
Programmable assembly/disassembly of metal–organic cage integrated 2D nanosheets†‡
Vellaiyadevan
Sivalingam
 and
Dillip Kumar
Chand
*
and
Dillip Kumar
Chand
*
IoE Center of Molecular Architecture, Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: dillip@zmail.iitm.ac.in
First published on 12th February 2025
Abstract
This work demonstrates a stepwise assembly process to prepare [Pd(Pd2L4)]n-type 2D nanosheets using Pd(II) and a stoichiometry-controlled metal–organic cage of Pd2L4 type. A convenient design strategy for dynamic interconversion between cage-based 2D nanosheets and well-defined monomeric cages is achieved.
A wide variety of nanostructured coordination assemblies were prepared to exploit their unique structural and functional properties. Among the nanostructured assemblies, coordination cages possessing confined nanospace were self-assembled through coordination-driven self-assembly of designer ligands and metal ions, that continue to open new possibilities.1 Coordination cage-based extended assemblies were prepared by aggregating the cage molecules via covalent crosslinking or non-covalent interactions.2 In particular, metal–ligand coordination was successfully employed to crosslink the coordination cages to construct dynamic supramolecular architectures owing to their reversible nature. Most coordination cage-based extended assemblies often result in three-dimensional (3D) nanostructures, including covalently crosslinked coordination cages containing well-defined cavity environments.3 Though 2D assemblies were observed during the self-assembly process of metallocages,4 the coordination cage integrated two-dimensional (2D) nanosheet assemblies that contain tailor-made confined nanospace remains a formidable challenge. Towards this, we envisioned employing an exohedral pyridine-functionalized coordination cage as a building block and then interconnecting the monomeric cage units utilizing the reversible metal–ligand coordination bond to construct two-dimensional nanoarchitectures that consist of a well-established cage cavity.
To construct the coordination cage-based 2D nanosheets, we presume that a highly symmetrical M2L4-type cage as a building block unit could be a better choice owing to its backbone functionalization ability5 to integrate secondary non-covalent interaction sites. Reversible interconversion between a well-defined monomeric cage and 2D nanosheets by utilizing the labile nature of the Pd(II)-ligand bond would be fascinating.
Herein, we report the preparation of a stoichiometry-controlled Pd2L4 cage, which bears additional non-coordinated pyridine units available further for Pd(II)-driven coordination polymerization. The monomeric Pd2L4 cage (nitrate-encapsulated) could bind chloride and bromide quantitatively. The monomeric Pd2L4 cage could be reversibly interconverted with [Pd(Pd2L4)]n-type 2D nanosheets by stoichiometry control.
At first, the design of a monomeric Pd2L4 cage with additional coordination sites available for secondary coordination and the preparation of a “stoichiometry-controlled, thermodynamically stable” cage is crucial for this study. Recently, we investigated the template-free Pd2L4 cages that bind halides and nitrate with estimated >105 M−1 affinity in DMSO and micromolar/nanomolar affinity measured for chloride in aqueous/aqueous-DMSO solutions, respectively.6 Furthermore, we envisage that embedding an exohedral pyridine unit (in a two-dimensionally constrained manner) could potentially transform the high-affinity halide-binding Pd2L4 cage into a 2D material possessing well-defined cavity environments upon the addition of Pd(II). Also, templating anions (e.g., nitrate, chloride, bromide, etc.) can provide additional stability to the cage framework in the proposed 2D nanosheet assembly. Thus, we first decided to integrate an exohedral 4-pyridyl group in the backbone, where the pyridine coordination vectors face outwards from the cage. With this rigid design, we expect a cage with additional diverging coordination sites that are precisely placed ∼90° from each other in a 2D array (Fig. 1A).
