII(O22−)]2− indicated a mixture of 2H NMR features at δpyrrole = 38.7 and −2.1 ppm (Fig. 6B), which closely parallels our EPR characterization. Furthermore, such δpyrrole2H NMR features below 0 ppm are indicative of low-spin heme ferric systems, as described in our previous work.10 These findings, therefore, suggest clear ‘activation’ of the electron-deficient heme–PO adduct upon the ligation of strong axial donor ligands. Oxime oxidation reactivity studies of this series of [(B)(F20TPP)FeIII(O22−)]− adducts nicely echo our spectroscopy-based interpretation, where [(4-MeIm)(F20TPP)FeIII(O22−)]− was found to be inert toward acetophenone oxime, while both [(DCHIm)(F20TPP)FeIII(O22−)]− and [(ArS−)(F20TPP)FeIII(O22−)]2− reacted (Fig. S20†), with the latter leading to a reaction rate of 0.03 s−1 at −40 °C with 500 equiv. of substrate (Fig. S20B†). Similar ‘turn-on’ of reactivity of [(F20TPP)FeIII(O22−)]− has previously been reported by Valentine and co-workers with regard to alkene epoxidation reactivity, where axial ligation of DMSO solvent was postulated.35
 | ||
| Fig. 6 (A) 7 K CW EPR spectra of [(ArS−)(F20TPP)FeIII(O22−)]2− (2 mM) dissolved in 9:1 DCM:THF (light green), and quantitative EPR simulations for high- and low-spin ferric sites (dashed blue). The isotropic feature (*) at g ∼2 is from residual (<1 μM) cobaltacene. (B) 2H NMR spectrum (in 9:1 DCM:THF at −40 °C) for [(ArS−)(F20TPP-d8)FeIII(O22−)]2− indicating the presence of both high- and low-spin heme–PO adducts (**peaks correspond to the solvent). | ||
Computational studies
To further clarify the precise geometric and electronic modifications imposed on heme–PO adducts upon axial ligand coordination, we carried out theoretical investigations using Density Functional Theory (DFT). Therein, optimized ground state geometries of [(THF)(TPP)FeIII(O22−)]−, and a series of axially ligated [(B)(TPP)FeIII(O22−)]− complexes (where B = THF, 4-MeIm, ArO− or ArS−; see Fig. 7 and S21†) over three spin surfaces (S = 5/2, 3/2, 1/2) were probed. Fig. S22† illustrates all spin state energies organized with respect to the energy of the S = 5/2 spin state in each case. For the parent peroxo species, [(THF)(TPP)FeIII(O22−)]−, the high-spin complex (i.e., S = 5/2) was found to be the clear ground state.117 Importantly, the calculated ground states for the axially ligated heme–PO complexes varied depending on the extent of σ- and/or π-donation from the axial ligand. In that, the neutral axial ligands (THF, 4-MeIm) were found to only weakly interact with the heme center (Fe⋯OTHF and Fe⋯NIm distances of 4.4 and 3.2 Å, respectively). Regardless, the energy disparity between the high-spin (i.e., S = 5/2) and low-spin (i.e., S = 1/2) states decreased dramatically upon the ligation of those axial ligands. For example, in the case of 4-MeIm, the high-spin state remains the ground state, however, the difference in energy for the high- and low-spin states shrinks to ∼2.9 kcal mol−1 (i.e., from ∼16.3 kcal mol−1 for [(THF)(TPP)FeIII(O22−)]−). With anionic ligands, the low-spin state becomes slightly more favored, and for [(ArS−)(TPP)FeIII(O22−)]2− the gap is only −1.8 kcal mol−1, supporting its experimentally observed equilibrium mixture of high- and low-spin states. | ||
| Fig. 7 Optimized geometries of (A) [(THF)(TPP)FeIII(O22−)]−, and (B) [(ArS−)(TPP)FeIII(O22−)]2− with calculated bond lengths shown in Å. | ||
The trend in theoretically estimated Fe–Operoxo bond lengths aligns well with the experimentally predicted (i.e., by resonance Raman data; Table 2), where ligation of 4-MeIm was observed to result in a shorter Fe–Operoxo bond compared to the parent complex (1.968 Å vs. 1.978 Å, respectively; see Table S4†).118 This change in bond length is accompanied by a minor decrease in the natural atomic charge at the iron center from 0.937 to 0.929 upon 4-MeIm coordination. Moreover, the trend in theoretically predicted ν(Fe–O) and ν(O–O) Raman stretching frequencies for the parent peroxo complex, [(THF)(TPP)FeIII(O22−)]−, and [(4-MeIm)(TPP)FeIII(O22−)]− are in agreement with the experimental observations (i.e., increased and decreased ν(Fe–O) and ν(O–O) frequencies, respectively; Table 2 and S5†). Furthermore, the calculated 18O isotope shifts, for (Fe–O) and ν(O–O), respectively, are in line with experimental observations (Table 2).
|
|
|
|
|
|---|---|---|---|
|
a
λ
max values in nm.
b
g
app-values measured at 7 K.
c
δ
pyrrole chemical shifts (in ppm) in 9:1 DCM:THF at −40 °C.
d In cm−1 at 77 K.
e Computed with DFT shown in Å.
f In M−1 s−1 at −40 °C.
g In kcal mol−1.
h In cal K−1 mol−1.
i In kcal mol−1 at −40 °C.
|
|||
| UV-visa | 436, 565 | 436, 565 | 436, 564 |
| EPRb | 4.2 | 4.2 | 4.2 & 2.28, 2.13, 1.97 |
| 2H NMRc | 38.5 | 9.1 | — |
| rRamand | ν(Fe–O) 472 (Δ18O = −21) | ν(Fe–O) 479 (Δ18O = −23) | ν(Fe–O) 484 (Δ18O = −21) |
| ν(O–O) 807 (Δ18O = −44) | ν(O–O) 803 (Δ18O = −47) | ||
| Fe–O lengthe | 1.978 | 1.968 | 1.898 |
| rRaman (DFT)d | ν(Fe–O) 407 (Δ18O = −14) | ν(Fe–O) 414 (Δ18O = −15) | ν(Fe–O) 465 (Δ18O = −14) |
| ν(O–O) 899 (Δ18O = −52) | ν(O–O) 896 (Δ18O = −51) | ν(O–O) 1015 (Δ18O = −63) | |
| k 2 | 0.26 ± 0.01 | 0.79 ± 0.06 | 1.53 ± 0.04 |
| ΔH≠g | 7.6 ± 0.7 | 9.0 ± 0.9 | — |
| ΔS≠h | −27.7 ± 3.0 | −20.2 ± 3.8 | — |
| ΔG≠i | 14.0 ± 0.8 | 13.7 ± 1.2 | — |

Our experimental (vide supra) and theoretical findings therefore in concert illustrate that upon ligating of anionic axial ligands, the accessibility of the low-spin heme–PO adducts increases. The calculated Fe–Operoxo bond lengths for [(ArO−)(TPP)FeIII(O22−)]2− and [(ArS−)(TPP)FeIII(O22−)]2− were shorter compared to other complexes, measuring up to 1.875 Å and 1.898 Å, respectively (Table S4†). This shortening of the Fe–Operoxo bond is further corroborated by the higher Fe–Operoxo Raman stretching frequencies observed/computed for [(ArO−)(TPP)FeIII(O22−)]2− and [(ArS−)(TPP)FeIII(O22−)]2− (Fig. S3; †Tables 2 and S5†). However, albeit our theoretical predictions suggest the formation of end-on heme–PO adducts upon anionic axial ligation, we see no experimental evidence to support that notion. For example, we see no evidence for an isotope-sensitive Fe–O feature within the 550–600 cm−1 region in resonance Raman that would be indicative of an end-on peroxo species. Nonetheless, we do observe increased decomposition products upon the addition of anionic ligands to the peroxo adducts (vide supra), and whether those result from a highly reactive but destabilized end-on peroxo adduct is unclear. Moreover, the enhanced electron-donation by these anionic ligands is evident by the dramatic decrease in the natural atomic charge of the iron center upon their ligation (0.214 and 0.047 for phenolate and thiophenolate, respectively, compared to 0.937 in the parent complex; see Table S4†). To this end, this work unequivocally demonstrates the fundamental reasonings behind Nature evolutionarily favoring a thiolate proximal ligand for NOS oxygenase domain, which effectively enhances the feasibility of rate-limiting events of its second mechanistic step.
Conclusions
Herein, we conduct a detailed discussion into how the primary coordination sphere, especially the proximal heme ligation, modulates the nucleophilic reactivity patterns of heme–PO species, with relevance to their proposed reactivity landscape in the second mechanistic step of NOS. We utilized acetophenone oxime as an NHA substrate mimic, and surveyed its reactivity toward a series of electronically divergent heme–PO adducts with and without the axial ligation of bioinspired neutral and anionic ligands. Spectroscopic and theoretical characterization reveal that axial ligation leads to the formation of high-spin/low-spin mixtures, while the heme–PO structure remains side-on bound. In support, DFT interrogations reveal a trend in the energy gap between the high-spin (S = 5/2) and low-spin (S = 1/2) heme–PO species decreasing with increased axial ligand donor ability (i.e., the “push” effect). The lack of transition into a low-spin, end-on heme–PO merely upon axial ligation (i.e., the “push” effect) can be attributed to the absence of a non-covalently interacting (e.g., hydrogen bonding) secondary sphere in these systems (i.e., the “pull” effect). The nucleophilic reactivities of these heme–PO systems vary as expected with respect to their axial ligand donor abilities; that is, in the order of anionic axial ligands > neutral axial ligands > parent complex (Table 1), which supports the claim that heme–PO nucleophilic attack on the oxime substrate is rate-limiting. Moreover, the nucleophilically inert, electron-deficient heme–PO adduct, [(THF)(F20TPP)FeIII(O22−)]−, could be competently activated toward oxime substrate oxidation upon the axial ligation of bioinspired anionic ligands. Variable temperature kinetic (Eyring) analyses of these systems illustrate that upon axial ligation both ΔH≠ and ΔS≠ increase for the oxime oxidation reaction (Table 2). This study, therefore, marks the first instance where modulation of kinetic and thermodynamic parameters of heme–PO mediated nucleophilic reactivities with respect to proximal ligation has been clearly demonstrated, wherein pivotal structure–activity relationships can be elucidated. The distinct structure–activity relations described in this work will not only aid in comprehending relevant biological implications, but will also unveil new knowledge to be integrated into the rational design of novel (e.g., mechanism-based) drug targets.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
G. B. W. conceived the project idea, and supervised the investigation; S. R. synthesized all compounds, and generated and analyzed experimental data. S. B. J. carried out DFT investigations. P. M. and A. S. assisted in compound characterization. Y. L. performed resonance Raman experiments and carried out data evaluation under the supervision of H. S. S., J. H. performed EPR characterization and data workup under the supervision of B. S. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation CAREER Award CHE-2045005 (to G. B. W.) G. B. W. also acknowledges The University of Alabama and The University of Alabama at Birmingham for startup funds, as well as the UAB FDGP program. S. R. would like to thank the Alabama EPSCoR Graduate Research Scholar Program. H. S. S. would like to acknowledge NIH GM128852 (to H. S. S. and Y. L.), and B. S. P. NIH 2R15GM117511-04 for support. The authors also thank Dr Ken Belmore and Dr Qiaoli Liang for assistance with 2H NMR and mass spectrometry/LC-MS experiments. EPR experiments were performed at the UA Chemistry & Biochemistry facility. Computational investigations were made possible in part by the high-performance computing resources and technical support provided by the Alabama Supercomputer Authority.Notes and references
- T. L. Poulos, Heme Enzyme Structure and Function, Chem. Rev., 2014, 114, 3919–3962 Search PubMed.
- S. M. Adam, G. B. Wijeratne, P. J. Rogler, D. E. Diaz, D. A. Quist, J. J. Liu and K. D. Karlin, Synthetic Fe/Cu Complexes: Toward Understanding Heme-Copper Oxidase Structure and Function, Chem. Rev., 2018, 118, 10840–11022 CrossRef PubMed.
- J. P. T. Zaragoza and D. P. Goldberg, in Dioxygen-dependent Heme Enzymes, ed. M. Ikeda-Saito and E. Raven, The Royal Society of Chemistry, 2018, vol. 13, pp. 3–36 Search PubMed.
- T. L. Poulos, The role of the proximal ligand in heme enzymes, JBIC, J. Biol. Inorg. Chem., 1996, 1, 356–359 CrossRef.
- J. Dawson, Probing structure-function relations in heme-containing oxygenases and peroxidases, Science, 1988, 240, 433–439 CrossRef PubMed.
- M. T. Green, J. H. Dawson and H. B. Gray, Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry, Science, 2004, 304, 1653–1656 CrossRef.
- T. H. Yosca, J. Rittle, C. M. Krest, E. L. Onderko, A. Silakov, J. C. Calixto, R. K. Behan and M. T. Green, Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450, Science, 2013, 342, 825–829 CrossRef PubMed.
- X. Huang and J. T. Groves, Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins, Chem. Rev., 2018, 118, 2491–2553 CrossRef PubMed.
- P. Mondal and G. B. Wijeratne, Modeling Tryptophan/Indoleamine 2,3-Dioxygenase with Heme Superoxide Mimics: Is Ferryl the Key Intermediate?, J. Am. Chem. Soc., 2020, 142, 1846–1856 CrossRef.
- P. Mondal, I. Ishigami, E. F. Gérard, C. Lim, S.-R. Yeh, S. P. de Visser and G. B. Wijeratne, Proton-coupled electron transfer reactivities of electronically divergent heme superoxide intermediates: a kinetic, thermodynamic, and theoretical study, Chem. Sci., 2021, 12, 8872–8883 RSC.
- G. B. Tolbert, S. B. Jayawardana, Y. Lee, J. Sun, F. Qu, L. M. Whitt, H. Shafaat and G. B. Wijeratne, Secondary Sphere Lewis Acid Activated Heme Superoxo Adducts Mimic Crucial Non-Covalent Interactions in IDO/TDO Heme Dioxygenases, Chem.–Eur. J., 2024, e202402310 CrossRef PubMed.
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Heme-Containing Oxygenases, Chem. Rev., 1996, 96, 2841–2888 CrossRef PubMed.
- A. P. Hunt, S. Samanta, M. R. Dent, M. W. Milbauer, J. N. Burstyn and N. Lehnert, Model Complexes Elucidate the Role of the Proximal Hydrogen-Bonding Network in Cytochrome P450s, Inorg. Chem., 2020, 59, 8034–8043 CrossRef PubMed.
- M. Couture, S. Adak, D. J. Stuehr and D. L. Rousseau, Regulation of the Properties of the Heme-NO Complexes in Nitric-oxide Synthase by Hydrogen Bonding to the Proximal Cysteine, J. Biol. Chem., 2001, 276, 38280–38288 CrossRef PubMed.
- J. Lang, J. Santolini and M. Couture, The Conserved Trp–Cys Hydrogen Bond Dampens the “Push Effect” of the Heme Cysteinate Proximal Ligand during the First Catalytic Cycle of Nitric Oxide Synthase, Biochemistry, 2011, 50, 10069–10081 CrossRef CAS PubMed.
- J. Tejero, A. Biswas, Z.-Q. Wang, R. C. Page, M. M. Haque, C. Hemann, J. L. Zweier, S. Misra and D. J. Stuehr, Stabilization and Characterization of a Heme-Oxy Reaction Intermediate in Inducible Nitric-oxide Synthase, J. Biol. Chem., 2008, 283, 33498–33507 CrossRef CAS.
- M. Aarabi, R. Omidyan, S. Soorkia, G. Grégoire, M. Broquier, M.-E. Crestoni, A. de la Lande, B. Soep and N. Shafizadeh, The dramatic effect of N-methylimidazole on trans axial ligand binding to ferric heme: experiment and theory, Phys. Chem. Chem. Phys., 2019, 21, 1750–1760 RSC.
- P. K. Das, S. Chatterjee, S. Samanta and A. Dey, EPR, Resonance Raman, and DFT Calculations on Thiolate- and Imidazole-Bound Iron(III) Porphyrin Complexes: Role of the Axial Ligand in Tuning the Electronic Structure, Inorg. Chem., 2012, 51, 10704–10714 CrossRef CAS.
- A. P. Green, T. Hayashi, P. R. E. Mittl and D. Hilvert, A Chemically Programmed Proximal Ligand Enhances the Catalytic Properties of a Heme Enzyme, J. Am. Chem. Soc., 2016, 138, 11344–11352 CrossRef CAS.
- R. Perera, M. Sono, H. L. Voegtle and J. H. Dawson, Molecular basis for the inability of an oxygen atom donor ligand to replace the natural sulfur donor heme axial ligand in cytochrome P450 catalysis. Spectroscopic characterization of the Cys436Ser CYP2B4 mutant, Arch. Biochem. Biophys., 2011, 507, 119–125 CrossRef CAS PubMed.
- S. Samanta, P. K. Das, S. Chatterjee and A. Dey, Effect of axial ligands on electronic structure and O2 reduction by iron porphyrin complexes: Towards a quantitative understanding of the “push effect”, J. Porphyrins Phthalocyanines, 2015, 19, 92–108 CrossRef CAS.
- M. P. Roach, A. E. Pond, M. R. Thomas, S. G. Boxer and J. H. Dawson, The Role of the Distal and Proximal Protein Environments in Controlling the Ferric Spin State and in Stabilizing Thiolate Ligation in Heme Systems: Thiolate Adducts of the Myoglobin H93G Cavity Mutant, J. Am. Chem. Soc., 1999, 121, 12088–12093 CrossRef.
- S. Yoshioka, S. Takahashi, K. Ishimori and I. Morishima, Roles of the axial push effect in cytochrome P450cam studied with the site-directed mutagenesis at the heme proximal site, J. Inorg. Biochem., 2000, 81, 141–151 CrossRef PubMed.
- Z. Gross, The effect of axial ligands on the reactivity and stability of the oxoferryl moiety in model complexes of Compound I of heme-dependent enzymes, JBIC, J. Biol. Inorg. Chem., 1996, 1, 368–371 CrossRef.
- A. Takahashi, T. Kurahashi and H. Fujii, Effect of Imidazole and Phenolate Axial Ligands on the Electronic Structure and Reactivity of Oxoiron(IV) Porphyrin π-Cation Radical Complexes: Drastic Increase in Oxo-Transfer and Hydrogen Abstraction Reactivities, Inorg. Chem., 2009, 48, 2614–2625 CrossRef PubMed.
- E. L. Onderko, A. Silakov, T. H. Yosca and M. T. Green, Characterization of a selenocysteine-ligated P450 compound I reveals direct link between electron donation and reactivity, Nat. Chem., 2017, 9, 623–628 CrossRef.
- T. H. Yosca, R. K. Behan, C. M. Krest, E. L. Onderko, M. C. Langston and M. T. Green, Setting an Upper Limit on the Myoglobin Iron(IV)Hydroxide pKa: Insight into Axial Ligand Tuning in Heme Protein Catalysis, J. Am. Chem. Soc., 2014, 136, 9124–9131 CrossRef PubMed.
- T. H. Yosca, A. P. Ledray, J. Ngo and M. T. Green, A new look at the role of thiolate ligation in cytochrome P450, JBIC, J. Biol. Inorg. Chem., 2017, 22, 209–220 CrossRef.
- W. Nam, M. H. Lim and S.-Y. Oh, Effect of Anionic Axial Ligands on the Formation of Oxoiron(IV) Porphyrin Intermediates, Inorg. Chem., 2000, 39, 5572–5575 CrossRef.
- J. Thomas, T. Mokkawes, L. Senft, A. Dey, J. B. Gordon, I. Ivanovic-Burmazovic, S. P. de Visser and D. P. Goldberg, Axial Ligation Impedes Proton-Coupled Electron-Transfer Reactivity of a Synthetic Compound-I Analogue, J. Am. Chem. Soc., 2024, 146, 12338–12354 CrossRef PubMed.
- N. Hessenauer-Ilicheva, A. Franke, M. Wolak, T. Higuchi and R. van Eldik, Spectroscopic and Mechanistic Studies on Oxidation Reactions Catalyzed by the Functional Model SR Complex for Cytochrome P450: Influence of Oxidant, Substrate, and Solvent, Chem.–Eur. J., 2009, 15, 12447–12459 CrossRef PubMed.
- Y. Kang, H. Chen, Y. J. Jeong, W. Lai, E. H. Bae, S. Shaik and W. Nam, Enhanced Reactivities of Iron(IV)-Oxo Porphyrin π-Cation Radicals in Oxygenation Reactions by Electron-Donating Axial Ligands, Chem.–Eur. J., 2009, 15, 10039–10046 CrossRef PubMed.
- M. A. Ehudin, L. B. Gee, S. Sabuncu, A. Braun, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, Tuning the Geometric and Electronic Structure of Synthetic High-Valent Heme Iron(IV)-Oxo Models in the Presence of a Lewis Acid and Various Axial Ligands, J. Am. Chem. Soc., 2019, 141, 5942–5960 CrossRef PubMed.
- M. Ortmayer, K. Fisher, J. Basran, E. M. Wolde-Michael, D. J. Heyes, C. Levy, S. L. Lovelock, J. L. R. Anderson, E. L. Raven, S. Hay, S. E. J. Rigby and A. P. Green, Rewiring the “Push-Pull” Catalytic Machinery of a Heme Enzyme Using an Expanded Genetic Code, ACS Catal., 2020, 10, 2735–2746 CrossRef.
- M. Selke and J. S. Valentine, Switching on the Nucleophilic Reactivity of a Ferric Porphyrin Peroxo Complex, J. Am. Chem. Soc., 1998, 120, 2652–2653 CrossRef.
- W. Nam, M. H. Lim, S.-Y. Oh, J. H. Lee, H. J. Lee, S. K. Woo, C. Kim and W. Shin, Remarkable Anionic Axial Ligand Effects of Iron(III) Porphyrin Complexes on the Catalytic Oxygenations of Hydrocarbons by H2O2 and the Formation of Oxoiron(IV) Porphyrin Intermediates by m-Chloroperoxybenzoic Acid, Angew. Chem., 2000, 112, 3792–3795 CrossRef.
- A. Franke, C. Fertinger and R. van Eldik, Axial Ligand and Spin-State Influence on the Formation and Reactivity of Hydroperoxo–Iron(III) Porphyrin Complexes, Chem.–Eur. J., 2012, 18, 6935–6949 CrossRef PubMed.
- S. Yokota, Y. Suzuki, S. Yanagisawa, T. Ogura, S. Nozawa, M. Hada and H. Fujii, How Do the Axial and Equatorial Ligands Modulate the Reactivity of a Metal-Bound Terminal Oxidant? An Answer from the Hypochlorite Adduct of Iron(III) Porphyrin, ACS Catal., 2022, 12, 10857–10871 CrossRef.
