
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
OH-stretching dynamics in trimethylamine monohydrate: what can we learn from three different direct absorption spectra?†
Taija L.
Fischer
 a,
Casper V.
Jensen
b,
Eaindra
Lwin
a,
Dhritabrata
Pal
b,
Henrik G.
Kjaergaard
*b and
Martin A.
Suhm
*a
a,
Casper V.
Jensen
b,
Eaindra
Lwin
a,
Dhritabrata
Pal
b,
Henrik G.
Kjaergaard
*b and
Martin A.
Suhm
*a
aInstitute of Physical Chemistry, University of Göttingen, Tammannstr. 6, 37077 Göttingen, Germany. E-mail: msuhm@gwdg.de
bDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark. E-mail: hgk@chem.ku.dk
First published on 14th May 2025
Abstract
The hydrogen-bonded binary complex between water and trimethylamine is characterised in jet expansions, in the room temperature gas phase and in frozen argon matrices via its OH-stretching fundamental. The spectral comparison reveals two bracketing resonance partners with weak environmental dependence of their wavenumber. They correspond to the water bending overtone alone and in combination with a stretching motion of the monomers relative to each other. The environment-sensitive OH-stretching mode moves from the combination band location at room temperature all the way down to the pure water bending overtone in cryomatrix isolation, sharing its infrared intensity in proportion to the spectral vicinity and coupling strength. The intermediate jet-cooled spectrum largely removes thermal excitation and embedding effects. It thus provides the easiest entry point for theoretical modelling. Chemical and isotope substitutions at the amine support the robustness of the assignment, whereas a switch from water to methanol removes the bending-based resonance opportunities. After correction for the resonances, the OH-stretching positions of the water complex follow those of the methanol complex quite closely. This shows that methanol and water undergo similar hydrogen bond interactions with the amine. Previous contradicting spectral interpretations of water trimethylamine mixtures in supersonic jets and in argon matrices are discussed and discouraged. For the dihydrate of trimethylamine, both hydrogen-bonded OH stretching vibrations are characterised in the jet expansion.
Hydrogen bonds to triply alkylated amines1,2 are among the strongest which water can form in binary neutral molecular complexes. It makes sense to investigate a particularly small representative, the monohydrate of trimethylamine,3 by vibrational spectroscopy2,4 to probe this interaction and its dynamics. Due to the potential importance for atmospheric processes, this has been done before in the room temperature gas phase,1 where the hydrogen-bonded complex coexists with a large excess of its molecular constituents due to the intermediate strength of the hydrogen bonding interaction.1,2 Because of the observed spectral width and complexity, complementary investigations at low temperature are helpful. Surprisingly, such studies appear to be limited to an argon matrix isolation investigation5 and a more recent supersonic jet investigation by VUV-IR double resonance.6 Both studies find different and unexpected spectral complexity in the OH-stretching range of the solvating water. The matrix isolation study concludes that the spectral pattern is incompatible with 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes (assigning a 4:2 complex instead) and the VUV-IR double resonance technique has produced controversial results for hydrogen-bonded complexes before,7 perhaps due to unexplained fragmentation. The two contradictory low temperature vibrational studies5,6 were accompanied by electronic structure calculations in combination with anharmonic vibrational analysis to support their unusual conclusions. In the present study we focus on a multifaceted experimental approach including three different media and support our spectral assignments by chemical and isotopic substitution.
:1 complexes (assigning a 4:2 complex instead) and the VUV-IR double resonance technique has produced controversial results for hydrogen-bonded complexes before,7 perhaps due to unexplained fragmentation. The two contradictory low temperature vibrational studies5,6 were accompanied by electronic structure calculations in combination with anharmonic vibrational analysis to support their unusual conclusions. In the present study we focus on a multifaceted experimental approach including three different media and support our spectral assignments by chemical and isotopic substitution.
In addition to the available room temperature gas phase spectra,1 we employ fragmentation-free, direct absorption spectroscopy in an argon cryomatrix and in a supersonic jet. These techniques are well-established8,9 and the jet technique has recently been applied to other triply alkylated amines.10,11 We can thus refer the reader to the ESI† (Section S1, Tables S1–S3) and to the references for technical details and start discussing Fig. 1, which compares the monohydrate of trimethyl amine in the hydrogen-bonded OH-stretching region for three states of aggregation (from top to bottom): thermally excited gas phase,1 supersonic jet expansion, and argon matrix isolation. Small signals due to water monomer bend overtone transitions (w) and a mixed trimer with two water molecules (T, not expected at room temperature) are observed in the jet spectrum (central trace), and can be easily assigned by concentration variation (Fig. S2 in the ESI†).
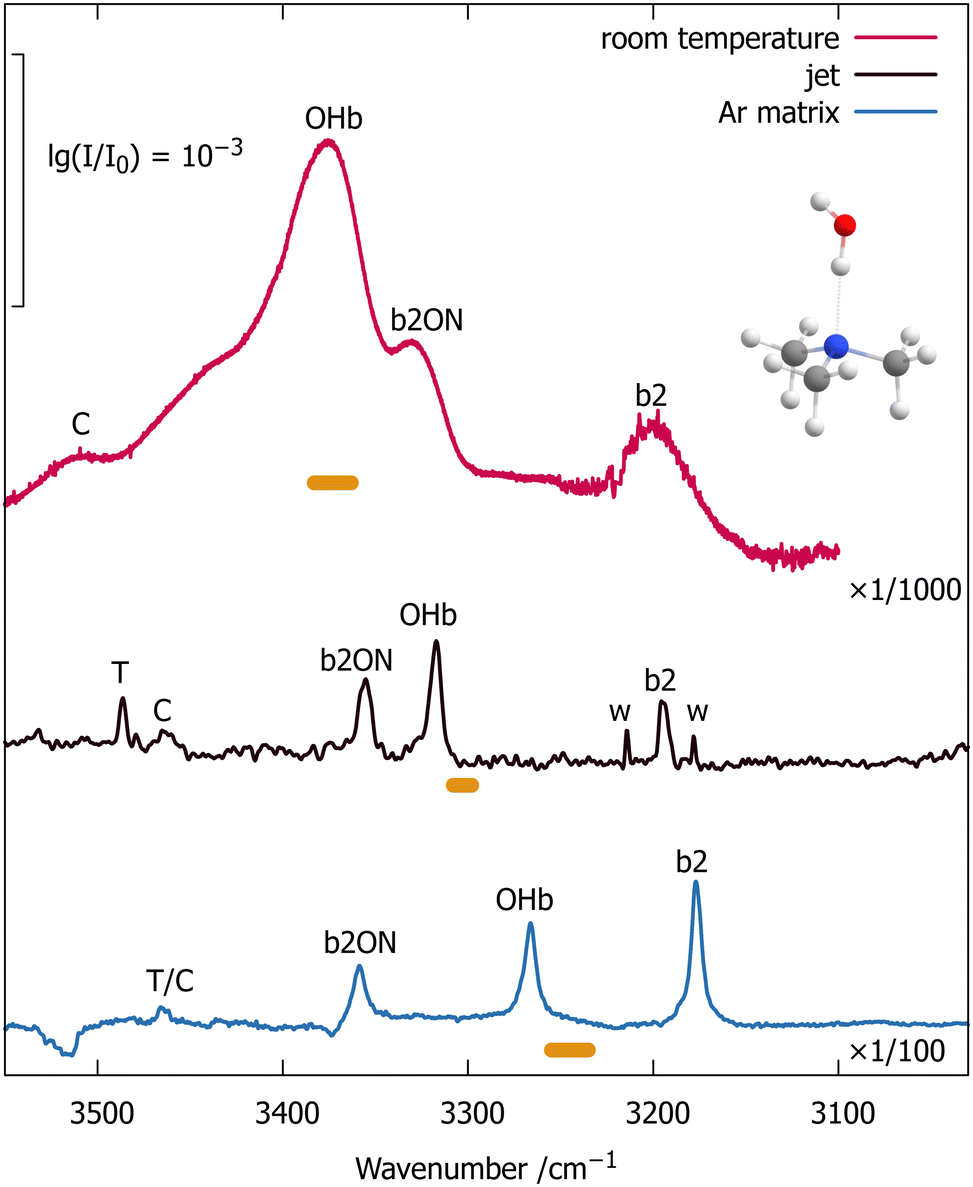 | ||
| Fig. 1 Evolution of the OH-stretching spectrum of jet-cooled trimethylamine monohydrate (center trace) with thermal excitation (red upper trace, room temperature1) and argon matrix embedding (blue lower trace) obtained by FTIR spectroscopy. The intensity scaling factors emphasize the limited sensitivity of the supersonic jet approach, but it provides a valuable anchor point for the thermal and matrix shifts of the intensity center (horizontal bars) and the closest contact to quantum chemical treatments at 0 K in vacuum. T is a 2:1 trimer (Fig. S4 in the ESI†), see text for further explanations of the key contributions b2, OHb, b2ON and the small C signal. | ||
We observe 3–4 transitions (marked b2, OHb, b2ON and C in the jet spectrum without implying that these correspond to pure modes) which scale similarly with varying conditions. The recent trialkylamine monohydrate study10 has revealed analogous spectral patterns, with the central transition (OHb) carrying most OHb oscillator strength and the flanking transitions (b2 and b2ON) stealing some of this intensity due to wavefunction mixing. For the water bending overtone b2, this is clearly due to a strong resonance with OHb (a Fermi or 1:2 resonance). For b2ON, it is best explained10 by a three-quantum combination of b2 with hydrogen bond stretching motion of the water unit (O) relative to the amine unit (N), which also gains intensity through anharmonic resonance with OHb. This assignment is supported by earlier room temperature experiments, where partial deuteration of the water removes both the b2 and b2ON resonance partners due to a downshift of the DOH-bending mode compared to the HOH-bending mode.1 We ignore the weak peak labelled C in the following analysis, but it can be plausibly assigned as the combination band of OHb and ON, profiting from vibrational Franck–Condon intensity redistribution. This assignment is also supported by the earlier room temperature experiments as partial deuteration of the coordinating water (see Fig. S8 in the ESI†) does not remove the peak.1 However, for HOH and in close analogy to OHb with b2, such an OHbON state would likely be in resonance with b2ON, thus complicating the interpretation of its position and intensity.11