 | ||
| Fig. 1 (A) Schematic for the stoichiometry-controlled synthesis of a building block cage, 1a; (B) 1H NMR spectra of (i) ligand L1, (ii) isolated NO3⊂1·3NO3, 1a, and (iii)/(iv) halide-encapsulated cages obtained upon addition of 1 equiv. of TBACl/TBABr to 1a. | ||
First, we synthesized ligand L1 (4′-(pyridin-4-yl)-3,2′:6′,3′′-terpyridine) in a one-pot synthesis from pyridine-4-carboxaldehyde and 3-acetylpyridine.7 The stoichiometry-controlled synthesis of NO3⊂1·3NO3, 1a was carried out by treating 4 equiv. of L1 and 2 equiv. of Pd(NO3)2 in DMSO-d6 at 90 °C for 2 h. The 1H NMR spectrum showed a single set of signals consistent with discrete assembly. All the 1H NMR signals corresponding to a 3,2′:6′,3′′-terpyridine unit displayed downfield shift upon complexation, consistent with the formation of stoichiometry-controlled Pd2L4-type assembly (Fig. 1B(ii)). In contrast, the 1H NMR signals corresponding to the 4-pyridyl unit showed a slight upfield shift, indicating the presence of an uncoordinated 4-pyridyl unit. High-resolution ESI mass spectral (HR-ESI-MS) analysis unambiguously confirmed the formation of Pd2L4-type assembly exhibiting the presence of a series of intense signals for [1·4NO3–nNO3]n+ (n = 1–3) at m/z = 1640.254, 789.133 and 505.425 (Fig. S16, ESI‡). However, attempts to prepare ‘empty’ cage 1·4BF4 resulted in the partial generation of F⊂1·3BF4 (∼21%, after 2 hours at 90 °C upon complexation) owing to the cleavage of the B−F bond (Fig. S31, ESI‡). This finding is consistent with our earlier observation, where a slight excess of ligand/DMAP to a strongly halide-binding Pd2L4 cage with BF4− as a counteranion induced the cleavage of the B−F bond in DMSO-d6.6 In this case, an extra pyridine unit is present in the backbone of the cage-forming 3,2′:6′,3′′-terpyridine moiety.
The central cavity of cage 1a was pre-occupied by NO3−, since Pd(NO3)2 was used as a Pd(II) source. Hence, we decided to explore whether the cage cavity is anion-exchangeable with halides (i.e., F−, Cl−, Br− and I−) in DMSO-d6. While cage 1a was associated with four NO3− ions, one can infer that there will be a competition between halides and nitrates. Addition of up to 1 equiv. of tetrabutylammonium chloride (TBACl) to the DMSO-d6 solution of 1a (i.e., NO3⊂1·3NO3) resulted in the appearance of a corresponding chloride-encapsulated cage, Cl⊂1·3NO3, displaying slow exchange with 1a in the NMR timescale. The 1H NMR titration revealed quantitative binding of Cl− even in the presence of 4 equiv. of NO3−. Similar observations in the titration of TBABr confirm ≥105 M−1 affinity for both chloride and bromide in DMSO (Fig. 1B(iii) and (iv)). Furthermore, we titrated TBAF/TBAI with cage 1a for up to 1.5 equiv. with respect to the cage. Upon 1 equiv. of F−/I− addition, 52%/72% (calculated with respect to L1) of NO3⊂1·3NO3 was converted to F⊂1·3NO3 and I⊂1·3NO3, respectively. 1H NMR signals corresponding to free L1 were also found (Fig. S27 and S30, ESI‡). From these observations, we infer that the binding affinity of cage 1a towards halides should be Cl−/Br− > I− > F−.
While attempting to synthesize cage 1a at higher concentrations (≥10 mM of Pd(II)), we observed gel formation even when the ratio between L1 and Pd(NO3)2 was maintained at 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in DMSO-d6. However, when we heated the metallogel at 70 °C for 3 days, it transformed to a solution phase upon the formation of 1a. This observation indicates that metallogel formation is a kinetic assembly due to the random coordination-driven polymerization of L1. Next, we mixed L1 and Pd(NO3)2 in a 4:3 ratio (to ensure that a sufficient quantity of Pd(II) was present for the complexation of exohedral pyridines) at 15 mM of Pd(II) in DMSO, which resulted in metallogel-1 (MG-1) by direct assembly approach. We observed Pd(II)-driven metallogel formation at conc. as low as 8 mM of Pd(II) upon thermal annealing (by heating at 70 °C for 1 h, followed by standing at room temperature for 2 h).
:2 in DMSO-d6. However, when we heated the metallogel at 70 °C for 3 days, it transformed to a solution phase upon the formation of 1a. This observation indicates that metallogel formation is a kinetic assembly due to the random coordination-driven polymerization of L1. Next, we mixed L1 and Pd(NO3)2 in a 4:3 ratio (to ensure that a sufficient quantity of Pd(II) was present for the complexation of exohedral pyridines) at 15 mM of Pd(II) in DMSO, which resulted in metallogel-1 (MG-1) by direct assembly approach. We observed Pd(II)-driven metallogel formation at conc. as low as 8 mM of Pd(II) upon thermal annealing (by heating at 70 °C for 1 h, followed by standing at room temperature for 2 h).
Next, we tested the gelation ability of a stoichiometrically pre-formed Pd2L4 cage, 1a, by the addition of up to 1 equiv. of Pd(NO3)2 with respect to the cage in DMSO-d6. This stepwise assembly approach resulted in the formation of metallogel-2 (referred as MG-2). All the 1H NMR signals disappeared upon the addition of 1 equiv. of Pd(NO3)2, and this is suggestive of the formation of an oligomeric assembly i.e., coordination cage-based 2D nanosheets (Fig. 2A).
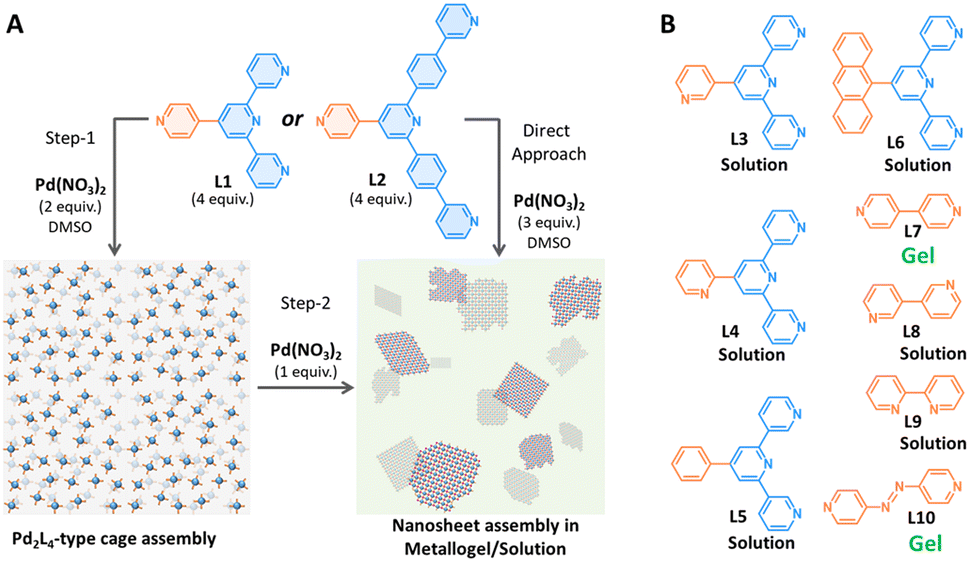 | ||
| Fig. 2 (A) Schematic illustration for nanosheet assembly by a stepwise or direct assembly approach; (B) chemical structures of other ligands (L3–L10) used in gelation study. | ||
The gelation ability of ligands upon the addition of Pd(II) could depend on multiple factors, e.g., counteranion, concentration, solvent, etc. that needs thorough optimization.8 Hence, we began to study the influence of the counteranion in metallogel formation in DMSO. We used a variety of freshly prepared PdX2 salts (where X = TsO−, BF4−, PF6−, TfO−, MsO− and ClO4−) in combination with L1 in 3:4 ratio. The treatment of L1 with Pd(PF6)2 (at 15 mM of Pd(II)) resulted in the formation of a gel. However, other samples remained in the solution phase for 24 hours at room temperature or after thermal annealing. This study confirmed the definitive role of the counteranion in gelation. Also, we observed disulfonate-induced gelation for the solution containing nanosheet assembly, probably due to the aggregation of 2D nanosheets into 3D assembly (Fig. S32, ESI‡). Subsequently, we attempted post-assembly gelation of cage 1a in different solvents like H2O, CH3CN, methanol, acetone and THF. However, the insoluble nature of 1a precluded the possibility of studying the gelation behaviour in different solvents. Later, the dissolution of cage 1a in a CH3CN:H2O (1:1) mixed solvent system, followed by the addition of Pd(NO3)2, lead to gel formation within 15 minutes.
Since we suspect Pd(II)-driven coordination polymerization as one of the prerequisite conditions for gelation in addition to the factors studied above, we modified the framework of L1 by incorporating a phenyl spacer between the core and terminal pyridine units, where the designed ligand L2 was synthesized in two steps (Scheme S1, ESI‡). Then, we treated 4 equiv. of L2 with 2 equiv. of Pd(NO3)2 in DMSO-d6. This invariably resulted in a single set of signals in 1H NMR spectroscopy after 2 hours at room temperature. 1H NMR spectral analysis indicated assembly similar to that of cage 1a, and it was termed as 2·4NO3, 2a (Scheme S4 and Fig. S15, ESI‡). Upon successful synthesis of cage 2a, we added calc. 1 equiv. of Pd(NO3)2 into the DMSO-d6 solution of 2a that caused the gel formation. Consequently, all the 1H NMR signals corresponding to the cage disappeared completely as observed earlier in the case of the stepwise assembly via cage 1a (Fig. S38, ESI‡).
Furthermore, we were also curious to understand the prerequisite conditions for the Pd(II)-driven metallogel formation using a series of pyridine-based ligands L3–L10 (Fig. 2B). Treatment of L3 or L4 (regioisomers of L1) with Pd(NO3)2 in 4:3 ratio could not form a gel in DMSO at 15 mM of Pd(II). The ligands L3 or L4 are 3-pyridyl/2-pyridyl appended at the backbone of the ligand structure, while the gel-forming L1 is 4-pyridyl appended. Subsequently, we decided to test the ligand systems consisting of π-surface (exohedrally) i.e., phenyl and 9-anthracenyl. Reacting 2 equiv. of Pd(NO3)2 with 4 equiv. of ligands L5 or L6 in DMSO at 15 mM conc. of Pd(II) afforded a clear solution only. Next, we used 4,4′-bipyridine (L7) to test the gelation ability. Though L7 forms a molecular square or a mixture of molecular square and triangle upon reaction with suitable cis-protected Pd(II),9 it was expected to undergo oligomeric assembly when reacted with a simple Pd(II).10 Mixing L7 with Pd(NO3)2 (at 15 mM) in 2:1 ratio in DMSO invariably led to gel formation. However, gel formation was not observed when L8 or L9 (regioisomers of L7) was mixed with Pd(NO3)2 (at 15 mM) in DMSO. Thermal annealing or allowing the solution to stand longer (up to 48 hours) did not alter the outcome. Since L7 possessing coordination vectors of 180° resulted in gelation upon complexation with Pd(II), we synthesized ligand L10 to examine the gelation ability. On treating L10 with Pd(NO3)2 (at 15 mM) in DMSO, it instantly formed a dark red gel. These investigations strongly suggest that the Pd(II)-driven coordination polymerization could be one of the important prerequisites for metallogel formation.
To study the mechanical properties of MG-2, we sought to perform strain and frequency sweep experiments using the thermally annealed gel that was prepared at a 15 mM concentration of Pd(II). Rheology experiments were performed to measure storage modulus (G′) and loss modulus (G′′) as a function of strain/frequency. The solid-like behaviour of the metallogel was retained at lower strain percentages, where the value of G′ is higher than G′′. The yield strain was measured to be around 88% (G′ = G′′). At higher strain percentages i.e. above yield strain (G′′ exceeds G′), the sample displayed sol–gel transition (Fig. 3A). Measurement of G′ and G′′ as a function of frequency at constant strain (∼5%) revealed that the average G′ (5618 Pa) was about one order of magnitude higher than G′′ (421 Pa). The higher ΔG (i.e., G′ − G′′) suggests that the metallogel formed by combining 1a and Pd(NO3)2 in DMSO is highly stable (Fig. 3B).
 | ||
| Fig. 3 (A) Strain sweep measurements (at a constant frequency of 1 rad s−1); (B) frequency sweep measurements (at a constant strain of 5%); (C)/(D) HRTEM of metallogel/solution phase nanosheet obtained through Pd(II)-directed coordination polymerization of cage 1a (at 15 mM/5 mM conc. of Pd(II)). | ||
Then, the thermally annealed gel samples were subjected to scanning electron microscopy (SEM) analysis. The microscopic images displayed less ordered assembly for MG-1, while the stepwise assembled MG-2 exhibited relatively well-ordered nanosheet formation (Fig. S33, ESI‡). Well-ordered nanosheet domains were also observed in high-resolution transmission electron microscopy (HRTEM) for MG-2. The acetonitrile dispersion of thermally annealed MG-2 (prepared at a 15 mM concentration of Pd(II)) was used for HRTEM analysis. The HRTEM images display the single/multi-layer assembly with steps as part of the nanosheet domains (Fig. 3C).11 Subsequently, we treated cage 1a with Pd(NO3)2 at 5 mM of Pd(II) to prepare the nanosheet in DMSO solution. Then, the DMSO solution in which the Pd(II)-driven coordination polymerization was supposed to have taken place was used for the HRTEM analysis. We have obtained similar results as the metallogel sample, containing the multilayer nanosheet assembly (Fig. 3D).
Having established the formation of nanosheet assembly in solution, we envisioned that the labile nature of the Pd−Npyridine bond could be utilised for the reversible interconversion between the metal–organic cage and 2D nanosheets. The gradual addition of up to 1 equiv. of Pd(NO3)2 in 2 mM DMSO-d6 solution of cage 1a resulted in the formation of an oligomeric assembly as indicated by 1H NMR spectral measurements. For the addition of 0.4 and 0.8 equiv. of Pd(NO3)2, we could clearly observe very broad 1H NMR signals (Fig. 4) adjacent to the sharp signals of the corresponding NO3−-bound cage, 1a. Also, the broad signal corresponding to the inwards pointed pyridine-αCH indicates that the anion-bound cage assembly is, in fact, intact in the oligomeric assembly. Hence, it is reasonable to assume that the broadened 1H NMR signals/baseline spectrum that appeared for the sub-stoichiometric addition of Pd(II) is most likely due to the nanosheet assembly in DMSO-d6. To verify the reversible nature, we added an additional 0.5 equiv. of L1 (required to seize the Pd(II), previously bound to exohedral 4-pyridines) to the DMSO-d6 solution containing the oligomerized sample and heated at 90 °C for 30 minutes. Ultimately, this led to the regeneration of cage 1a, as confirmed by using 1H NMR spectroscopy. Thus, the ability to reversibly interconvert between the metal–organic cage and 2D nanosheet was successfully achieved by exploiting the reversibility of the Pd−Npyridine bond. Alternatively, the 2D nanosheet could be interconverted with the metal–organic cage using 4-(dimethylamino)pyridine and p-TsOH, consecutively (Fig. S37, ESI‡).
 | ||
| Fig. 4 1H NMR spectra for the interconversion between cage 1a and oligomeric/nanosheet assembly in DMSO-d6 solution (at 2 mM conc. of the cage). | ||
In conclusion, we successfully synthesized a coordination cage-based nanosheet that can be reversibly interconverted with a Pd2L4-type cage bearing secondary coordination sites. Microscopic analysis using TEM revealed multilayer nanosheet assembly in both gel and solution. Pd(II)-driven coordination polymerisation was found to play a vital role in the metallogel assembly. This convenient design strategy demonstrating the programmable assembly/disassembly of cage-based 2D materials could open new possibilities for the development of functional 2D materials.
D. K. C. thanks the Science and Engineering Research Board (SERB), Government of India (CRG/2022/004413) and Institute of Eminence program, IIT Madras offered by Ministry of Education, Government of India (IoE Phase-II) for financial support.
Data availability
The data supporting this article have been included as part of the ESI.‡Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111(11), 6810–6918 CrossRef CAS PubMed; (c) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- (a) J. Liu, Z. Wang, P. Cheng, M. J. Zaworotko, Y. Chen and Z. Zhang, Nat. Rev. Chem., 2022, 6, 339–356 CrossRef PubMed; (b) Y. Gu, E. A. Alt, H. Wang, X. Li, A. P. Willard and J. A. Johnson, Nature, 2018, 560, 65–69 CrossRef CAS PubMed; (c) M. L. Schneider, J. A. Campbell, A. D. Slattery and W. M. Bloch, Chem. Commun., 2022, 58, 12122–12125 RSC; (d) J. A. Foster, R. M. Parker, A. M. Belenguer, N. Kishi, S. Sutton, C. Abell and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 9722–9729 CrossRef CAS PubMed.
- (a) E. Sanchez-Gonzalez, M. Y. Tsang, J. Troyano, G. A. Craig and S. Furukawa, Chem. Soc. Rev., 2022, 51, 4876–4889 RSC; (b) Y. Wang, Y. Gu, E. G. Keeler, J. V. Park, R. G. Griffin and J. A. Johnson, Angew. Chem., Int. Ed., 2017, 56, 188–192 CrossRef CAS PubMed; (c) I. Jahovic, Y.-Q. Zou, S. Adorinni, J. R. Nitschke and S. Marchesan, Matter, 2021, 4, 2123–2140 CrossRef CAS; (d) C. Hu and K. Severin, Angew. Chem., Int. Ed., 2024, 63, e20240383 Search PubMed.
- (a) L. H. Foianesi-Takeshige, S. Takahashi, T. Tateishi, R. Sekine, A. Okazawa, W. Zhu, T. Kojima, K. Harano, E. Nakamura, H. Sato and S. Hiraoka, Commun. Chem., 2019, 2, 128 CrossRef; (b) S. Kai, T. Tateishi, T. Kojima, S. Takahashi and S. Hiraoka, Inorg. Chem., 2018, 57, 13083–13086 CrossRef CAS PubMed.
- (a) J. E. M. Lewis, Chem. Commun., 2022, 58, 13873–13886 RSC; (b) J. E. M. Lewis, C. J. McAdam, M. G. Gardiner and J. D. Crowley, Chem. Commun., 2013, 49, 3398–3400 RSC; (c) J. E. M. Lewis, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Sci., 2014, 5, 1833–1843 RSC; (d) S.-P. Zheng, Y.-W. Xu, P.-Y. Su, C.-H. Liu, Y.-H. Huang, Y.-L. Lu, Z.-W. Wei, Z. Jiao, H.-S. Xu and C.-Y. Su, Chin. Chem. Lett., 2024, 35, 108477 CrossRef CAS; (e) Z. T. Avery, M. G. Gardiner and D. Preston, Angew. Chem., Int. Ed., 2024, e202418079 Search PubMed.
- V. Sivalingam, S. Krishnaswamy and D. K. Chand, Chem. – Eur. J., 2023, 29, e202300891 CrossRef CAS PubMed.
- J. Wang and G. S. Hanan, Synlett, 2005, 1251–1254 CAS.
- S. Ganta and D. K. Chand, Inorg. Chem., 2018, 57, 3634–3645 CrossRef CAS PubMed.
- M. Fujita, O. Sasaki, T. Mitsuhashi, T. Fujita, J. Yazaki, K. Yamaguchi and K. Ogura, Chem. Commun., 1996, 1535–1536 RSC.
- S. Prusty, K. Yazaki, M. Yoshizawa and D. K. Chand, Chem. – Eur. J., 2017, 23, 12456–12461 CrossRef CAS PubMed.
- D. P. August, R. A. W. Dryfe, S. J. Haigh, P. R. C. Kent, D. A. Leigh, J.-F. Lemonnier, Z. Li, C. A. Muryn, L. I. Palmer, Y. Song, G. F. S. Whitehead and R. J. Young, Nature, 2020, 588, 429–435 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. G. Ranga Rao on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06557c |
| This journal is © The Royal Society of Chemistry 2025 |