
Donor–acceptor semiconducting polymers as superior photocatalysts in the homogeneous phase and recycled via an antisolvent precipitation strategy†
Sibu
Manna
 ,
Krishnendu
Maity
,
Flora
Banerjee
,
Soumitra
Sau
and
Suman Kalyan
Samanta
*
,
Krishnendu
Maity
,
Flora
Banerjee
,
Soumitra
Sau
and
Suman Kalyan
Samanta
*
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: sksamanta@chem.iitkgp.ac.in
First published on 26th May 2025
Abstract
Donor–acceptor conjugated polymer PBDT-AQ outperformed PBDT-AM in homogeneous photocatalytic oxidation of phenyl methyl sulfides and phenylboronic acids with excellent yields due to superior oxygen adsorption and lower charge transfer resistance, along with excellent solubility in suitable solvents, strong visible-light absorption, optimal band gaps, and appropriate LUMO levels, and can be recycled over five times by heterogenization via an antisolvent precipitation method.
Traditional organic transformations often require harsh conditions and high energy consumption, which could pose environmental risks. To address these challenges, green and sustainable alternatives have gained interest, and therefore, photocatalytic organic transformation is emerging as a promising approach.1 By utilizing renewable solar energy, these reactions enable efficient and atom-economic synthesis while reducing the environmental impact.2–4 Extensive studies have explored the utility of homogeneous photocatalysts, such as organic dyes and metal-based complexes, demonstrating excellent performance.5,6 However, to overcome the drawbacks associated with their limited scope of reuse, conjugated organic polymers (CPs), graphitic carbon nitride (g-C3N4), conjugated microporous polymers (CMPs), and covalent organic frameworks (COFs) have been introduced as heterogeneous photocatalysts.7–9 Despite the advantages of reusability, their insolubility in organic solvents limits their use in homogeneous phase reactions, in which a higher catalytic activity is often observed due to a better substrate interaction with the active sites of the catalysts.10 This has led to a growing interest in designing solution-processable CPs for homogeneous catalysis in ‘good’ solvents, and their insolubility in a ‘poor’ solvent like methanol allows easy recovery via solvent-induced precipitation. Additionally, donor–acceptor (D–A) conjugated polymers offer superior optical and electronic properties that are suitable for various optoelectronic device applications.11,12 Their visible-light absorption, tunable band gap, photochemical stability, long exciton lifetime, and efficient charge separation make them highly effective photocatalysts.13 However, only a few reports have demonstrated the photocatalytic potential of solution-processable D–A CPs in homogeneous phase reactions, where they enable enhanced catalytic activity and easy recovery in the heterogeneous phase.14
Herein, we present a sustainable approach combining the enhanced photocatalytic activity in homogeneous phase reactions and the easy recovery in the heterogeneous phase via the antisolvent precipitation method. Anthraquinone (AQ) and its dimalononitrile derivative (AM) are incorporated as acceptors of varying strengths within the benzodithiophene (BDT)-based D–A linear, soluble copolymers that are capable of broad visible-light absorption and D–A charge separation. These polymers enable reactive oxygen species (ROS)-mediated oxidative hydroxylation of phenylboronic acids and the photooxidation of toxic phenyl methyl sulfides to less toxic sulfoxides with high yields over multiple cycles.
The D–A copolymers PBDT-AQ and PBDT-AM were synthesized via Stille polymerization between BDT-(SnBu3)2 with AQ-Br2 and AM-Br2,15 respectively (Fig. 1a, Experimental section, ESI†).16 Both polymers exhibited good solubility in suitable organic solvents such as chloroform, dichloromethane (DCM), chlorobenzene, o-dichlorobenzene, toluene, and tetrahydrofuran (THF), making them ideal for homogeneous catalysis. However, they were insoluble in methanol, acetone, ethylacetate, and acetonitrile, allowing their easy recovery by precipitation upon adding these solvents (Fig. 1b).