Assuming that the three major features arise predominantly from wavefunction mixing of the bright OHb-stretching state and the two dark overtone/combination states, one can extract the original, unmixed position of the OHb bright state by computing the centroid or center of intensity of the three IR signals (see data in the ESI,† Section S2). This centroid is marked by a horizontal bar underneath each spectral trace which describes the error margins due to baseline and noise limitations (Table S4, ESI†). It falls close to the dominant signal in all three cases, because the perturbation effects from the other two states tend to act in opposite direction. As in many related cases,11 the net effect is a slight downshift from the main peak to the centroid.
In the argon matrix environment (lowest trace), the centroid is downshifted in wavenumber compared to the jet-cooled centroid by an amount which is typical for isolated OH stretching states in argon matrices.8 Despite the high sensitivity of OH stretching wavenumbers and intensities to different matrix environments,12 no site splittings in the resonance triad are observed. Because the OHb vibration is most sensitive to hydrogen bonding11 and downshifted in the matrix, it transfers more OHb character to the lowest wavenumber band which now becomes comparable in intensity. Its position is not upshifted relative to the jet spectrum, as one might have expected for an isolated bending mode enclosed in a matrix,8 because the downshift due to intensified Fermi resonance dominates. The combination band (b2ON) on the other side is actually slightly upshifted, which can be explained by higher wavenumber ON-stretching and HOH-bending vibrations under matrix confinement.8 The observed matrix shifts can also be tuned to some degree by experimentally building argon nanoshells13 around the molecular complex (Fig. S7 in the ESI†), thus offering a new dimension to theoretical modelling.
The room temperature gas phase spectrum (upper trace of Fig. 1) shows thermal effects which oppose the matrix embedding effects. While the intensity is still derived from OHb excitation, this excitation starts in a very large number of thermally populated intermolecular states,14 each of which modulates the resonance triplet observed at low temperature. OHb is upshifted towards the monomer position (symmetric stretch9,15 at 3657 cm−1), because thermal excitation of the intermolecular modes weakens the hydrogen bond and reduces the downshift.16 In an adiabatic picture, the OHb downshift due to hydrogen bonding corresponds to the increase in hydrogen bond strength upon OHb excitation17 and therefore is reduced by thermal excitation of modes which emerge from hydrogen bonding. A recent quantum-mechanical model accounts for the observed upshift and broadening of the OHb band by directly considering the increase in energy of the thermally populated states associated with the intermolecular modes upon exciting the OHb oscillator.14 The resulting upshift of the hot transitions (see also Fig. S6 in the ESI,† for the onset of this upshift) attenuates the Fermi resonance with b2. Therefore, b2 in the complex has two diverging thermal influences – its thermal downshift towards the asymptotic value for free water monomer15 (3152 cm−1) is counteracted by a decreasing Fermi resonance downward push from the thermally upshifted OHb state. The net effect is a slight overall upshift of the band maximum. The combination of b2 with ON is actually downshifted compared to the cold jet spectrum. This is the result of thermal weakening (the hydrogen bond softens with increasing temperature) and of the avoided crossing with the intensity-providing upshifted OHb transition.
The centroid position of b2, OHb, and b2ON is now less certain (wider horizontal bar in Fig. 1) because of the extension of the overlapping thermally broadened bands14 into the (C) spectral range which we neglect in the resonance analysis, but it is clearly higher in wavenumber than in the jet spectrum. This is expected for a thermally weakened interaction between water and the trimethylamine, when averaging over the competing resonance effects. The onset of this spectral evolution from a cold jet expansion to the room temperature gas phase can be observed in a soft expansion at very low stagnation pressure (Fig. S6 in the ESI†). While the exact (vibrational) temperature remains unknown, the crossover between OHb and b2ON is demonstrated by similar infrared intensities already for moderate warming.
We thus have a sequence of decreasing OHb stretching wavenumber from the thermal gas phase over different degrees of jet-cooling to the matrix-embedded situation, irrespective of whether one looks at the transition with the strongest intensity or the centroid of the three strongly coupled states.
To further support the emerging picture, Fig. 2 shows analogous spectra for the 1:1 complex of trimethylamine with methanol. This homolog of the trimethylamine monohydrate has been used repeatedly for comparison purposes.1,6,18–20 The HOC bending overtone in methanol is much lower in wavenumber than for HOH (see Fig. S4, ESI†) and therefore no more available for an anharmonic resonance with OHb. This also removes the resonance condition for the combination with ON, such that in analogy to the HOD case1 a single OHb transition is now expected. Apart from a minor contribution T, which we neglect for the centroid (also due to a different concentration dependence in the jet than the 1:1 complex, see Fig. S3 and S4 in the ESI†) and apart from a contribution (O) due to oligomers in the matrix isolation spectrum, there is now a single dominant transition in each state of aggregation. Within error bars, the gas phase and jet centroid positions overlap between water1 and methanol18 coordination, whereas the matrix shift is clearly larger for water than for methanol. This is an observation which matches other recent cases of jet-matrix comparison.21 The thermal shift of the gas phase band relative to the jet-cooled band varies with temperature20,22 and is largely due to hot transitions.14 The weaker absorptions flanking the thermally broadened OHb profile are likely due to combination and difference bands involving intermolecular modes. While the difference bands must disappear in the jet due to depopulation, the combination bands should survive. However, the signal-to-noise ratio is not sufficient to reveal them in the jet spectrum and as shown in the ESI† (Fig. S3), even the weak signal marked T is mostly due to a jet-cooled mixed trimer impurity, rather than an OHb + ON combination band in the 1:1 complex. In the matrix spectrum, combination bands may perhaps be assigned and in the room temperature gas phase spectrum there is a notable secondary band to the left of the main transition. Therefore it remains unclear whether the IR transition moment of OHbON in the absence of b2-based resonances and thermal excitation is substantial. This is somewhat in contrast to other hydrogen-bonded complexes,23,24 where pronounced vibrational Franck–Condon intensity redistribution to intermolecular combination bands happens.25
 | ||
| Fig. 2 As Fig. 1, but for methanol instead of water. The room temperature spectrum was obtained before.18 See text for further explanations. | ||
The drastic simplification when moving from water to methanol has been qualitatively observed before in the VUV-IR jet study,6 but only two of the four jet cooled band positions reported in the present work (C and b2ON) formally match the previous reports, whereas the main OHb transition is absent (Table S6 in the ESI†) and the intensity pattern is completely different. Even for the methanol case, where both techniques suggest a dominant OHb peak and at best a minor combination band, the band maxima and widths differ quite significantly, with the linear spectra yielding more narrow, more OHb accentuated and higher wavenumber signals. For the water case, the three dominant peaks observed in the linear spectrum must be compared to at least six significantly broader features in the action spectrum6 spread over a much wider range. Whatever the VUV-IR spectrum reflects in terms of OH-stretching dynamics in amine–water complexes, it is not the dynamics of two dominant resonance partners in the 1:1 complex which follows from the cold linear FTIR spectra. Note that the anharmonic simulations presented together with the VUV-IR spectra6 fit the dominant features and intensity patterns of the linear spectra presented in this work reasonably well (including small details for methanol), significantly better than the VUV-IR observations, which they aim to support.
