
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectricity-driven organic hydrogenation using water as the hydrogen source
Bidyut Kumar
Kundu
 and
Yujie
Sun
*
and
Yujie
Sun
*
Department of Chemistry, University of Cincinnati, Cincinnati, Ohio 45221, USA. E-mail: yujie.sun@uc.edu
First published on 26th September 2024
Abstract
Hydrogenation is a pivotal process in organic synthesis and various catalytic strategies have been developed in achieving effective hydrogenation of diverse substrates. Despite the competence of these methods, the predominant reliance on molecular hydrogen (H2) gas under high temperature and elevated pressure presents operational challenges. Other alternative hydrogen sources such as inorganic hydrides and organic acids are often prohibitively expensive, limiting their practical utility on a large scale. In contrast, employing water as a hydrogen source for organic hydrogenation presents an attractive and sustainable alternative, promising to overcome the drawbacks associated with traditional hydrogen sources. Integrated with electricity as the sole driving force under ambient conditions, hydrogenation using water as the sole hydrogen source aligns well with the environmental sustainability goals but also offers a safer and potentially more cost-effective solution. This article starts with the discussion on the inherent advantages and limitations of conventional hydrogen sources compared to water in hydrogenation reactions, followed by the introduction of representative electrocatalytic systems that successfully utilize water as the hydrogen source in realizing a large number of organic hydrogenation transformations, with a focus on heterogeneous electrocatalysts. In summary, transitioning to water as a hydrogen source in organic hydrogenation represents a promising direction for sustainable chemistry. In particular, by exploring and optimizing electrocatalytic hydrogenation systems, the chemical industry can reduce its reliance on hazardous and expensive hydrogen sources, paving the way for safer, greener, and less energy-intensive hydrogenation processes.
1. Introduction
Hydrogenation, a cornerstone of modern chemical synthesis, holds immense significance in various sectors including petrochemical, coal chemical, fine chemical, and environmental industries. It was estimated that approximately 25% of all chemical processes employ at least one hydrogenation step.1 For instance, hydrodenitrogenation and hydrodesulfurization are vital for purifying crude oil in petrochemical refineries,2 while selective processes like asymmetric hydrogenation provide efficient routes to synthesize high-value fine chemicals.3–5 Consequently, the pursuit of straightforward hydrogenation methods under mild conditions has become a paramount focus in contemporary research, which also aligns with the need of enhanced sustainability and efficiency for the chemical industry in general.While industrial hydrogenation largely depends on thermocatalytic methods using H2 as the primary hydrogen source (Fig. 1A),6 recent shifts in focus have illuminated the potential of electrocatalysis as viable alternatives. These new methods diverge from the conventional high-temperature, energy-intensive thermocatalytic processes, which pose significant sustainability concerns.3,7,8 Electrocatalytic hydrogenation (Fig. 1B) has gained attraction due to its simplicity, safety, and environmental benefits. Since electricity can be produced from photovoltaics, the potential integration of solar energy in electrocatalytic hydrogenation further enhance its greenness and sustainability.9–11
 | ||
| Fig. 1 General representations of (A) thermocatalytic hydrogenation using H2 and (B) electrocatalytic hydrogenation using water as the hydrogen source. | ||
Besides energy input, hydrogen sources are equally critical in hydrogenation reactions. Even though H2 and other molecular hydrogen donors (e.g., formic acid, alcohols, amines, hydrides, etc.) have been widely employed in hydrogenation reactions, their own limitations/restrictions call for a greener and inexpensive alternative. Arguably, the greenest while lowest-cost hydrogen source is water. In fact, incorporated in electrocatalysis, water has been successfully employed as a versatile hydrogen source in various organic hydrogenation applications. This perspective article aims to delve into the principles underlying hydrogenation reactions utilizing water as the primary hydrogen source, a promising direction in the field of green hydrogenation using heterogeneous electrocatalysts. Emphasizing the distinct advantages of water over conventional hydrogen sources, we will conduct a comparative analysis of different hydrogenation strategies, followed by the introduction of representative electrocatalytic systems using water as the hydrogen source for a variety of hydrogenation reactions. By examining these emerging methodologies, we aim to highlight the potential of water-based hydrogenation as a sustainable and efficient approach for the chemical industry.
2. Comparison of different hydrogen sources for organic hydrogenation
In hydrogenation processes, aside from catalysts, the essential element required is the hydrogen source. The following sections will briefly discuss the advantages and limitations of commonly employed hydrogen sources, including H2, inorganic hydrides, and organic hydrogen donors.2.1 Molecular H2
H2 plays a dominant role in the industrial thermocatalytic hydrogenation processes, offering several distinct advantages. The high reactivity of H2 is a primary benefit, enabling efficient hydrogenation of various functional groups. This reactivity allows for the hydrogenation of alkenes, alkynes, and carbonyl compounds, among others. Another advantage is the simplicity and cleanliness of these reactions using H2, because it often results in fewer byproducts compared to other reducing agents. This aspect is particularly advantageous in terms of product purity and ease of post-reaction purification. Additionally, H2 can be paired with various heterogeneous and homogeneous catalysts,12–25 making the whole processes economically and environmentally more favorable.26–28 Furthermore, H2's ability to be employed in asymmetric hydrogenation provides a pathway to chiral molecules, crucial in pharmaceutical synthesis.29–31 Lastly, the use of H2 aligns well with green chemistry principles, especially when derived from renewable resources such as biomass,32 contributing to sustainable chemical practices.33Despite its advantages, H2 also presents certain limitations in hydrogenation reactions. Because hydrogenation requires either hydrogen atom or hydride, homolytic or heterolytic cleavage of the H–H bond H2 is thus a prerequisite in H2-involved hydrogenation reactions, which could be energy demanding. In addition, one of the primary challenges is the safety concern associated with its use. H2 is highly flammable and explosive, especially at high pressure, necessitating stringent safety protocols and specialized equipment. This can lead to increased operational cost and complexity. Additionally, the requirement for high pressure in many hydrogenation processes can limit the scalability of reactions and require expensive reactors. Another disadvantage is the selectivity issue. Even though H2 is effective for broad hydrogenation, its non-specificity can lead to over-hydrogenation or reduction of multiple functional groups, which is undesirable in complex molecule synthesis. The reliance on H2, predominantly derived from fossil fuels in the current market, also raises environmental concerns. In fact, since industrial H2 is primary produced from steam methane reforming, which may contain a certain amount of CO impurity, catalyst poisoning is not a negligible issue.
2.2 Inorganic hydrides
Hydrogenation reactions can also be carried out using a number of inorganic hydrides, especially for relatively small-scale applications. Inorganic hydrides, such as LiAlH4 and NaBH4, are widely used in organic hydrogenation reactions in research laboratories, each presenting unique advantages and limitations.34,35 LiAlH4 is highly effective in reducing a variety of functional groups, such as carbonyls and carboxylic acids, under relatively mild conditions. However, its extreme reactivity and moisture sensitivity necessitate anhydrous conditions and careful handling. NaBH4 is less reactive than LiAlH4, offering safer handling and compatibility with some protic solvents, but it is less powerful, primarily reducing aldehydes and ketones. The use of these inorganic hydrides generally requires stoichiometric amounts, leading to waste disposal concerns, especially with aluminum salts. Their non-selectivity can also be an issue in complex syntheses where more controlled reduction is desired. Each of these hydrides offers a balance of reactivity and selectivity, tailored to specific applications to maximize their benefits while minimizing drawbacks in organic synthesis.2.3 Organic hydrogen donors
In addition to H2 and inorganic hydrides, there are a large number of organic molecules, such as alcohols, aldehydes, formic acid, amines, etc., have been utilized as hydrogen sources in various organic hydrogenation reactions.36–42Fig. 2A presents those representative organic molecules used as hydrogen sources in hydrogenation reactions, each exhibiting their own merits and limitations. | ||
| Fig. 2 (A) Selected organic molecules used as the hydrogen sources in organic hydrogenation processes. (B) Benzothiazoline in combination with chiral phosphoric acid exhibits reducing ability by releasing hydrogen to form more stable benzothiazole.67 | ||
For instance, the use of alcohols in hydrogenation processes presents a unique combination of both being hydrogen donors and solvents, making the effects of these aspects intertwined. Various alcohols, such as methanol,43 ethanol,38,40,44,45 iso-propanol,46–52 2-butanol,38 glycerol,36 and benzyl alcohol,53 offer several advantages, including wide availability, affordability, potentially renewable nature, convenient transport and storage.54 The use of alcohols can often lead to more selective hydrogenation processes due to their specific reactivity. However, there are also notable disadvantages. The efficiency of hydrogen release from these alcohols is typically lower compared to pure H2, potentially leading to higher energy consumption and lower reaction rates. This inefficiency can be exacerbated in the case of larger alcohols like 2-butanol due to their more complex molecular structure.55,56 Furthermore, the use of alcohols often requires more complex catalyst systems, which can increase the overall cost of the reaction. There is also the challenge of byproduct formation, which can complicate the purification process of the desired product and impact the overall sustainability of the process.