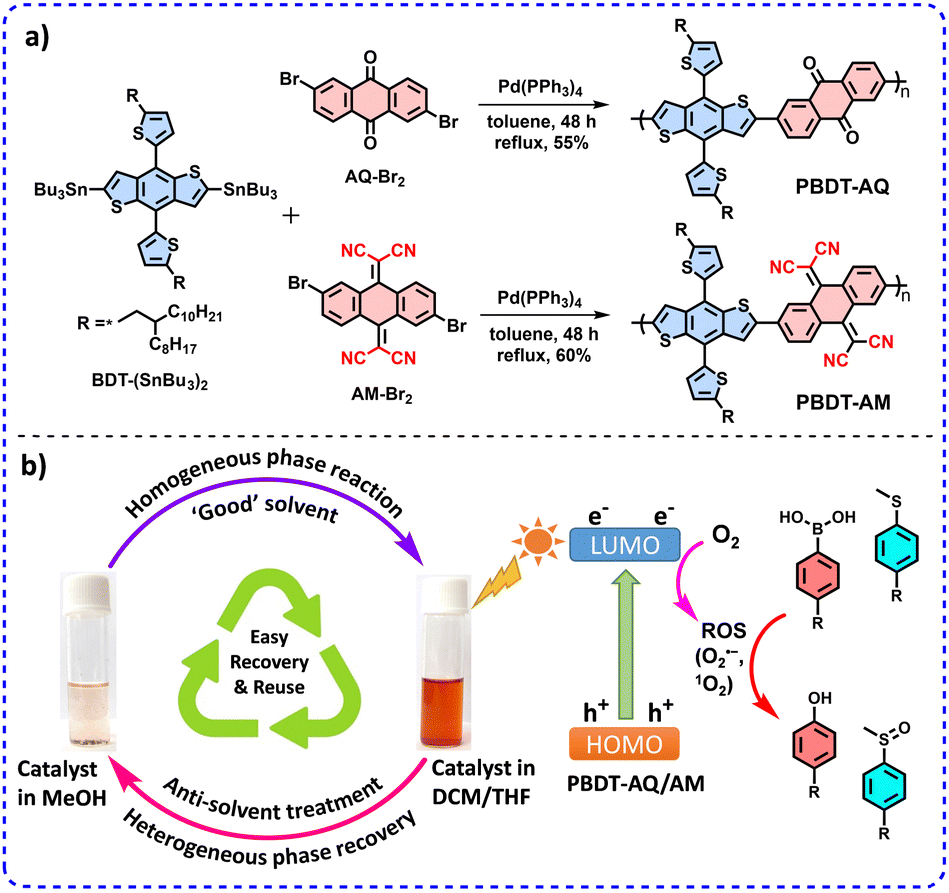 | ||
| Fig. 1 (a) Synthetic scheme of PBDT-AQ and PBDT-AM. (b) Schematic representation of photocatalysis in the homogeneous phase and recovery via the anti-solvent treatment precipitation method. | ||
The chemical structures of both polymers were characterized using 1H NMR and FT-IR spectroscopy (Fig. S1–S3, ESI†). Additionally, X-ray photoelectron spectroscopy (XPS) analysis of PBDT-AQ and PBDT-AM upon deconvolution displayed C 1s, N 1s, O 1s, and S 2p spectra, confirming the chemical composition and oxidation states of the elements present in the polymers (Fig. 2a and Fig. S4, ESI†). Notably, the deconvoluted C 1s spectrum of PBDT-AQ revealed three distinct peaks at 283.41 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 284.31 eV (C–C), and 286.03 eV (C–S and CO), confirming the coexistence of BDT and AQ within the polymer. The gel permeation chromatography (GPC) measurements provided the weight-average molecular weight (Mw) and polydispersity index (PDI) values, which are 11.4 kDa and 1.81 for PBDT-AQ, and 20.7 kDa and 2.72 for PBDT-AM, respectively, indicating their sufficient polymer growth (Table S1, ESI†). Both polymers demonstrated excellent thermal stability, up to 392 °C for PBDT-AQ and 332 °C for PBDT-AM, with a 5% weight loss (Fig. S5, ESI†).
C), 284.31 eV (C–C), and 286.03 eV (C–S and CO), confirming the coexistence of BDT and AQ within the polymer. The gel permeation chromatography (GPC) measurements provided the weight-average molecular weight (Mw) and polydispersity index (PDI) values, which are 11.4 kDa and 1.81 for PBDT-AQ, and 20.7 kDa and 2.72 for PBDT-AM, respectively, indicating their sufficient polymer growth (Table S1, ESI†). Both polymers demonstrated excellent thermal stability, up to 392 °C for PBDT-AQ and 332 °C for PBDT-AM, with a 5% weight loss (Fig. S5, ESI†).
 | ||
| Fig. 2 (a) XPS deconvoluted spectra for C 1s of PBDT-AQ. (b) UV-vis absorption spectra, (c) energy level diagram, and (d) EIS studies of PBDT-AQ and PBDT-AM. | ||
The UV-vis spectra of PBDT-AQ and PBDT-AM were recorded in chloroform solution. Both the polymers showed a broad absorption band from 300–460 nm attributed to π–π* transitions, accompanied by comparatively weaker absorption bands in the ranges of 470–650 nm for PBDT-AQ and 490–670 nm for PBDT-AM, corresponding to intramolecular charge transfer (ICT) between the donor and acceptor segments (Fig. 2b). Notably, a red-shift of 10 nm in the absorption maxima for PBDT-AM indicated a stronger donor–acceptor conjugation as compared to PBDT-AQ. The optical band gaps (Eg) calculated from the absorption onset are 2.06 and 1.92 eV for PBDT-AQ and PBDT-AM, respectively (Table S1, ESI†). The nitrile group's electron-withdrawing effect lowers PBDT-AM's band gap compared to PBDT-AQ.8 From cyclic voltammetry (CV) measurements, the lowest unoccupied molecular orbital (LUMO) energy levels were determined to be −0.80 V for PBDT-AQ and −0.82 V for PBDT-AM (Fig. 2c and Fig. S5, ESI†). The higher LUMO energy levels of the polymers, relative to the O2/O2˙− couple (−0.48 V vs. Ag/AgCl),17 indicate an efficient reduction of molecular oxygen (O2) to the superoxide radical anion (O2˙−) under visible light irradiation. The corresponding highest occupied molecular orbital (HOMO) energy levels of the polymers were calculated as 1.26 and 1.10 V for PBDT-AQ and PBDT-AM, respectively, using the equation Eg = ELUMO − EHOMO.18 Furthermore, electrochemical impedance spectroscopy (EIS) showed a smaller arc radius for PBDT-AQ, indicating a lower charge transfer resistance and enhanced conductivity (Fig. 2d). These factors promote increased charge carrier mobility, boosting photocatalytic activity and overall performance.
The molecular orbital distributions of PBDT-AQ and PBDT-AM from density functional theory (DFT) revealed the favorable ICT from the BDT donor to AQ or AM acceptor units that facilitate better electron/hole separation, leading to superior photocatalytic properties (Fig. S6, ESI†). Additionally, oxygen adsorption studies on both polymer model units identified AQ/AM as the primary site for oxygen reduction, with an O–O bond length of 1.46 Å in the case of PBDT-AQ. The O2 adsorption energy was significantly higher for PBDT-AQ (−3.26 kcal mol−1) compared to PBDT-AM (−0.82 kcal mol−1), indicating a better photocatalytic performance of PBDT-AQ in ROS-mediated photocatalytic reactions (Fig. 3).
 | ||
| Fig. 3 Oxygen adsorption study of (a) PBDT-AQ and (b) PBDT-AM by DFT. | ||
The strong absorption in the visible region, optimal band gap, excellent solubility in suitable solvents, and well-positioned LUMO levels have inspired us to utilize these materials as highly effective homogeneous photocatalysts for ROS-mediated reactions. To produce phenols and sulfoxides, photocatalytic hydroxylation of phenylboronic acids and photooxidation of phenyl methyl sulfides offer safer, environmentally friendly, and effective methods that avoid toxic metals and oxidants while maintaining excellent selectivity and sustainability.19,20 In this study, these two model reactions were chosen to evaluate the photocatalytic performance of PBDT-AQ and PBDT-AM. Initially, the reactions were performed in both homogeneous and heterogeneous phases, which revealed that the product conversion rate was 2–5 fold faster in the homogeneous phase (DCM or 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, THF/H2O) compared to the heterogeneous phase, owing to the greater availability of the active sites on the photocatalysts, facilitating more efficient interactions with the reactant molecules in the homogeneous phase (Table S2 and S3, ESI†).14 In both reactions, PBDT-AQ demonstrated superior photocatalytic activity over PBDT-AM, which can be attributed to its optimal band gap, lower charge transfer resistance, and higher oxygen adsorption energy, leading to the activation of O2.
:1, THF/H2O) compared to the heterogeneous phase, owing to the greater availability of the active sites on the photocatalysts, facilitating more efficient interactions with the reactant molecules in the homogeneous phase (Table S2 and S3, ESI†).14 In both reactions, PBDT-AQ demonstrated superior photocatalytic activity over PBDT-AM, which can be attributed to its optimal band gap, lower charge transfer resistance, and higher oxygen adsorption energy, leading to the activation of O2.
In the homogenous phase (DCM solvent), the oxidative hydroxylation of phenylboronic acid to phenol gave a 90% yield for the conversion after 8 hours using PBDT-AQ. To investigate the mechanistic pathway of this reaction, a series of scavenger studies were conducted under optimized reaction conditions using various scavengers (Table S4, ESI†). After adding sodium azide (1O2 scavenger), isopropanol (˙OH scavenger), and potassium iodide (h+ scavenger) separately to the reaction, only a slight decrease in product yield was observed. However, in the presence of p-benzoquinone (p-BQ, a scavenger for O2˙−), only a trace amount of the product was formed, highlighting the critical role of O2˙− in the phenol formation process. The generation of O2˙− was confirmed through electron paramagnetic resonance (EPR) studies using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as an O2˙− trapping reagent, with a characteristic DMPO-O2˙− peak observed after light irradiation (Fig. S7, ESI†). The higher EPR signal intensity indicated that PBDT-AQ exhibits a greater O2˙− generation capacity than PBDT-AM, resulting in superior photocatalytic activity. Additionally, the absence of photocatalysts, O2, light, and triethylamine (TEA) significantly reduced the reaction yield, underscoring the importance of these components in the reaction (Table S5, ESI†). The plausible reaction mechanism, depicted in Fig. S8, ESI† begins with light irradiation, generating photoexcited electrons and holes in the polymer that are located in the LUMO and HOMO, respectively. Electrons available at the LUMO reduce oxygen to O2˙−, while holes react with TEA to produce a radical intermediate. Then, O2˙− reacts with phenylboronic acid, leading to rearrangement and hydroxylation, ultimately producing phenol.
Using the optimized reaction conditions, several para-substituted phenylboronic acids were tested in a homogeneous phase reaction to expand the substrate scope of our catalyst, PBDT-AQ (Table 1). It was observed that phenylboronic acids containing electron-withdrawing groups (–COOH, –CN, –NO2) reacted significantly faster, achieving the highest yield of 93% within 3.5 h, compared to those with electron-donating groups (–Me, –OMe). This trend supports the proposed reaction mechanism, where the electron-withdrawing groups at the p-position of the phenyl ring in the substrate provide greater stabilization to the intermediate (i) (Table 1 and Fig. S8, ESI†).
| a Reaction conditions: 1 mol% PBDT-AQ, 0.2 mmol phenylboronic acid, dichloromethane (CH2Cl2, 2 mL), triethylamine (0.3 mmol), 20 W white LED (400 mW cm−2), O2, room temperature (rt), and isolated yields. |
|---|
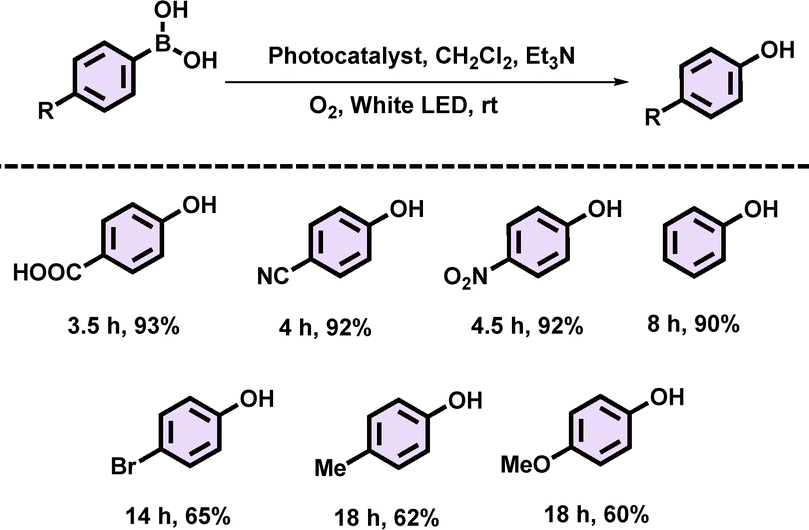
|
On the other hand, using our synthesized photocatalyst, PBDT-AQ, a 90% yield of sulfoxide was achieved from phenyl methyl sulfide after 9 hours in the homogeneous phase (10:1, THF:H2O) (Tables S6 and S7, ESI†). In the scavenger studies of the above reaction using PBDT-AQ, a significant reduction in product yield was observed (Table S8, ESI†). Specifically, the yield dropped to 40% in the presence of p-BQ, a scavenger of the superoxide anion (O2˙−), and to 60% when sodium azide, a scavenger of singlet oxygen (1O2), was added, demonstrating that both O2˙− and 1O2 played a crucial role in the reaction path. Based on the results obtained from scavenger studies, a plausible reaction mechanism was proposed (Fig. 4a), where photoexcited electrons react with oxygen to produce ROS (O2˙− and 1O2), while the holes interact with phenyl methyl sulfide to generate a cationic radical intermediate (intermediate 1). This intermediate subsequently reacts with ROS, leading to the final product formation.18 Furthermore, the generation of possible key intermediates (1O2, O2˙−) with PBDT-AQvia triplet oxygen activation was confirmed with EPR. The generation of 1O2, as evidenced by a prominent peak (1:1:1) corresponding to the 2,2,6,6-tetramethylpiperidine (TEMP)–1O2 adduct and O2˙− was confirmed with a DMPO–O2˙− characteristic signal (Fig. 4b and Fig. S9, ESI†). Table S9 (ESI†) further highlights the essential roles of the catalyst, oxygen, and light in this photooxidation process. Our photocatalyst PBDT-AQ demonstrates a broad substrate tolerance, yielding up to 90% of the product across various para-substituted phenyl methyl sulfides (Table 2). Phenyl methyl sulfides containing electron-donating groups, such as –OH and –OMe, react more rapidly, furnishing sulfoxides with better yields (up to 90%) compared to those with electron-withdrawing groups, like –CN and –NO2, giving 42% and 40% yields, respectively. This difference in reactivity can be attributed to the stabilization of intermediate 1 by the electron-donating groups, as depicted in Fig. 4.
 | ||
| Fig. 4 (a) Proposed mechanistic pathway of photo-oxidation of phenyl methyl sulfide. (b) ROS (O2˙− and 1O2) generation by using PBDT-AQ. (c) Recyclability of PBDT-AQ for oxidative hydroxylation of phenylboronic acid to phenol and photo-oxidation of phenyl methyl sulfide to sulfoxides. | ||
| a Reaction conditions: 1 mol% PBDT-AQ, 0.2 mmol phenyl methyl sulfides, THF (2 mL) and H2O (0.2 mL), 20 W white LED, O2, rt, and isolated yields. |
|---|

|
Our approach combines the advantages of homogeneous phase reactions for efficient photocatalysis with heterogeneous phase separation for easy recovery and recyclability, promoting sustainability. After the photocatalytic reaction in ‘good’ solvents (DCM, THF:H2O), the polymers were easily recovered via simple precipitation in methanol (anti-solvent) (Fig. 1b). The recovered catalysts were reused for five cycles with minimal activity loss and excellent recovery efficiency (Fig. 4c and Table S10, ESI†). FT-IR spectra and GPC data confirmed the catalyst stability after five cycles, demonstrating the exceptional durability of these polymers (Fig. S10 and Table S11, ESI†) in photocatalytic reactions (Fig. S11–S24, ESI†).
In summary, two solution-processable D–A copolymers, PBDT-AQ and PBDT-AM, were synthesized using the Stille coupling reaction. Their π-conjugated backbones endowed strong visible-light absorption, while branched alkyl chains enhanced solubility for seamless photocatalysis. PBDT-AQ outperformed PBDT-AM with remarkable yields of 93% for the photoproduction of phenol and 90% for sulfoxide formation, attributed to the superior O2˙− and 1O2 generation ability of PBDT-AQ. The experimental and theoretical insights suggested that PBDT-AQ comprised suitable oxygen adsorption sites and improved charge carrier mobility. Additionally, these photocatalysts can be easily recovered via antisolvent precipitation, allowing for efficient reuse. This practical approach offers a sustainable and high-performing photocatalytic system for oxidative transformations.
S. K. S. acknowledges CSIR (22(0828)/19/EMR-II) for funding this work. S. M. is thankful to CSIR for the fellowship. K. M. and S. S are thankful to IIT Kharagpur for the fellowships. F. B. is thankful to MoE, Govt. of India for the PMRF fellowship.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- J. Xiao, X. Liu, L. Pan, C. Shi, X. Zhang and J.-J. Zou, ACS Catal., 2020, 10, 12256–12283 CrossRef.
- F. Parrino, M. Bellardita, E. I. García-López, G. Marcì, V. Loddo and L. Palmisano, ACS Catal., 2018, 8, 11191–11225 CrossRef.
- S. Zhang, Y. Zhao, R. Shi, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2020, 10, 1901973 CrossRef.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef.
- S. K. Samanta, E. Preis, C. W. Lehmann, R. Goddard, S. Bag, P. K. Maiti, G. Brunklaus and U. Scherf, Chem. Commun., 2015, 51, 9046–9049 RSC.
- S. Sau and S. K. Samanta, Chem. Commun., 2023, 59, 635–638 RSC.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- C. Huang, J. Wen, Y. Shen, F. He, L. Mi, Z. Gan, J. Ma, S. Liu, H. Ma and Y. Zhang, Chem. Sci., 2018, 9, 7912–7915 RSC.
- S. K. Samanta, I. Song, J. H. Yoo and J. H. Oh, ACS Appl. Mater. Interfaces, 2018, 10, 32444–32453 CrossRef.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef PubMed.
- S.-H. Wang, F. Khurshid, P.-Z. Chen, Y.-R. Lai, C.-W. Cai, P.-W. Chung, M. Hayashi, R.-J. Jeng, S.-P. Rwei and L. Wang, Chem. Mater., 2022, 34, 4955–4963 CrossRef CAS.
- K. Maity, S. Sau, F. Banerjee and S. K. Samanta, ACS Appl. Mater. Interfaces, 2024, 16, 50834–50845 CrossRef CAS.
- F. Bureš, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht, M. Gross and F. Diederich, Eur. J. Org. Chem., 2008, 994–1004 CrossRef.
- J. Wu, Z. Jin and Q. Zhang, Polym. Bull., 2015, 72, 2553–2560 CrossRef CAS.
- S. Sau, S. Bera, B. Mondal, N. Kumar and S. K. Samanta, J. Catal., 2024, 437, 115658 CrossRef CAS.
- S. Sau, S. Karmakar, F. Banerjee and S. K. Samanta, Nanoscale, 2024, 16, 21096–21105 RSC.
- G. W. Burton, T. Doba, E. Gabe, L. Hughes, F. Lee, L. Prasad and K. U. Ingold, J. Am. Chem. Soc., 1985, 107, 7053–7065 CrossRef.
- J. Wen, X. Yang, Z. Sun, J. Yang, P. Han, Q. Liu, H. Dong, M. Gu, L. Huang and H. Wang, Green Chem., 2020, 22, 230–237 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, synthetic procedures, characterization, and photocatalytic performances. See DOI: https://doi.org/10.1039/d5cc01869b |
| This journal is © The Royal Society of Chemistry 2025 |