When comparing the current argon matrix isolation spectra of trimethylamine monohydrate with the previous literature finding,5 the situation is somewhat different (Table S7 in the ESI†). Considering the width of the peaks, the agreement for the three prominent signals is reasonable, also for the most diluted literature spectra. However, the additional peaks observed in the previous study, in particular at higher concentrations, have distracted from a simple 1:1 complex interpretation. Although the possibility of a Fermi resonance between OHb and b2 was mentioned by the authors, it was not considered in their assignment. Furthermore, prior to the systematic experimental study of amine monohydrates10 and the theoretical anharmonic treatment of the 1:1 complex,6 a second resonance partner involving ON-stretching had not been prominently proposed in the literature to the best of our knowledge.
Because we do not want to lightheartedly refute the literature findings for trimethylamine monosolvate OH-stretching dynamics, the ESI† (Section S3) contains further pieces of supportive evidence. This includes figures which compare the jet spectra of water and methanol complexes of perdeuterated trimethylamine (Fig. S1 and S4, ESI†) to those without deuteration. They confirm the absence of significant 1:1 complex absorptions with OHb character in the CH-stretching range and a subtle downshift in particular for the ON combination band (Table S5, ESI†), in line with expectations for an increased mass of the amine. For the OHb signal, there is a somewhat larger downshift with methanol than with water and some evidence for subtle spectral splittings with water, possibly due to further weak resonances. The shoulder seen with methanol is partly due to water impurities in the expansion, as evidenced by the direct comparison of methanol and water spectra (Fig. S1, ESI†). Fig. S5 in the ESI† compares the effect of trimethylamine perdeuteration with the effect of chemical substitution, which at the same time freezes methyl internal rotation. For this purpose, DABCO (1,4-diazabicyclo[2,2,2]octane) is chosen, which is formally obtained when removing a hydrogen from each methyl group of two trimethylamine units and forming three C–C bonds between the two radical species. The chemical substitution effect is as subtle as that of perdeuteration and supports the robustness of the coupling scenario. Perhaps a VUV-IR study of DABCO monohydrate could shed further light on the features of this action spectroscopy technique.
The vibrational spectra reported in this work finally raise the prototype hydrogen-bonded complex of trimethylamine with water to vibrational benchmark level. After the decade-old structural benchmark work on the system,3 this step was overdue. Now, theoretical methods can be systematically consulted to reproduce and explain the resonance details and thus demonstrate their power. These methods can range from simple n × n coupling models10,11,26 to high-dimensional treatments6,14,27 including hot transitions.14 While weakly and moderately bound water complexes are already well characterized,2,9,28,29 there is still a scarcity of environment-free experimental benchmarks for the vibrational dynamics of strong hydrogen bonds in the XH-stretching range for neutral complexes. Perhaps most prominently, the OH-stretching modes of simple carboxylic acid dimers23,30 still pose considerable assignment challenges in detail and the complex of formic acid with trimethylamine31 also remains to be characterised vibrationally.
The experimental benchmark potential,32 which we introduce for hydrate complexes of trimethylamine, is multifacetted. Scaled harmonic prediction methods for OH-stretching spectra can be better calibrated in the 3 μm range by comparing to the centroid of IR intensity in the single bright state approximation. Reduced-dimensionality1,27 and perturbational anharmonic approaches9 can now be explored closer to the water Fermi resonance regime. If large amplitude methyl rotors cause a problem, one can resort to the analogous DABCO (ESI,† Fig. S5) or quinuclidine10 cases. The training set for future HyDRA blind challenges9 can now be extended (Table S8 in the ESI†). This includes the dihydrates of TMA and TMA-d9, where the hydrogen-bonded water from the second solvation shell gives rise to a sharp transition and the amine-contacting water leads to a broader transition (Fig. 1 and ESI,† Fig. S2 and S4) indicative of faster energy redistribution. The latter water stretching transition for TMA-d9 at 3014 cm−1 is among the most down-shifted for neutral water complexes without resonance splitting, safely expanding the HyDRA training range. Attempts to model distant deuterium isotope effects can now target a simple model complex. Attempts to model matrix isolation shifts21 can now build on a particularly elementary pair of cold reference data deep in the hydrogen bonding regime. In the future, this modelling can extend to nanomatrices13 (Fig. S7, ESI†) as a compromise between isolated molecular complexes and infinite solids. Attempts to model gas phase thermal shifts14 can go beyond the hot band model and incorporate resonances as a function of thermal excitation. All this was not the case for the previous interpretations of the vibrational spectra of trimethylamine hydration,5,6 which may still point the way to a better understanding of larger aggregate spectra and dynamical processes upon VUV excitation.
Author contributions
TLF: investigation, visualization, writing – review and editing; CVJ: formal analysis, investigation, visualization, writing – review and editing; EL: formal analysis, investigation, visualization, writing – review and editing; DP: formal analysis, investigation, visualization, writing – review and editing; HGK: conceptualization, funding acquisition, methodology, supervision, writing – review and editing; MAS: conceptualization, funding acquisition, methodology, supervision, writing – original draft, writing – review and editing.Data availability
Extensive data supporting the claims are available as part of the ESI.† Raw jet and matrix spectra are provided in a data repository (https://doi.org/10.25625/IVHUHR). A compact summary of benchmarking-relevant quantities will be made available33 and linked to this publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 389479699/GRK2455 and by the Novo Nordisk Foundation (Grant No. NNF19OC0057374 and NNF22OC0080193). We thank M. Bödecker for help with the DABCO spectra and valuable discussions. We thank E. Vogt and N. Lüttschwager for helpful discussions and programming support.Notes and references
- A. Kjaersgaard, E. Vogt, A. S. Hansen and H. G. Kjaergaard, J. Phys. Chem. A, 2020, 124, 7113–7122 CrossRef CAS PubMed.
- A. S. Hansen, E. Vogt and H. G. Kjaergaard, Int. Rev. Phys. Chem., 2019, 38, 115–148 Search PubMed.
- M. J. Tubergen and R. L. Kuczkowski, J. Am. Chem. Soc., 1993, 115, 9263–9266 Search PubMed.
- D. J. Millen and G. W. Mines, J. Chem. Soc., Faraday Trans. 2, 1977, 73, 369–377 Search PubMed.
- M. Rozenberg, A. Loewenschuss and C. J. Nielsen, J. Phys. Chem. A, 2012, 116, 4089–4096 Search PubMed.
- S. Jiang, M. Su, S. Yang, C. Wang, Q.-R. Huang, G. Li, H. Xie, J. Yang, G. Wu, W. Zhang, Z. Zhang, J.-L. Kuo, Z.-F. Liu, D. H. Zhang, X. Yang and L. Jiang, J. Phys. Chem. Lett., 2021, 12, 2259–2265 CrossRef CAS PubMed.
- I. León, R. Montero, A. Longarte and J. A. Fernández, J. Phys. Chem. Lett., 2021, 12, 1316–1320 Search PubMed.
- A. J. Barnes, J. Mol. Struct., 2018, 1163, 77–85 CrossRef CAS.
- T. L. Fischer, M. Bödecker, S. M. Schweer, J. Dupont, V. Lepère, A. Zehnacker-Rentien, M. A. Suhm, B. Schröder, T. Henkes, D. M. Andrada, R. M. Balabin, H. K. Singh, H. P. Bhattacharyya, M. Sarma, S. Käser, K. Töpfer, L. I. Vazquez-Salazar, E. D. Boittier, M. Meuwly, G. Mandelli, C. Lanzi, R. Conte, M. Ceotto, F. Dietrich, V. Cisternas, R. Gnanasekaran, M. Hippler, M. Jarraya, M. Hochlaf, N. Viswanathan, T. Nevolianis, G. Rath, W. A. Kopp, K. Leonhard and R. A. Mata, Phys. Chem. Chem. Phys., 2023, 25, 22089–22102 RSC.
- E. Lwin, T. L. Fischer and M. A. Suhm, J. Phys. Chem. Lett., 2023, 14, 10194–10199 CrossRef CAS PubMed.
- E. Lwin, N. O. B. Lüttschwager and M. A. Suhm, Phys. Chem. Chem. Phys., 2025, 27, 5808–5820 RSC.
- P. D. Cooper, H. G. Kjaergaard, V. S. Langford, A. J. McKinley, T. I. Quickenden and D. P. Schofield, J. Am. Chem. Soc., 2003, 125, 6048–6049 CrossRef CAS PubMed.
- T. N. Wassermann, P. Zielke, J. J. Lee, C. Cézard and M. A. Suhm, J. Phys. Chem. A, 2007, 111, 7437–7448 CrossRef CAS.
- E. Vogt, C. V. Jensen and H. G. Kjaergaard, J. Phys. Chem. A, 2024, 128, 392–400 CrossRef CAS.
- N. O. B. Lüttschwager, Phys. Chem. Chem. Phys., 2024, 26, 10120–10135 Search PubMed.
- J. Bournay and G. Robertson, Mol. Phys., 1980, 39, 163–174 Search PubMed.
- R. Knochenmuss, R. K. Sinha and S. Leutwyler, Annu. Rev. Phys. Chem., 2020, 71, 189–211 Search PubMed.
- A. Kjaersgaard, E. Vogt, N. F. Christensen and H. G. Kjaergaard, J. Phys. Chem. A, 2020, 124, 1763–1774 Search PubMed.
- X.-Q. Tan, I. I. Ioannou, K. B. Foltz and R. L. Kuczkowski, J. Mol. Spectrosc., 1996, 177, 181–193 Search PubMed.
- L. Du, K. Mackeprang and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2013, 15, 10194–10206 Search PubMed.
- M. Bödecker, D. Mihrin, M. A. Suhm and R. Wugt Larsen, J. Phys. Chem. A, 2024, 128, 7124–7136 Search PubMed.
- M. Fild, M. F. Swiniarski and R. R. Holmes, Inorg. Chem., 1970, 9, 839–843 Search PubMed.
- J. Dreyer, J. Chem. Phys., 2005, 122, 184306 CrossRef PubMed.
- A. Nejad, A. F. Pérez Mellor, M. Lange, I. Alata, A. Zehnacker and M. A. Suhm, Phys. Chem. Chem. Phys., 2023, 25, 10427–10439 RSC.
- K. Heyne, E. T. J. Nibbering, T. Elsaesser, M. Petković and O. Kühn, J. Phys. Chem. A, 2004, 108, 6083–6086 CrossRef CAS.
- Q.-R. Huang, Y.-C. Li, T. Nishigori, M. Katada, A. Fujii and J.-L. Kuo, J. Phys. Chem. Lett., 2020, 11, 10067–10072 CrossRef CAS.
- E. Vogt, I. Simkó, A. G. Császár and H. G. Kjaergaard, J. Chem. Phys., 2022, 156, 164304 CrossRef CAS PubMed.
- T. S. Zwier, J. Phys. Chem. A, 2001, 105, 8827–8839 CrossRef CAS.
- A. Potapov and P. Asselin, Int. Rev. Phys. Chem., 2014, 33, 275–300 Search PubMed.
- K. Mackeprang, Z.-H. Xu, Z. Maroun, M. Meuwly and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2016, 18, 24654–24662 RSC.
- R. B. Mackenzie, C. T. Dewberry and K. R. Leopold, J. Phys. Chem. A, 2016, 120, 2268–2273 CrossRef CAS PubMed.
- R. A. Mata and M. A. Suhm, Angew. Chem., Int. Ed., 2017, 56, 11011–11018 CrossRef CAS.
- Hydrate Donor Redshift Anticipation, a database maintained by the BENCh DFG research training group in Göttingen, 2024-2025, available under https://qmbench.net/databases/hydra.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and band positions, deperturbation approach, literature comparison, isotope and chemical substitution, soft expansion, benchmarking database. See DOI: https://doi.org/10.1039/d5cp01424g |
| This journal is © the Owner Societies 2025 |