- P. K. Das, K. Mittra and A. Dey, Spectroscopic characterization of a phenolate bound FeII–O2 adduct: gauging the relative “push” effect of a phenolate axial ligand, Chem. Commun., 2014, 50, 5218–5220 RSC.
- S. Sivaramakrishnan, H. Ouellet, H. Matsumura, S. Guan, P. Moënne-Loccoz, A. L. Burlingame and P. R. Ortiz de Montellano, Proximal Ligand Electron Donation and Reactivity of the Cytochrome P450 Ferric–Peroxo Anion, J. Am. Chem. Soc., 2012, 134, 6673–6684 CrossRef.
- R. L. Khade, Y. Yang, Y. Shi and Y. Zhang, HNO-Binding in Heme Proteins: Effects of Iron Oxidation State, Axial Ligand, and Protein Environment, Angew. Chem., Int. Ed., 2016, 55, 15058–15061 CrossRef.
- D. Singh, H. Wasan and K. H. Reeta, Heme oxygenase-1 modulation: A potential therapeutic target for COVID-19 and associated complications, Free Radicals Biol. Med., 2020, 161, 263–271 CrossRef PubMed.
- L. Hornyák, N. Dobos, G. Koncz, Z. Karányi, D. Páll, Z. Szabó, G. Halmos and L. Székvölgyi, The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy, Front. Immunol., 2018, 9, 1–8 CrossRef.
- J. A. Locke, L. Fazli, H. Adomat, J. Smyl, K. Weins, A. A. Lubik, D. B. Hales, C. C. Nelson, M. E. Gleave and E. S. Tomlinson Guns, A novel communication role for CYP17A1 in the progression of castration-resistant prostate cancer, The Prostate, 2009, 69, 928–937 CrossRef PubMed.
- S. Chumsri, T. Howes, T. Bao, G. Sabnis and A. Brodie, Aromatase, aromatase inhibitors, and breast cancer, J. Steroid Biochem. Mol. Biol., 2011, 125, 13–22 CrossRef PubMed.
- T. Togo, O. Katsuse and E. Iseki, Nitric oxide pathways in Alzheimer's disease and other neurodegenerative dementias, Neurol. Res., 2004, 26, 563–566 CrossRef PubMed.
- J. J. Woodward, M. M. Chang, N. I. Martin and M. A. Marletta, The Second Step of the Nitric Oxide Synthase Reaction: Evidence for Ferric-Peroxo as the Active Oxidant, J. Am. Chem. Soc., 2009, 131, 297–305 CrossRef.
- S. L. Gantt, I. G. Denisov, Y. V. Grinkova and S. G. Sligar, The critical iron–oxygen intermediate in human aromatase, Biochem. Biophys. Res. Commun., 2009, 387, 169–173 CrossRef.
- K. D. McCarty, Y. Tateishi, T. Y. Hargrove, G. I. Lepesheva and F. P. Guengerich, Oxygen-18 Labeling Reveals a Mixed Fe−O Mechanism in the Last Step of Cytochrome P450 51 Sterol 14α-Demethylation, Angew. Chem., Int. Ed., 2024, 63, e202317711 CrossRef.
- P. J. Mak, M. C. Gregory, I. G. Denisov, S. G. Sligar and J. R. Kincaid, Unveiling the crucial intermediates in androgen production, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15856–15861 Search PubMed.
- S.-i. J. Takayama, S. A. Loutet, A. G. Mauk and M. E. P. Murphy, A Ferric–Peroxo Intermediate in the Oxidation of Heme by IsdI, Biochemistry, 2015, 54, 2613–2621 Search PubMed.
- H. Li, C. S. Raman, C. B. Glaser, E. Blasko, T. A. Young, J. F. Parkinson, M. Whitlow and T. L. Poulos, Crystal Structures of Zinc-free and -bound Heme Domain of Human Inducible Nitric-oxide Synthase: implications for dimer stability and comparison with endothelial nitric-oxide synthase, J. Biol. Chem., 1999, 274, 21276–21284 CrossRef PubMed.
- G. Fermi, M. F. Perutz, B. Shaanan and R. Fourme, The crystal structure of human deoxyhaemoglobin at 1.74 Å resolution, J. Mol. Biol., 1984, 175, 159–174 CrossRef PubMed.
- M. Alfonso-Prieto, A. Borovik, X. Carpena, G. Murshudov, W. Melik-Adamyan, I. Fita, C. Rovira and P. C. Loewen, The Structures and Electronic Configuration of Compound I Intermediates of Helicobacter pylori and Penicillium vitale Catalases Determined by X-ray Crystallography and QM/MM Density Functional Theory Calculations, J. Am. Chem. Soc., 2007, 129, 4193–4205 CrossRef.
- M. Gajhede, D. J. Schuller, A. Henriksen, A. T. Smith and T. L. Poulos, Crystal structure of horseradish peroxidase C at 2.15 Å resolution, Nat. Struct. Mol. Biol., 1997, 4, 1032–1038 CrossRef.
- O.-M. Iova, G.-E. Marin, I. Lazar, I. Stanescu, G. Dogaru, C. A. Nicula and A. E. Bulboacă, Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders—An Overview, Antioxidants, 2023, 12, 753 Search PubMed.
- R. Kavya, R. Saluja, S. Singh and M. Dikshit, Nitric oxide synthase regulation and diversity: Implications in Parkinson's disease, Nitric Oxide, 2006, 15, 280–294 CrossRef PubMed.
- A. W. Deckel, Nitric oxide and nitric oxide synthase in Huntington's disease, J. Neurosci. Res., 2001, 64, 99–107 CrossRef.
- M. K. Tripathi, M. Kartawy and H. Amal, The role of nitric oxide in brain disorders: Autism spectrum disorder and other psychiatric, neurological, and neurodegenerative disorders, Redox Biol., 2020, 34, 101567 CrossRef PubMed.
- D. A. Drechsel, A. G. Estévez, L. Barbeito and J. S. Beckman, Nitric Oxide-Mediated Oxidative Damage and the Progressive Demise of Motor Neurons in ALS, Neurotox. Res., 2012, 22, 251–264 CrossRef PubMed.
- D. Basudhar, V. Somasundaram, G. A. de Oliveira, A. Kesarwala, J. L. Heinecke, R. Y. Cheng, S. A. Glynn, S. Ambs, D. A. Wink and L. A. Ridnour, Nitric Oxide Synthase-2-Derived Nitric Oxide Drives Multiple Pathways of Breast Cancer Progression, Antiox. Redox Sign, 2016, 26, 1044–1058 Search PubMed.
- S. Korde Choudhari, M. Chaudhary, S. Bagde, A. R. Gadbail and V. Joshi, Nitric oxide and cancer: a review, World J. Surg. Oncol., 2013, 11, 118 CrossRef PubMed.
- Y. Soni, K. Softness, H. Arora and R. Ramasamy, The Yin Yang Role of Nitric Oxide in Prostate Cancer, Am. J. Men. Health., 2020, 14, 1557988320903191 CrossRef.
- K. Zafirellis, A. Zachaki, G. Agrogiannis and K. Gravani, Inducible nitric oxide synthase expression and its prognostic significance in colorectal cancer, APMIS, 2010, 118, 115–124 CrossRef PubMed.
- W.-C. Hung, T.-F. Wu, S.-C. Ng, Y. C. Lee, H.-P. Shen, S.-F. Yang and P.-H. Wang, Involvement of endothelial nitric oxide synthase gene variants in the aggressiveness of uterine cervical cancer, J. Cancer, 2019, 10, 2594–2600 CrossRef CAS PubMed.
- G. G. Chen, T. W. Lee, H. Xu, J. H. Y. Yip, M. Li, T. S. K. Mok and A. P. C. Yim, Increased inducible nitric oxide synthase in lung carcinoma of smokers, Cancer, 2008, 112, 372–381 CrossRef CAS.
- A. Bakshi, T. C. Nag, S. Wadhwa, A. K. Mahapatra and C. Sarkar, The expression of nitric oxide synthases in human brain tumours and peritumoral areas, J. Neurol. Sci., 1998, 155, 196–203 CrossRef CAS PubMed.
- C. S. Cobbs, J. E. Brenman, K. D. Aldape, D. S. Bredt and M. A. Israel, Expression of Nitric Oxide Synthase in Human Central Nervous System Tumors1, Cancer Res., 1995, 55, 727–730 CAS.
- F. Cianchi, C. Cortesini, O. Fantappiè, L. Messerini, N. Schiavone, A. Vannacci, S. Nistri, I. Sardi, G. Baroni, C. Marzocca, F. Perna, R. Mazzanti, P. Bechi and E. Masini, Inducible Nitric Oxide Synthase Expression in Human Colorectal Cancer: Correlation with Tumor Angiogenesis, Am. J. Pathol., 2003, 162, 793–801 CrossRef CAS PubMed.
- R. S. Nomelini, L. C. d. A. Ribeiro, B. M. Tavares-Murta, S. J. Adad and E. F. C. Murta, Production of Nitric Oxide and Expression of Inducible Nitric Oxide Synthase in Ovarian Cystic Tumors, Mediators Inflammation, 2008, 2008, 186584 CrossRef PubMed.
- U. Förstermann and W. C. Sessa, Nitric oxide synthases: regulation and function, Eur. Heart J., 2012, 33, 829–837 CrossRef.
- B. Salimian Rizi, A. Achreja and D. Nagrath, Nitric Oxide: The Forgotten Child of Tumor Metabolism, Trends Cancer, 2017, 3, 659–672 CrossRef CAS PubMed.
- S. Daff, NO synthase: structures and mechanisms, Nitric Oxide, 2010, 23, 1–11 CrossRef CAS PubMed.
- N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, The Biologically Relevant Coordination Chemistry of Iron and Nitric Oxide: Electronic Structure and Reactivity, Chem. Rev., 2021, 121, 14682–14905 CrossRef CAS PubMed.
- A. Aicardo, D. M. Martinez, N. Campolo, S. Bartesaghi and R. Radi, in Biochemistry of Oxidative Stress: Physiopathology and Clinical Aspects, ed. R. J. Gelpi, A. Boveris and J. J. Poderoso, Springer International Publishing, Cham, 2016, vol. 16, pp. 49–77 Search PubMed.
- M. A. Cinelli, H. T. Do, G. P. Miley and R. B. Silverman, Inducible nitric oxide synthase: Regulation, structure, and inhibition, Med. Res. Rev., 2020, 40, 158–189 CrossRef CAS.
- S. Tong, M. A. Cinelli, N. S. El-Sayed, H. Huang, A. Patel, R. B. Silverman and S. Yang, Inhibition of interferon-gamma-stimulated melanoma progression by targeting neuronal nitric oxide synthase (nNOS), Sci. Rep., 2022, 12, 1701 CrossRef CAS PubMed.
- A. D. Vaz, S. J. Pernecky, G. M. Raner and M. J. Coon, Peroxo-iron and oxenoid-iron species as alternative oxygenating agents in cytochrome P450-catalyzed reactions: switching by threonine-302 to alanine mutagenesis of cytochrome P450 2B4, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4644–4648 CrossRef CAS PubMed.
- R. Minhas, Y. Bansal and G. Bansal, Inducible nitric oxide synthase inhibitors: A comprehensive update, Med. Res. Rev., 2020, 40, 823–855 CrossRef CAS PubMed.
- J. T. Groves and C. C. Y. Wang, Nitric oxide synthase: models and mechanisms, Curr. Opin. Chem. Biol., 2000, 4, 687–695 CrossRef.
- H. Huang, J.-M. Hah and R. B. Silverman, Mechanism of Nitric Oxide Synthase. Evidence that Direct Hydrogen Atom Abstraction from the O–H Bond of NG-Hydroxyarginine Is Not Relevant to the Mechanism, J. Am. Chem. Soc., 2001, 123, 2674–2676 CrossRef.
- Y. Zhu and R. B. Silverman, Revisiting Heme Mechanisms. A Perspective on the Mechanisms of Nitric Oxide Synthase (NOS), Heme Oxygenase (HO), and Cytochrome P450s (CYP450s), Biochemistry, 2008, 47, 2231–2243 CrossRef.
- T. Doukov, H. Li, M. Soltis and T. L. Poulos, Single Crystal Structural and Absorption Spectral Characterizations of Nitric Oxide Synthase Complexed with Nω-Hydroxy-l-arginine and Diatomic Ligands, Biochemistry, 2009, 48, 10246–10254 CrossRef PubMed.
- R. Davydov, J. Sudhamsu, N. S. Lees, B. R. Crane and B. M. Hoffman, EPR and ENDOR Characterization of the Reactive Intermediates in the Generation of NO by Cryoreduced Oxy-Nitric Oxide Synthase from Geobacillus stearothermophilus, J. Am. Chem. Soc., 2009, 131, 14493–14507 CrossRef.
- J. Santolini, The molecular mechanism of mammalian NO-synthases: A story of electrons and protons, J. Inorg. Biochem., 2011, 105, 127–141 CrossRef PubMed.
- R. Davydov, K. J. Labby, S. E. Chobot, D. A. Lukoyanov, B. R. Crane, R. B. Silverman and B. M. Hoffman, Enzymatic and Cryoreduction EPR Studies of the Hydroxylation of Methylated Nω-Hydroxy-l-arginine Analogues by Nitric Oxide Synthase from Geobacillus stearothermophilus, Biochemistry, 2014, 53, 6511–6519 CrossRef PubMed.
- K. Jansen Labby, H. Li, L. J. Roman, P. Martásek, T. L. Poulos and R. B. Silverman, Methylated Nω-Hydroxy-l-arginine Analogues as Mechanistic Probes for the Second Step of the Nitric Oxide Synthase-Catalyzed Reaction, Biochemistry, 2013, 52, 3062–3073 CrossRef.
- S. Bhattacharya, T. R. Lakshman, S. Sutradhar, C. K. Tiwari and T. K. Paine, Bioinspired oxidation of oximes to nitric oxide with dioxygen by a nonheme iron(II) complex, JBIC, J. Biol. Inorg. Chem., 2020, 25, 3–11 CrossRef.
- C. C. Y. Wang, D. M. Ho and J. T. Groves, Models of Nitric Oxide Synthase: Iron(III) Porphyrin-Catalyzed Oxidation of Fluorenone Oxime to Nitric Oxide and Fluorenone, J. Am. Chem. Soc., 1999, 121, 12094–12103 CrossRef.
- A. Maldotti, A. Molinari, I. Vitali, E. Ganzaroli, P. Battioni, D. Mathieu and D. Mansuy, Oxidation of N-(4-Chlorophenyl)-N′-hydroxyguanidine to N-(4-Chlorophenyl)urea and Nitric Oxide by Photoexcited Iron Porphyrins, Eur. J. Inorg. Chem., 2004, 2004, 3127–3135 CrossRef.
- N. Jain, A. Kumar and S. M. S. Chauhan, Metalloporphyrin and heteropoly acid catalyzed oxidation of C=NOH bonds in an ionic liquid: biomimetic models of nitric oxide synthase, Tetrahedron Lett., 2005, 46, 2599–2602 CrossRef.
- P. Mondal, I. Ishigami, S.-R. Yeh and G. B. Wijeratne, The Role of Heme Peroxo Oxidants in the Rational Mechanistic Modeling of Nitric Oxide Synthase: Characterization of Key Intermediates and Elucidation of the Mechanism, Angew. Chem., Int. Ed., 2022, 61, e202211521 CrossRef.
- K. D. Karlin, Model offers intermediate insight, Nature, 2010, 463, 168 CrossRef PubMed.
- The addition of axial ligands did not result in significant alterations in self-decay rates of heme-PO adducts (Fig. S1†)..
- J.-G. Liu, T. Ohta, S. Yamaguchi, T. Ogura, S. Sakamoto, Y. Maeda and Y. Naruta, Spectroscopic Characterization of a Hydroperoxo–Heme Intermediate: Conversion of a Side-On Peroxo to an End-On Hydroperoxo Complex, Angew. Chem., Int. Ed., 2009, 48, 9262–9267 CrossRef PubMed.
- I. Garcia-Bosch, S. M. Adam, A. W. Schaefer, S. K. Sharma, R. L. Peterson, E. I. Solomon and K. D. Karlin, A “Naked” FeIII–(O22−)−CuII Species Allows for Structural and Spectroscopic Tuning of Low-Spin Heme-Peroxo-Cu Complexes, J. Am. Chem. Soc., 2015, 137, 1032–1035 CrossRef PubMed.
- H. Kim, P. J. Rogler, S. K. Sharma, A. W. Schaefer, E. I. Solomon and K. D. Karlin, Ferric Heme Superoxide Reductive Transformations to Ferric Heme (Hydro)Peroxide Species: Spectroscopic Characterization and Thermodynamic Implications for H-Atom Transfer (HAT), Angew. Chem., Int. Ed., 2021, 60, 5907–5912 CrossRef PubMed.
- J.-G. Liu, Y. Shimizu, T. Ohta and Y. Naruta, Formation of an End-On Ferric Peroxo Intermediate upon One-Electron Reduction of a Ferric Superoxo Heme, J. Am. Chem. Soc., 2010, 132, 3672–3673 CrossRef PubMed.
- S. Panda, H. Phan and K. D. Karlin, Heme-copper and Heme O2-derived synthetic (bioinorganic) chemistry toward an understanding of cytochrome c oxidase dioxygen chemistry, J. Inorg. Biochem., 2023, 249, 112367 CrossRef.
- E. M. Arnett and C. Y. Wu, Base Strengths of Some Saturated Cyclic Ethers in Aqueous Sulfuric Acid, J. Am. Chem. Soc., 1962, 84, 1684–1688 CrossRef.
- J. D. Vaughan, V. L. Vaughan, S. S. Daly and W. A. Smith, Jr., Kinetics of iodination of 4-methylimidazole and 2-methylimidazole, J. Org. Chem., 1980, 45, 3108–3111 CrossRef.
- Calculated using Advanced Chemistry Development (ACD/Labs) Software (© 1994-2024 ACD/Labs)..
- J. J. Christensen, L. D. Hansen and R. M. Izatt, Handbook of proton ionization heats and related thermodynamic quantities, Wiley, 1976 Search PubMed.
- P. Bolton, F. Hall and J. Kudrynski, Thermodynamic functions of ionization of t-butyl-substituted and methoxysubstituted phenols, Aust. J. Chem., 1972, 25, 75–80 CrossRef.
- F. G. Bordwell and D. L. Hughes, Thiol acidities and thiolate ion reactivities toward butyl chloride in dimethyl sulfoxide solution. The question of curvature in Broensted plots, J. Org. Chem., 1982, 47, 3224–3232 CrossRef.
- K. Nakamoto and H. Oshio, Effect of thiolate vs. nitrogen-base ligands on oxygen stretching frequencies of (oxytetraphenylporphyrinato)cobalt, J. Am. Chem. Soc., 1985, 107, 6518–6521 CrossRef.
- P. Anzenbacher, J. H. Dawson and T. Kitagawa, Towards a unified concept of oxygen activation by heme enzymes: the role of the proximal ligand, J. Mol. Struct., 1989, 214, 149–158 CrossRef.
- H. M. Neu, M. G. Quesne, T. Yang, K. A. Prokop-Prigge, K. M. Lancaster, J. Donohoe, S. DeBeer, S. P. de Visser and D. P. Goldberg, Dramatic Influence of an Anionic Donor on the Oxygen-Atom Transfer Reactivity of a MnV–Oxo Complex, Chem.–Eur. J., 2014, 20, 14584–14588 CrossRef.
- G. Palmer, in The Porphyrins, Academic Press, 1979, pp. 313–353 Search PubMed.
- J. Peisach, W. E. Blumberg, S. Ogawa, E. A. Rachmilewitz and R. Oltzik, The Effects of Protein Conformation on the Heme Symmetry in High Spin Ferric Heme Proteins as Studied by Electron Paramagnetic Resonance, J. Biol. Chem., 1971, 246, 3342–3355 CrossRef.
- J. Peisach, W. E. Blumberg, B. A. Wittenberg, J. B. Wittenberg and L. Kampa, Hemoglobin A: An Electron Paramagnetic Resonance Study Of The Effects Of Interchain Contacts On The Heme Symmetry Of High-Spin And Low-Spin Derivatives Of Ferric Alpha Chains, Proc. Natl. Acad. Sci. U. S. A., 1969, 63, 934–939 CrossRef.
- H. C. Lee, J. Peisach, Y. Dou and M. Ikeda-Saito, Electron-Nuclear Coupling to the Proximal Histidine in Oxycobalt-Substituted Distal Histidine Mutants of Human Myoglobin, Biochemistry, 1994, 33, 7609–7618 CrossRef PubMed.
- G. Roelfes, V. Vrajmasu, K. Chen, R. Y. N. Ho, J.-U. Rohde, C. Zondervan, R. M. la Crois, E. P. Schudde, M. Lutz, A. L. Spek, R. Hage, B. L. Feringa, E. Münck and L. J. Que, End-On and Side-On Peroxo Derivatives of Non-Heme Iron Complexes with Pentadentate Ligands: Models for Putative Intermediates in Biological Iron/Dioxygen Chemistry, Inorg. Chem., 2003, 42, 2639–2653 CrossRef.
- N. J. York, M. M. Lockart and B. S. Pierce, Low-Spin Cyanide Complexes of 3-Mercaptopropionic Acid Dioxygenase (MDO) Reveal the Impact of Outer-Sphere SHY-Motif Residues, Inorg. Chem., 2021, 60, 18639–18651 CrossRef CAS PubMed.
- J. S. Griffith, Theory of E.P.R. in low-spin ferric haemoproteins, Mol. Phys., 1971, 21, 135–139 CrossRef CAS.
- C. P. S. Taylor, The EPR of Low Spin Heme Complexes: relation of the t2g hole model to the directional properties of the g-tensor and a new method for calculating the ligand field parameters, Biochim. Biophys. Acta, 1977, 491, 137–149 CrossRef CAS PubMed.
- K. Duerr, J. Olah, R. Davydov, M. Kleimann, J. Li, N. Lang, R. Puchta, E. Hubner, T. Drewello, J. N. Harvey, N. Jux and I. Ivanovic-Burmazovic, Studies on an iron(iii)-peroxo porphyrin. Iron(iii)-peroxo or iron(ii)-superoxo?, Dalton Trans., 2010, 39, 2049–2056 RSC.
- The change in O–Operoxo bond length is negligible.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure, spectroscopic characterization, and computational procedures. See DOI: https://doi.org/10.1039/d4sc08701a |
| This journal is © The Royal Society of Chemistry 2025 |