Similarly, formaldehyde and formic acid41,42,57–64 have also been widely utilized as hydrogen sources in hydrogenation reactions. As a readily available and cost-effective commodity chemical, formaldehyde is well regarded for its high hydrogen content. However, its toxicity nature requires stringent safety protocols and specialized equipment, increasing operational complexity and cost. Moreover, the reactivity of formaldehyde can lead to side reactions and challenges in selectivity, potentially complicating the purification process of the desired products. On the other hand, formic acid is substantially less toxic, making it a safer alternative for laboratory and industrial hydrogenation use. In addition, formic acid is compatible with a variety of catalysts and can be used in different types of hydrogenation reactions, including the reduction of alkenes, alkynes, and carbonyl groups. Nevertheless, its low reactivity typically requires large amounts or more rigorous reaction conditions to achieve comparable results as other hydrogen sources. In some cases, formic acid can participate in side reactions, potentially leading to undesirable byproducts.57,65 The risk of catalyst deactivation by formic acid and its derived species (e.g., CO) can not be overlooked.
In the realm of catalytic hydrogenation, the utilization of organic hydrogen donors such as benzothiazoline (Fig. 2B),66,67 benzimidazoline,68,69 5,6-dihydrophenanthridine,70 hydrazine,71–73 thiophenol,74 9,10-dihydrophenanthrene,75 HBpin,76 and Hantzsch ester (a molecular mimic of NAD(P)H)77–81 offers a nuanced balance of advantages and disadvantages. These compounds are recognized for their ability to provide controlled and selective hydrogen transfer, akin to enzymatic processes in biological systems. This selectivity is particularly advantageous for reactions requiring selective hydrogenation at specific sites without disturbing other sensitive functional groups. However, high cost and availability can be significant limiting factors for their large-scale application, especially for compounds like Hantzsch ester and HBpin. Even though several recent reports have demonstrated the electrocatalytic regeneration of some of these molecular hydrogen donors,82 the potential generation of byproducts is another critical concern, often necessitating additional purification steps that increase the complexity and cost of the overall process. Thus, while the use of these organic hydrogen donors presents an innovative approach to catalytic hydrogenation, it is essential to weigh their benefits against their economic, operational, and environmental implications.
As carbon-free compounds, ammonia (NH3) and ammonium salts have also been widely employed as hydrogen sources in various hydrogenation reactions.40,83 NH3 is a particularly dense source of hydrogen (∼17.6 wt%), making it efficient for hydrogen storage and transport. It is also relatively easy to liquefy under mild conditions, which enhances its practicality for industrial applications. Additionally, NH3 and ammonium salts are widely available and can be produced on a large scale, often at lower costs compared to pure H2. Within this context, NH3 has been successfully utilized as a hydrogen source in the electrocatalytic hydrogenation of alkenes, alkynes, and ketones.84–86 For instance, Fig. 3 presents the electrocatalytic hydrogenation scheme of alkenes at the cathode using NH3 as the hydrogen source.84 Besides NH3, organic secondary amines87 and ammonium salts (e.g., triethylammonium formate88) have also been utilized as hydrogen sources in various hydrogenation reactions.39,89 However, the use of NH3 and ammonium salts in hydrogenation also comes with significant challenges. Under thermocatalytic conditions, the release of hydrogen from these compounds often requires high temperature and pressure, leading to increased energy consumption and restriction in reaction scope. The catalytic decomposition of NH3 and ammonium salts can produce undesirable byproducts, leading to challenging product purification and potential environmental concerns. Given their toxic and corrosive nature, specialized equipment and safety protocols are often mandated.
 | ||
| Fig. 3 Possible mechanism for the electrochemical hydrogenation of olefins using NH3 as the hydrogen source using 0.1 M LiClO4 in CH3CN as the electrolyte with NH3 (1 atm) as the nitrogen source.83 | ||
2.4 Different reactive hydrogen species (H+, H˙, and H−)
As indicated in previous discussions, different hydrogen species could be involved in organic hydrogenation reactions. In fact, proton (H+), hydrogen atom (H˙), and hydride ion (H−) are all possible hydrogen species dependent on the specific hydrogenation mechanism. As shown in Fig. 4, electricity-driven hydrogenation reactions primarily follow two pathways: (i) hydrogen atom transfer (HAT) and (ii) proton-coupled electron transfer (PCET). Following the HAT mechanism, an adsorbed hydrogen atom is first formed on the cathode surface, which will be subsequently added to the substrate,90–92 resembling the HAT step in organometallic catalysis.34 In contrast, following the PCET mechanism, both H+ and e− are transferred concurrently to the substrate.93–96 The specific hydrogenation pathway will be indicated by many factors, including electrocatalyst, microenvironment, potential, pH, etc.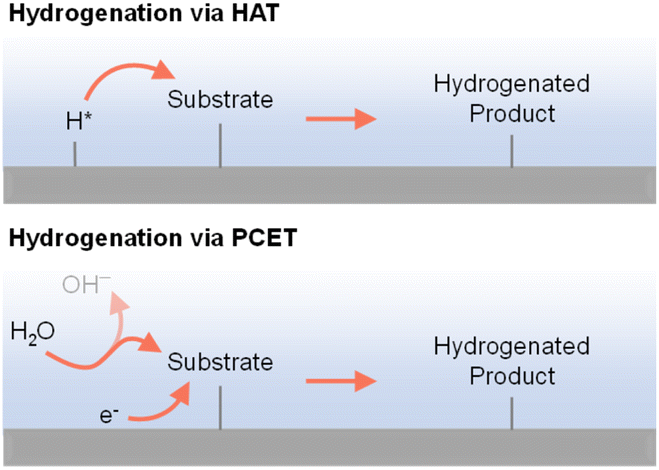 | ||
| Fig. 4 Electricity-driven hydrogenation via hydrogen atom transfer (top) or proton-coupled electron transfer (bottom) processes.97 | ||
3. Electrocatalytic hydrogenation using water as the hydrogen source
Given the aforementioned limitations of H2 and various inorganic/organic hydrogen sources, there is a growing interest in exploring greener alternatives, among which water stands out as an environmentally and economically viable option. In fact, electrocatalytic hydrogenation using water as the hydrogen source has emerged as an exemplary alternative to traditional thermocatalytic hydrogenation, offering an efficient and sustainable pathway for synthesizing hydrogenated products under ambient conditions using electricity as the primary driving force.For organic substrates which are easy to be directly reduced, their hydrogenation reactions can occur in the absence of any electrocatalyst. For instance, Werz et al. reported the site-selective hydrogenation of benzylic olefins through electroreduction, employing water as the hydrogen donor.98 As depicted in Fig. 5, following a single electron transfer (SET) process, benzylic olefin is directly reduced at the cathode to form a radical anion, which is consequently quenched via protonation by H2O. Another SET step at the cathode delivers an anion from the radical intermediate that leads to the desired product after protonation. On the other side, the anodic oxidation of water can directly generate O2 and H+.
 | ||
| Fig. 5 Hydrogenation of different olefins using water as the hydrogen source (GC = glassy carbon) using 0.02 M TBAPF6 in CH3CN/H2O (v/v = 4/1) as the electrolyte (TBA = tetra-n-butyl ammonium).98 | ||
Nevertheless, most electrochemical hydrogenation reactions require electrocatalysts to facilitate the processes. In most cases, it is critical to split water to yield a certain active form of hydrogen (e.g., atomic hydrogen, proton, hydride, and molecular dihydrogen), prior to the hydrogenation step. To achieve this, researchers have employed a variety of electrocatalytic strategies. The following paragraphs will discuss the factors that will affect the electrocatalytic hydrogenation performance and also introduce several promising strategies that are able to reduce energy input while double the reaction rate in electrocatalytic hydrogenation.
3.1 Influence of electrocatalyst in hydrogenation
Many reported electrocatalysts actually consist of metals with known thermocatalytic hydrogenation activity, such as Pd, Pt, Ru, Cu, and Ni, all of which are able to form active hydrogen species under reductive conditions.97,99–101 Highly porous electrocatalysts are always preferred because of the observed electrocatalytic activity is usually linearly proportional to the active surface area of an electrocatalyst. Therefore, nanostructuring to achieve small particle size of the electrocatalyst is a common strategy in electrocatalytic hydrogenation. The extreme case is the so-called single atom electrocatalysts which not only exhibit maximized utilization of electrocatalyst atoms, especially for expensive noble metals, but may also show unique activities.102–104Because of the critical role played by catalyst, understanding the origins of an electrocatalyst's intrinsic activity is essential for developing competent electrocatalytic hydrogenation systems. A representative example comes from the Koper group in which they thoroughly compared the activities of three different crystalline facets of a Pt catalyst for the electrocatalytic hydrogenation of acetone.105 Single-crystal Pt electrodes were utilized in their investigation. DFT calculations suggested that the interaction of acetone with Pt(111) and Pt(110) is energetically unfavorable because of the high coordination number of their surface Pt atoms. Instead, acetone reduction takes place at the step sites of Pt[(n − 1)(111) × (110)] and Pt[(n + 1)(100) × (110)], albeit with different selectivities towards either isopropanol or propane. As shown in Fig. 6, the two pathways of acetone reduction proceed through the formation of a common intermediate *CH3COHCH3 but differ for its subsequent conversion. In other words, the two pathways bifurcate before the formation of isopropanol. Once isopropanol is formed, no further hydrogenation to propane will be possible. In fact, selectivity stems from the relative ease of the protonation of *CH3COHCH3 (top route in Fig. 6) versus its C–O bond scission (bottom route in Fig. 6). It was found that acetone is predominantly reduced to isopropanol at the steps of Pt[(n − 1)(111) × (110)], whereas the step sites of Pt[(n + 1)(100) × (110)] result in propane. The guidance of this study can be applied to the electrocatalytic hydrogenation of other higher aliphatic ketones as well.
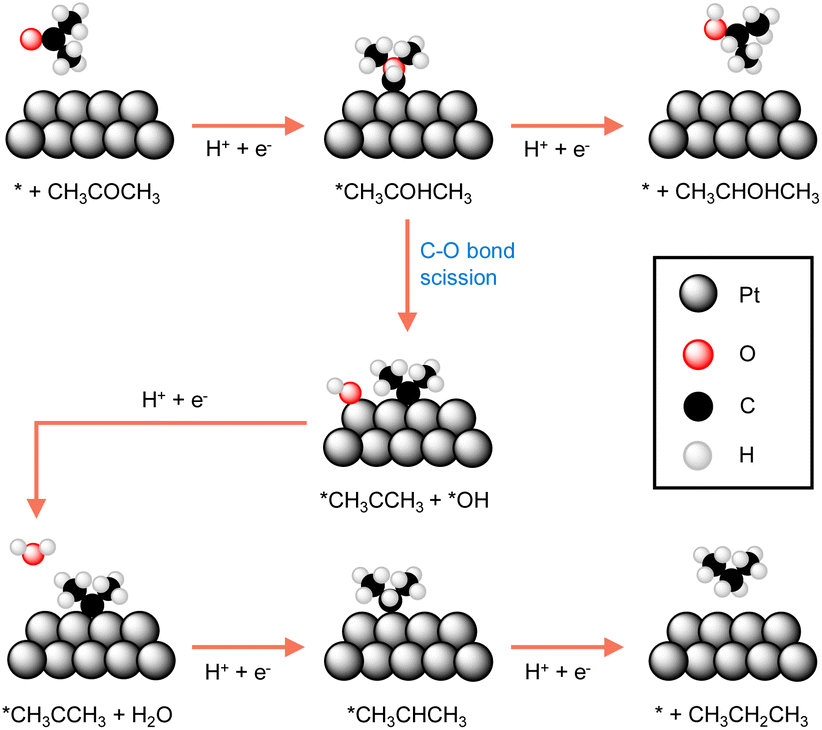 | ||
| Fig. 6 The most favourable reaction pathways computed for acetone reduction to 2-propanol and propane at Pt electrodes.105 | ||
Besides noble metals like aforementioned Pt, earth-abundant 1st-row transition metals have also found popular applications in electrocatalytic hydrogenation of organics in water. For instance, Cu-based composites, which are widely used as electrocatalysts for electrocatalytic CO2 and CO hydrogenation, also exhibit excellent performance for the semihydrogenation of acetylene to ethylene, the foremost olefin in the petrochemical industry. Unfortunately, ∼1% acetylene is inevitably generated as an impurity in naphtha crackers used for ethylene production and the co-produced acetylene impurity in the ethylene feedstock will seriously poison the polymerization catalysts, adversely degrading the quality of the target polymers. Compared to thermocatalytic acetylene semihydrogenation, electrocatalytic semihydrogenation presents a promising alternative because of its environmental friendliness and economic efficiency. An ideal electrocatalyst for the semihydrogenation of acetylene should exhibit appropriate adsorption energy of acetylene but poor HER performance.106–108 In the meantime, the resulting ethylene should be desorbed from the electrocatalyst surface easily, otherwise over-hydrogenation to ethane will occur. Deng et al. demonstrated that Cu was a competent electrocatalyst for the highly selective semihydrogenation of acetylene.109In situ X-ray absorption fine structure experiments were performed to investigate the electronic properties of the Cu catalyst in the absence and presence of acetylene adsorption. The obtained results indicate that the acetylene adsorption on the Cu surface leads to increased oxidation state of the catalyst owing to the electron transfer from Cu to the antibonding π orbital of the adsorbed acetylene. This result is also confirmed by in situ Raman spectroscopy characterizations of adsorbed acetylene, wherein weakened C–C bond after adsorption on Cu was observed. DFT calculations were also carried out to corroborate these experimental results. Three crystal facets of Cu including (111), (100), and (110) were considered and it was found that acetylene was strongly adsorbed on the hollow sites on all the facets with much higher adsorption free energies compared with hydrogen adsorption. Based on the Bader charge analysis, a significant decrease in the valence electrons of Cu with an increase of up to 0.65 electrons on the adsorbed acetylene, confirming the electron transfer from the Cu surface to acetylene. Fig. 7 presents the differential charge density of the adsorption structure, showing charge depletion areas around the Cu adsorption sites and charge accumulation areas around the adsorbed acetylene on the Cu surface.
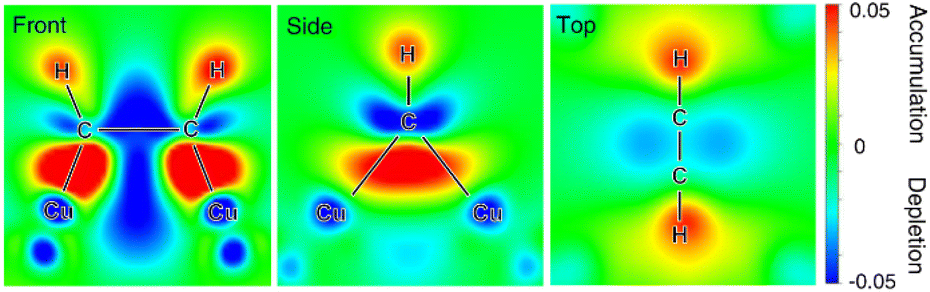 | ||
| Fig. 7 Charge density difference of the adsorbed acetylene on the Cu surface at front, side and top views. Charge accumulation and depletion regions are shown in red and blue, respectively. This figure has been adapted/reproduced from ref. 109, with permission from Nature Publishing Group, copyright 2021. | ||
Other Cu-based electrocatalytic systems with modified preparation methods achieved further improved performance. For example, electrochemically deposited Cu dendrites could reduce the acetylene impurity level in an ethylene flow from 1 × 104 ppm to 4 ppm.110 Zhang et al. reported a Cu nanoparticle electrocatalyst synthesized on gas diffusion layer coated carbon paper which achieved a high C2H4 yield rate of 70.15 mmol mg−1 h−1 and a Faraday efficiency of 97.7% at an industrially relevant current density of 0.5 A cm−2, which is industrially profitable based on their techno-economic analysis.111
More challenging hydrogenation of aromatics have also been reported for electrocatalysts modified by various dopants. For example, a fluorine-doped cobalt (Co–F) electrocatalyst was able to facilitate the electrocatalytic hydrogenation of quinoline (1a) to form 1,2,3,4-tetrahydroquinoline (2a) using water as the hydrogen source.112Fig. 8A reveals that the presence of surface F can significantly enhance the adsorption of 1a, which will facilitate electron transfer between 1a and the electrocatalyst. In addition, the much lower adsorption energy of 2a than that of 1a implies that the product is easier to desorb from the Co cathode (Fig. 8B). Furthermore, the Gibbs free energy for H* formation on Co–F is more negative than that on pure Co (−0.46 vs. −0.38 eV, Fig. 8C). This indicates that the Co–F cathode is favorable for generating H* via H2O dissociation, which will benefit the hydrogenation of 1a.
 | ||
| Fig. 8 (A and B) Comparisons of adsorption energies of 1a and 2a on pure Co (A) and Co–F (B), respectively (inset: the stable adsorption configurations of 1a and 2a). (C) The calculated free energy for H* formation on Co versus CoF. This figure has been adapted/reproduced from ref. 112 with permission from Nature Publishing Group, copyright 2022. | ||
A bimetallic PtRu electrocatalyst was reported for the efficient hydrogenation of benzoic acid to cyclohexanecarboxylic acid in acidic electrolytes.113 The ring structures of many benzoic acid derivatives could be hydrogenated in a similar fashion, albeit with mediocre faradaic efficiencies. In fact, a large number of electrocatalytic systems primarily composed of earth-abundant elements have been explored for the electrocatalytic hydrogenation of various organic substrates, specifically biomass-derived intermediate compounds, in aqueous electrolytes in which water is the main hydrogen source.114–118
3.2 Effect of microenvironment in hydrogenation
In addition to the judicious selection of competent electrocatalysts, the fine controlling of the microenvironment surrounding the active sites of an electrocatalyst is equally important in dictating the hydrogenation efficiency and selectivity.97,119 The microenvironment can be tuned by varying pH, solvent, supporting electrolyte, additive, etc.Since electrocatalytic hydrogenation requires the electrochemical generation of adsorbed hydrogen on the electrode surface through proton or water reduction, there exists a possible competing reaction which is the direct reduction of the organic reactant. In the latter scenario, electron transfer takes place between the reactant and cathode, followed by protonation occurring in solution which may lead to side-products. In order to minimize the direct electroreduction of organic substrates, it is of critical importance to control the interfacial interaction between organic reactant, proton, and electrode. Using the electrocatalytic hydrogenation of furfural on copper electrodes as a model reaction, Li et al. performed a thorough investigation to distinguish the mechanisms of electrocatalytic hydrogenation and direct electroreduction.120 Copper electrodes modified with the self-assembled monolayers of organothiols (e.g., 3-mercaptopropionic acid (MPA) and 2-mercaptobenzothiazole (MBT)) were utilized to determine the necessity of direct interaction between organic reactants and the cathode surface, and the nature of heterogeneous electron transfer. It was found that the formation of 2-furfuryl alcohol and 2-methylfuran were largely inhibited on electrodes covered with MPA and MBT (Fig. 9), suggesting that the direct interaction between furfural and the copper electrode is required for its electrocatalytic hydrogenation and hydrogenolysis. However, the formation of hydrofuroin was unaffected, indicating that the first electron transfer in the electroreduction mechanism is an out-sphere process, insensitive to the electrode surface properties or catalytic activity. Surfactant modification on electrode has been utilized to achieve high selectivity for the electrocatalytic semihydrogenation of alkynols to alkenols as well, whereas water is the hydrogen source.121
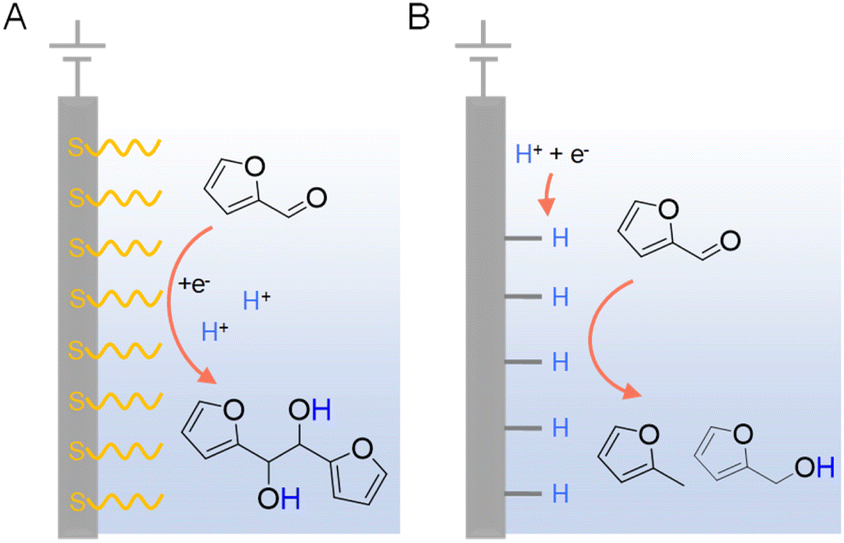 | ||
| Fig. 9 Two distinct mechanisms of electrocatalytic furfural reduction on Cu electrodes in acidic electrolytes: (A) direct electroreduction and (B) electrocatalytic hydrogenation. Typical electrolytes were 0.5 M sulfuric acid (pH = 0.5) or 0.5 M sulfate solutions (pH = 1.4–3.0) with 25% v/v CH3CN.120 | ||
Besides the direct interaction of organic substrates with electrode, the specific adsorption configuration of organic substrate on the electrode surface will also impact the consequence of electrocatalytic hydrogenation. In fact, due to the requirement of proton source during electrocatalytic hydrogenation, the local concentration of H+ plays a critical role not only in controlling the competing HER but also affecting the adsorption configuration of organic reactant. For instance, Qiao et al. simply varied the supporting electrolyte H2SO4 concentration from 0.1 to 1.0 M, a striking difference in product distribution was observed for the electrocatalytic hydrogenation and hydrogenolysis of acetone to isopropanol and propane using Pt-based electrocatalysts.122 A thorough electrochemical investigation together with in situ FTIR spectroscopic studies and operando DFT computation was carried out to elucidate the observed results (Fig. 10). It was concluded that the local pH environment largely determines the adsorption configuration of acetone and hence the efficiency of acetone to propane hydrogenation (APH). When the local H+ concentration is low (e.g., 0.1 M H2SO4), the adsorption configuration of acetone is potential dependent, being vertically adsorbed through a Pt@O bond when the potential is positive. At this positive potential, the activity of APH is quite low due to the limited amount of adsorbed H*. Once a more negative potential is applied, a flat adsorption configuration of acetone is preferred on Pt, which is not beneficial to APH. Instead, HER becomes the dominant reaction. However, in a highly acidic environment (1.0 M H2SO4) with high H coverage on Pt, acetone will adopt a vertical configuration because of its strong adsorption strength compared to that of flat configuration. Consequently, a much higher selectivity towards APH was achieved. Even though HER is usually very active in highly acidic environment, the competing H2 evolution is severely suppressed, in that when acetone is vertically adsorbed, the majority of the adsorbed H* participate in APH instead of HER. It is envisioned that such a new strategy of altering the adsorption configuration of reactants/intermediates by varying local pH could be applicable to the selectivity optimization of other electrocatalytic reactions involving multiple species.
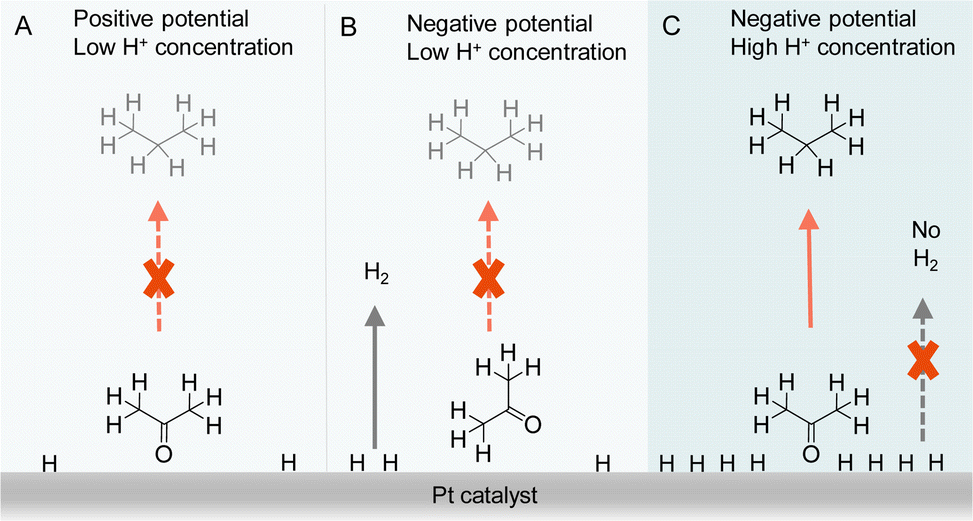 | ||
| Fig. 10 Representations of acetone to propane hydrogenation (A) under positive potential in electrolyte with low H+ (0.1 M H2SO4) concentration, (B) under negative potential in electrolyte with low H+ (0.1 M H2SO4) concentration, and (C) under negative potential in electrolyte with high H+ (1 M H2SO4) concentration.122 | ||
3.3 Advantages of paired electrolysis in hydrogenation
In conventional electrochemical hydrogenation reactions conducted in water, the common counter reaction taking place at the anode is the O2 evolution reaction (OER), which demands high voltage input while only produces O2 as a low-value product. Hence, paired electrolysis has emerged as a promising strategy to couple the target electrocatalytic hydrogenation reaction with a value-added anodic reaction that can replace OER. Paired electrolysis represents a burgeoning field at both the laboratory and industrial scales, attributed to its enhanced energy efficiency derived from conducting simultaneous and productive reactions at both electrodes.123–125 In particular, oxidative valorization of biomass-derived intermediate compounds (e.g., 5-hydroxymethylfurfural (HMF), furfural, etc.) has been widely employed as viable options for the anodic counter reactions.126–133For example, Sun et al. were inspired by the active Ni3+ intermediates in electrocatalytic OER on Ni-based composites to develop NiBx as a competent electrocatalyst for the oxygenation of HMF to 2,5-furandicarboxylic acid (FDCA) under alkaline aqueous conditions (Fig. 11A).134 A thorough electrochemical and spectroscopic study was conducted to confirm that electrochemically generated Ni3+ (as NiOOH) on NiBx was the active species for the oxidation of HMF to FDCA. The NiBx electrocatalyst was also employed at the cathode for the hydrogenation of p-nitrophenol to p-aminophenol. Hence, during the entire paired electrolysis, two value-added products (i.e., FDCA and p-aminophenol) were obtained using water as both the oxygen and hydrogen sources, respectively. Such a paired electrolysis cell was also integrated with a solar cell as a stand-alone reactor for sunlight-driven production with high conversion and selectivity (>90%).
 | ||
| Fig. 11 (A) HMF oxidation paired with the hydrogenation of p-nitrophenol to p-aminophenol.134 (B) Production of amino acids from biomass-derived α-hydroxyl acids in a cascade manner. For the selective oxidation of α-hydroxyl acids, N-hydroxyphthalimide (NHPI) or (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) was used as the redox mediator and carbon paper as the anode with 0.1 M LiClO4 CH3CN/H2O (v/v = 2/1) as the electrolyte. For the electroreductive amination of α-keto acids, Ti foil was used as the cathode. The cathode electrolyte was formed by stirring 20 mM α-keto acids with 1.2–1.5 equivalents of NH3 (aq.) or NH2OH·HCl in 0.1 M LiClO4 CH3CN/H2O (v/v = 2/1) overnight at room temperature prior to electrochemical experiments.135 | ||
Different from the above parallel paired electrolysis, which utilizes two reactants to produce two value-added products at both electrodes, cascade paired electrolysis can also be adopted to minimize the voltage input while generate highly valuable products, in which electrocatalytic hydrogenation is one of the two consecutive steps. For instance, starting from biomass-derived α-hydroxyl acids, selective oxidation using NHPI (N-hydroxyphthalimide) as a redox mediator produced α-keto acids, which could be directly pumped into the cathodic chamber (Fig. 11B).135 Subsequent reductive amination at a Ti cathode was able to produce amino acids as the final products. Such a cascade strategy utilizes water as the hydrogen source and yields a variety of amino acids (e.g., glycine, alanine, and leucine) with decent yields and faradaic efficiencies.
In addition to value-added oxidation reactions taking place in the anodic chamber, which will inevitably require the separation of products from electrolyte, on-site halogenation could be coupled with on-site hydrogenation that enable the two desirable reactions to proceed in reactors outside of the electrochemical cell.136 As shown in Fig. 12, by taking advantage of the phase separation of H2 and halogen gases (i.e., Br2 and Cl2) from the aqueous electrolyte, H2 produced from water reduction at the cathode can be transported to a reactor physically isolated from the cathode chamber to perform the desirable hydrogenation reactions. Meanwhile, halide oxidation in the anode chamber will generate halogen gases to carry out the target halogenation reactions in another separate reactor. Instead of utilizing water oxidation as the counter reaction which only produces O2 as a low-value product, on-site halogenation not only incorporates halogen atoms into the more valuable oxidation products but also requires much less voltage input than OER. Since both hydrogenation and halogenation reactions take place in reactors physically separated from the electrochemical cell, such an on-site hydrogenation/halogenation strategy not only improves the atom economy and energy efficiency, but also bypasses the energy-intensive product separation from electrolytes.
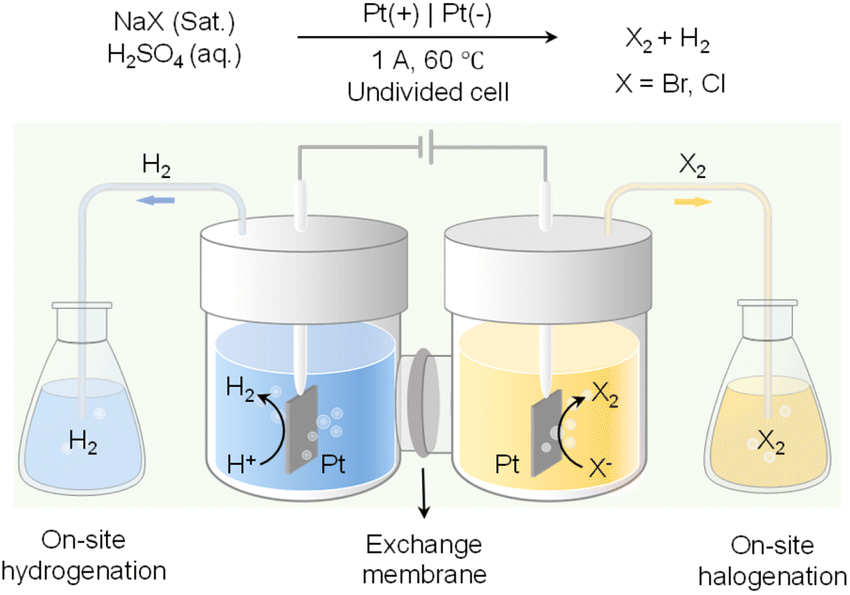 | ||
| Fig. 12 Paired electrolysis consisting of on-site hydrogenation coupled with on-site halogenation (X: Br or Cl).136 | ||
3.4 Hydrogenation using Pd membrane electrodes
The phenomenon of hydrogen absorption by Pd was first observed by Graham in 1866 and more recent studies indicate that when Pd was employed as a cathode, the absorbed H could push the H/Pd ratio above 0.9, in which water was the hydrogen source. Owing to the excellent permeation of hydrogen atoms in the face-centered cubic lattice of Pd, Iwakura et al. reported in 1996 that a Pd foil could be used as a cathode to carry out the hydrogenation of styrene to ethylbenzene in a chamber outside of the electrochemical cell (Fig. 13).137 Different from conventional electrocatalytic hydrogenation, this Iwakura's design employed the Pd foil as a membrane electrode (Pdm) to serve dual roles: a cathode for the electrochemical generation of adsorbed H* from water reduction (Pd + xH+ + xe− → PdHx) as well as a physical barrier separating the hydrogenation chamber (PdHx + R → PdHx−1 + R–H) from the electrochemical cell. Following this seminal work, many substrates have been explored for hydrogenation using Pdm as the membrane electrode and water as the hydrogen source.138 More recently, Berlinguette's group performed several systematic studies to elucidate the impacts of Pd crystalline facet,139 co-catalyst,140 support,141 solvent,142–144 cell design,145 and applied current146,147 on the outcomes of various hydrogenation reactions, further expanding the application scope and also providing deep insights in electricity-driven hydrogenation using Pdm. Electricity-driven hydrogenation using Pdm also found application in removing butadiene impurity from alkene feedstocks.148 | ||
| Fig. 13 Hydrogenation of styrene to ethylbenzene using a Pd sheet electrode.137 | ||
In most reported hydrogenation systems employing Pdm, the target hydrogenation reaction was typically performed in a reactor positioned outside of the cathodic Pdm. Inspired by the “dual” H2 evolution using low-potential formaldehyde oxidation as the anodic reaction,149 Sun et al. explored the adoption of Pdm electrodes as both anode and cathode, enabling concurrent hydrogenation processes outside of both Pdm electrodes.150 Such a “dual” hydrogenation system doubles the hydrogenation rate and hence the overall reaction efficiency. In addition, because of the extremely small oxidation potential requirement for formaldehyde oxidation versus water oxidation, the overall voltage input for this dual hydrogenation system is also much smaller than conventional electrocatalytic hydrogenation systems. In this novel dual hydrogenation design, H* are generated at the Pdm cathode through water reduction, similar to conventional hydrogenation systems. The striking difference comes from the anode side, where the Pdm anode facilitates the low-potential oxidation of formaldehyde to formate and also generates H* at the Pdm anode. Instead of being oxidized to H+, these H* atoms can permeate through the Pdm anode to catalyze hydrogenation reactions in a separate chemical chamber situated outside the anode compartment. Despite the dissimilar electrochemical processes at the Pdm anode and cathode, both contribute to the generation of H*, effectively doubling the faradaic efficiency. Utilizing a four-compartment assembly, when maleic acid served as the substrate, its hydrogenation product succinic acid was obtained in both external chambers adjacent to the Pdm electrodes. Remarkably, this process exhibits a theoretical maximum faradaic efficiency of 200%, in that each passed electron can generate two H* species, thereby doubling the faradaic efficiency of hydrogenation. Indeed, an overall faradaic efficiency of 184% was achieved at an applied current of 10 mA. In the meantime, the cell voltage was reduced by over 1 V in comparison to that of traditional single-side hydrogenation systems that require water oxidation at the anode. Besides alkene hydrogenation, alkyne substrates such as 4-ethynylaniline could also be used. In fact, when normal formaldehyde in D2O was employed in the electrochemical cell, deuterated product was obtained in the chamber outside of the Pdm cathode while normal hydrogenation product was produced in the chamber outside of the Pdm anode (Fig. 14). These results confirmed that water was the hydrogen source in the cathode chamber while formaldehyde was the hydrogen source in the anode chamber. In short, this work demonstrates that it is feasible to simultaneously perform hydrogenation and deuteration in two different chemical chambers.
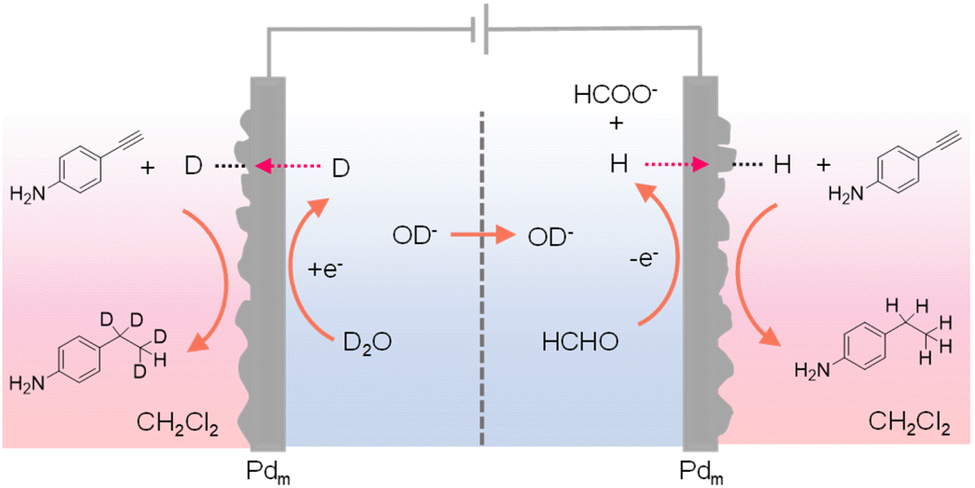 | ||
| Fig. 14 A four-compartment assembly for the hydrogenation of 4-ethynylaniline in the chamber outside of the anode chamber coupled with its deuteration in a chamber next to the cathode chamber.150 | ||
4. Conclusions and future outlook
In this Perspective article, we have delved into the promising role of water as a hydrogen source in the latest developments of advanced organic hydrogenation. Our discussion started with the comparison of the advantages and limitations of more conventional hydrogen sources versus water, followed by the presentation of several representative electrocatalytic hydrogenation reactions utilizing water as the sole hydrogen source. Despite the great progress made in this area, there remain several challenges and opportunities for further exploration. For instance, the current electrocatalytic hydrogenation reactions using water as the hydrogen source are still quite limited, compared to thermocatalytic hydrogenation. It is anticipated that taking advantage of the ambient conditions of electrocatalysis, more diverse and challenging hydrogenation reactions should be explored. Within this context, electricity/light-driven biocatalysis is a particularly appealing direction that awaits more attention, especially for asymmetric hydrogenation reactions.In addition, the use of heavy water (D2O) in generating deuterated compounds and drug molecules represents a noteworthy application.151 By incorporating deuterium into pharmaceutical molecules, it's possible to modify their metabolic profiles and enhance their stability through the kinetic isotope effect. The application of hydrogenation principles to deuteration reactions, particularly through D2O electrolysis, promises to expedite the development of deuterated molecules. In fact, a few promising electrocatalytic systems have been reported for the deuteration of alkenes, aldehydes, etc.152 It is anticipated that more research results will emerge in this promising area.
In conclusion, this Perspective article aims to provide a succinct overview of the state-of-the-art in electrocatalytic hydrogenation using water as the sole hydrogen source. The advancements highlighted here underscore a promising future for catalytic reactions that harness water as a green, abundant, and inexpensive hydrogen source, pointing towards sustainable and innovative solutions in organic synthesis.
Data availability
No new data was generated during the writing of this perspective.Author contributions
Y. S. conceived the idea and supervised the project. Y. S. and B. K. K. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. S. acknowledges the support of the US National Science Foundation (CHE-2328176 and CHE-2102220) and the ACS Petroleum Research Fund (66635-ND3).References
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- S. T. Oyama, T. Gott, H. Zhao and Y.-K. Lee, Catal. Today, 2009, 143, 94–107 CrossRef CAS.
- X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS PubMed.
- J. Mas-Roselló, T. Smejkal and N. Cramer, Science, 2020, 368, 1098–1102 CrossRef PubMed.
- B. Li, J. Chen, D. Liu, I. D. Gridnev and W. Zhang, Nat. Chem., 2022, 14, 920–927 CrossRef CAS PubMed.
- M. Guo, M. Zhang, R. Liu, X. Zhang and G. Li, Adv. Sci., 2022, 9, 2103361 CrossRef CAS PubMed.
- N. Garg, A. Sarkar and B. Sundararaju, Coord. Chem. Rev., 2021, 433, 213728 CrossRef CAS.
- W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC.
- N. Ishida, Y. Kamae, K. Ishizu, Y. Kamino, H. Naruse and M. Murakami, J. Am. Chem. Soc., 2021, 143, 2217–2220 CrossRef CAS PubMed.
- Z. Li, X. Zhang, J. Liu, R. Shi, G. I. N. Waterhouse, X.-D. Wen and T. Zhang, Adv. Mater., 2021, 33, 2103248 CrossRef CAS PubMed.
- C. Lv, X. Bai, S. Ning, C. Song, Q. Guan, B. Liu, Y. Li and J. Ye, ACS Nano, 2023, 17, 1725–1738 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- X. Guo, P. Hong, L. Yao, X. Liu, Z. Jiang and B. Shi, Fuel, 2023, 353, 129231 CrossRef CAS.
- C. Ortiz-Cervantes, M. Flores-Alamo and J. J. García, ACS Catal., 2015, 5, 1424–1431 CrossRef CAS.
- J. L. Fiorio, R. V. Gonçalves, E. Teixeira-Neto, M. A. Ortuño, N. López and L. M. Rossi, ACS Catal., 2018, 8, 3516–3524 CrossRef CAS.
- Y. Liu, Z. Sun, C. Huang and T. Tu, Chem.–Asian J., 2017, 12, 355–360 CrossRef CAS PubMed.
- H. Heeres, R. Handana, D. Chunai, C. Borromeus Rasrendra, B. Girisuta and H. Jan Heeres, Green Chem., 2009, 11, 1247–1255 RSC.
- A. Wittstock and M. Bäumer, Acc. Chem. Res., 2014, 47, 731–739 CrossRef CAS PubMed.
- R. Ciriminna, E. Falletta, C. Della Pina, J. H. Teles and M. Pagliaro, Angew. Chem., Int. Ed., 2016, 55, 14210–14217 CrossRef CAS PubMed.
- J. L. Fiorio, N. López and L. M. Rossi, ACS Catal., 2017, 7, 2973–2980 CrossRef CAS.
- J. L. Fiorio, L. R. Borges, T. Neves-Garcia, D. K. Kikuchi, R. R. G. Guerra and L. M. Rossi, Catal. Sci. Technol., 2023, 13, 3205–3215 RSC.
- Y. Yao and K. P. Giapis, Angew. Chem., Int. Ed., 2016, 55, 11595–11599 CrossRef CAS PubMed.
- M. Trivedi, A. Kumar, A. Husain and N. P. Rath, Inorg. Chem., 2021, 60, 4385–4396 CrossRef CAS PubMed.
- Z. Shao, Y. Li, C. Liu, W. Ai, S.-P. Luo and Q. Liu, Nat. Commun., 2020, 11, 591 CrossRef CAS PubMed.
- S. Nandi, A. Saha, P. Patel, N.-u. H. Khan, R. I. Kureshy and A. B. Panda, ACS Appl. Mater. Interfaces, 2018, 10, 24480–24490 CrossRef CAS PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- F. A. Westerhaus, R. V. Jagadeesh, G. Wienhöfer, M.-M. Pohl, J. Radnik, A.-E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Brückner and M. Beller, Nat. Chem., 2013, 5, 537–543 CrossRef CAS PubMed.
- F. Chen, C. Kreyenschulte, J. Radnik, H. Lund, A.-E. Surkus, K. Junge and M. Beller, ACS Catal., 2017, 7, 1526–1532 CrossRef CAS.
- R. Noyori, M. Kitamura and T. Ohkuma, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5356–5362 CrossRef CAS PubMed.
- D. Zhang, E.-Z. Zhu, Z.-W. Lin, Z.-B. Wei, Y.-Y. Li and J.-X. Gao, Asian J. Org. Chem., 2016, 5, 1323–1326 CrossRef CAS.
- Y. Liu, M. A. Mellmer, D. M. Alonso and J. A. Dumesic, ChemSusChem, 2015, 8, 3983–3986 CrossRef CAS PubMed.
- S. Manna and A. P. Antonchick, ChemSusChem, 2019, 12, 3094–3098 CrossRef CAS PubMed.
- E. C. Ra, K. Y. Kim, E. H. Kim, H. Lee, K. An and J. S. Lee, ACS Catal., 2020, 10, 11318–11345 CrossRef CAS.
- S. J. Bonyhady, D. Collis, G. Frenking, N. Holzmann, C. Jones and A. Stasch, Nat. Chem., 2010, 2, 865–869 CrossRef CAS PubMed.
- S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Chem. Sci., 2020, 11, 12401–12422 RSC.
- Y. Zhang, W. Yu, S. Cao, Z. Sun, X. Nie, Y. Liu and Z. Zhao, ACS Catal., 2021, 11, 13408–13415 CrossRef CAS.
- A. Kumar, R. Bhardwaj, S. K. Mandal and J. Choudhury, ACS Catal., 2022, 12, 8886–8903 CrossRef CAS.
- Y. Wang, X. Cao, L. Zhao, C. Pi, J. Ji, X. Cui and Y. Wu, Adv. Synth. Catal., 2020, 362, 4119–4129 CrossRef CAS.
- M. N. A. Fetzer, G. Tavakoli, A. Klein and M. H. G. Prechtl, ChemCatChem, 2021, 13, 1317–1325 CrossRef CAS.
- S. Lau, D. Gasperini and R. L. Webster, Angew. Chem., Int. Ed., 2021, 60, 14272–14294 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- J. Everaert, K. Leus, H. Rijckaert, M. Debruyne, K. Van Hecke, R. Morent, N. De Geyter, V. Van Speybroeck, P. Van Der Voort and C. V. Stevens, Green Chem., 2023, 25, 3267–3277 RSC.
- T. S. Hansen, K. Barta, P. T. Anastas, P. C. Ford and A. Riisager, Green Chem., 2012, 14, 2457–2461 RSC.
- N. Castellanos-Blanco, A. Arévalo and J. J. García, Dalton Trans., 2016, 45, 13604–13614 RSC.
- P. Weingart and W. R. Thiel, ChemCatChem, 2018, 10, 4844–4848 CrossRef.
- L. Deng, J. Li, D.-M. Lai, Y. Fu and Q.-X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529–6532 CrossRef CAS PubMed.
- D. Scholz, C. Aellig and I. Hermans, ChemSusChem, 2014, 7, 268–275 CrossRef CAS PubMed.
- Z. Yang, Y.-B. Huang, Q.-X. Guo and Y. Fu, Chem. Commun., 2013, 49, 5328–5330 RSC.
- J. Jae, E. Mahmoud, R. F. Lobo and D. G. Vlachos, ChemCatChem, 2014, 6, 508–513 CrossRef CAS.
- I. Gandarias, P. L. Arias, S. G. Fernández, J. Requies, M. El Doukkali and M. B. Güemez, Catal. Today, 2012, 195, 22–31 CrossRef CAS.
- M. G. Musolino, L. A. Scarpino, F. Mauriello and R. Pietropaolo, Green Chem., 2009, 11, 1511–1513 RSC.
- Y. Liu, W. Miao, W. Tang, D. Xue, J. Xiao, C. Wang and C. Li, Chem.–Asian J., 2021, 16, 1725–1729 CrossRef CAS PubMed.
- M.-Y. Qi, Y.-H. Li, M. Anpo, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2020, 10, 14327–14335 CrossRef CAS.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- M. Trincado, J. Bösken and H. Grützmacher, Coord. Chem. Rev., 2021, 443, 213967 CrossRef CAS.
- I. Borthakur, S. Kumari and S. Kundu, Dalton Trans., 2022, 51, 11987–12020 RSC.
- R. Nie, Y. Tao, Y. Nie, T. Lu, J. Wang, Y. Zhang, X. Lu and C. C. Xu, ACS Catal., 2021, 11, 1071–1095 CrossRef CAS.
- A. Osatiashtiani, A. F. Lee and K. Wilson, J. Chem. Technol. Biotechnol., 2017, 92, 1125–1135 CrossRef CAS.
- S. Dutta, I. K. M. Yu, D. C. W. Tsang, Y. H. Ng, Y. S. Ok, J. Sherwood and J. H. Clark, Chem. Eng. J., 2019, 372, 992–1006 CrossRef CAS.
- G. K. Zieliński, J. Majtczak, M. Gutowski and K. Grela, J. Org. Chem., 2018, 83, 2542–2553 CrossRef PubMed.
- J. Du, J. Zhang, Y. Sun, W. Jia, Z. Si, H. Gao, X. Tang, X. Zeng, T. Lei, S. Liu and L. Lin, J. Catal., 2018, 368, 69–78 CrossRef CAS.
- G. Li, H. Yang, H. Zhang, Z. Qi, M. Chen, W. Hu, L. Tian, R. Nie and W. Huang, ACS Catal., 2018, 8, 8396–8405 CrossRef CAS.
- J. Patel and A. Patel, Renewable Energy, 2022, 201, 190–201 CrossRef CAS.
- M. Ruiz-Castañeda, L. Santos, B. R. Manzano, G. Espino and F. A. Jalón, Eur. J. Inorg. Chem., 2021, 2021, 1358–1372 CrossRef.
- C. Guo, W. Zhou, X. Lan, Y. Wang, T. Li, S. Han, Y. Yu and B. Zhang, J. Am. Chem. Soc., 2022, 144, 16006–16011 CrossRef CAS PubMed.
- C. Zhu and J. R. Falck, ChemCatChem, 2011, 3, 1850–1851 CrossRef CAS.
- C. Zhu, K. Saito, M. Yamanaka and T. Akiyama, Acc. Chem. Res., 2015, 48, 388–398 CrossRef CAS PubMed.
- R. Matsubara, T. Harada, W. Xie, T. Yabuta, J. Xu and M. Hayashi, J. Org. Chem., 2023, 88, 12276–12288 CrossRef CAS PubMed.
- C.-H. Lim, S. Ilic, A. Alherz, B. T. Worrell, S. S. Bacon, J. T. Hynes, K. D. Glusac and C. B. Musgrave, J. Am. Chem. Soc., 2019, 141, 272–280 CrossRef CAS PubMed.
- G.-B. Shen, Y.-H. Fu and X.-Q. Zhu, J. Org. Chem., 2020, 85, 12535–12543 CrossRef CAS PubMed.
- W.-J. Kang, B. Li, M. Duan, G. Pan, W. Sun, A. Ding, Y. Zhang, K. N. Houk and H. Guo, Angew. Chem., Int. Ed., 2022, 61, e202211562 CrossRef CAS PubMed.
- M. Zheng, J. Zhang, P. Wang, H. Jin, Y. Zheng and S.-Z. Qiao, Adv. Mater., 2024, 36, 2307913 CrossRef CAS PubMed.
- M. K. Jana, U. Gupta and C. N. R. Rao, Dalton Trans., 2016, 45, 15137–15141 RSC.
- Z.-W. Xi, L. Yang, D.-Y. Wang, C.-D. Pu, Y.-M. Shen, C.-D. Wu and X.-G. Peng, J. Org. Chem., 2018, 83, 11886–11895 CrossRef CAS PubMed.
- T. Shen, J. Wu, Q. Liu and Z. Liu, Ind. Eng. Chem. Res., 2023, 62, 10721–10728 CrossRef CAS.
- Q. Guo, J. Chen, G. Shen, G. Lu, X. Yang, Y. Tang, Y. Zhu, S. Wu and B. Fan, J. Org. Chem., 2022, 87, 540–546 CrossRef CAS PubMed.
- M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781–3783 CrossRef CAS PubMed.
- S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424–7427 CrossRef CAS PubMed.
- R. I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84–86 CrossRef CAS PubMed.
- J. W. Yang, M. T. Hechavarria Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 6660–6662 CrossRef PubMed.
- S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32–33 CrossRef CAS PubMed.
- C. Trotta, G. Menendez Rodriguez, C. Zuccaccia and A. Macchioni, ACS Catal., 2024, 14, 10334–10343 CrossRef CAS.
- J. Yang, H. Qin, K. Yan, X. Cheng and J. Wen, Adv. Synth. Catal., 2021, 363, 5407–5416 CrossRef CAS.
- J. Li, L. He, X. Liu, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2019, 58, 1759–1763 CrossRef CAS PubMed.
- X. Zhang, R. Jiang and X. Cheng, J. Org. Chem., 2021, 86, 16016–16025 CrossRef CAS PubMed.
- F. Zhou, C. Jehoulet and A. J. Bard, J. Am. Chem. Soc., 1992, 114, 11004–11006 CrossRef CAS.
- D. Pu, Y. Zhou, F. Yang, G. Shen, Y. Gao, W. Sun, R. Khan and B. Fan, Org. Chem. Front., 2018, 5, 3077–3082 RSC.
- S. D. Dempsey, A. A. Ryan, M. Smyth, T. S. Moody, S. Wharry, K. Fahey, A. M. Beale, S. Mediavilla Madrigal, P. Dingwall, D. W. Rooney, P. C. Knipe, M. J. Muldoon and J. M. Thompson, React. Chem. Eng., 2023, 8, 1559–1564 RSC.
- M. Pang, J.-Y. Chen, S. Zhang, R.-Z. Liao, C.-H. Tung and W. Wang, Nat. Commun., 2020, 11, 1249 CrossRef CAS PubMed.
- F. Harnisch and M. C. Morejón, Chem. Rec., 2021, 21, 2277–2289 CrossRef CAS PubMed.
- S. Zhang and M. Findlater, ACS Catal., 2023, 13, 8731–8751 CrossRef CAS PubMed.
- S. Gnaim, A. Bauer, H.-J. Zhang, L. Chen, C. Gannett, C. A. Malapit, D. E. Hill, D. Vogt, T. Tang, R. A. Daley, W. Hao, R. Zeng, M. Quertenmont, W. D. Beck, E. Kandahari, J. C. Vantourout, P.-G. Echeverria, H. D. Abruna, D. G. Blackmond, S. D. Minteer, S. E. Reisman, M. S. Sigman and P. S. Baran, Nature, 2022, 605, 687–695 CrossRef CAS PubMed.
- H. An, G. Sun, M. J. Hülsey, P. Sautet and N. Yan, ACS Catal., 2022, 12, 15021–15027 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- R. E. Warburton, A. V. Soudackov and S. Hammes-Schiffer, Chem. Rev., 2022, 122, 10599–10650 CrossRef CAS PubMed.
- C. Liu, F. Chen, B.-H. Zhao, Y. Wu and B. Zhang, Nat. Rev. Chem, 2024, 8, 277–293 CrossRef CAS PubMed.
- S. Kolb and D. B. Werz, Chem.–Eur. J., 2023, 29, e202300849 CrossRef CAS PubMed.
- C. Xing, Y. Xue, X. Zheng, Y. Gao, S. Chen and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202310722 CrossRef CAS PubMed.
- C. Han, J. Zenner, J. Johny, N. Kaeffer, A. Bordet and W. Leitner, Nat. Catal., 2022, 5, 1110–1119 CrossRef CAS.
- X. Xu, J. Ma, B. Kui, G. Zhu, G. Jia, F. Wu, P. Gao and W. Ye, ACS Appl. Nano Mater., 2023, 6, 5357–5364 CrossRef CAS.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed.
- Y. Wang, D. Wang and Y. Li, Adv. Mater., 2021, 33, 2008151 CrossRef CAS PubMed.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Pérez-Ramírez and J. Lu, Adv. Mater., 2021, 33, 2003075 CrossRef CAS PubMed.
- C. J. Bondue, F. Calle-Vallejo, M. C. Figueiredo and M. T. M. Koper, Nat. Catal., 2019, 2, 243–250 CrossRef CAS.
- Y. Wu, C. Liu, C. Wang, Y. Yu, Y. Shi and B. Zhang, Nat. Commun., 2021, 12, 3881 CrossRef CAS PubMed.
- Z. Song, C. Cheng, C. Wang, J. Yao, C. Liu, B. Zhang and Y. Wu, ACS Mater. Lett., 2023, 5, 3068–3073 CrossRef CAS.
- K. Zhu, J. Ma, L. Chen, F. Wu, X. Xu, M. Xu, W. Ye, Y. Wang, P. Gao and Y. Xiong, ACS Catal., 2022, 12, 4840–4847 CrossRef CAS.
- S. Wang, K. Uwakwe, L. Yu, J. Ye, Y. Zhu, J. Hu, R. Chen, Z. Zhang, Z. Zhou, J. Li, Z. Xie and D. Deng, Nat. Commun., 2021, 12, 7072 CrossRef CAS PubMed.
- J. Bu, Z. Liu, W. Ma, L. Zhang, T. Wang, H. Zhang, Q. Zhang, X. Feng and J. Zhang, Nat. Catal., 2021, 4, 557–564 CrossRef CAS.
- B.-H. Zhao, F. Chen, M. Wang, C. Cheng, Y. Wu, C. Liu, Y. Yu and B. Zhang, Nat. Sustain., 2023, 6, 827–837 CrossRef.
- S. Guo, Y. Wu, C. Wang, Y. Gao, M. Li, B. Zhang and C. Liu, Nat. Commun., 2022, 13, 5297 CrossRef CAS PubMed.
- Y. Du, X. Chen, W. Shen, H. Liu, M. Fang, J. Liu and C. Liang, Green Chem., 2023, 25, 5489–5500 RSC.
- R. J. Dixit, K. Bhattacharyya, V. K. Ramani and S. Basu, Green Chem., 2021, 23, 4201–4212 RSC.
- S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS PubMed.
- Y. Wang, Q. Wang, L. Wu, K. Jia, M. Wang and Y. Qiu, Nat. Commun., 2024, 15, 2780 CrossRef CAS PubMed.
- K. Ji, M. Xu, S.-M. Xu, Y. Wang, R. Ge, X. Hu, X. Sun and H. Duan, Angew. Chem., Int. Ed., 2022, 61, e202209849 CrossRef CAS PubMed.
- S. Wu, X. Huang, H. Zhang, Z. Wei and M. Wang, ACS Catal., 2022, 12, 58–65 CrossRef CAS.
- C. Liu, Y. Wu, B. Zhao and B. Zhang, Acc. Chem. Res., 2023, 56, 1872–1883 CrossRef CAS PubMed.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J.-P. Tessonnier and W. Li, J. Am. Chem. Soc., 2017, 139, 14120–14128 CrossRef CAS PubMed.
- Y. Zhao, J. Xu, K. Huang, W. Ge, Z. Liu, C. Lian, H. Liu, H. Jiang and C. Li, J. Am. Chem. Soc., 2023, 145, 6516–6525 CrossRef CAS PubMed.
- X. Wang, Y. Jiao, L. Li, Y. Zheng and S.-Z. Qiao, Angew. Chem., Int. Ed., 2022, 61, e202114253 CrossRef CAS PubMed.
- C. Briens, J. Piskorz and F. Berruti, Int. J. Chem. Eng., 2008, 6, 1–49 Search PubMed.
- J. C. Vantourout, Org. Process Res. Dev., 2021, 25, 2581–2586 CrossRef CAS.
- A. Devi, S. Bajar, H. Kour, R. Kothari, D. Pant and A. Singh, Bioenergy Res., 2022, 15, 1820–1841 CrossRef CAS PubMed.
- N. Jiang, B. You, R. Boonstra, I. M. Terrero Rodriguez and Y. Sun, ACS Energy Lett., 2016, 1, 386–390 CrossRef CAS.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- N. Jiang, X. Liu, J. Dong, B. You, X. Liu and Y. Sun, ChemNanoMat, 2017, 3, 491–495 CrossRef CAS.
- K. Li and Y. Sun, Chem.–Eur. J., 2018, 24, 18258–18270 CrossRef CAS PubMed.
- W. Li, N. Jiang, B. Hu, X. Liu, F. Song, G. Han, T. J. Jordan, T. B. Hanson, T. L. Liu and Y. Sun, Chem, 2018, 4, 637–649 CAS.
- M. Yang, Z. Yuan, R. Peng, S. Wang and Y. Zou, Energy Environ. Mater., 2022, 5, 1117–1138 CrossRef CAS.
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1904780 CrossRef CAS.
- P. Zhang, X. Sheng, X. Chen, Z. Fang, J. Jiang, M. Wang, F. Li, L. Fan, Y. Ren, B. Zhang, B. J. J. Timmer, M. S. G. Ahlquist and L. Sun, Angew. Chem., Int. Ed., 2019, 58, 9155–9159 CrossRef CAS PubMed.
- K. Yan, M. L. Huddleston, B. A. Gerdes and Y. Sun, Green Chem., 2022, 24, 5320–5325 RSC.
- X. Shang, X. Liu and Y. Sun, Green Chem., 2021, 23, 2037–2043 RSC.
- H. Inoue, T. Abe and C. Iwakura, Chem. Commun., 1996, 55–56, 10.1039/CC9960000055.
- G. Han, G. Li and Y. Sun, JACS Au, 2024, 4, 328–343 CrossRef CAS PubMed.
- A. Huang, Y. Cao, R. S. Delima, T. Ji, R. P. Jansonius, N. J. J. Johnson, C. Hunt, J. He, A. Kurimoto, Z. Zhang and C. P. Berlinguette, JACS Au, 2021, 1, 336–343 CrossRef CAS PubMed.
- A. Kurimoto, R. P. Jansonius, A. Huang, A. M. Marelli, D. J. Dvorak, C. Hunt and C. P. Berlinguette, Angew. Chem., Int. Ed., 2021, 60, 11937–11942 CrossRef CAS PubMed.
- R. S. Sherbo, R. S. Delima, V. A. Chiykowski, B. P. MacLeod and C. P. Berlinguette, Nat. Catal., 2018, 1, 501–507 CrossRef CAS.
- R. S. Delima, R. S. Sherbo, D. J. Dvorak, A. Kurimoto and C. P. Berlinguette, J. Mater. Chem. A, 2019, 7, 26586–26595 RSC.
- R. S. Sherbo, A. Kurimoto, C. M. Brown and C. P. Berlinguette, J. Am. Chem. Soc., 2019, 141, 7815–7821 CrossRef CAS PubMed.
- R. S. Delima, M. D. Stankovic, B. P. MacLeod, A. G. Fink, M. B. Rooney, A. Huang, R. P. Jansonius, D. J. Dvorak and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 215–224 RSC.
- R. P. Jansonius, A. Kurimoto, A. M. Marelli, A. Huang, R. S. Sherbo and C. P. Berlinguette, Cell Rep. Phys. Sci., 2020, 1, 1–12 Search PubMed.
- C. Hunt, A. Kurimoto, G. Wood, N. LeSage, M. Peterson, B. R. Luginbuhl, O. Horner, S. Issinski and C. P. Berlinguette, J. Am. Chem. Soc., 2023, 145, 14316–14323 CrossRef CAS PubMed.
- M. D. Stankovic, J. F. Sperryn, R. S. Delima, C. C. Rupnow, M. B. Rooney, M. Stolar and C. P. Berlinguette, Energy Environ. Sci., 2023, 16, 3453–3461 RSC.
- Y.-Q. Yan, Y. Chen, Z. Wang, L.-H. Chen, H.-L. Tang and B.-L. Su, Nat. Commun., 2023, 14, 2106 CrossRef CAS PubMed.
- G. Li, G. Han, L. Wang, X. Cui, N. K. Moehring, P. R. Kidambi, D.-e. Jiang and Y. Sun, Nat. Commun., 2023, 14, 525 CrossRef CAS PubMed.
- G. Han, G. Li and Y. Sun, Nat. Catal., 2023, 6, 224–233 CrossRef CAS.
- A. Kurimoto, R. S. Sherbo, Y. Cao, N. W. X. Loo and C. P. Berlinguette, Nat. Catal., 2020, 3, 719–726 CrossRef CAS.
- Y.-T. Xia, X.-T. Sun, L. Zhang, K. Luo and L. Wu, Chem.–Eur. J., 2016, 22, 17151–17155 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |