
Recent insights of PROTAC developments in inflammation-mediated and autoimmune targets: a critical review
Mary Sravani
Galla
,
Nitika
Sharma†
,
Priyanka
Mishra†
and
Nagula
Shankaraiah
 *
*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Hyderabad, 500037, India. E-mail: shankar@niperhyd.ac.in; shankarnbs@gmail.com
First published on 8th May 2024
Abstract
According to the mounting evidence in the literature, pro-inflammatory mediators/targets activate multiple signalling pathways to trigger illnesses that are ultimately responsible for acute pain, chronic inflammatory diseases, and several auto-immune disorders. Conventional drugs have been ruled out since proteolysis-targeting chimeras (PROTACs) are poised to overcome the limitations of traditional therapies. These heterobifunctional molecules help to degrade the targeted proteins of interest through ubiquitination. This review encompasses current and future aspects of PROTACs in inflammation-mediated and autoimmune targets. Different key points are highlighted and discussed, such as why PROTACs are preferred in this disease area, drawbacks and lessons learnt from the past, the role of linkers in establishing crucial degradation, in vitro findings, pharmacokinetics, in silico parameters, limitations of PROTACs in clinical settings, and future outcomes.
1. Introduction
The dynamic interplay of the synthesis and degradation of the cellular components in living organisms serves as a cornerstone in maintaining homeostasis.1 The strategy of leveraging the degradation mechanism has surfaced a novel paradigm for treating multiple disease conditions, representing a substantial milestone in modern drug discovery. In inhibitor-based strategies, there is always a setback when it comes to undruggable proteins, their accessible binding sites, or the specific quantifiable functions of these target proteins.2 Driving these anomalies, intracellular targeted protein degradation (TPD) encompasses autophagy-lysozyme systems [ALS], ubiquitin–proteasome systems [UPS], molecular glues and chaperone-mediated autophagy, with all of them adhering to the disposable mechanism.3 The advent of these bivalent degradations increases the scope for rendering the undruggable events.4 TPD could be the next frontier in treating inflammatory and autoimmune diseases.5 Unfortunately, drugs such as corticosteroids are still being used in the treatment of many inflammatory conditions for which they are not specifically intended. This is one of the main reasons for the downfall of treatment options in this area. For example, TNF inhibitors were often used to treat various diseases such as psoriasis, IBD, and arthritis.6 However, they failed as a strategy upon identification of the distinct mechanism of these diseases. Consequently, the small molecule inhibitors or biologics, which are targeted in specific areas, are also limited by low organ penetration, attributed to high molecular weights, complicated routes of administration, toxicity, and off-target effects (Fig. 1).7,8 | ||
| Fig. 1 Illustration of inflammatory/autoimmune disorders that need to be addressed by targeting through PROTACs. | ||
According to the WHO assessment, “chronic inflammatory illness is considered the most significant health hazard to the human population”. To date, many diseases or disorders have no cure and only management is being perpetuated. Emphasizing this, we have identified a list of potential targets, with their diseases highlighted in Table 1. It has been indisputably acknowledged that treating immune-related disorders requires multifaceted approaches since their mechanisms are quite complex and clinically heterogeneous.9 In the pathophysiology of various immune checkpoints or endosomal receptors, when activated by pathogens or external/internal stimuli, they undergo an interconnected downstream pathway to awaken the pro-inflammatory genes and cytokines responsible for triggering the disease. Some of the well-known targets and receptors responsible for inflammatory signalling pathways are mentioned in Fig. 2.10–12
| S. no | Disease | Target | Disadvantages of conventional therapy |
|---|---|---|---|
| 1. | Asthma and COPD | HDAC-VIII | Toxicities are related to gastrointestinal or constitutional symptoms and haematological abnormalities |
| 2 | Neuroinflammation | HDAC-VI | Inhibitors failed to achieve acute and reversible changes in genes |
| 3 | Diabetes-induced inflammation | HDAC-III | Limited isoenzyme selectivity, underprivileged pharmacokinetics properties |
| 4 | Asthma, rheumatoid arthritis, hidradenitis suppurativa | IRAK3/4 | Traditional small-molecule inhibitors are unable to block kinase activity and scaffold signalling of IRAK |
| 5 | Rheumatoid arthritis, Alzheimer's neuroinflammation | JAK/STAT | Lipid profile disturbances, thromboembolic complications |
| 6 | Asthma, atopic dermatitis | IKT | Poor pharmacokinetics, insufficient target inhibition |
| 7 | Crohn's disease, IBD, multiple sclerosis | RIPK2 | Toxicity and safety issues during clinical trials; intolerance to therapy over time |
| 8 | IBD, autoimmune uveitis | BRD4 | Short half-life, off-target effects, development of resistance |
| 9 | Systemic lupus erythematosus, Aicardi-Goutières, psoriasis | STING | Poor solubility profile, limited selectivity |
| 10 | Inflammatory bowel syndrome, hepatitis | MIF | Low water solubility, pulmonary interstitial fibrosis |
| 11 | Asthma, arthritis | MAFF | Limited oral bioavailability |
| 12 | Acute lung injury, allergy | H-PGDS | Differences in pharmacokinetics and pharmacodynamics between preclinical animals and humans |
| 13 | Alzheimer's and Parkinson's diseases | GSK-3β | Small therapeutic window, low selectivity |
| 14 | Neurodegenerative diseases | Keap 1 | Off-target effects unpredicted reactions |
| 15 | Neuroinflammation, Alzheimer's disease | SHP 2 | Higher dose |
| Insulin-induced endothelial inflammation COPD, asthma | Drug resistance, less efficacy |
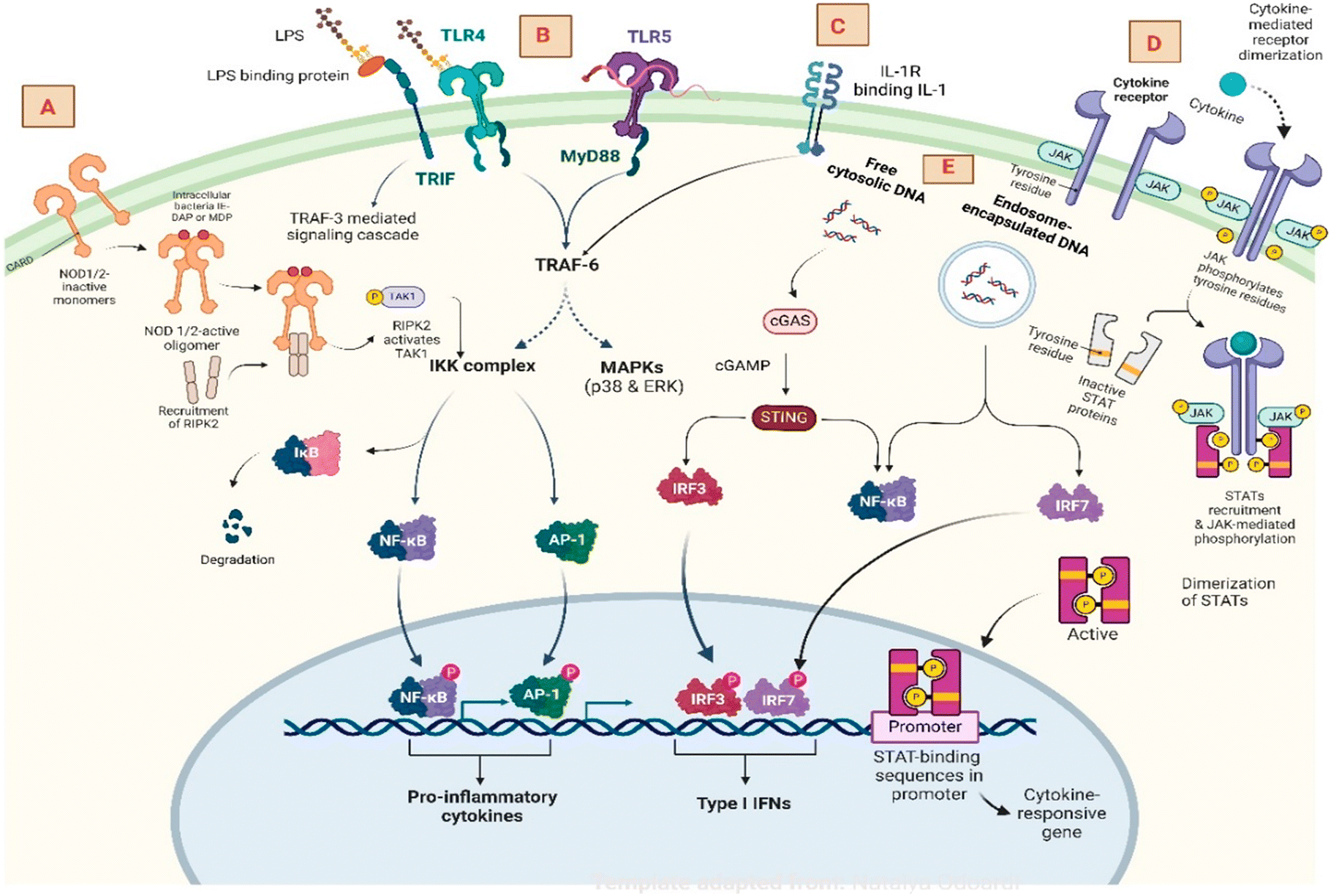 | ||
| Fig. 2 The inflammatory signalling pathway and key components: A) NOD1 and NOD2 receptors involving RIPK1&2 to activate NF-κB for gene translation. B) Toll-like receptors activate the MyD88 complex, followed by IKK and MAPK induction to produce an inflammatory response. C) IL-1R signalling pathway to TRAF-6 and NF-κB. D) The JAK–STAT pathway activates the tyrosine kinase receptor recruiting JAK-mediated pathway to produce cytokine-responsive genes. E) Cytosolic DNA pathway in activating cGAS STING generating interferon regulating factor. | ||
PRTOACs (proteolysis targeting chimeras) represent a revolutionary breakthrough advancement in TPD. They are also called heterobifunctional molecules meant to hijack the ubiquitin–proteasome system, thus overcoming all the glitches in traditional chemotherapy in terms of selectivity, catalytic nature,13 and potency.14,15 Currently, the creation of tailored protein degradation techniques for treating multiple malignancies has been gaining much attention.16,17 This golden gateway has diminished nearly all the major drawbacks of drug discovery using autophagy as a pivotal tool, i.e., a slight modification of the body's destruction machinery, causing the target approach and thus facilitating its degradation through ubiquitination. Perhaps, implying this technique in inflammatory-mediated diseases would herald a new era in drug innovation.
1.1 Timeline of PROTAC
The Crews and Deshaies group pioneered the ground-breaking idea of PROTAC in 2001.18 In 2008, Schneekloth et al. introduced the first small-molecule PROTAC.19 The timeline of two decades of PROTACs demonstrates how briskly the technology has progressed from academia to industry to clinical candidates.201.2 Mechanism of action
Since the mechanism of PROTACs has been described in numerous reports, its pathway is elucidated in short as follows.21 The highly conserved ubiquitin–proteasome system (UPS) is meant for degrading misfolded, non-functional, and other undesirable proteins in the body. It also reported that it is crucially engaged in a diverse array of cellular processes, such as gene transcription,22 protein transport,23 synaptic plasticity and memory functions.24 The degradation pathway is initiated with ubiquitin-activating enzyme (E1) activating the free ubiquitin, concurrently transferring the ubiquitin to the conjugating enzyme E2.25 The E3 ligase enzyme then catalyses the final step of ubiquitination, which allows the transfer of the ubiquitin molecule from E2 to the target protein via forming an iso-peptide bond between the lysine residue of the targeted protein and glycine of ubiquitin. Subsequent ubiquitination leads the target protein to be polyubiquitinated.26 Finally, the 26S proteasomal degradation pathway easily recognizes this polyubiquitinated protein for destruction. Ubiquitin molecules are utilised for the subsequent cycle of degradation in this overall process.27The E3 ligases/degrons are designated as crucial elements in PROTACs, which are responsible for the ubiquitination. Although more than 600 E3 ligases are known to exist in the human body, only a few are being utilized in the development of PROTACs. Von Hippel–Lindau (VHL) and cereblon (CRBN) are the most used E3 ligases. Inhibitors of apoptosis protein (IAP), Kelch-like ECH-associated protein (KEAP1) and mouse double minute 2 (MDM2) are also being explored.28,29 In this review, we briefly summarize the status of anti-inflammatory targets being explored as PROTACs and their key findings. In the discussion section, we focus on the crucial topics relevant to the growth needed for anti-inflammatory PROTACs from the perspective of achieving success in bringing the molecule from the bench to the clinic.
2. HDAC
Histone and non-histone proteins present in DNA via acetylation and deacetylation govern various functions at the cellular level, such as gene expression and cellular proliferation.30 Blocking of HDAC can lead to cell death and anti-inflammatory activities.31,32 A non-selective HDAC inhibitor, trichostatin A (TSA), showed elevation in the GM-CSF (granulocyte-macrophage colony-stimulating factor) and interleukin-8 (ref. 33) Eventually, SAHA (suberoylanilide hydroxamic acid) and TSA, well-known potent HDAC inhibitors used in cancer treatment, were proven to reduce swelling, pannus formation and bone erosion in rats with induced arthritis.34 Hendrix et al. reported HDAC VIII inhibition with reduced pro-inflammatory and anti-inflammatory mediators.35,36 Likewise, HDAC VI is accountable for the regulation of inflammation and is used in the treatment of rheumatoid arthritis for the improvement of synovial inflammation.37 NLRP3 (NLR family pyrin domain-containing protein 3 inflammasome) is expressed predominantly in macrophages, and the mutation of this gene is associated with the pathophysiology of many immune-related diseases such as rheumatoid arthritis, gout, diabetes mellitus, etc.38Dekker and co-workers detailed the PROTACs approach over HDAC inhibitors in controlling inflammatory diseases like asthma, COPD (chronic obstructive pulmonary disorder), etc. With o-amino anilide CI994 (tacedinaline, ligand for class-1 HDAC I, II and III) as a warhead and cereblon as the E3 ligase, a series of HD-TACs (HDAC-PROTACs) has been developed by varying linker lengths. Compound 1 (Fig. 3), having o-aminoanilide substituted with the fluorine atom, showed improved selectivity resulting in an IC50 value of 1.1 μM ± SD against HDAC III degradation in RAW 264.7 cells, and a DC50 value of 0.32 μM, using western blot analysis.39
 | ||
| Fig. 3 HDAC-targeted PROTACs. | ||
To address a variety of inflammatory conditions linked to the NLRP3 inflammasome, Cao et al. investigated the HDAC VI protein. They introduced the concept of ‘click’ chemistry to synthesize PROTAC molecules comprised of different linker lengths by joining the alkyne moiety at the 1-position of indirubin with pomalidomide. Compound 2 exhibited a DC50 of 108.9 nM against K562 cell lines, along with a significant reduction in the release of IL-1β, as well as nigericin-induced caspase-1 processing (p20). In vivo studies of compound 2 revealed that it leads to a decrease in IL-1β levels and has no role in affecting tumor necrosis factor-α (TNF-α) levels, indicating the inhibition of the NLRP3 inflammasome pathway activation.40
Yang et al. optimized a cell-based assay method to evaluate the binding affinity for modified CRBN ligands with thalidomide, pomalidomide and lenalidomide. Triazoles were incorporated as linkers, and among them, lenalidomide exhibited maximum binding affinity, proving its promising ability to be incorporated as an E3 ligase for PROTAC molecules. Further analysis of compound 3 resulted in the significant HDAC VI degradation of 1.9 nM, which is comparable to PROTACs synthesised based on nextruastat-A.41
Chotitumnavee and co-workers performed in silico studies upon obtaining the X-ray crystal structure of HDAC VIII with NCC149 (N-hydroxy-3-[1-(phenylthio)methyl-1H-1,2,3-triazol-4-yl]benzamide) to determine a suitable docking position for the inhibition of HDAC VIII. Among the synthesized PROTACs, compound 4 showed dose-dependent HDAC VIII degradation with 11 carbon linker length and exhibited a DC50 of 0.702 μM. In vitro HDAC assay studies revealed that the compound has an IC50 of 0.372 μM against HDAC VIII, irrespective of other isoforms of HDACs.42
3. IRAK3 and 4 (interleukin-1 receptor-associated kinase)
Interleukin-1, interleukin-18, and toll-like receptors (TLRs) are critical immune factors involved in the pathway of interleukin-1-associated kinases. IRAK4 protein is currently the subject of intensive research for treating immunological disorders as it is involved in rheumatoid arthritis and hidradenitis suppurativa (HS) or atopic dermatitis (AD). Presently one lead molecule, KT-474, has entered the phase II clinical trials targeting IRAK4 through TLR/IL-1R mediated pathways.43Due to the high degree of similarity between the ATP binding sites of IRAK1 and IRAK4, selective inhibitors of IRAK4 could be valuable for therapeutic strategy in inflammation-mediated diseases like systemic lupus erythematosus. Kargbo et al. reasoned that it is particularly challenging to study the catalytic domain of IRAK4's protein since it was highly conserved where the scope of exploration in IRAK4 was delayed. Since the PROTAC could overcome all these limitations, the team developed compounds 5 and 6 (Fig. 4) to provide potent degradation for IRAK4 resulting in IC50 and DC50 values below 30 nM.44 Later, many groups focused on achieving a suitable linker for IRAK4 degradation, employing known inhibitors as warheads. Firstly, Nunes et al. employed isoquinoline warheads (IRAK4 inhibitor) and various E3 ligands, namely VHL (Von Hippel–Lindau), CRBN (cereblon) or IAP (inhibitor of apoptosis protein). Among them, compound 7, possessing a fluoro-group, revealed a DC50 of 259 nM through western blotting analysis. Further structural refinement of 7 into 8 exhibited a DC50 value of 151 nM, which includes a spirocyclic pyrimidine linker, more rigid and polar than a flexible linear carbon chain.45 Similarly, Chen and co-workers also investigated shorter linker length PROTACs for IRAK4 observing no significant protein degradation, whereas compound 9 with a polyethylene glycol linker was effective in lowering IRAK4 levels.46
 | ||
| Fig. 4 IRAK3 and 4-targeted PROTACs. | ||
Zhang et al. explained the inhibitory action of compound 10 by decreased IL-6 production; docking studies revealed that the molecule's cyclopropyl group in the solvent-exposed region is a convenient site for linker conjugation. By structurally altering the cyclopropyl group to a linker, a set of linkers was synthesized and evaluated, of which 10c and 10d showed DC50 of 190 nM and 405 nM by immunoblotting.47
We know that IRAK is a large group of families, involving IRAK1, IRAK2, and IRAK4 stimulating myeloid differentiation primary response 88 (MyD88)-dependent activation of nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) and thus pro-inflammatory cytokine production. Degorce et al. encountered serendipitous results in the discovery of potent IRAK3 inhibitors while studying the IRAK4 protein. The cis-isomer of pyrrolotriazine has been employed as an efficient POI ligand with VHL or CRBN as an E3 ligase. CRBN E3 ligases containing PEG linkers showed better degradation than VHL ligands. It was noted that the length of the linker is directly proportional to degradation in the case of CRBN. Compounds 11 and 12 showed selective degradation profiles with DC50 values of 0.052 μM and 0.002 μM for IRAK3 by 74% and 98% Dmax, respectively.48
Similar efforts were made by Rowley et al., who studied the IRAK3 protein for controlling the dendritic cell-mediated production of interleukin-12 (IL-12). Crizotinib as the warhead, VHL as the E3 ligase ligand, and compound 13 with trans-cyclohexylamide linker resulted in the preferential degradation of IRAK3. Treatment of 13 in THP-1 cell lines resulted in a DC50 value of 94 nM and 95% Dmax. Treatment of dendritic cells with 13 resulted in IRAK3 degradation both in the presence and absence of LPS, along with the significant elevation in IL-12p levels with LPS stimulation.49
4. PCAF and GCN5
PCAF (p300/CBP-associated factor) and GCN5 (general control nonderepressible 5) are closely related epigenetic proteins that regulate immune functions in various cellular pathways. According to Zheng et al. and Zhuang et al., PCAF and GCN5 are potential targets for treating autoimmune disorders as they cause a reduction in H3K18, H3K9 acetylation, accompanied by NF-κB, IFN-β (interferon β), TBK1 (TANK binding kinase-1), which are overexpressed in injury, regulating inflammatory action, and modulating immune signaling.50–52Bassi et al. focused on the PCAF/GCN5 PROTACs and employed GSK4027, an active inhibitor of PCAF and GCN5, with CRBN as an E3 ligase for enhancing the activity. Compound 14 (Fig. 5) showed a DC50 value of 1.5 nM for PCAF degradation and 3 nM DC50 for GCN5 degradation in THP-1 cell lines, and 80% degradation of proteins was observed within 10 min at a concentration of 30 nM. The cis (R,R) enantiomer of the parent compound GSK4027 is more active than the other enantiomer.53
 | ||
| Fig. 5 PCAF-GCN5 targeted PROTACs. | ||
5. JAK–STAT (Janus kinase-signal transducers and activators of transcription)
Another leading potential target in cytokine signaling is the JAK/STAT pathway involving interleukin-2 and other inflammatory factors, such as γ-interferons. Cytokines binding to JAK receptors initiate STAT transphosphorylation, leading to gene expression for activities like proliferation and differentiation,54 which are responsible for numerous disorders, such as rheumatoid arthritis (RA), Alzheimer's disease (AD), inflammatory bowel disease (IBD), and multiple sclerosis (MS). JAK inhibitors such as tofacitinib 15, ruxolitinib 16, baricitinib 17, (Fig. 6), etc., provided benefits for the improvement of rheumatoid arthritis by altering T-cell activation and natural killer (NK) cell activity.55 Although JAK/STAT targets were disclosed decades ago, there are still challenges such as the bulky dose of the drug with a short half-life, side effects like thromboembolism and cross-talk between the JAK/STAT pathway and non-canonical signal transductions.56 PROTACs could be a simple answer to the limitations.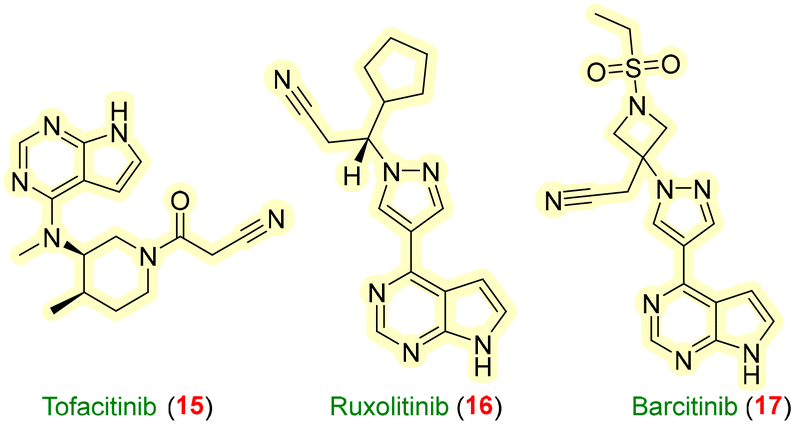 | ||
| Fig. 6 Drugs used as JAK–STAT inhibitors. | ||
Shah et al. disclosed PROTACs with pyrimidine and quinoxaline cores as warheads. The primary aim was to develop PROTAC molecules with suitable physicochemical properties to permeate the cells. Among them, the quinoxaline motif showed better permeability than pyrimidine motifs and exhibited more selectivity towards JAK 2. The PBMC (peripheral blood mononuclear cells) assay has been performed to identify the trends regarding cell permeability. The molecules were tested against THP-1 cell lines bearing JAK 1 and 2 proteins, out of which six PROTACs (21–26, Fig. 7) were proven to show appropriate degradation.57
 | ||
| Fig. 7 JAK/STAT targeted PROTACs. | ||
In 2021, Kargbo filed a patent unravelling PROTAC-mediated degradation targeting Janus kinase (JAK). Pyrrolo-pyrimidines were evaluated for the Caco-2 cell permeability experiment, CRISPER-Cas9 editing, and other biological assays, which revealed that compound 28 has the lowest inhibitory concentration of EC50 = 0.0002 μM for JAK protein degradation. A simple modification of the linker pattern resulted in a drastic increase in EC50 values. The team evaluated PROTACs 29 and 30 containing ruxolitinib and baricitinib (potent JAK inhibitors) as POI ligands with benzamide-containing linkers providing EC50 values of 0.00054 μM and 0.0039 μM, respectively.58
6. ITK (interleukin (IL)-2-inducible T cell kinase)
Jiang et al. designed a potent, target-selective ITK (IL-2-inducible T-cell kinase) PROTAC 31 (Fig. 8) to help in degrading inflammatory responses with the inhibition of STAT3 and STAT5 signaling. Compound 31 showed >90% degradation of ITK selectively at a 1 μM concentration. To determine its effect on immunology, they treated against the Hut78 cell line and observed a significant decrease in the GATA-3 (transcription factor binding to DNA sequence-3) response.59 | ||
| Fig. 8 ITK targeted PROTACs. | ||
7. RIPK2 (receptor-interacting serine/threonine protein kinase 2)
RIPK2 functions as a downstream protein for the detection of the NOD1 (nucleotide-binding oligomerization domain 1) and NOD2 (nucleotide-binding oligomerization domain 2) receptors, and further aids in the detection of bacterial protein and regulates the immune system by activating pro-inflammatory signaling in the body.60 A pyridinyl imidazole inhibitor and gefitinib showed significant RIPK2 inhibition and led to improvement in Crohn's disease.61 RIPK2 is a potential target for treating inflammatory illnesses such as IBD (inflammatory bowel disease), pulmonary sarcoidosis, and multiple sclerosis.62 To date, there are no RIPK2 molecules available for clinical use, and reported molecules might not be suitable for further application due to their pan-inhibitions.63Mares et al. developed RIPK2 PROTACs to envision in vivo pharmacokinetics and pharmacodynamic profiles by employing aminobenzothiazole-quinoline warheads with IAP E3 ligase. Molecule 32 (Fig. 9) showed a pDC50 of 9.4 ± 0.2 in THP-1 cell lines but had a poor solubility profile. Therefore, it was structurally refined to 33, consisting of pyrazole-quinazoline, to enhance its solubility by adding non-lipophilic groups. Along with reduced lipophilicity and increased solubility, it showed a pDC50 of 7.9. Consequently, to address the large dose dependency, for PROTAC 34, methylene groups increased its binding and potency by ten-fold, with a pIC50 of 8.0.64
 | ||
| Fig. 9 RIPK2-targeted PROTACs. | ||
Bondeson et al. developed a RIPK2-based PROTAC for treating autoinflammatory diseases such as Blau syndrome, which is involved in the NF-κB and MAPK activation. Compound 35 resulted in a DC50 of 1.4 nM and a Dmax of 95% in THP-1 cell lines at a concentration >10 nM.65
8. BRD4 (bromodomain-containing proteins)
Bromodomain-containing proteins expressed by the BET (bromodomain and extra-terminal domain) family have been proven to significantly affect the inflammatory genes through NF-κB or Nrf2 (nuclear factor erythroid 2-related factor 2) pathways.66 JQ1 and I-BET are two potent and selective BET inhibitors that have shown their capability in anti-inflammatory activities. As the literature suggests, JQ1 binds with greater affinity to BRD4 than other BET proteins and modulates histone acetylation function, which is responsible for producing inflammatory genes.67 In PROTACs for BRD4, the initial efforts of employing JQ1 as warheads, focusing on the optimal E3 ligases, have been discussed.Tong et al. employed a terpene-based derivative such as bardoxolone, an E3 ligase recruiter, as an electrophilic moiety to establish reversible covalent bonds with the cysteine residue of the KEAP1 (Kelch-like ECH-associated protein) and JQ1 (BRD4 inhibitor), which was employed as a POI to explore the activity of bardoxolone as an E3 ligase. The paper highlights the critical need to explore diverse E3 ligases to withstand the emerging resistance of PROTACs. The descyano derivative 37 (Fig. 10) resulted in the loss of activity to degrade BRD4 in 231 MFP cells, indicating the significance of the α-cyanoenone moiety for the inhibitory action towards BRD4. In general, it has been found that any alteration to the bardoxolone methyl group would significantly impair both its anti-inflammatory efficacy and the activation of the Nrf2 pathway.68
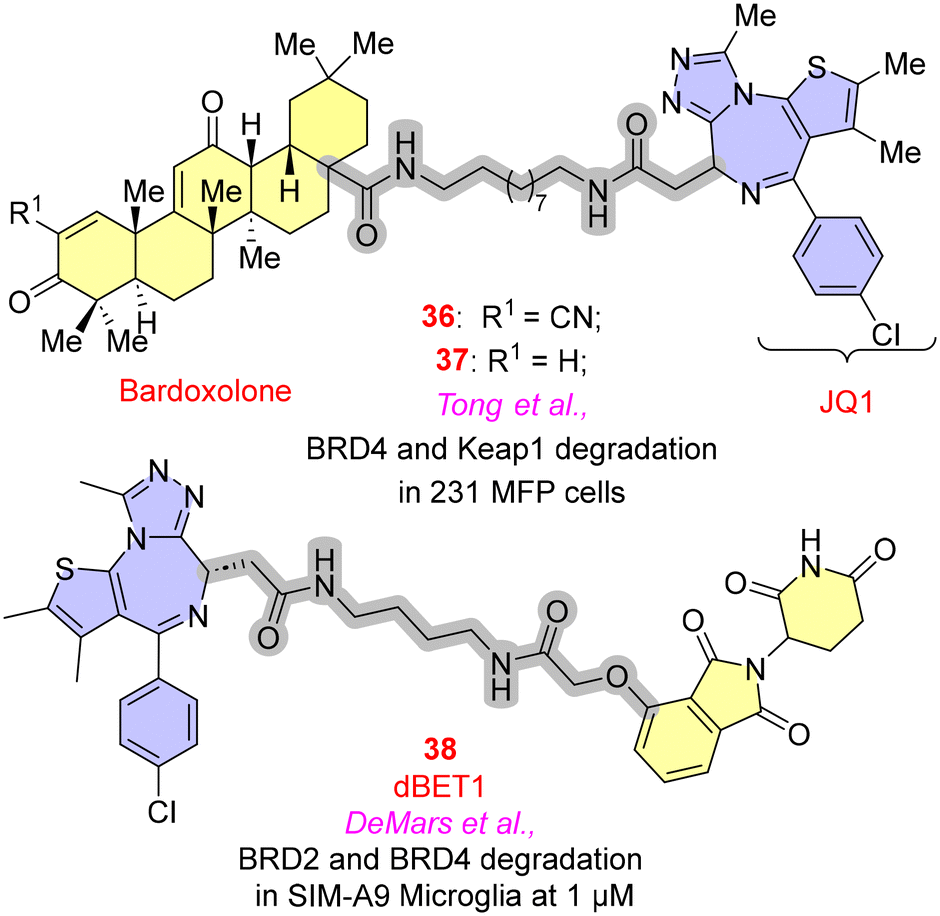 | ||
| Fig. 10 BRD4-targeted PROTACs. | ||
iNOS (inducible nitric oxide synthase), COX-2 (cyclo-oxygenase-2), IL-1β, TNF-α, IL-6, CCL2 and MMP-9 (matrix metalloproteinases) are pro-inflammatory factors that are modulated with BET proteins. A significant reduction in the expression of these proteins was observed with the degradation of BRD2 and BRD4. DeMars et al. studied dBET1 in the treatment of neuroinflammatory diseases. JQ1 was a warhead with CRBN E3 ligases. In SIM-A9 microglia cell lines, time-dependent degradation of BRD2 and BRD4 of 38 was observed at the concentration of 1 μM, blocking the production of pro-inflammatory genes.69
9. BTK (Bruton's tyrosine kinase)
Many pro-inflammatory factor modulations were correlated with Bruton's tyrosine kinase (BTK) to maintain immune haemostasis. X-linked agammaglobulinemia (XLA) is an immunodeficiency disorder caused by the suppression of BTK signalling. Targeting BTK protein would reduce various inflammatory factors such as IL-6, IL-1, TNF-α, type 1-IFN, etc. Robert B. Kargbo and co-workers designed and patented BTK degraders; compound 39 (Fig. 11), containing a piperazine linker with CRBN E3 ligase, and compound 40, containing constrained butane and piperidine rings with MDM2 E3 ligase, showed minimum DC50 values of 0.0029 μM and 0.0072 μM, respectively, resulting in excellent activity.70 | ||
| Fig. 11 BTK-targeted PROTACs. | ||
Given the linker modifications, Tinworth et al. compared the degradation profile of covalent binding and reversible binding PROTAC molecules against the BTK target. Halo-PROTACs are a suitable example of covalent binding PROTACs with profound degradation. Compound 42 resulted in concentration-dependent BTK degradation with a DC50 of 200 nM for recruiting the VHL E3 ligase. Covalent PROTACs 41, 43 resulted in no BTK degradation. In contrast, 44 was slightly active towards the degradation. This suggests that non-covalent interactions primarily mediated the degradation as compared to the covalent binding PROTAC. The researchers proposed the reason for this as the blockade of the site by the covalently bound ligand for ubiquitin transfer or the inability of the proteasome to recognize the modified region required for degradation.71
10. STING (stimulator of interferon genes)
Stimulators of interferon genes (STING) present in the human body aid in the detection of bacterial nucleotides synthesized by the cyclic GMP-AMP synthase enzyme (cGAS) present in bacteria. Under normal circumstances, the human body has a cGAS-STING pathway to regulate innate immune responses. In abnormal conditions, this STING pathway activates pro-inflammatory mediators like cytokines and the release of IFNs to fight against bacterial infections. Liu and co-workers designed PROTACs to target the STING pathway to treat various autoimmune and inflammatory conditions like Aicardi-Goutieres syndrome and systemic lupus erythematosus. Compounds 45–50 (Fig. 12) were synthesized using C-170 as a warhead that interacts with the STING protein through its nitro and furan moieties. In contrast, the benzene ring plays a crucial role by providing a tethering site for the CRBN E3 ligase through a suitable linker. It was observed that the compounds exhibiting α, β-unsaturated carbonyl linkers are biologically active towards degrading the STING protein; compound 47 showed the most satisfactory results with a DC50 of 3.2 μM with time-dependent degradation. In vivo studies by ELISA in the kidney tissues of mice showed that 47 could reduce the level of IFN-β, IL-6, and CXCL-10 in a dose-dependent manner.72 | ||
| Fig. 12 STING-targeted PROTACs. | ||
11. MIF (macrophage migration inhibitory factor)
Although MIF is known for its anti-cancer potential, it is also associated with immune responses. In 2021, Xiao et al. studied the effects of macrophage migration inhibitory factor (MIF) degradation by employing 7-hydroxycoumarin and 7-hydroxy-3,4-dihydrobenzoxazin-2-one cores as warheads, with cereblon as the E3 ligase ligand. Compound 54 (Fig. 13) was found to be the most potent, with 91% degradation at 2 μM and 71% degradation against A549 cells. HEK293 cell lines had also been tested with 54, which resulted in 90% degradation at 200 nM. The cellular uptake of 54 was also confirmed using intrinsic fluorescence microscopy studies.73 | ||
| Fig. 13 MIF-targeted PROTACs. | ||
12. IKZF (Ikaros zinc-finger transcriptional factors)
Ikaros zinc-finger transcriptional factors (encoded by gene IKZF1) and Aiolos (encoded by gene IKZF3) are critical for immunomodulatory activity and their polymorphism can lead to systemic lupus erythematosus. Two degronimers, 57 and 58 (Fig. 14), have been developed to target the E3 ligase cereblon, which may cause the potential degradation of Aiolos or Ikaros through the ubiquitin–proteasome pathway. The investigation of these compounds for binding to the CRBN-DDB1 (DNA damage binding protein-1) complex using fluorescence polarization assay resulted in values less than 10 μM Kd.74 | ||
| Fig. 14 IKZF-targeted PROTACs. | ||
13. MAFF (v-maf musculoaponeurotic fibrosarcoma oncogene family)
To date, there have been many reports on the anti-inflammatory activity of lathyrane diterpenoids. Wu et al. studied PROTACs as a potential strategy for identifying the targets for natural products. ZCY-001 was employed as the warhead for the development of ZCY-PROTAC 59 (Fig. 15) with thalidomide as the E3 ligase linked by a 3-PEG linker, which resulted in the profound inhibition of LPS-stimulated NO (nitric oxide) release. It also showed that MAFF degradation is responsible for inflammatory gene expression. However, individually, lathyrol and thalidomide treatment resulted in no degradation of MAFF, thus confirming that the degradation is induced through the ternary complex formation by PROTAC.75 | ||
| Fig. 15 MAFF-targeted PROTACs. | ||
As an extension of this work, the same group designed 15 new PROTACs based on 59 to establish the anti-inflammatory action of lathyrol PROTACs. Different E3 ligase ligands (VHL, cIAP and MDM2) were employed with variations in the linker chains. Compound 60, having a PEG linker chain and cIAP ubiquitin ligase ligand, showed maximum inhibitory activity with an IC50 value of 5.30 ± 1.23 μM against LPS-induced NO production in RAW264.7 cells.76
14. H-PGDS (hematopoietic prostaglandin D synthase)
For the treatment of allergic disorders, targeting H-PGDS (hematopoietic prostaglandin D synthase) would be beneficial because some allergy disorders are linked to excessive prostaglandin D2 production. However, H-PGDS inhibitors have not shown adequate clinical efficacy. With the advancing research, Yokoo et al. approached PROTACs and SNIPERs (specific and nongenetic IAP-dependent protein erasers) as potential treatments (Fig. 16). TFC-007 (a potent inhibitor of H-PGDS with IC50 of 83 nM) was employed as a warhead and PROTAC (H-PGDS)-1 61 was synthesized by employing pomalidomide as a E3 ligase. Significant reduction in H-PGDS was observed with maximum degradation between 100–1000 nM in Human KU812 and MEG-01 cell lines. For better outcomes in degradation and to improve pharmacokinetics, in silico simulations were conducted. A library of compounds was designed to dock and understand the binding pattern of the stable ternary complex of PROTAC (H-PGDS). A deeper analysis revealed that the length of the linker is inversely proportional to the stability of the ternary complex. Astonishingly, compound 62 without any linker showed maximum protein degradation with a DC50 value of 17.3 pM against KU812 cell lines. The co-crystal structure of the H-PGDS protein indicates that the N-carbonyl piperazine–piperidine moiety of TFC-007 serves as a linker rather than a warhead because it does not bind to the protein. This probably reduced its molecular weight and number of rotatable bonds and rendered compound 62 more druggable with the rule of five chemical spaces.77 | ||
| Fig. 16 H-PGDS-PROTACs molecules. | ||
15. IDO1 (indoleamine 2,3-dioxygenase 1)
IDO1 has been identified as a possible candidate for cancer immunotherapy due to its substantial role in the tumor immune escape mechanism through T-cell regulation. Considering this, Hu et al. developed a PROTAC molecule based on Epa (highly selective IDO1 inhibitor) that was employed as a warhead with E3 ligases like lenalidomide and pomalidomide. Compound 63 (Fig. 17), containing seven PEG units, resulted in the degradation of IDO1 with a DC50 value and Dmax of 2.84 μM L−1 and 93%, respectively.78 | ||
| Fig. 17 IDO1-PROTACs molecules. | ||
Later, Bollu et al. reported a PROTAC molecule-degrading IDO1 protein. A library of PROTACs was synthesized based on CRBN and VHL-based E3 ligase ligands and with the employment of linrodostat. CRBN-based PROTAC molecules with PEG linkers (<10 carbon atoms) showed a favourable pharmacokinetic profile. Compound 64 with a para-attachment to the linker was the most potent, with 94% degradation of IDO1 with a DC50 of 0.3290 μM in U87 cell lines and 0.5438 μM in GBM43 cell lines.79
16. GSK-3β (glycogen synthase kinase 3β)
GSK-3β is regarded as a critical target for treating several neurodegenerative disorders, most notably Alzheimer's disease. GSK-3β demonstrates its pronounced pro-inflammatory effect, resulting in the loss of neurons. Jiang et al. conjugated the GSK-3β inhibitor with E3 ligases with variable chain length linkers. Through click chemistry, compound 65 (Fig. 18) showed 65.8% degradation of the GSK-3β at 10 μM. To examine the inhibitory activity of compound 65 towards the TNF-α, neuroinflammation was induced in the microglia BV2 cells of the mouse. As anticipated, 65 reduced the TNF-α to its basal value, attenuating several inflammatory responses.80 | ||
| Fig. 18 GSK-3β-PROTACs molecules. | ||
17. Keap 1 (Kelch-like ECH associated protein 1)
Lu et al. designed a peptide-based PROTAC that acts on keap1 (Kelch-like ECH associated protein 1) that served as a substrate protein for the Cul3/Rbx1 (cullin-3/RING box protein-1)-dependent E3 ligase complex. The PROTAC facilitates the degradation of tau protein by binding with Keap1, which regulates the Nrf2 signalling pathway related to inflammatory diseases. Ac-LDPETGEYL-OH, a peptide-based PROTAC 66 (Fig. 19), was synthesized and it had a strong affinity for both Keap1 and Tau proteins, with Kd values of 22.8 nM and 763 nM, respectively. PROTAC 66 was crucial for tau degradation, as evidenced by CO-IP, flow cytometry, and western blotting assays in SH-SY5Y, Neuro-2a, and PC-12 cells. Targeting tau breakdown through Keap1 recruitment may be a better way to treat neurodegenerative diseases than suppressing the Nrf2 pathway.81 | ||
| Fig. 19 Peptide PROTAC inhibiting keap1 protein. | ||
Keap1 protein is associated with Nrf2 pathway activation and mediates the oxidative response. Wang et al. designed PROTAC 67, which includes a chimeric keap1-keap1 peptide to recruit keap1 target proteins and connect them to the E3 ligase. The peptide sequence was conjugated to the cell-penetrating peptide hPP10 through the C-terminal to obtain 67, which resulted in 25% of keap1 protein degradation when treated in HSC-T6 cells (hepatic stellate cells). The degradation of keap1 protein was found to be associated with the activation of ARE (antioxidant release elements) through the Nrf2 pathway, ultimately leading to the downregulation of TNF-α, IL-1β, and IL-6. The degradation was confirmed to be induced via the ubiquitin–proteasome pathway.82
Ji et al. designed PROTACs based on the ARE (antioxidant response elements) sequence as the Nrf2 ligand. The double-stranded oligonucleotide sequence 5′-TCACAGTGACTCAGCAGAATC-3′ from the NQO1 (NADPH quinone oxidoreductase 1) promoter was recruited as a warhead. Two series of ARE-PROTAC molecules were synthesized based on CRBN and VHL E3 ligands using a CuAAC (copper-catalyzed azide-alkyne cycloaddition) reaction for conjugation with the Nrf2-DNA ligand. CRBN-based ARE-PROTACs showed favourable depletion levels when tested in A549 cells. Among them, 68 (Fig. 20) is the most potent compound exhibiting a DC50 of 1.85 nM against Nrf2 protein and a DC50 value of 66 nM against MafG (musculoaponeurotic fibrosarcoma transcription factor G) in A549 cells. Antioxidant activity was also lowered, leading to oxidative damage to cancer cells.83
 | ||
| Fig. 20 Nrf2-targeted PROTACs. | ||
18. SHP2 (Src homology 2 domain-containing phosphatase 2)
SHP2 is a protein tyrosine phosphate encoded by the PTPN11 gene and was considered difficult to target because of its highly positively charged nature. In this regard, Wang et al. put efforts into designing a PROTAC molecule with a potent SHP2 inhibitor (IC50 = 98.7 nM). With the recruitment of the VHL E3 ligand, a series of PROTAC molecules have been designed with varying lengths of alkyl, PEG and piperazinyl linker chains. SHP2-D26 (69, Fig. 21) showed maximum SHP2 degradation with DC50 values of 6.0 and 2.6 nM in KYSE520 and MV4 cell lines with good aqueous solubility.84 | ||
| Fig. 21 SHP2-targeted PROTACs. | ||
19. Perspectives and findings
Undeniably, the complexity of the immune signalling pathways and their underlying mechanisms have made the interventions more challenging. Being the next big therapeutic area after oncology, their advancements are of utmost importance to the research community. In most of the scenarios, the severity of diseases aberrantly depends upon the individual's genetic makeup, making their treatment limited for the targeted therapy. Existing protein therapeutics like monoclonal antibodies (mABs) bind straightforwardly to the targets, yet their potency is limited towards in vivo aspects like bulky molecular weight, poor bioavailability, and high in terms of cost. Comparatively, small molecules have showcased better pharmacokinetic profiles compared to mABs; nevertheless, their drawbacks in binding to the target, and in protein accumulation resulted in extending the half-life, which is detrimental to the efficacy of drugs. Furthermore, it enabled only 20–25% of druggable targets in the entire human body to be covered. Amalgamating all the instances underscores a pressing need for an innovative approach. The quest was fulfilled with the advent of PROTACs as valuable therapeutic tools surpassing the drawbacks of the aforementioned strategies. Undoubtedly, PROTACs exemplify excellent in vitro potency in explored targets like cancer, autoimmune diseases, etc., with established clinical proof of concept. However, there are hurdles to reaching clinical prospects, narrowing the way regarding the solubility and low pharmacokinetic profile of the molecules. However, research efforts have made it possible by the fundamental design, such as optimizing the linker length to stabilize the ternary complex, adding non-lipophilic groups to increase solubility, and nano-formulations for sustained drug delivery. Given the treatment strategies, the clinical exposure of PROTACs could be confined to a limited duration because long-term use could result in complete loss of the proteins, prompting the risk of developing additional immune diseases. Given that our body system is intricately designed with counter-current mechanisms, only in the case of overexpression or encroachments of a protein due to the loss of self-recognizability or the entry of pathogens is the implication of treatment tactics necessary. Additionally, since they are not fond of inhibition but ultimately degrade the target, the escalation of doses and tolerance buildup might be less/not observed in PROTACs. Given the formulation aspects, the nano-formulations with the extended release of the drug make it possible to prolong the half-life through oral/IV administration.At present, two degraders, ARV-110 and ARV-471, entered phase II clinical trials for treating prostate and breast cancers. IRAK 4 PROTAC has reached phase II clinical trials (KT-474) to treat hidradenitis suppurativa (HS) and atopic dermatitis (AD). The phase I trial revealed that PROTACs are well tolerated in healthy volunteers at multiple dose frequencies and cope with the safety profile without any adverse effects. A 14 day trial of KT-474 showed reduced IRAK4 levels by over 95% in PBMCs from patients or healthy volunteers. It also dampened the elevated levels of proinflammatory cytokines in the patients' plasma, explaining the drug's high selectivity for IRAK4.43 Another blockbuster clinical candidate in the phase I trial was NX-2127, a degrader of BTK with immunomodulatory activity by IKZF1 degradation. Preclinical evaluation of this molecule ensured the safety parameters conducted by repeated-dose toxicology studies, and cross-species pharmacokinetics.85
20. Future perspectives
A more intensive focused study on the anti-inflammatory targets will allow promising clinical candidates to be the next generation of therapeutics. Unlike small molecules, PROTACs offer unlimited benefits like binding, which is specifically a secondary concern for degradation, only relevant groups are mandatory for binding with the protein. This will reduce the molecular weight of the compounds and additional covalent interaction/irreversible bindings, leading to toxicity. These unnecessary groups can be modified as linkers to reduce the additional bulkiness for PROTACs. From the origin of PROTACs, it was majorly confined to anticancer drug development, while other targets received less attention in recognising their potential. In inflammatory/autoimmune diseases, the breach between the existing therapies like NSAIDS and the discovery rate of novel entities is very large. It was observed that the incidence rates of rare/uncurable disorders have been abruptly increasing irrespective of age, gender, species, etc. For example, international attention towards irritable bowel syndrome (IBS) is being carried forward as the condition depends on the individual genetic makeup enclosing minimal treatment options. Gratifyingly, given the progress of BioPROTACs and hybrid PROTACs, the solutions to POI that lack the ligand binding site could be found. The footsteps of immuno-oncology will be an important modality for treating resistance-mediated cancers. All the concerns consolidate to highlight the immediate need for the development of novel PROTACs for future generations.Author contributions
M. S. Galla: writing – review & editing – original draft. P. Mishra, N. Sharma: literature survey, writing N. Shankaraiah: conceptualization, investigation, writing – review & editing, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Authors are thankful to DoP, Ministry of Chemicals & Fertilizers, Govt. of India, New Delhi, for the award of NIPER fellowship.References
- Z. Yin, H. Popelka, Y. Lei, Y. Yang and D. J. Klionsky, Cells, 2020, 9, 2025 CrossRef CAS PubMed.
- Y. Ding, Y. Fei and B. Lu, Trends Pharmacol. Sci., 2020, 41, 464–474 CrossRef CAS PubMed.
- S. J. Hughes, A. Testa, N. Thompson and I. Churcher, Drug Discovery Today, 2021, 26, 2889–2897 CrossRef CAS PubMed.
- Q. Q. Zhou, H. T. Xiao, F. Yang, Y. D. Wang, P. Li and Z. G. Zheng, Pharmacol. Res., 2023, 188, 106627 CrossRef CAS PubMed.
- R. Ma, T. P. Ortiz Serrano, J. Davis, A. D. Prigge and K. M. Ridge, FASEB J., 2020, 34, 13156–13170 CrossRef CAS PubMed.
- I. B. McInnes and E. M. Gravallese, Nat. Rev. Immunol., 2021, 21, 680–686 CrossRef CAS PubMed.
- J. Zheng, D. Chen, J. Xu, X. Ding, Y. Wu, H. C. Shen and X. Tan, Bioorg. Med. Chem. Lett., 2021, 48, 128229 CrossRef CAS PubMed.
- P. Li, Y. Zheng and X. Chen, Front. Pharmacol., 2017, 8, 460 CrossRef PubMed.
- D. S. Pisetsky, Nat. Rev. Nephrol., 2023, 19, 509–524 CrossRef CAS PubMed.
- K. A. Zarember and P. J. Godowski, J. Immunol., 2002, 168, 554–561 CrossRef CAS PubMed.
- K. Murphy, P. Travers and M. Walport, Janeway's I., Garland Science, 8th edn, 2012 Search PubMed.
- G. George, G. L. Shyni and K. G. Raghu, Inflammopharmacology, 2020, 28, 1457–1476 CrossRef CAS PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2018, 25, 78–87 CrossRef CAS PubMed.
- K. M. Riching, S. Mahan, C. R. Corona, M. McDougall, J. D. Vasta, M. B. Robers, M. Urh and D. L. Daniels, ACS Chem. Biol., 2018, 13, 2758–2770 CrossRef CAS PubMed.
- P. Ottis and C. M. Crews, ACS Chem. Biol., 2017, 12, 892–898 CrossRef CAS PubMed.
- M. Toure and C. M. Crews, Angew. Chem., Int. Ed., 2016, 55, 1966–1973 CrossRef CAS PubMed.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- A. R. Schneekloth, M. Pucheault, H. S. Tae and C. M. Crews, Bioorg. Med. Chem. Lett., 2008, 18, 5904–5908 CrossRef CAS PubMed.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- (a) P. Ottis and C. M. Crews, ACS Chem. Biol., 2017, 12, 892–898 CrossRef CAS PubMed; (b) M. Toure and C. M. Crews, Angew. Chem., Int. Ed., 2016, 55, 1966–1973 CrossRef CAS PubMed; (c) J. Salami and C. M. Crews, Science, 2017, 355, 1163–1167 CrossRef CAS PubMed.
- M. Muratani and W. P. Tansey, Nat. Rev. Mol. Cell Biol., 2003, 4, 192–201 CrossRef CAS PubMed.
- R. C. Aguilar and B. Wendland, Curr. Opin. Cell Biol., 2003, 15, 184–190 CrossRef CAS PubMed.
- T. J. Jarome and F. J. Helmstetter, Neurobiol. Learn. Mem., 2013, 105, 107–116 CrossRef CAS PubMed.
- A. Ocaña and A. Pandiella, J. Exp. Clin. Cancer Res., 2020, 39, 189 CrossRef PubMed.
- D. Nandi, P. Tahiliani, A. Kumar and D. Chandu, J. Biosci., 2006, 31, 137–155 CrossRef CAS PubMed.
- N. Myeku, C. L. Clelland, S. Emrani, N. V. Kukushkin, W. H. Yu, A. L. Goldberg and K. E. Duff, Nat. Med., 2016, 22, 46–53 CrossRef CAS PubMed.
- A. Bricelj, C. Steinebach, R. Kuchta, M. Gütschow and I. Sosič, Front. Chem., 2021, 9, 707317 CrossRef CAS PubMed.
- X. Han and Y. Sun, MedComm, 2023, 4, e290 CrossRef CAS PubMed.
- H. K. Namballa, P. Anchi, K. L. Mansa, J. P. Soni, C. Godugu, N. Shankaraiah and A. Kamal, Bioorg. Chem., 2021, 117, 105461 CrossRef CAS PubMed.
- I. M. Adcock, Br. J. Pharmacol., 2007, 150, 829–831 CrossRef CAS PubMed.
- P. J. Barnes, I. M. Adcock and K. Ito, Eur. Respir. J., 2005, 25, 552–563 CrossRef CAS PubMed.
- K. Tomita, P. J. Barnes and I. M. Adcock, Biochem. Biophys. Res. Commun., 2003, 301, 572–577 CrossRef CAS PubMed.
- Y. L. Chung, M. Y. Lee, A. J. Wang and L. F. Yao, Mol. Ther., 2003, 8, 707–717 CrossRef CAS PubMed.
- S. Hendrix, S. Sanchez, E. Ventriglia and S. Lemmens, Int. J. Mol. Sci., 2020, 21, 4539 CrossRef CAS PubMed.
- J. Ran and J. Zhou, Thorac. Cancer, 2019, 10, 405–412 CrossRef PubMed.
- B. R. Oh, D. H. Suh, D. Bae, B. Lee and Y. W. Song, Arthritis Res. Ther., 2017, 19, 154 CrossRef PubMed.
- H. M. Blevins, Y. Xu, S. Biby and S. Zhang, Front. Aging Neurosci., 2022, 14, 879021 CrossRef CAS PubMed.
- F. Cao, S. de Weerd, D. Chen, M. R. H. Zwinderman, P. E. van der Wouden and F. J. Dekker, Eur. J. Med. Chem., 2020, 208, 112800 CrossRef CAS PubMed.
- Z. Cao, Z. Gu, S. Lin, D. Chen, J. Wang, Y. Zhao, Y. Li, T. Liu, Y. Li, Y. Wang, H. Lin and B. He, ACS Chem. Biol., 2021, 16, 2746–2751 CrossRef CAS PubMed.
- K. Yang, Y. Zhao, X. Nie, H. Wu, B. Wang, C. M. Almodovar-Rivera, H. Xie and W. Tang, Cell Chem. Biol., 2020, 27, 866–876 CrossRef CAS PubMed.
- J. Chotitumnavee, Y. Yamashita, Y. Takahashi, Y. Takada, T. Iida, M. Oba, Y. Itoh and T. Suzuki, Chem. Commun., 2022, 58, 4635–4638 RSC.
- L. Ackerman, G. Acloque, S. Bacchelli, H. Schwartz, B. J. Feinstein, P. La Stella, A. Alavi, A. Gollerkeri, J. Davis, V. Campbell, A. McDonald, S. Agarwal, R. Karnik, K. Shi, A. Mishkin, J. Culbertson, C. Klaus, B. Enerson, V. Massa, E. Kuhn, K. Sharma, E. Keaney, R. Barnes, D. Chen, X. Zheng, H. Rong, V. Sabesan, C. Ho, N. Mainolfi, A. Slavin and J. A. Gollob, Nat. Med., 2023, 29, 3127–3136 CrossRef CAS PubMed.
- R. B. Kargbo, ACS Med. Chem. Lett., 2019, 10, 1251–1252 CrossRef CAS PubMed.
- J. Nunes, G. A. McGonagle, J. Eden, G. Kiritharan, M. Touzet, X. Lewell, J. Emery, H. Eidam, J. D. Harling and N. A. Anderson, ACS Med. Chem. Lett., 2019, 10, 1081–1085 CrossRef CAS PubMed.
- Y. Chen, Y. Ning, G. Bai, L. Tong, T. Zhang, J. Zhou, H. Zhang, H. Xie, J. Ding and W. Duan, ACS Med. Chem. Lett., 2021, 12, 82–87 CrossRef CAS PubMed.
- J. Zhang, L. Fu, B. Shen, Y. Liu, W. Wang, X. Cai, L. Kong, Y. Yan, R. Meng, Z. Zhang, Y. N. P. Chen, Q. Liu, Z. K. Wan, T. Zhou, X. Wang, P. Gavine, A. del Rosario, K. Ahn, U. Philippar, R. Attar, J. Yang, Y. Xu, J. P. Edwards and X. Dai, Cell Chem. Biol., 2020, 27, 1500–1509 CrossRef CAS PubMed.
- S. L. Degorce, O. Tavana, E. Banks, C. Crafter, L. Gingipalli, D. Kouvchinov, Y. Mao, F. Pachl, A. Solanki, V. Valge-Archer, B. Yang and S. D. Edmondson, J. Med. Chem., 2020, 63, 10460–10473 CrossRef CAS PubMed.
- A. Rowley, B. S. Brown, M. Stofega, H. Hoh, R. Mathew, V. Marin, R. X. Ding, R. A. McClure, F. M. Bittencourt, J. Chen, T. Gururaja, T. Kinoshita, X. Wang, A. Rivkin and K. R. Woller, ACS Chem. Biol., 2022, 17, 1315–1320 CrossRef CAS PubMed.
- J. Huang, D. Wan, J. Li, H. Chen, K. Huang and L. Zheng, Epigenetics, 2015, 10, 62–72 CrossRef PubMed.
- T. Agalioti, G. Chen and D. Thanos, Cell, 2002, 111, 381–392 CrossRef CAS PubMed.
- Q. Jin, L. Zhuang, B. Lai, C. Wang, W. Li, B. Dolan, Y. Lu, Z. Wang, K. Zhao, W. Peng, S. Y. Dent and K. Ge, EMBO Rep., 2014, 15, 92–1201 CrossRef PubMed.
- Z. I. Bassi, M. C. Fillmore, A. H. Miah, T. D. Chapman, C. Maller, E. J. Roberts, L. C. Davis, D. E. Lewis, N. W. Galwey, K. E. Waddington, V. Parravicini, A. L. Macmillan-Jones, C. Gongora, P. G. Humphreys, I. Churcher, R. K. Prinjha and D. F. Tough, ACS Chem. Biol., 2018, 13, 2862–2867 CrossRef CAS PubMed.
- R. Morris, N. J. Kershaw and J. J. Babon, Protein Sci., 2018, 27, 1984–2009 CrossRef CAS PubMed.
- C. J. Malemud, Ther. Adv. Musculoskeletal Dis., 2018, 10, 117–127 CrossRef CAS PubMed.
- X. Hu, J. Li, M. Fu, X. Zhao and W. Wang, Signal Transduction Targeted Ther., 2021, 6, 402 CrossRef PubMed.
- R. R. Shah, J. M. Redmond, A. Mihut, M. Menon, J. P. Evans, J. A. Murphy, M. A. Bartholomew and D. M. Coe, Bioorg. Med. Chem., 2020, 28, 115326 CrossRef CAS PubMed.
- R. B. Kargbo, ACS Med. Chem. Lett., 2021, 12, 945–946 CrossRef CAS PubMed.
- B. Jiang, D. M. Weinstock, K. A. Donovan, H. W. Sun, A. Wolfe, S. Amaka, N. L. Donaldson, G. Wu, Y. Jiang, R. A. Wilcox, E. S. Fischer, N. S. Gray and W. Wu, Cell Chem. Biol., 2023, 30, 383–393 CrossRef CAS PubMed.
- D. J. Philpott, M. T. Sorbara, S. J. Robertson, K. Croitoru and S. E. Girardin, Nat. Rev. Immunol., 2014, 14, 9–23 CrossRef CAS PubMed.
- G. M. Argast, N. Fausto and J. S. Campbell, Mol. Cell. Biochem., 2005, 268, 129–140 CrossRef CAS PubMed.
- P. Canning, Q. Ruan, T. Schwerd, M. Hrdinka, J. L. Maki, D. Saleh, C. Suebsuwong, S. Ray, P. E. Brennan, G. D. Cuny, H. H. Uhlig, M. Gyrd-Hansen, A. Degterev and A. N. Bullock, Chem. Biol., 2015, 22, 1174–1184 CrossRef CAS PubMed.
- Y. Lai, X. Wang, X. Sun, S. Wu, X. Chen, C. Yang, W. Zhang, X. Yu, Y. Tong, F. Ma, H. Zheng, X. Zhang and S. He, Biochem. Pharmacol., 2023, 214, 115647 CrossRef CAS PubMed.
- A. Mares, A. H. Miah, I. E. D. Smith, M. Rackham, A. R. Thawani, J. Cryan, P. A. Haile, B. J. Votta, A. M. Beal, C. Capriotti, M. A. Reilly, D. T. Fisher, N. Zinn, M. Bantscheff, T. T. MacDonald, A. Vossenkamper, P. Dace, I. Churcher, A. B. Benowitz, G. Watt, J. Denyer, P. Scott-Stevens and J. D. Harling, Commun. Biol., 2020, 3, 140 CrossRef CAS PubMed.
- D. P. Bondeson, A. Mares, I. E. D. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, B. W. Den, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed.
- A. C. Belkina and G. V. Denis, Nat. Rev. Cancer, 2012, 12, 465–477 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- B. Tong, M. Luo, Y. Xie, J. N. Spradlin, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Sci. Rep., 2020, 10, 1–8 CrossRef PubMed.
- K. M. DeMars, C. Yang, C. I. Castro-Rivera and E. Candelario-Jalil, Biochem. Biophys. Res. Commun., 2018, 497, 410–415 CrossRef CAS PubMed.
- R. B. Kargbo, ACS Med. Chem. Lett., 2021, 12, 688–689 CrossRef CAS PubMed.
- C. P. Tinworth, H. Lithgow, L. Dittus, Z. I. Bassi, S. E. Hughes, M. Muelbaier, H. Dai, I. E. D. Smith, W. J. Kerr, G. A. Burley, M. Bantscheff and J. D. Harling, ACS Chem. Biol., 2019, 14, 342–347 CrossRef CAS PubMed.
- J. Liu, L. Yuan, Y. Ruan, B. Deng, Z. Yang, Y. Ren, L. Li, T. Liu, H. Zhao, R. Mai and J. Chen, J. Med. Chem., 2022, 65, 6593–6611 CrossRef CAS PubMed.
- Z. Xiao, S. Song, D. Chen, R. van Merkerk, P. E. van der Wouden, R. H. Cool, W. J. Quax, G. J. Poelarends, B. N. Melgert and F. J. Dekker, Angew. Chem., Int. Ed., 2021, 60, 17514–17521 CrossRef CAS PubMed.
- R. B. Kargbo, ACS Med. Chem. Lett., 2019, 10, 276–277 CrossRef CAS PubMed.
- Y. Wu, Y. Yang, W. Wang, D. Sun, J. Liang, M. Zhu, H. Li and L. Chen, Acta Pharm. Sin. B, 2022, 12, 4262–4265 CrossRef CAS PubMed.
- W. Wang, Y. Liu, L. Xiong, D. Sun, H. Wang, Z. Song, Y. Li, H. Li and L. Chen, J. Nat. Prod., 2023, 86, 67–781 Search PubMed.
- H. Yokoo, N. Shibata, M. Naganuma, Y. Murakami, K. Fujii, T. Ito, K. Aritake, M. Naito and Y. Demizu, ACS Med. Chem. Lett., 2021, 12, 236–241 CrossRef CAS PubMed.
- M. Hu, W. Zhou, Y. Wang, D. Yao, T. Ye, Y. Yao, B. Chen, G. Liu, X. Yang, W. Wang and Y. Xie, Acta Pharm. Sin. B, 2020, 10, 1943–1953 CrossRef CAS PubMed.
- L. R. Bollu, P. V. Bommi, P. J. Monsen, L. Zhai, K. L. Lauing, A. Bell, M. Kim, E. Ladomersky, X. Yang, L. C. Platanias, D. E. Matei, M. G. Bonini, H. G. Munshi, R. Hashizume, J. D. Wu, B. Zhang, C. D. James, P. Chen, M. Kocherginsky, C. Horbinski, M. D. Cameron, A. A. Grigorescu, B. Yamini, R. V. Lukas, G. E. Schiltz and D. A. Wainwright, J. Med. Chem., 2022, 65, 15642–15662 CrossRef CAS PubMed.
- X. Jiang, J. Zhou, Y. Wang, X. Liu, K. Xu, J. Xu, F. Feng and H. Sun, Eur. J. Med. Chem., 2021, 210, 112949 CrossRef CAS PubMed.
- M. Lu, T. Liu, Q. Jiao, J. Ji, M. Tao, Y. Liu, Q. You and Z. Jiang, Eur. J. Med. Chem., 2018, 146, 251–259 CrossRef CAS PubMed.
- F. Wang, Y. Zhan, L. Manman, L. Wang, A. Zheng, C. Liu, H. Wang and T. Wang, ACS Pharmacol. Transl. Sci., 2022, 6, 76–87 CrossRef PubMed.
- J. Ji, S. Ma, Y. Zhu, J. Zhao, Y. Tong, Q. You and Z. Jiang, J. Med. Chem., 2023, 66, 6070–6081 CrossRef CAS PubMed.
- M. Wang, J. Lu, M. Wang, C. Y. Yang and S. Wang, J. Med. Chem., 2020, 63, 7510–7528 CrossRef CAS PubMed.
- D. W. Robbins, M. A. Noviski, Y. S. Tan, Z. A. Konst, A. Kelly, P. Auger, N. Brathaban, R. Cass, M. L. Chan, G. Cherala, M. C. Clifton, S. Gajewski, T. G. Ingallinera, D. Karr, D. Kato, J. Ma, J. McKinnell, J. McIntosh, J. Mihalic, B. Murphy, J. R. Panga, G. Peng, J. Powers, L. Perez, R. Rountree, A. Tenn-McClellan, A. T. Sands, D. R. Weiss, J. Wu, J. Ye, C. Guiducci, G. Hansen and F. Cohen, J. Med. Chem., 2024, 67(4), 2321–2336 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |