
Ultrafast Joule heating-induced formation of amorphous CoFeNi phosphate for efficient and stable oxygen evolution reaction†
Junhao
Ma‡
,
Chonghan
Xia‡
,
Teddy
Salim
,
Yee Yan
Tay
,
Lydia H.
Wong
 and
Kwan W.
Tan
*
and
Kwan W.
Tan
*
School of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore. E-mail: kwtan@ntu.edu.sg
First published on 7th August 2024
Abstract
Developing efficient and durable non-precious metal-based catalysts for the oxygen evolution reaction (OER) in water electrolysis is crucial for large-scale and affordable hydrogen production. Many transition metal-based OER catalysts have been explored, but controlling their transformation into active oxyhydroxides during reconstruction, with fast reaction kinetics and low energy barriers, remains challenging. This article reports an ultrafast Joule heating strategy to synthesize efficient and stable amorphous transition metal phosphate electrocatalysts from metal chlorides and phytic acid precursors within 100 milliseconds at around 420 °C. The resulting amorphous CoFeNi phosphate coated on a superhydrophilic activated carbon cloth (CoFeNiPi@ACC) delivered a low overpotential of 235 mV at 10 mA cm−2, a small Tafel slope of 32.2 mV dec−1, and high stability for OER in 1.0 M KOH solution over 100 hours. The transient Joule heating process facilitates the rapid formation of an amorphous metal phosphate structure with abundant active sites, higher oxidation states for metal cations, self-reconstruction into active metal oxyhydroxides, and enhanced charge carrier diffusion rates, resulting in outstanding OER performance. This approach could be extended to other advanced material combinations for sustainable and efficient renewable energy production and storage.
 Kwan W. Tan | Kwan W. Tan is an assistant professor in the School of Materials Science and Engineering at Nanyang Technological University, Singapore (NTU). He earned his bachelor's degree at NTU and his master's degrees from the Massachusetts Institute of Technology and the National University of Singapore. He completed his PhD in 2014 under the guidance of Professors Uli Wiesner and Mike Thompson at Cornell University. Following this, he conducted postdoctoral research at Cornell and with the Singapore-MIT Alliance for Research and Technology. His current research focuses on advanced self-assembling nanocomposites, porous materials, and non-equilibrium phase behaviors, and their applications in clean energy and environmental sustainability. |
1. Introduction
Hydrogen, the lightest element, is a promising alternative energy storage and carrier agent due to its high gravimetric energy density of 120 MJ kg−1 at 298 K.1 Generating hydrogen from water is an environmentally conscious approach for energy usage and storage, mitigating climate change and supporting economic and technological developments.1–4 In water electrolysis, two simultaneous half-reactions occur: hydrogen evolution reaction (HER) and oxygen evolution reaction (OER). However, OER is more complex, requiring more reaction steps to transfer four electrons (compared to two for HER), and has sluggish kinetics and higher energy barriers for forming oxygen-containing intermediates.4–8 Improving OER performance enhances the efficiency of water electrolysis and benefits clean energy applications, such as metal–air batteries and fuel cells.5–7 Typical benchmark OER catalysts include precious metal-based IrO2 and RuO2 that exhibit excellent catalytic performance. However, transition metal oxide catalysts, such as Co, Ni and Fe, are more stable in alkaline media as well as affordable.6,7,9–15 Other transition metal compounds, such as nitrides,16 sulfides,17 phosphides,18 and phosphates,8,19–24 have shown outstanding electrocatalytic properties for OER, attributed to facilitating the formation of active metal oxides or hydroxides and enabling long term stability.25Various strategies have been implemented to improve OER catalytic activities, including combining different metals and anion species, phase engineering, and exploiting synergistic effects with the catalyst support, such as enhanced electron and ion transport and improved wettability with the electrolyte.6–8 For example, mixing Fe with more electronegative Co and Ni lowers the oxygen binding energies, resulting in smaller reaction overpotential values,6,26 and facilitates the reconstruction into oxyhydroxides, increasing the number of electrocatalytically active sites and improving electronic conductivity.14,15
Xu et al. demonstrated that substituting Fe into the otherwise inactive spinel CoAl2O4 formed CoFe0.25Al1.75O4, a highly active and stable catalyst that expedited surface reconstruction into Co oxyhydroxides and stimulated a two-step deprotonation process at lower overpotentials, thereby augmenting oxygen generation.15 In another study, Grimaud et al. revealed that incorporating Fe into nickel oxyhydroxide altered the interactions between active oxygen species and OH− groups, influencing the OER reaction pathway.27 Particularly notable is the role of the phosphate group (Pi), a bulky, oppositely charged entity with nucleophilic properties that acts as an effective proton acceptor.6 This functionality facilitates key proton transfer steps in OER, influencing water adsorption dynamics and stabilizing reaction intermediates such as HOO* by modifying the electronic and geometric properties of the metal adsorption sites.28,29 Additionally, Pi enhances chemical stability, increases the metal oxidation state, leading to enhanced electron transfer kinetics, boosts the total number active sites, and facilitates the reconstruction process into oxyhydroxides.19,30
Many studies of transition metal Pi catalysts for OER typically involve crystalline materials prepared through high-temperature processes such as hydrothermal and furnace annealing, which require extended periods, or through rapid thermal shocks occurring over short durations.8,21,24 For instance, Hu et al. described the synthesis of multicomponent transition metal Pi particles using an aerosol fly-through process, wherein particles passed through a furnace held at 900 °C for subsecond timeframes.24 The resulting CoFeNiMnMoPi particles exhibited low overpotential and Tafel slope values of 227 mV at 10 mA cm−2 and 74 mV dec−1, respectively, showing better performance than commercial IrO2 anode for OER.24 However, an amorphous transition metal Pi anode is generally preferred because its abundant defects provide more potential active sites.7,22,23,31,32 Moreover, the intrinsic structural disorder enhances ion diffusion and facilitates self-reconstruction into an amorphous oxyhydroxide structure.7,22,23,31,32 Yan et al. described an oil-phase method to form ultrathin flakes of amorphous mesoporous NiCoPi, using a NiCo oleate precursor and tetradecylphosphonic acid as the P source, heated at 300 °C for about 1.5 h under inert argon.22 Although the amorphous mesoporous NiCoPi catalyst exhibited a decent overpotential of 327 mV at 10 mA cm−2 and Tafel slope of 73.7 mV dec−1 for OER, the multistep synthesis approach is complex, and the NiCo oleate precursor must be prepared separately.22
Herein, we present a rapid and facile approach for synthesizing amorphous CoFeNi phosphate on activated carbon cloth (CoFeNiPi@ACC), achieved by briefly heating metal chloride salts and phytic acid to approximately 420 °C for just 100 ms using transient Joule heating. The resulting CoFeNiPi@ACC exhibited excellent performance as an OER electrode, with a low overpotential of 235 mV at 10 mA cm−2 and a small Tafel slope of 32.2 mV dec−1 in an alkaline 1.0 M KOH electrolyte. Long-term chronopotentiometry experiments further revealed that the CoFeNiPi@ACC electrode exhibited remarkable stability, with minimal degradation in overpotential at 10 mA cm−2 over 100 hours. Detailed structural, chemical, and electrochemical analyses, including energy-dispersive X-ray spectroscopy, X-ray photoelectron spectroscopy, Raman spectroscopy, and cyclic voltammetry, revealed that transient Joule heating facilitates several key features contributing to efficient OER performance. These features include the direct formation of an amorphous metal phosphate structure, the pre-oxidation of Ni2+ and Co2+ to higher oxidation states, fast charge transfer kinetics, and the facilitation of metal oxyhydroxide species formation through self-reconstruction during the OER.
2. Experimental section
2.1 Chemicals and materials
All chemicals were used as received. Iron(III) chloride (FeCl3·6H2O, reagent grade, 97%), nickel(II) chloride hexahydrate (NiCl2·6H2O, ACS reagent, 98%), cobalt(II) chloride hexahydrate (CoCl2·6H2O, ReagentPlus), commercial RuO2 (99.9% trace metals basis), phytic acid solution (50% (w/w) in water) and tetramethylammonium hydroxide solution (TMAOH, 25 wt% in water) were purchased from Sigma-Aldrich. Absolute ethanol (EtOH) and hydrochloric acid fuming (HCl, 37%) were purchased from Supelco. Potassium hydroxide (KOH, 99.99%) was supplied by Aladin. Commercial AvCarb carbon cloth (AvCarb 1071 HCB) was purchased from Fuel Cell Earth and used after activation treatment.2.2 Activation of carbon cloth
The activation procedure was reported elsewhere.33 The carbon cloth was first immersed in 7 M KOH solution at room temperature for 24 h and then dried in a vacuum oven at 60 °C. Subsequently, the KOH-infused carbon cloth was pyrolyzed under nitrogen at 800 °C for 2 h (ramp rate of 5 °C min−1) and naturally cooled. The heat-treated KOH-infused carbon cloth was rinsed thoroughly with deionized water until a pH of ∼7 was reached. Finally, the activated carbon cloth (ACC) was rinsed with EtOH and dried at ambient temperature in a vacuum oven overnight before use.2.3 Deposition of CoFeNi chloride salt and phytic acid precursor solutions
119 mg of CoCl2·6H2O, 88 mg of FeCl3·6H2O, and 149 mg of NiCl2·6H2O were separately dissolved in a mixed solvent comprising 9 mL of EtOH and 1 mL 37% HCl in individual vials. Subsequently, 0.23 mL of phytic acid was added to each metal salt solution to form the metal salt/phytic acid precursor solution. By combining 1 mL aliquots of the respective metal salt/phytic acid precursor solutions, a ternary metal salt/phytic acid precursor solution with total volume of 3 mL was obtained. Next, 0.3 mL of the ternary metal salt/phytic acid precursor solution was dropcast onto the ACC (25 mm length × 10 mm width), followed by drying at 60 °C on a hotplate. The precursor loading on the ACC substrate was approximately 6.8 ± 1.3 mg, based on measurements from 5 samples.2.4 Joule heating of amorphous CoFeNiPi@ACC
Joule heating experiments were conducted as described elsewhere.34,35 Briefly, a piece of the ACC deposited with the metal salt/phytic acid precursor was placed between two metal clips that were connected to a DC power source. The precursor samples were then electrically heated for 100 ms at ∼420 °C, measured with a Raytek Raynger 3i Plus thermal infrared sensor (Fig. S1†). The loading of CoFeNiPi on the ACC substrate was approximately 4.6 ± 1.1 mg, based on measurements from 5 samples, after Joule heating.2.5 Electrochemical measurements
All electrochemical measurements were performed using a CHI760E electrochemical workstation under ambient conditions with a standard three-electrode configuration. The setup consisted of the CoFeNiPi@ACC sample as the working electrode, a platinum mesh as the counter electrode, and a calibrated Hg/HgO electrode as the reference electrode. All ACC-based working electrodes were sandwiched between two pieces of hydrophobic carbon papers in order to minimize the influence of capillary wetting effects and to isolate them from the platinum clamp holder. RuO2 ink as a control was prepared by dispersing 5 mg of commercial RuO2 in 1.0 mL of a solution mixture consisting of 0.75 mL EtOH, 0.23 mL deionized water, and 0.02 mL of 5 wt% Nafion solution. The mixture was ultrasonicated for 1 h to obtain a homogenous ink. Approximately 0.2 mL of the RuO2 ink was then dropcast onto the ACC substrate (∼1 cm2 area) and allowed to dry under ambient conditions prior to electrochemical measurements. To activate the electrocatalysts, cyclic voltammetry (CV) measurements in the potential range of 1.2 to 2.0 V versus reversible hydrogen electrode (RHE) were conducted at a scan rate of 100 mV s−1 for 60 cycles.All potentials were recorded with respect to the RHE and iR-compensated according to eqn (1),
| ERHE = EHg/HgO + 0.0592 × pH − iR | (1) |
Linear sweep voltammetry experiments were conducted in an alkaline aqueous medium (1.0 M KOH) with a scan rate of 1 mV s−1. The overpotential (η) was determined by η = ERHE − 1.23 (V). The Tafel slope values were determined from the polarization curves using eqn (2),
 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j).
j).
Electrochemical impedance spectroscopy (EIS) measurements were performed over a frequency range from 1 Hz to 100 kHz. Electrochemical surface area (ECSA) values were obtained using the double-layer capacitance method.36 The double-layer capacitance (Cdl) is calculated from CV measurements in non-faradaic regions at scan rates ranging from 20 to 120 mV s−1 based on eqn (3),
| ic = νCdl | (3) |
| ic = Δj/2 = janodic − jcathodic/2 | (4) |
| ECSA = Cdl/Cs, | (5) |
2.6 Characterization
Wide-angle X-ray scattering (WAXS) measurements were performed using a Xenocs NanoinXider instrument in transmission mode using Cu Kα radiation source and Dectris Pilatus 3 detectors. All samples were mounted on the sample holder using Kapton tape. Scanning electron microscopy (SEM) images were taken on a JEOL 7600F field emission scanning electron microscope equipped with a half-in-lens detector. Samples were mounted on carbon tape for SEM characterization. Transmission electron microscopy (TEM), selected area electron diffraction (SAED), high-angle annular dark field scanning TEM (HAADF-STEM), and energy-dispersive spectroscopy (EDS) were conducted using a JEOL-2100F electron microscope operating at 200 kV, equipped with a 2K × 2K Gatan Ultrascan 1000XP CCD camera, Gatan Digiscan and STEM detectors, and an energy dispersive spectroscopy (EDS) detector. The metal phosphate@ACC sample was ground with an agate mortar and pestle and redispersed in 1 mL of EtOH. After ultrasonication for 30 s, the ethanolic solution was dropcast on a Mo-based grid for TEM characterization. The EDS mapping was obtained with a windowless 100 mm2 Oxford Ultim Max silicon drift detector. X-ray photoelectron spectroscopy (XPS) measurements were conducted using an AXIS Supra spectrometer (Kratos Analytical, UK) equipped with a hemispherical analyzer and a monochromatic Al Kα source (1487 eV) operating at 15 mA and 15 kV. The XPS data were obtained from an analysis area of 700 × 300 μm2 at a take-off angle of 90°. Pass energies of 160 and 20 eV were used for survey and high resolutions scans, respectively, with a 3.1 V bias applied to prevent charge build-up on the samples. Raman spectra were obtained using a WITec Alpha300 RS with a 488 nm excitation laser source. Water contact angle measurements were performed using a Dataphysics OCA15 system. Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis was conducted using an Avio 550 Max ICP-OES (PerkinElmer). Sample solutions were measured after being filtered through a 0.22 μm PTFE filter. An ICP multi-element standard solution IV (Supelco) was used for calibration.3 Results and discussion
3.1 Joule heating of amorphous CoFeNiPi@ACC samples
Transient thermal shock treatments have been widely employed to form highly crystalline multicomponent metal alloy and oxide nanostructures with high electrocatalytic performance through rapid high temperature heating and quenching.35,37–43 Hereinafter, we instead applied transient Joule heating to form amorphous CoFeNiPi coated on activated carbon cloth fibers (CoFeNiPi@ACC), as depicted in the schematic (Fig. 1a). Chloride salts of Co, Ni and Fe were mixed with phytic acid (as the P source) in EtOH and HCl acid (providing excess Cl− for solubilization), and then deposited on the superhydrophilic ACC by drop-casting (Fig. S2†). The precursors underwent rapid thermal treatment via resistive Joule heating at ∼420 °C for a dwell time of 100 ms, leading to interdiffusion of metal cations and the formation of metal–phosphate (M–Pi) bonds. As a result of the moderate heating temperature, short heating time and rapid quenching, the resulting CoFeNiPi@ACC exhibited a uniform coating thickness of ∼165 nm (Fig. 1b and S3†) with an amorphous structure (Fig. 1c and d). It should be noted that the superhydrophilic nature of ACC not only improves the precursor coating but also enhances wetting properties with the electrolyte, potentially leading to enhanced mass transport of charged ionic species during electrocatalysis.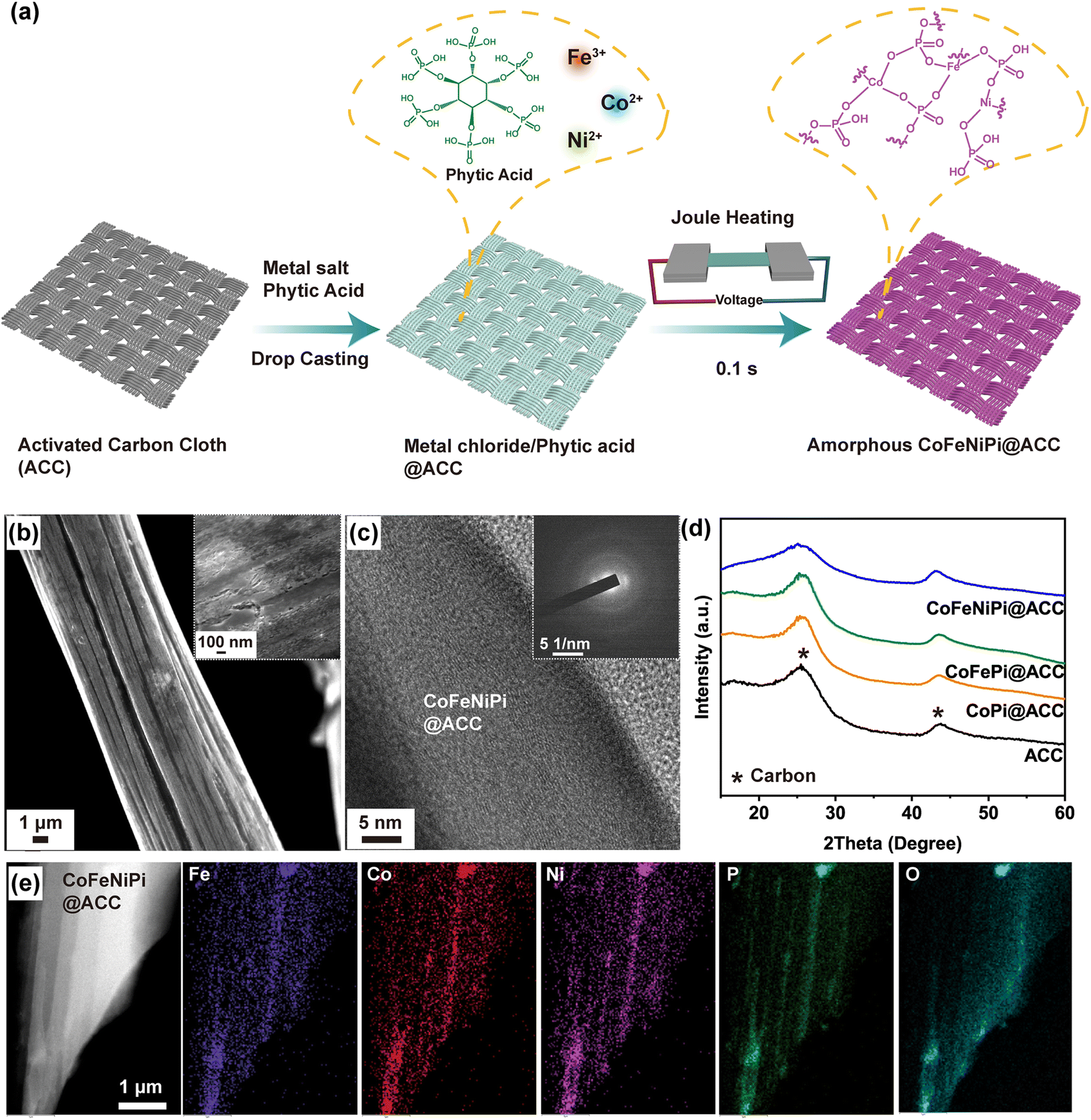 | ||
| Fig. 1 (a) Schematic representation of Joule heating of amorphous CoFeNiPi@ACC. (b) SEM, (c) HR-TEM, and SAED (inset) micrographs of CoFeNiPi@ACC. (d) WAXS spectra of uncoated ACC, CoPi@ACC, CoFePi@ACC and CoFeNiPi@ACC as indicated. (e) HAADF-STEM and corresponding EDS maps of amorphous CoFeNiPi@ACC after Joule heating. | ||
We employed electron microscopy and X-ray scattering to evaluate the film morphology, structure, and composition of CoFeNiPi@ACC. Scanning electron microscopy (SEM) images in Fig. 1b show a relatively smooth CoFeNiPi surface morphology, albeit with regular pits and short cracks (see inset), likely due to some degree of thermal-induced contraction after quenching. The high-resolution transmission electron microscopy (HR-TEM) image in Fig. 1c reveals only long-range, well-aligned graphitic carbon sheets, with the notable absence of other crystalline lattice fringes. This observation was corroborated by the selected area electron diffraction (SAED) pattern (inset in Fig. 1c), exhibiting two diffuse rings ascribed to the ACC fibers and indicated the amorphous nature of the resulting CoFeNiPi layer. As a control experiment, we observed similar surface morphologies for single metal Co phosphate (CoPi@ACC) and binary metal CoFe phosphate (CoFePi@ACC) after Joule heating (Fig. S4†). The wide-angle X-ray scattering (WAXS) spectra of uncoated ACC, CoPi@ACC, CoFePi@ACC and CoFeNiPi@ACC were remarkably similar, showing only two broad peaks around the angular positions of 25° and 43°, attributed to graphitic carbon (Fig. 1d). Taken together, HR-TEM, SAED and WAXS unambiguously confirmed the amorphous nature of metal phosphates on ACC fibers after Joule heating.44 Energy-dispersive spectroscopy (EDS) mapping analysis in high-angle annular dark field scanning TEM (HAADF-STEM) mode, shown in Fig. 1e, confirmed the homogeneous elemental distribution of Co, Fe, Ni, O and P on the ACC fiber. Co, Fe and Ni each has nearly equal atomic concentration, ranging from 1 to 2 at%, while the ratio of P to O is about 1:4 (Table S1†).
X-ray photoelectron spectroscopy (XPS) provides further insights into the valence and chemical bonding of all elements in the CoFeNiPi@ACC samples (Fig. 2). The 2p core level spectra for Co (Fig. 2a), Fe (Fig. 2b), and Ni (Fig. 2c) were deconvoluted by applying appropriate constraints on their 2p3/2 and 2p1/2 spin–orbit components. This included accounting for any overlapping LMM Auger signals from the other metals and the shake-up satellite features. Distinct peaks were observed for Co2+/Co3+–O–P at binding energies of 783.5 and 781.9 eV, for Fe2+/Fe3+–O–P at 710.5 and 713.5 eV, and for Ni2+/Ni3+–O–P at 856.7 and 857.7 eV in the 2p3/2 region of Co 2p, Fe 2p, and Ni 2p, respectively.23,45,46 These observations confirmed the formation of a mixed-metal phosphate structure. It is interesting to note that the presence of phosphates with mixed oxidation states (2+ and 3+) indicates the oxidation of Co2+, Fe2+, and Ni2+ species from the salt precursors during the Joule heating treatment.
 | ||
| Fig. 2 XPS analysis of CoFeNiPi@ACC and CoFeNiPi@ACC-24 h samples showing the spectra of (a) Co 2p, (b) Fe 2p, (c) Ni 2p, (d) O 1s, and (e) P 2p, as well as (f) the atomic concentrations. | ||
The O 1s spectrum in Fig. 2d shows a peak at 531.9 eV, suggesting the combined effect of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and non-bridging PO groups, while a peak at 533 eV indicates the contribution of both C–O and P–O groups.47 The P 2p spectrum in Fig. 2e displays peaks at 134.5 eV and 135.4 eV, assigned to the P–O–metal (metal phosphate) bond and PO3− ion, respectively.23,47 Notably, the peak at ∼129 eV, associated with the metal–P (metal phosphide) bond, is absent, indicating that phosphate was the only species formed after the Joule heating treatment.23,47 The atomic concentrations of all elements closely matched those found in the EDS analysis, with metals ranging between 1 to 2 at%, and the ratio of P to O is approximately 1:3.2 (Fig. 2f). Finally, the carbon–oxygen bonds at binding energies of 285.5 eV, 286.7 eV and 288.9 eV were observed in the C 1s spectrum (Fig. S5†). These may be attributed to oxygen-containing hydrophilic groups (such as epoxide, carbonyl, and carboxyl) on the carbon fiber or to adventitious carbon contamination.48
O and non-bridging PO groups, while a peak at 533 eV indicates the contribution of both C–O and P–O groups.47 The P 2p spectrum in Fig. 2e displays peaks at 134.5 eV and 135.4 eV, assigned to the P–O–metal (metal phosphate) bond and PO3− ion, respectively.23,47 Notably, the peak at ∼129 eV, associated with the metal–P (metal phosphide) bond, is absent, indicating that phosphate was the only species formed after the Joule heating treatment.23,47 The atomic concentrations of all elements closely matched those found in the EDS analysis, with metals ranging between 1 to 2 at%, and the ratio of P to O is approximately 1:3.2 (Fig. 2f). Finally, the carbon–oxygen bonds at binding energies of 285.5 eV, 286.7 eV and 288.9 eV were observed in the C 1s spectrum (Fig. S5†). These may be attributed to oxygen-containing hydrophilic groups (such as epoxide, carbonyl, and carboxyl) on the carbon fiber or to adventitious carbon contamination.48
3.2 Electrocatalytic OER Experiments
After confirming the amorphous structure and chemical states of the Joule heating-induced CoFeNiPi@ACC, we evaluated their electrocatalytic performance for OER in a 1.0 M KOH electrolyte using a standard three-electrode configuration. Fig. 3a displays the polarization curves of the ternary metallic phosphate CoFeNiPi@ACC and, for comparison, the amorphous metal phosphates of CoFePi@ACC and CoPi@ACC, both prepared by Joule heating, alongside the benchmark RuO2@ACC and uncoated ACC support. CoFeNiPi@ACC exhibited the best OER performance, requiring only an overpotential of 235 mV to drive a current density of 10 mA cm−2. CoFePi@ACC followed closely with an overpotential of 257 mV, and CoPi@ACC was significantly higher at 389 mV. In contrast, the control RuO2@ACC required an overpotential of 332 mV to achieve the same current density. This indicates that amorphous CoFeNiPi improved the overpotential by almost 30% at 10 mA cm−2 compared to RuO2. The summary bar charts in Fig. 3b consistently show that CoFeNiPi@ACC required lower overpotentials of 258 and 267 mV to drive current densities of 50 and 100 mA cm−2, respectively, compared to CoFePi@ACC (280 and 296 mV) and CoPi@ACC (410 and 430 mV). Although the uncoated ACC substrate exhibited negligible OER activity in the alkaline medium, its superhydrophilicity and high porosity can facilitate mass transport and fast elimination of generated bubbles under high current densities, preventing charged ionic species from being blocked from the active catalytic sites. Moreover, we observed an oxidation peak at around 1.35 V in the polarization curve of amorphous CoFeNiPi@ACC attributed to the oxidation of either Co2+ or Ni2+. | ||
| Fig. 3 (a) iR-corrected OER polarization curves of amorphous samples of CoFeNiPi@ACC, CoFePi@ACC, CoPi@ACC, RuO2@ACC, and uncoated ACC in 1.0 M KOH. (b) Overpotential bar charts of amorphous metal phosphate catalysts at current densities of 10, 50, and 100 mA cm−2, as indicated. (c) Tafel plots of CoFeNiPi@ACC, CoFePi@ACC, CoPi@ACC, and RuO2@ACC. (d) Nyquist plots and the equivalent resistance circuit (inset) of amorphous metal phosphate catalysts as indicated. (e) Plots of Δj/2 versus CV scan rate to obtain Cdl values for the indicated amorphous metal phosphate catalysts. (f) Plot of overpotential required to drive 10 mA cm−2 against Tafel slope, comparing OER performance in alkaline media extracted from literature and this work: TiO2|Co2P4O12 (ref. 51), NiCoPi (ref. 22), CoNiPi@V-Co4N (ref. 52), NiCoFeMnMoPi (ref. 24), V-FeP2/FePO4 (ref. 53), NiFePi (ref. 23), Ni1.5Sn@triMPO4 (ref. 54), Co2(OH)2(HPO4)2/NF (ref. 55), Co1.6Ni0.4P4O12–C (ref. 56). (g) Chronopotentiometry curve of CoFeNiPi@ACC over 100 h at a constant current density of 10 mA cm−2. | ||
Fig. 3c displays Tafel slope values extracted from the Tafel region of the linear sweep voltammetry curves, which were used to assess the OER activity kinetics. Both amorphous CoFeNiPi@ACC and CoFePi@ACC catalysts exhibited similar, low Tafel slopes of 32.2 and 32.3 mV dec−1, respectively, indicating rapid OER surface kinetics. In contrast, the unary metallic CoPi@ACC showed a higher Tafel slope of 45.8 mV dec−1, suggesting that the inclusion of Fe and Ni metal cations synergistically enhances the OER kinetics. Notably, the benchmark RuO2@ACC catalyst had the highest Tafel slope value of 118.9 mV dec−1. Fig. S6† presents the polarization curves of other amorphous unary and binary metallic phosphate combinations prepared via Joule heating, among which the amorphous CoFeNiPi@ACC catalyst consistently exhibited the highest OER activity. We further conducted control experiments by Joule heating ternary metal precursors for extended dwell times of 250 and 500 ms at higher temperatures of ∼820 and 1140 °C, respectively, resulting in the formation of crystalline materials (Fig. S7†). The polarization curves and Tafel slope plots presented in Fig. S8† clearly show that the CoFeNiPi@ACC catalyst, when Joule-heated for a shorter dwell of 100 ms, exhibited the lowest overpotential (267 mV at 100 mA cm−2) and Tafel slope (32.2 mV dec−1) for OER. These excellent performance metrics are attributed to its amorphous structure, in contrast to its crystalline counterparts, which were heated for 250 ms (298 mV and 37.8 mV dec−1) and 500 ms (319 mV and 47.9 mV dec−1).
Electrochemical impedance spectroscopy (EIS) provides further insights into the surface reaction kinetics and charge carrier transfer of the amorphous metal phosphate catalysts. Fig. 3d shows the Nyquist plots and the equivalent circuit resistance (inset) for all amorphous metal phosphate catalysts over the frequency range from 1 Hz to 100 kHz. The ternary metallic phosphate FeCoNiPi@ACC exhibited the lowest charge transfer resistance (Rct) at 0.22 Ω, compared to CoFePi@ACC at 0.57 Ω and CoPi@ACC at 0.89 Ω. This underscores the significant roles of Fe and Ni in enhancing electroconductivity and facilitating surface charge transfer kinetics.49
To corroborate that the enhanced OER performance of amorphous metal phosphates is due to intrinsic activity improvement, we calculated the electrochemical active surface areas (ECSA) using cyclic voltammetry (CV) curves in the non-faradaic region measured at scan rates ranging from 20 to 120 mV s−1 (Fig. S9†).50Fig. 3e displays the double-layer capacitance (Cdl) values of amorphous metal phosphate catalysts on the ACC substrate, obtained from the plots of Δj/2 versus CV scan rate. Both CoFeNiPi@ACC and CoFePi@ACC exhibited similar Cdl values of 17.2 and 15.7 mF cm−2, respectively, indicating comparable ECSA values of 430 and 392.5 cmECSA2 and similar numbers of active sites. Although CoPi@ACC had the highest Cdl at 65.9 mF cm−2, corresponding to the highest ECSA of 1648 cmECSA2 and largest number of active sites, it exhibited the lowest OER activity. This underscores that the enhanced intrinsic activities of the amorphous multi-metallic phosphate combinations, particularly CoFeNiPi@ACC, were responsible for their remarkable OER performance. Fig. S10† presents the specific activities normalized by ECSA (jECSA) of the amorphous metal phosphate catalysts. CoFeNiPi@ACC exhibited the highest specific activity of 0.129 mA cmECSA−2 at the OER overpotential of 270 mV, which was 3.5 and 117.3 times higher than CoFePi@ACC (0.037 mA cmECSA−2) and CoPi@ACC (0.0011 mA cmECSA−2), respectively. We further compared the OER performance of the Joule-heated amorphous CoFeNiPi@ACC from this study with other phosphorous-containing OER catalysts reported in the past five years, demonstrating its leading performance as characterized by low overpotential and small Tafel slope values (Fig. 3f and Table S2†).22–24,51–56
Finally, we conducted long-term stability tests on the Joule-heated amorphous metal phosphates. Fig. 3g shows the chronopotentiometry experiment of the amorphous CoFeNiPi@ACC at a current density of 10 mA cm−2, which maintained a relatively stable OER overpotential of 250 mV at the 100 h timepoint. The robustness of the catalyst may have benefited from the formation of the amorphous metal phosphate structure and the oxidation of Co2+ and Ni2+ to higher valence states during Joule heating. To test this hypothesis, we performed linear sweep voltammetry and chronopotentiometry experiments on the metal salts/phytic acid precursor coated on the ACC substrate (precursor@ACC). Fig. S11a† displays the polarization curves for precursor@ACC samples in two states: upon activation and after immersion in 1 M KOH for 3 h. Both samples showed pronounced oxidation peaks at 1.44 V, indicating the oxidation of cations from Ni2+ to Ni3+. In contrast, this anodic peak is significantly less evident in the polarization curve of the amorphous CoFeNiPi@ACC sample, suggesting that most metal cations were already in the 3+ state due to prior Joule heating process. While the precursor@ACC exhibited comparable polarization behavior to the amorphous metal phosphate catalyst (Fig. S11a†), the performance of the precursor working electrode degraded rapidly after the first 3 h of chronopotentiometry, indicating poor stability (Fig. S12†). This degradation is attributed to the weak interaction forces within the precursor coating, causing physical disintegration during continuous OER operation in 1 M KOH. To test this assumption, we immersed both precursor@ACC and amorphous CoFeNiPi@ACC samples in 1 M KOH for 3 h. ICP-OES analysis detected leached Fe species solely from the precursor@ACC sample (Fig. S11b†), confirming that Joule heating is essential for forming the amorphous phosphate structure, oxidizing metal cations, and stabilizing the coating through heat treatment at 420 °C for a brief 100 ms.
3.3 Surface reconstruction in Joule-heating-induced amorphous metal phosphate OER electrocatalysts
The preoxidation of Co2+ and Ni2+ to higher valence states and the formation of Co and Ni oxyhydroxides are crucial steps in OER.15,57 We investigated the pseudocapacitive behaviors of amorphous metal phosphates by analyzing their CV profiles to determine the influence of the transient Joule heating process and composition on oxyhydroxide generation. Fig. 4a illustrates the first and second CV cycles conducted on CoPi@ACC, CoFePi@ACC, CoNiPi@ACC, and CoFeNiPi@ACC over the range from 0.85 to 1.55 V. Initially, all samples displayed larger pseudocapacitive charges in the first cycle than in the second, suggesting irreversible surface reconstruction and oxyhydroxide evolution. In addition, the pseudocapacitive charges were notably similar between CoPi@ACC and CoFePi@ACC, as well as between CoNiPi@ACC and CoFeNiPi@ACC. | ||
| Fig. 4 (a) Pseudocapacitive behaviors in the first and second CV cycles for CoPi@ACC, CoFePi@ACC, CoNiPi@ACC and CoFeNiPi@ACC catalysts at a scan rate of 10 mV s−1. (b) Raman spectra of CoFeNiPi@ACC in the states of Joule-heated as-made, upon activation, and after chronopotentiometry for 24 and 100 h. (c) EDS line scan analysis of the CoFeNiPi@ACC catalyst after chronopotentiometry for 24 h. | ||
For the CoPi@ACC catalyst, an anodic peak at 1.02 V observed in the first cycle indicates the oxidation of Co2+ to CoOOH. Notably, this transformation occurred at a significantly lower potential compared to other reported Co-based oxides,15,22 indicating that Joule heating facilitated the preoxidation of Co2+ to higher oxidation states. A second, smaller anodic peak at 1.01 V in the subsequent cycle suggested a lesser degree of surface reconstruction in CoPi@ACC. The addition of other metals shifted the anodic peaks for Co2+ oxidation to 1.09 V in CoFePi@ACC and 1.05 V in CoNiPi@ACC. However, the absence of the Co oxidation peak in the second cycles of both CoFePi@ACC and CoNiPi@ACC suggested complete surface reconstruction involving Co. Additionally, in CoNiPi@ACC, another anodic peak at 1.38 V observed in the first cycle was attributed to the oxidation of Ni2+ and formation of NiOOH, with a smaller subsequent peak at 1.32 V in the second cycle indicating a lesser degree of surface reconstruction involving Ni.
Unlike the single and binary metal-based phosphate catalysts, CoFeNiPi@ACC exhibited only the 1.33 V and 1.40 V anodic peaks in the first cycle, attributed to Ni2+ oxidation and no detectable peak for Co oxidation. This absence is consistent with previous studies showing that substituting Co with Ni or Fe facilitates Co2+ preoxidation.15,22 Our result was further corroborated by the presence of Co3+ in the XPS spectrum of Co 2p (Fig. 2a), confirming that Joule heating and the addition of Ni and Fe enhanced the preoxidation of Co2+. Similarly, the presence of smaller anodic peaks at 1.32 V and 1.36 V in the second cycle indicated reduced surface reconstruction.
Raman spectroscopy provides further insights into structural and compositional changes in the catalysts. Fig. 4b shows the Raman spectra of the amorphous CoFeNiPi@ACC in various states: as-made immediately right after Joule heating, upon activation, and after chronopotentiometry for 24 and 100 h. The Raman spectrum of the as-made CoFeNiPi@ACC (blue curve) exhibited a strong reflection at 1020 cm−1 and another broad, low-intensity peak around 630 cm−1, attributed to the symmetric stretching mode of PO4 and P–O–P symmetric-stretching mode, respectively.58 However, the activated CoFeNiPi@ACC sample (green curve) displayed a significantly broader band in the 400 to 700 cm−1 range, featuring component reflections corresponding to γ-CoOOH (461, 514 and 661 cm−1) and β-CoOOH (501 cm and 602 cm−1),59–61 FeOOH (696 cm−1),62 and NiOOH (475 and 557 cm−1).63–65 The presence of γ-CoOOH and β-CoOOH signals suggests some locally structured regions within the activated catalyst.
After chronopotentiometry for 24 and 100 h, both the CoFeNiPi@ACC-24 h and CoFeNiPi@ACC-100 h exhibited a smoother broad band in the 450 to 700 cm−1 range (see pink and violet curves in Fig. 4b). The Raman reflection at 1020 cm−1 had almost completely disappeared, indicating the breakdown of phosphate groups and surface restructuring into the respective oxyhydroxides. Fig. S13† shows that the Raman spectra of the unary and binary metal phosphates, CoPi and CoFePi, exhibited similar signals of γ-CoOOH, β-CoOOH and FeOOH. Notably, the signal intensity of γ-CoOOH at 661 cm−1 is significantly reduced in the CoFePi and CoFeNiPi spectra, as Fe has been reported to inhibit the transformation from the OER-active β-CoOOH to the less active γ-CoOOH structure.66
To determine the changes in elemental compositions and distributions, we conducted EDS line scan analysis on the amorphous CoFeNiPi@ACC catalyst in its Joule-heated as-made form and after chronopotentiometry at a constant current density of 10 mA cm−2 for 24 and 100 h. The EDS line scan of the as-made CoFeNiPi@ACC revealed that oxygen varied between 60 to 70 at%, and phosphorous between 20 to 30 at% at the surface (Fig. S14a†). After 24 h of chronopotentiometry, the oxygen content increased to 80 to 90 at%, and phosphorous decreased significantly to less than 10 at%; indicating surface reconstruction and oxyhydroxide formation (Fig. 4c). Similar chemical compositions were observed after 100 h of chronopotentiometry (Fig. S14b†). WAXS analysis confirmed that the catalysts remained amorphous (Fig. S15†), and SEM showed some surface roughening (Fig. S16†).
XPS analysis of the CoFeNiPi catalyst after 24 h of chronopotentiometry verified the formation of CoOOH, FeOOH and NiOOH species on the electrocatalyst surface. High-resolution XPS spectra displayed in Fig. 2 reveal notable shifts to lower binding energies in the component peaks of Co 2p, Ni 2p and Fe 2p. Specifically, peaks at 779.8 eV, 712.6 eV and 857.0 eV are assigned to Co 2p3/2 for Co3+–OOH (Fig. 2a), Fe 2p3/2 for Fe3+–OOH (Fig. 2b), and Ni 2p3/2 for Ni3+–OOH (Fig. 2c), respectively.23,67–69 In the O 1s spectrum, peaks at 532.6 eV, 531.0 eV and 528.9 eV correspond to C–O, M–OH, and M–O, respectively,70 indicating the predominant formation of active oxyhydroxides, NiOOH and CoOOH (Fig. 2d). In addition, a weak P–O signal at 133.2 eV was observed in the P 2p spectrum, likely from residual oxidized phosphorus species doped into the ACC fiber (Fig. 2e). Quantitative XPS analysis showed that the phosphorus content on the ACC fiber surface decreased significantly to ∼0.43 at% (Fig. 2f), contrasting with EDS results that showed a bulk phosphorous composition of ∼4.12 at% (Table S1†). This reduction in surface phosphate groups, accompanied by the formation of a substantial amount of active NiOOH and CoOOH, is attributed to dynamic surface reconstruction during OER.23 The changes in surface composition also suggest the release of some metal ions into the electrolyte during the reconstruction process. Previous studies have indicated that the phosphate group can promote the formation of active oxyhydroxides, NiOOH and CoOOH, consistent with our findings.71 It should be mentioned that FeOOH is unlikely to actively participate in the OER, as FePi@ACC is the least active catalyst among all unary metallic phosphates as shown in Fig. S6.† Further XPS analysis of CoFeNiPi catalyst after 100 h of chronopotentiometry confirmed that the signals of Co, Fe, Ni, O and P remained almost unchanged compared to the data of the 24 h chronopotentiometry sample (Fig. S17†).
Finally, we subjected the amorphous phosphate catalyst to linear sweep voltammetry experiments in tetramethylammonium hydroxide (TMAOH) electrolyte to determine the OER pathway. The TMA+ cation acts as a chemical probe to detect the presence of O22− species, which are considered key intermediates in the lattice oxygen mechanism (LOM) pathway.72 Fig. S18† shows the OER polarization curves and Tafel slopes of CoFeNiPi@ACC in 1.0 M TMAOH compared to that in 1 M KOH. Both CoFeNiPi@ACC samples exhibited highly similar OER polarization activities and kinetics, indicating negligible lattice oxygen contribution and suggesting that the OER pathway followed the adsorbate evolution mechanism (AEM).
4 Conclusions
In summary, we have developed a rapid and simple approach to fabricate a highly efficient and stable amorphous CoFeNi-based phosphate catalyst on activated carbon cloth via an ultrafast Joule heating process at moderate temperatures. The CoFeNiPi@ACC catalyst exhibited exceptional OER performance in 1.0 M KOH electrolyte, with a low overpotential of 235 mV at 10 mA cm−2 and a Tafel slope of 32.2 mV dec−1, indicating favorable reaction kinetics and robust electrocatalytic activity. Additionally, CoFeNiPi@ACC maintained a stable low overpotential during chronopotentiometry at 10 mA cm−2 for up to 100 h, highlighting its remarkable stability. Characterization methods, including CV, HAADF-STEM, EDS, XPS, and Raman spectroscopy, corroborated that Joule heating directly formed a multimetallic phosphate with an amorphous structure and promoted the preoxidation of active cation species, facilitating surface reconstruction and oxyhydroxide formation during electrolysis. This transient thermal processing approach can be readily adapted to create amorphous structures of various functional materials, including metal alloys,35,38 oxides,15,73,74 nitrides,75–77 sulfides,78,79 and other phosphorous-based structures on alternative 2D substrates,79–82 presenting significant potential for advanced energy storage and conversion applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Junhao Ma: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, visualization. Chonghan Xia: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, visualization. Teddy Salim: formal analysis, investigation, writing – review & editing, visualization. Yee Yan Tay: formal analysis, investigation, writing – review & editing. Lydia H. Wong: formal analysis, writing – review & editing. Kwan W. Tan: conceptualization, methodology, validation, formal analysis, data curation, writing – review & editing, visualization, supervision, project administration, funding acquisition.Conflicts of interest
A provisional patent application related to this research has been filed by Nanyang Technological University, Singapore.Acknowledgements
This work was supported by the Singapore Ministry of Education AcRF Tier 2 grants (MOE-T2EP50221-0017 and MOE-T2EP50120-0008). This work made use of research facilities at the Facility for Analysis, Characterization, Testing and Simulation (FACTS), Nanyang Technological University, Singapore. We also appreciate the kind experimental assistance and constructive discussions provided by Pio J. S. Buenconsejo, Tianshu Gao and Zihao Li of Nanyang Technological University, as well as Jianze Zhang of Nankai University.References
- M. D. Allendorf, V. Stavila, J. L. Snider, M. Witman, M. E. Bowden, K. Brooks, B. L. Tran and T. Autrey, Nat. Chem., 2022, 14, 1214–1223 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- H. Ishaq, I. Dincer and C. Crawford, Int. J. Hydrogen Energy, 2022, 47, 26238–26264 CrossRef CAS.
- Z.-Y. Yu, Y. Duan, X.-Y. Feng, X. Yu, M.-R. Gao and S.-H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- T. Guo, L. Li and Z. Wang, Adv. Energy Mater., 2022, 12, 2200827 CrossRef CAS.
- X. Zhang, Q. Hou, S. Cao, X. Lin, X. Chen, Z. Wang, S. Wei, S. Liu, F. Dai and X. Lu, Green Chem., 2023, 25, 7883–7903 RSC.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- M. D. Merrill and R. C. Dougherty, J. Phys. Chem. C, 2008, 112, 3655–3666 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- A. Sivanantham, P. Ganesan and S. Shanmugam, Adv. Funct. Mater., 2016, 26, 4661–4672 CrossRef CAS.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- H. Kim, J. Park, I. Park, K. Jin, S. E. Jerng, S. H. Kim, K. T. Nam and K. Kang, Nat. Commun., 2015, 6, 8253 CrossRef CAS PubMed.
- C.-Z. Yuan, Y.-F. Jiang, Z. Wang, X. Xie, Z.-K. Yang, A. B. Yousaf and A.-W. Xu, J. Mater. Chem. A, 2016, 4, 8155–8160 RSC.
- L. Yang, H. Ren, Q. Liang, K. N. Dinh, R. Dangol and Q. Yan, Small, 2020, 16, 1906766 CrossRef CAS PubMed.
- X. Liu, Q. Yin, C. Dai, G. Li, J. Lian, Y. Zhao, S. Yang and H. Li, ACS Sustainable Chem. Eng., 2021, 9, 5345–5355 CrossRef CAS.
- H. Qiao, X. Wang, Q. Dong, H. Zheng, G. Chen, M. Hong, C.-P. Yang, M. Wu, K. He and L. Hu, Nano Energy, 2021, 86, 106029 CrossRef CAS.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- P. Liao, J. A. Keith and E. A. Carter, J. Am. Chem. Soc., 2012, 134, 13296–13309 CrossRef CAS PubMed.
- C. Yang, O. Fontaine, J. Tarascon and A. Grimaud, Angew. Chem., Int. Ed., 2017, 56, 8652–8656 CrossRef CAS PubMed.
- K. Jin, J. Park, J. Lee, K. D. Yang, G. K. Pradhan, U. Sim, D. Jeong, H. L. Jang, S. Park, D. Kim, N.-E. Sung, S. H. Kim, S. Han and K. T. Nam, J. Am. Chem. Soc., 2014, 136, 7435–7443 CrossRef CAS PubMed.
- C. Yang, C. Laberty-Robert, D. Batuk, G. Cibin, A. V. Chadwick, V. Pimenta, W. Yin, L. Zhang, J.-M. Tarascon and A. Grimaud, J. Phys. Chem. Lett., 2017, 8, 3466–3472 CrossRef CAS PubMed.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dincă, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- G. Chen, Y. Zhu, H. M. Chen, Z. Hu, S.-F. Hung, N. Ma, J. Dai, H.-J. Lin, C.-T. Chen, W. Zhou and Z. Shao, Adv. Mater., 2019, 31, 1900883 CrossRef PubMed.
- Y.-J. Ye, N. Zhang and X.-X. Liu, J. Mater. Chem. A, 2017, 5, 24208–24216 RSC.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- L. Wang, G. L. Seah, Y. Li, W. H. Tu, W. Manalastas Jr, M. J.-H. Reavley, E. W. Corcoran Jr, A. K. Usadi, Z. Du, S. Madhavi, J. M. McConnachie, H. G. Ong and K. W. Tan, Adv. Mater. Interfaces, 2022, 9, 2200151 CrossRef CAS.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef CAS PubMed.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS PubMed.
- A. Abdelhafiz, B. Wang, A. R. Harutyunyan and J. Li, Adv. Energy Mater., 2022, 12, 2200742 CrossRef CAS.
- Y. Li, Y. Y. Tay, P. J. S. Buenconsejo, W. Manalastas Jr, W. H. Tu, H. K. Lim, T. Salim, M. O. Thompson, S. Madhavi, C. Y. Tay and K. W. Tan, Adv. Funct. Mater., 2023, 33, 2211279 CrossRef CAS.
- Y. Zou, L. Jing, J. Zhang, S. Luo, L. Wang, Y. Li, R. Goei, K. W. Tan and A. I. Y. Tok, J. Mater. Chem. A, 2024, 12, 5668–5678 RSC.
- L. Jing, Y. Zou, R. Goei, L. Wang, J. A. Ong, A. Kurkin, Y. Li, K. W. Tan and A. I. Y. Tok, Langmuir, 2023, 39, 3142–3150 CrossRef CAS PubMed.
- Y. Zou, L. Jing, L. Wang, H. T. Tan, R. Goei, S. K. R. Yong, A. J. Ong, K. W. Tan and A. I. Y. Tok, Adv. Eng. Mater., 2023, 25, 2201263 CrossRef CAS.
- C. Wang, W. Ping, Q. Bai, H. Cui, R. Hensleigh, R. Wang, A. H. Brozena, Z. Xu, J. Dai, Y. Pei, C. Zheng, G. Pastel, J. Gao, X. Wang, H. Wang, J.-C. Zhao, B. Yang, X. (Rayne) Zheng, J. Luo, Y. Mo, B. Dunn and L. Hu, Science, 2020, 368, 521–526 CrossRef CAS PubMed.
- D. X. Luong, K. V. Bets, W. A. Algozeeb, M. G. Stanford, C. Kittrell, W. Chen, R. V. Salvatierra, M. Ren, E. A. McHugh, P. A. Advincula, Z. Wang, M. Bhatt, H. Guo, V. Mancevski, R. Shahsavari, B. I. Yakobson and J. M. Tour, Nature, 2020, 577, 647–651 CrossRef CAS PubMed.
- F. N. I. Sari and J. Ting, ChemSusChem, 2018, 11, 897–906 CrossRef CAS PubMed.
- J. Xu, Q. Li, D. Sui, W. Jiang, F. Liu, X. Gu, Y. Zhao, P. Ying, L. Mao, X. Cai and J. Zhang, Nanomaterials, 2023, 13, 420 CrossRef CAS PubMed.
- R. Zhang, G. Van Straaten, V. Di Palma, G. Zafeiropoulos, M. C. M. Van De Sanden, W. M. M. Kessels, M. N. Tsampas and M. Creatore, ACS Catal., 2021, 11, 2774–2785 CrossRef CAS PubMed.
- K. P. Singh, E. J. Bae and J.-S. Yu, J. Am. Chem. Soc., 2015, 137, 3165–3168 CrossRef CAS PubMed.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes Carbon Nanostruct., 2020, 28, 1048–1058 CrossRef CAS.
- Y. Luo, L. Tang, U. Khan, Q. Yu, H.-M. Cheng, X. Zou and B. Liu, Nat. Commun., 2019, 10, 269 CrossRef PubMed.
- X. Wang, C. Xing, Z. Liang, P. Guardia, X. Han, Y. Zuo, J. Llorca, J. Arbiol, J. Li and A. Cabot, J. Mater. Chem. A, 2022, 10, 3659–3666 RSC.
- C. Lv, S. Xu, Q. Yang, Z. Huang and C. Zhang, J. Mater. Chem. A, 2019, 7, 12457–12467 RSC.
- T. I. Singh, A. Maibam, D. C. Cha, S. Yoo, R. Babarao, S. U. Lee and S. Lee, Adv. Sci., 2022, 9, 2201311 CrossRef CAS PubMed.
- Q. P. Ngo, T. T. Nguyen, M. Singh, R. Balaji, N. H. Kim and J. H. Lee, Appl. Catal., B, 2023, 331, 122674 CrossRef CAS.
- S. Li, Z. Li, R. Ma, C. Gao, L. Liu, L. Hu, J. Zhu, T. Sun, Y. Tang, D. Liu and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 3773–3780 CrossRef CAS PubMed.
- P. W. Menezes, C. Panda, C. Walter, M. Schwarze and M. Driess, Adv. Funct. Mater., 2019, 29, 1808632 CrossRef.
- Y. Li, Z. Wang, J. Hu, S. Li, Y. Du, X. Han and P. Xu, Adv. Funct. Mater., 2020, 30, 1910498 CrossRef CAS.
- J. Zhang, H. Yu, J. Yang, X. Zhu, M. Hu and J. Yang, J. Alloys Compd., 2022, 924, 166613 CrossRef CAS.
- L. Zhang and R. K. Brow, J. Am. Ceram. Soc., 2011, 94, 3123–3130 CrossRef CAS.
- Y. Li, J. Cai, J. Zhang, Z. Chen, G. Wang, Q. Chen and M. Chen, Adv. Energy Mater., 2023, 13, 2204114 CrossRef CAS.
- S. Wang, Q. Jiang, S. Ju, C.-S. Hsu, H. M. Chen, D. Zhang and F. Song, Nat. Commun., 2022, 13, 6650 CrossRef CAS PubMed.
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- B. Wu, S. Gong, Y. Lin, T. Li, A. Chen, M. Zhao, Q. Zhang and L. Chen, Adv. Mater., 2022, 34, 2108619 CrossRef CAS PubMed.
- Z. Qiu, Y. Ma and T. Edvinsson, Nano Energy, 2019, 66, 104118 CrossRef CAS.
- L. Bai, S. Lee and X. Hu, Angew. Chem., Int. Ed., 2021, 60, 3095–3103 CrossRef CAS PubMed.
- J. Huang, Y. Li, Y. Zhang, G. Rao, C. Wu, Y. Hu, X. Wang, R. Lu, Y. Li and J. Xiong, Angew. Chem., Int. Ed., 2019, 58, 17458–17464 CrossRef CAS PubMed.
- L. Ge, H. Yang, J. Guan, B. Ouyang, Q. Yu, H. Li and Y. Deng, Inorg. Chem., 2023, 62, 15664–15672 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. St. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- T. Yang, L. Pei, S. Yan, Z. Yu, T. Yu and Z. Zou, Dalton Trans., 2019, 48, 11927–11933 RSC.
- M. Ma, L. Cao, H. Qi, K. Yao, J. Huang, Z. Xu, S. Chen and J. Li, J. Alloys Compd., 2019, 808, 151657 CrossRef CAS.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- Y. Li, J. Liu, S. Li and S. Peng, ACS Catal., 2024, 14, 4807–4819 CrossRef CAS.
- C. Yang, O. Fontaine, J.-M. Tarascon and A. Grimaud, Angew. Chem., Int. Ed., 2017, 56, 8652–8656 CrossRef CAS PubMed.
- P. Edalati, Q. Wang, H. Razavi-Khosroshahi, M. Fuji, T. Ishihara and K. Edalati, J. Mater. Chem. A, 2020, 8, 3814–3821 RSC.
- M. V. Kante, M. L. Weber, S. Ni, I. C. G. van den Bosch, E. van der Minne, L. Heymann, L. J. Falling, N. Gauquelin, M. Tsvetanova, D. M. Cunha, G. Koster, F. Gunkel, S. Nemšák, H. Hahn, L. Velasco Estrada and C. Baeumer, ACS Nano, 2023, 17, 5329–5339 CrossRef CAS PubMed.
- T. Jin, X. Sang, R. R. Unocic, R. T. Kinch, X. Liu, J. Hu, H. Liu and S. Dai, Adv. Mater., 2018, 30, 1707512 CrossRef PubMed.
- Y. Li, R. Horia, W. X. Tan, N. Larbaram, W. A. Sasangka, W. Manalastas, S. Madhavi and K. W. Tan, Langmuir, 2020, 36, 10803–10810 CrossRef CAS PubMed.
- P. Edalati, X.-F. Shen, M. Watanabe, T. Ishihara, M. Arita, M. Fuji and K. Edalati, J. Mater. Chem. A, 2021, 9, 15076–15086 RSC.
- T. X. Nguyen, Y.-H. Su, C.-C. Lin and J.-M. Ting, Adv. Funct. Mater., 2021, 31, 2106229 CrossRef CAS.
- R. Mohili, N. R. Hemanth, H. Jin, K. Lee and N. Chaudhari, J. Mater. Chem. A, 2023, 11, 10463–10472 RSC.
- C. Yang, M. Cui, N. Li, Z. Liu, S. Hwang, H. Xie, X. Wang, Y. Kuang, M. Jiao, D. Su and L. Hu, Nano Energy, 2019, 63, 103855 CrossRef CAS.
- J. Zhao, N. Liao and J. Luo, J. Mater. Chem. A, 2023, 11, 9682–9690 RSC.
- Y. Ding, K.-W. Cao, J.-W. He, F.-M. Li, H. Huang, P. Chen and Y. Chen, Chin. J. Catal., 2022, 43, 1535–1543 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03130j |
| ‡ These authors contribute equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |