
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of the aminoperoxyl radical in the atmospheric oxidation of ammonia†
Vili-Taneli
Salo
 ab,
Jing
Chen
b and
Henrik G.
Kjaergaard
*b
ab,
Jing
Chen
b and
Henrik G.
Kjaergaard
*b
aDepartment of Chemistry, University of Helsinki, P.O. Box 55, Helsinki 00014, Finland
bDepartment of Chemistry, University of Copenhagen, Copenhagen 2100, Denmark. E-mail: hgk@chem.ku.dk
First published on 10th June 2025
Abstract
Atmospheric oxidation of ammonia is initiated by its reaction with the hydroxyl radical, producing the aminyl radical (NH2). Thus far, it has been believed that the subsequent fate of NH2 is to react bimolecularly with other atmospheric trace gases like NO, NO2, or O3. Its reaction with O2 has been considered insignificant under atmospheric conditions. However, this is based on a rate coefficient that is orders of magnitude smaller than those known for analogous reactions of O2 with carbon-, sulfur-, and other nitrogen-centered radicals. We demonstrate by multireference calculations and kinetic modelling that the reaction of NH2 and O2 leading to the formation of the aminoperoxyl radical (NH2O2) occurs with a rate coefficient similar to those of the aforementioned analogous radicals. We show that the previously estimated small rate coefficient is due to an unimolecular rate limiting step in the formation of measured products rather than the initial NH2 + O2 reaction. The lack of experimental detection of NH2O2 in the existing literature is likely due to the experiments being conducted at either high temperature or low pressure. We show that the atmospheric presence of NH2O2 depends greatly on atmospheric conditions. Its formation is an important, yet previously overlooked pathway in atmospheric ammonia oxidation, especially at low temperatures.
1 Introduction
Ammonia (NH3) is one of the most abundant nitrogen-containing compounds in the atmosphere. It is emitted into the atmosphere from agricultural activities, wild fires, and in polar regions also from penguin and seabird colonies, with a current estimated annual global emission of 58 Tg N.1–5 Ammonia has an atmospheric lifetime from hours to a few days.6–8 Its most significant loss mechanisms are acid–base reactions, wet deposition, and gas-phase oxidation reactions.7,9 The branching of these routes is uncertain, but the lifetime of NH3 with respect to oxidation is estimated to be ten times longer than its other loss mechanisms.10 This suggests that only a minor fraction of ammonia is oxidized in the gas phase. However, at high altitudes, above the boundary layer, this oxidation is likely more important. Moreover, because the transient concentration of NH3 is high, NH3 oxidation may lead to significant production of NOx and other nitrogen-containing species in the atmosphere.The atmospheric oxidation reactions of ammonia are known to be initiated via hydrogen abstraction by the OH radical11,12 or by halogen radicals,13 yielding the aminyl radical (NH2). The NH2 radical is known to react bimolecularly with NO2, NO, O3, and O2.14 The reactions with NO2 and NO are believed to be dominant since the IUPAC recommended rate coefficient for its reaction with O2 is smaller than 6 × 10−21 cm3 per molecule per s.14 It has been argued in earlier literature that if the rate coefficient of the NH2 + O2 reaction is larger than 3 × 10−18 cm3 per molecule per s, then it would be the dominant bimolecular reaction of NH2 under atmospheric conditions.15
A wide range of rate coefficients for the NH2 + O2 reaction (10−15–10−21 cm3 per molecule per s) have been determined using different experimental setups.15–21 The largest rate coefficient (10−15 cm3 per molecule per s) was determined in 1972 by measuring the NH2 decay, which was revised in 1979 to 10−17 cm3 per molecule per s by the same authors.19 The basis of the current IUPAC recommendation is a more recent experimental study from 1991,15 where the rate coefficient was inferred indirectly by measuring the NOx and N2O formation in experiments of NH2 in excess molecular oxygen with and without added NO. The product distributions were explained by the known kinetics of other competing reactions in their reaction system; therefore, the rate coefficient was given as an upper limit.
For methane, CH4, the reaction with OH and O2 leads to the formation of CH3O2, which is the most abundant alkylperoxyl radical in the atmosphere.22 This peroxyl radical has been detected in laboratory experiments by e.g. cavity ring down spectroscopy.23 As far as we know, clear evidence of the formation of the corresponding peroxyl radical, NH2O2, has not been reported in any of the experimental studies of NH3 oxidation.15–21
The thermostability of the aminoperoxyl radical, NH2O2, has been investigated theoretically in multiple studies.24–28 These previous studies give a somewhat conflicting picture of the reaction enthalpy for the NH2O2 radical formation. According to earlier studies, the formation of NH2O2 is endothermic by 10 kcal mol−1, while later studies found it to be exothermic by 3–6 kcal mol−1.24–28 The latter assessments are likely more reasonable for the radical addition reaction. These types of reactions are usually exoergic processes, because the reaction only involves the formation of a bond and does not require breaking of any covalent bonds. Despite the discrepancies between the available values, the NH2 + O2 → NH2O2 reaction appears to be close to thermoneutral. This suggests that unless the reaction is prevented by an insurmountable barrier, the reaction is reversible; thus, the branching between NH2 + O2 and NH2O2 is sensitive to the accuracy of the value of the reaction enthalpy and also likely to the specific reaction conditions.
In this work, we study the NH2 + O2 ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) NH2O2 reaction using multireference electronic structure methods. The interacting NH2 + O2 system consists of three unpaired electrons, coupled to an overall doublet state, so multireference methods are necessary to obtain reasonable predictions of the shape of the reaction potential. We assess the reversibility of this reaction and estimate the branching ratios between NH2 + O2 and NH2O2 under a broad set of relevant atmospheric conditions. We estimate the high pressure limit reaction rate coefficient using canonical variational theory (CVT)29 and model the temperature- and pressure-dependent rate coefficients with Rice–Ramsberger–Kassel–Marcus (RRKM/ME)30 and inverse Laplace transform (ILT/ME) master equation models.31 We also apply the multireference methodologies to the analogous CH3 + O2 → CH3O2 reaction for comparison.
NH2O2 reaction using multireference electronic structure methods. The interacting NH2 + O2 system consists of three unpaired electrons, coupled to an overall doublet state, so multireference methods are necessary to obtain reasonable predictions of the shape of the reaction potential. We assess the reversibility of this reaction and estimate the branching ratios between NH2 + O2 and NH2O2 under a broad set of relevant atmospheric conditions. We estimate the high pressure limit reaction rate coefficient using canonical variational theory (CVT)29 and model the temperature- and pressure-dependent rate coefficients with Rice–Ramsberger–Kassel–Marcus (RRKM/ME)30 and inverse Laplace transform (ILT/ME) master equation models.31 We also apply the multireference methodologies to the analogous CH3 + O2 → CH3O2 reaction for comparison.
2 Methods
We obtained the starting geometries for various multireference calculations by carrying out geometry optimizations at the ωB97X-D3/aug-cc-pVTZ level.32–34 In some instances, we also used B3LYP(D3BJ),35–37 M06-2X,38 and CAM-B3LYP39 functionals for comparison purposes. All DFT calculations were done using ORCA version 5.0.3.,40,41 in which the used DFT functionals were implemented with analytical energy gradients for geometry optimizations, while the second derivatives of the potentials (Hessian) were calculated numerically.In all multireference calculations, we used the complete active space self-consistent field (CASSCF) method for constructing the zeroth-order wave functions for the subsequent multireference calculations.42 The used active spaces are denoted using the general n-electrons in m-orbitals, CAS(ne,mo) notation, where the configuration state function space is constructed from the Full-CI expansion within the CAS subspace. We used the perturbation-based Super-CI SCF optimizer [SuperCI(PT)] in the CASSCF calculations.43 The natural orbitals corresponding to the optimized CAS orbitals for each relevant obtained stationary structure are visualized in the ESI (Section S8).† All CASSCF and subsequent multireference (MR) calculations were done using the aug-cc-pVTZ basis set.
In the calculations with the NH2O2 molecule and related bimolecular reactants, we used two active spaces. The first, which was used for all the geometry optimizations and frequency calculations, is a CAS(13e,11o) space, which includes all valence orbitals, except the 2s orbitals of the oxygen atoms and the lone pair of the nitrogen atom. The second active space CAS(19e,14o) corresponds to the full-valence CAS of NH2O2 and was used in single-point energy correction calculations. Calculations concerning the NH2 and O2 separately were done using full-valence CAS, which are CAS(7e,6o) and CAS(12e,8o), respectively.
In the calculations of further unimolecular reactions of NH2O2, the geometries and frequencies of all intermediate species were obtained using the CAS(13e,11o) active space, but the electronic energies were corrected with the full-valence CAS(19e,14o) active space. The HNO, OH, NO, and H2O calculations were done with their respective full-valence active spaces.
For geometry optimizations and frequency calculations concerning the CH3O2 molecule and related bimolecular reactants, we used a CAS(13e,9o) active space, which consists of all oxygen valence orbitals, as well as the C–O bonding and antibonding orbitals, and the C–H bonding and antibonding orbitals are not included in the CAS. The geometry and frequencies of the isolated CH3 molecule were calculated using the full-valence CAS(7e,7o) active space.
We used N-electron valence state second-order perturbation theory (NEVPT2)44,45 to treat the dynamical electron correlation in the studied systems. All NEVPT2 calculations in this work were done using the fully internally contracted (FIC) variants of theory (in earlier literature, they were referred to as partial contraction, PC). Strict pre-screening criteria were used for higher order reduced density matrices, 3-RDM and 4-RDM (1 × 10−16 cutoff for configuration weights), to prevent false intruder states arising from approximated RDM.46,47 NEVPT2 geometry optimizations and frequency calculations were done with ORCA-5.0.3,41 using numerical gradients and numerical Hessians. We studied the NH2 + O2 → NH2O2 reaction also with CASPT2 and CASPT2-IPEA methods.48,49 The results obtained with these methods are discussed in the ESI Section S6.†
We also carried out benchmark calculations with the complete basis set extrapolated CCSD(T),50–53 using two-point extrapolation with cc-pVTZ and cc-pVQZ basis sets,54 denoted as CCSD(T)/CBS. Additionally, we conducted W2X and W3X-L composite method calculations,55 using Molpro and MRCC programs.56,57 The W2X extends the CCSD(T)/CBS method by also considering core–valence correlation and scalar-relativistic effects, upon which the W3X-L method further adds post-CCSD(T) contributions up to CCSDT(Q).
2.1 Thermodynamics and kinetics
In the thermochemical analyses, standard approaches were used for obtaining rigid-rotor, translation, and electronic contributions to their respective thermodynamic quantities. By default, all vibrational analyses were based on the harmonic approximation, but at non-stationary points of potential energy surfaces the vibrations were obtained orthogonal to the gradient of the energy (see ESI Section S2.1.1† for details). The entropy contributions from low-frequency modes were scaled with the qRRHO method,58 using 100 cm−1 reference value for the weighting function between vibrational and rotational entropies. Furthermore, the vibration corresponding to the internal rotation around the N–O bond in NH2⋯O2 was treated with a one-dimensional rigid hindered rotor model (details are provided in the ESI Section S2.1.2†).We used three methodologies to assess the kinetics of the studied reaction systems: canonical variational theory for the high-pressure limit thermal rate coefficients, kCVT∞(T) (CVT details are provided in ESI Section S2†), and Rice–Ramsberger–Kassel–Marcus (RRKM/ME) and inverse Laplace transform (ILT/ME) master equation models to calculate temperature- and pressure-dependent rate coefficients k(p, T).30,31 The CVT rate coefficient equation is given by
 | (1) |
 and the Gibbs energy of the reactants (Greac):
and the Gibbs energy of the reactants (Greac): | (2) |
The Gibbs energy of activation for the 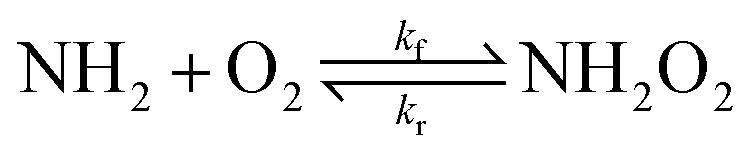 association reaction (ΔG‡,CVTf) was calculated indirectly from the reaction Gibbs energy change (ΔG) and the reverse dissociation Gibbs energy of activation (ΔG‡,CVTr):
association reaction (ΔG‡,CVTf) was calculated indirectly from the reaction Gibbs energy change (ΔG) and the reverse dissociation Gibbs energy of activation (ΔG‡,CVTr):
| ΔG‡,CVTf = ΔG + ΔG‡,CVTr | (3) |
| ΔG = G(NH2O2) − G(NH2 + O2) | (4) |
| ΔG‡,CVTr = G‡,CVTTS − G(NH2O2) | (5) |
We believe that W3X-L is the most accurate method in the present study for estimating the reaction energy [E(NH2) + E(O2) − E(NH2O2)]. Thus, we calculated the ΔG from the W3X-L electronic energies of NH2, O2, and NH2O2 and combined with the thermodynamic corrections obtained at full-valence NEVPT2 for NH2 and O2 and at NEVPT2(13e,11o) for NH2O2. We were not able to calculate the W3X-L energy in the transition state geometry, so the ΔG‡,CVTr was calculated using NEV-PT2(19e,14o) electronic energies with thermodynamic corrections obtained at the NEVPT2(13e,11o) level.
In both the RRKM/ME and ILT/ME calculations, we used a temperature independent collisional energy transfer model, where the average energy lost due to collisions with the N2 bath was set to 〈ΔE〉d = 100 cm−1. The high-pressure limit Arrhenius parameters, Ea and A, which are required for the ILT/ME simulations, were derived from Arrhenius plots of the kCVT∞(T) values at different temperatures (see ESI Section S3†). We used the standard Arrhenius equation with a temperature independent pre-exponential factor in the fitting of the kCVT∞(T) against the temperature:
| kCVT∞(T) = A × e−Ea/RT | (6) |
In the RRKM/ME calculations, the NH2 + O2 reaction is assumed to form a pre-reactive complex NH2⋯O2 with a collision limited rate coefficient (10−10 cm3 per molecule per s in our calculations), whereafter the isomerization of the complex through a transition state to the NH2O2 minimum was calculated with RRKM. In RRKM/ME and ILT/ME simulations, the rate coefficients of the reverse dissociation (kr) were obtained from the equilibrium constant (Keq) and the forward reaction pseudo-first order rate coefficient  with the following detailed balance condition:
with the following detailed balance condition:
 | (7) |
All ILT/ME and RRKM/ME simulations were done using the MESMER program version 7.0.60 The MESMER simulations are discussed in more detail in the ESI Section S3.†
3 Results and discussion
The formation of peroxyl radicals from alkyl radicals reacting with O2 is well known.61,62 Small peroxyl radicals like CH3O2 have been observed with cavity ring-down experiments.23,63 In contrast, the observation of the analogous NH2O2 radical has been elusive. In light of this, we have investigated the NH2 + O2 → NH2O2 reaction. First, the potential energy surface (PES) is explored, second the kinetics of the reaction, third the formation of NH2O2 under different atmospheric conditions, and finally we discuss the lack of detection of NH2O2 in previous experiments.3.1 Potential energy surface of NH2 + O2
In Fig. 1, we show that formation of the peroxyl radical is exoergic for both the CH3 and NH2 radicals reacting with O2. Both the CH3O2 and NH2O2 formation reactions proceed via a barrierless potential, with the alkyl peroxyl radical bound much stronger. Thus, it appears that NH2O2 formation is not hindered or prevented by unfavorable reaction energetics. A similar potential has also been found for the CH3S + O2 → CH3SO2 reaction.64 | ||
Fig. 1 Electronic potential energy curves of the R + O2 → RO2 reactions as a function of the R–O2 distance for R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH3 (dashed) and RNH2 (solid), obtained with NEVPT2(13e,9o)/aug-cc-pVTZ and NEVPT2(19e,14o)/aug-cc-pVTZ, respectively. CH3 (dashed) and RNH2 (solid), obtained with NEVPT2(13e,9o)/aug-cc-pVTZ and NEVPT2(19e,14o)/aug-cc-pVTZ, respectively. | ||
In Fig. 2, we show the reaction potential of the NH2 + O2 → NH2O2 reaction with different methods. The potential energy surface of the NH2 + O2 → NH2O2 reaction was obtained in reverse via dissociation of NH2O2 to NH2 and O2 by using the N–O distance as the reaction coordinate and carrying out a relaxed scan with respect to this coordinate from 1.45 Å to 3.00 Å distance with a step size of 0.05 Å (Fig. 2a). The geometries along the reaction potential were optimized at the multireference NEVPT2(13e,11o) level (black line in Fig. 2a), which were further corrected with full-valence NEVPT2(19e,14o) single-point energy calculations (red line in Fig. 2a). The relative energy difference between NH2 + O2 and NH2O2 was obtained by increasing the distance between NH2 and O2 to 30 Å and optimizing the geometry of the resulting structure with respect to this distance constraint and then comparing the energy with that of the minimum. In addition, the relative energy of the reactants with respect to NH2O2 was calculated with a range of other methods. These results are shown on the left-side of Fig. 2a by the scattered symbols at the ∞ mark on the horizontal axis.
 | ||
| Fig. 2 Minimum energy path of the NH2 + O2 → NH2O2 reaction as a function of the H2N–O2 distance. (a) Electronic potential energy surface with NEVPT2(13e,11o) geometry optimizations (black) and NEVPT2(19e,14o) single-point energy corrections (red). The black ★ symbols correspond to stationary points obtained at the NEVPT2(13e,11o) level, and the blue ★ is calculated with W3X-L. (b) The Gibbs energy surface of the reaction around the Gibbs energy saddle point, at 298 K and 1 atm, with electronic energies calculated at the NEVPT2(19e,14o) level and thermodynamic corrections at the NEVPT2(13e,11o) level. The orange ★ highlights the position of the NEVPT2(13e.11o) saddle point. | ||
The relative energy at the NEVPT2(19e,14o) level was obtained by optimizing the NH2 and O2 geometries separately at their respective full-valence active space NEVPT2 levels, summing up their energies, and then subtracting the NEVPT2(19e,14o)//NEVPT2(13e,11o) energy of NH2O2. The NEVPT2(13e,11o) and NEVPT2(19e,14o) reaction potentials show ca. 3 kcal mol−1 difference in the estimated association energies (Fig. 2a). The relative energies obtained with other methods are scattered between the NEVPT2(13e,11o) and NEVPT2(19e,14o) relative energies. W3X-L may be regarded as the most accurate method used in the present work, and therefore the NEVPT2(19e,14o) relative energies and PES appear more reasonable than the NEVPT2(13e,11o) energies.
The NEVPT2(13e,11o) reaction curve (black line in Fig. 2a) shows a pre-reactive van der Waals complex and a shallow submerged barrier at around 2.65 Å and 2.2 Å N–O distances, respectively (★ symbols in Fig. 2a). Despite the presence of the saddle point in the NEVPT2(13e,11o) PES, the height of the barrier is very low and is not present in the NEVPT2(19e,14o) corrected surface (red line in Fig. 2a). This means that using this point as a transition state may be inaccurate. The region around this point was inspected more closely in the corresponding Gibbs energy surface (Fig. 2b) to better define the location of the transition state of the reaction. Details of how the Gibbs energy surface was obtained are discussed in the ESI Section S2.1.† There are three distinct features in the Gibbs energy surface compared to the PES: the saddle point is more pronounced, it is shifted to a shorter N–O distance, and the pre-reactive complex is absent from the Gibbs energy surface.
Overall, the NH2 + O2 → NH2O2 reaction does not appear to be prevented by potential energy barriers; in contrast, the reaction PES shows either a negligible barrier or no barrier. The reaction is exoergic but considerably less than the analogous CH3 + O2 → CH3O2 reaction (Fig. 1), so a smaller fraction of the NH2O2 product is expected to be present under ambient conditions compared to CH3O2, due to a faster back reaction. At finite temperature, the entropy penalty associated with two molecules reacting to form one molecule introduces a clear saddle point in the Gibbs energy surface of the NH2 + O2 → NH2O2 reaction, which controls the rate of the reaction.
3.2 Kinetics of NH2 + O2
We have estimated the kinetics of the reaction with CVT, ILT/ME, and RRKM/ME (Table 1). The thermal high-pressure limit (HPL) rate coefficients k∞(T) for the forward association (kf) and reverse dissociation (kr) were calculated with the CVT method, using the maximum of the Gibbs energy surface as the transition state structure (Fig. 2b). The k∞(298 K) value, 1.1 × 10−13 cm3 per molecule per s, is eight orders of magnitude larger than the current IUPAC recommended value (10−21 cm3 per molecule per s). However, reactions involving small molecules often do not exhibit HPL kinetics at ambient pressure.65 Therefore, we studied the pressure and temperature dependence of the reaction system with the ILT and RRKM master equation models. As expected, the ILT/ME and RRKM/ME rate coefficients at 298 K and 1 atm are smaller than the HPL values obtained with CVT. The pressure dependence of kf at varying temperatures, obtained with the ILT/ME method, is illustrated in Fig. S7.† At 298 K and 1 atm, the ILT/ME and RRKM/ME rate coefficients are roughly 10−15 cm3 per molecule per s, suggesting the rapid formation of NH2O2 under atmospheric conditions.
NH2O2 reaction at 298 K
reaction with CVT, ILT/ME, and RRKM/ME (Table 1). The thermal high-pressure limit (HPL) rate coefficients k∞(T) for the forward association (kf) and reverse dissociation (kr) were calculated with the CVT method, using the maximum of the Gibbs energy surface as the transition state structure (Fig. 2b). The k∞(298 K) value, 1.1 × 10−13 cm3 per molecule per s, is eight orders of magnitude larger than the current IUPAC recommended value (10−21 cm3 per molecule per s). However, reactions involving small molecules often do not exhibit HPL kinetics at ambient pressure.65 Therefore, we studied the pressure and temperature dependence of the reaction system with the ILT and RRKM master equation models. As expected, the ILT/ME and RRKM/ME rate coefficients at 298 K and 1 atm are smaller than the HPL values obtained with CVT. The pressure dependence of kf at varying temperatures, obtained with the ILT/ME method, is illustrated in Fig. S7.† At 298 K and 1 atm, the ILT/ME and RRKM/ME rate coefficients are roughly 10−15 cm3 per molecule per s, suggesting the rapid formation of NH2O2 under atmospheric conditions.
NH2O2 reaction at 298 K
|
|
CVT (HPL) | ILT/ME (1 atm) | RRKM/ME (1 atm) |
|---|---|---|---|
| k f (cm3 per molecule per s) | 1.1 × 10−13 | 1.7 × 10−15 | 2.5 × 10−15 |
| k r (s−1) | 8.6 × 106 | 1.3 × 105 | 1.8 × 105 |

Based on our ME calculations, we find that the rate coefficient at 1 atm pressure is about two orders of magnitude lower, and at 0.1 atm pressure about 3 orders of magnitude lower, than the HPL value (Table 1) (ESI Section S3).†
Independent of the methodology used, our calculated rate coefficients of NH2O2 formation are larger than those obtained by experimentally measuring the NH2 decay rate18–21 and several orders of magnitude larger than the currently recommended rate coefficient value for the NH2 + O2 reaction by IUPAC.15,66 We hesitate to give absolute recommendations for the rate coefficient because the methods and approximations we have used also have their uncertainties. However, we carried out various sensitivity tests (ESI Section S3†) and found the rate coefficients to vary by less than two orders of magnitude.
In addition, we performed similar multireference electronic structure calculations and ME simulations for the analogous CH3 + O2 → CH3O2 system (see ESI Sections S1 and S3†), for which a lot of experimental kinetics data are available (see ref. 66 for example). Our k(p, T) values for that system are in reasonable agreement with the currently recommended values.66–68 The experimental rate coefficient for the CH3 + O2 → CH3O2 reaction at 298 K and 1 atm is 8.1 × 10−13 cm3 per molecule per s,68 while our calculated values under these conditions are 2.1 × 10−13 and 1.1 × 10−12 cm3 per molecule per s, with ILT/ME and RRKM/ME, respectively (ESI Table S2†), both of which are in good agreement with the experimental value.
3.3 Atmospheric presence of NH2O2
Whether formation of NH2O2 has any effect on atmospheric processes depends on the stability of NH2O2 with respect to back-dissociation, i.e. its fraction at equilibrium compared with the loss rates of NH2 and NH2O2 with respect to other competing reactions.The competition of bimolecular reactions of NH2 with NO, NO2, and O3 can be estimated by comparing the pseudo-first order rate coefficients of these reactions. An upper limit for these competing bimolecular reactions would be that they occur with a rate coefficient near the collision limit of around 10−10 cm3 per molecule per s and that the combined concentration of these reactants is 40 ppb, which leads to a pseudo-first order rate coefficient of 100 s−1.69 The actual pseudo-first order rate coefficient is likely much smaller than this upper limit.
The reactions of NH2 with NOx and O3 are considerably faster than the reaction with O2, but the concentration of O2 is substantially higher than the NOx and O3 concentrations. With our calculated rate coefficients in Table 1 (2 × 10−15 cm3 per molecule per s) and the atmospheric O2 concentration, the pseudo-first order rate coefficient of NH2 + O2 → NH2O2 is 104 s−1, thus the reaction is much faster than other bimolecular reactions. The back reaction NH2O2 → NH2 + O2 is even faster than the forward reaction (Table 1). This suggests that the NH2 + O2 NH2O2 equilibrium is established before the competing bimolecular reactions start consuming either NH2 or NH2O2. Even if the calculated rate coefficients in Table 1 were 1–2 orders of magnitude too high, this would still hold.
Then, if the NH2 + O2 NH2O2 reaction has reached equilibrium and there are no reactions affecting the equilibrium, the fraction of NH2O2 can be easily calculated. At equilibrium, kf[NH2][O2] = kr[NH2O2], hence the ratio [NH2O2]/[NH2] = (kf/kr)[O2]. Our calculations with ILT/ME and RRKM/ME suggest that at 298 K and 1 atm (with 21% O2 partial pressure), the fraction of NH2O2 is 6.4%. We carried out ILT/ME simulations of NH2 + O2 NH2O2 with a broad set of conditions to show how the equilibrium fraction of NH2O2 varies with temperature and pressure. In Fig. 3a, we show the fraction of NH2O2 as a function of temperature (230–330 K) at four total pressures: 700, 570, 400, and 200 torr, with 21% O2 partial pressure. Fig. 3b shows the NH2O2 equilibrium fraction as a function of altitude (0–10 km), for global (288 K, solid)70 and polar (263 K, dashed) average surface temperatures. The temperatures and pressures at various altitudes were calculated with the barometric formula, using the temperature lapse rate of −6.5 K km−1 (ESI Section S4†).
 | ||
| Fig. 3 Fraction of NH2O2 under typical tropospheric conditions: (a) at total pressures of 200 torr (black dotted line), 400 torr (blue dotted line), 570 torr (red dotted line), and 760 torr (orange dotted line), in the temperature range 230–330 K; (b) as a function of altitude of 0–10 km, with the global average surface temperature 288 K (solid line) and polar average surface temperature 263 K (dashed line). All calculations were carried out with 21% O2 concentration, using the ILT/ME method. | ||
The formation of NH2O2 is more pronounced at lower temperatures and higher pressures, which is explained by the entropy effect (Fig. 3a). The average temperature of Earth's atmosphere at the ground level is 288 K, where 10% of formed NH2 will add O2 forming NH2O2 (Fig. 3b). In polar regions, where the mean surface temperature is 263 K, the NH2O2 fraction is much larger at the ground level (32%); therefore, it is likely an important pathway in NH3 oxidation. At high altitudes (Fig. 3b), where both the temperature and pressure are low, the fraction of NH2O2 becomes substantial.
3.4 Unimolecular reactions of NH2O2
It is critical to assess whether there are any unimolecular reactions of NH2O2 that would affect the NH2 + O2 NH2O2 equilibrium. In previous literature, it has been demonstrated that the most exothermic products of the NH2 + O2 reaction would be NO + H2O and HNO + OH, the prior being the thermodynamically preferred product channel.15 We calculated the unimolecular reaction pathways that connect the NH2O2 to these product channels. The Gibbs energy profile of these reactions at 298 K and 1 atm is shown in Fig. 4. We also validated some of the calculated energy asymptotes in Fig. 4 against reaction enthalpies in the Active Thermochemical Tables database71 (see ESI Section S7† for details).
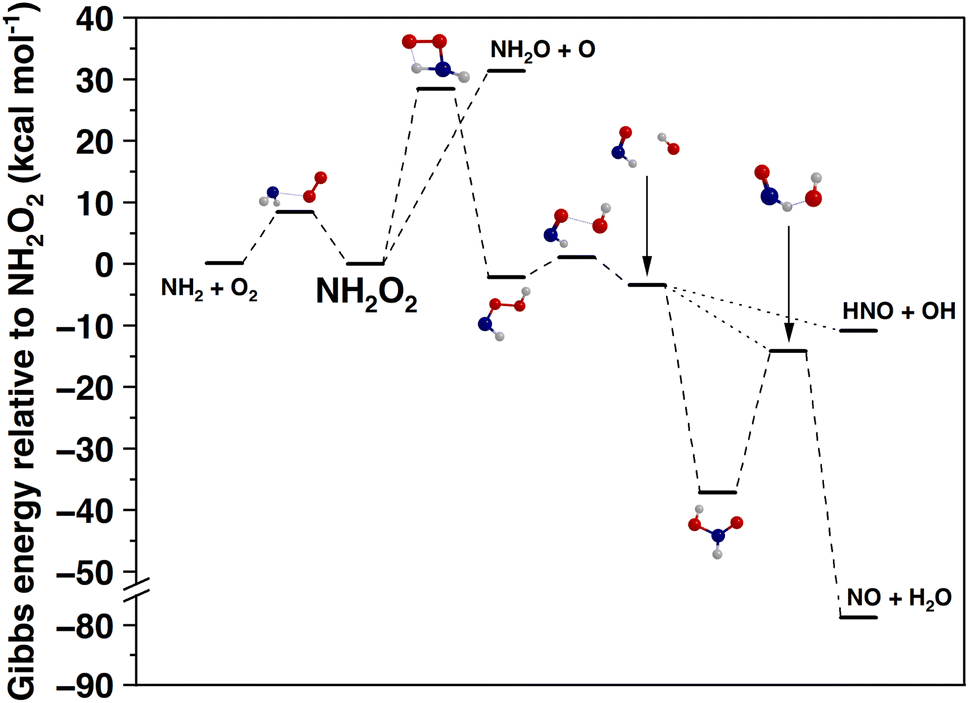 | ||
| Fig. 4 Gibbs energy diagram of the unimolecular reactions of NH2O2, including those leading to HNO and NO formation, calculated at the NEVPT2 level (see details in the Methods section), at 298 K and 1 atm. Red = oxygen, blue = nitrogen, and gray = hydrogen. Zero energy corresponds to the Gibbs energy of NH2O2. | ||
After the initial association of NH2 and O2, the formed NH2O2 has three accessible unimolecular reactions: the reverse dissociation to NH2 + O2, isomerization to the HNOOH radical via intramolecular hydrogen atom transfer, and dissociation to the aminoxyl radical and atomic oxygen (NH2O + O). The last reaction has been deemed unlikely to be competitive under ambient conditions,28 which is also supported by our findings: the reaction is endergonic by 31.4 kcal mol−1 at 298 K and 1 atm (Fig. 4). The isomerization of NH2O2 to HNOOH, which ultimately leads to the formation of NO and HNO, occurs via a tight transition state, with a Gibbs energy barrier height of 28.5 kcal mol−1 (Fig. 4).
The hydrogen atom transfer reaction (H-shift) is accelerated by quantum mechanical tunneling, which we estimated using the Eckart potential approximation.59 We simulated the hydrogen atom transfer reaction at 298 K and 1 atm pressure using RRKM/ME and determined a rate coefficient of 7.6 × 10−6 s−1 including tunneling. The effective rate coefficient for the overall reaction from NH2 + O2 to HNOOH, which we use as a proxy for NO production, is calculated by combining ILT/ME for the NH2 + O2 → NH2O2 reaction with a RRKM/ME for the NH2O2 → HNOOH reaction. This gives 5.8 × 10−25 cm3 per molecule per s at 298 K and 1 atm. Thus, this pathway is insignificant under ambient conditions. This small rate coefficient is in qualitative agreement with the IUPAC recommendation.15,66
It is evident that under atmospheric conditions, the only plausible reactions of NH2O2 are the reverse dissociation back to NH2 + O2 or further bimolecular reactions. In this work, we did not explicitly model any of these further bimolecular reactions. Because NH2O2 is a peroxyl radical, its most likely further bimolecular reactions are with NO, NO2, HO2, and other RO2. If we estimate that the bimolecular rate coefficients of NH2O2 are comparable to those of alkyl RO2, which are known to be in the range of 10−10–10−13 cm3 per molecule per s,72 the lifetimes of NH2 (excluding the reaction with O2) and NH2O2 are similar.
It is known that peroxyl radicals (RO2) can react bimolecularly with NO2, NO, HO2 and other RO2 to form e.g. peroxy nitrates (ROONO2), organic nitrates (RONO2), hydroperoxides (ROOH), alcohols (ROH), carbonyl compounds (R–HO), alkoxyl radicals (RO), and organic peroxides (ROOR).72 For the aminoperoxyl radical NH2O2, the corresponding products would be NH2OONO2, NH2ONO2, NH2OOH, NH2OH, and NH2OOR, which are different compound classes to those currently expected as products of NH3 oxidation (NO, N2O, and N2).9 The bimolecular reactions of NH2O2 will likely also produce the NH2O radical, which is also known to form in the bimolecular reactions of NH2 with O3, NO2, and HO2.14,21,73 Therefore, the substantial branching ratio of NH2O2 may lead to previously undiscovered nitrogen-containing compounds in the atmosphere and influence the current atmospheric modeling.
3.5 Previous experiments
There have been several experimental investigations of the reaction between NH2 and O2 by measuring the NH2 decay rate in the presence of O2, and the results suggest that the reaction between NH2 and O2 is negligible. However, these experiments have been conducted largely under either low-pressure or high-temperature conditions, where the reaction between NH2 and O2 forming NH2O2 is not expected to play a substantial role based on our calculations. The calculated fraction of NH2O2 under different experimental conditions is given in the ESI Section S5.†The pulse radiolysis experiments of NH3 by Pagsberg et al.19 showed that, at 350 K, the NH2 decay rate is independent of O2 concentration, even at their highest O2 concentration experiment (42% O2 at 705 torr). In agreement, our results show that the fraction of NH2O2 is 1.5% under these conditions (ESI Table S4†), and thus the decay of NH2 would be largely independent of O2.
Cheskis and Sarkisov18 carried out room temperature flash photolysis experiments of NH3 at 100 and 570 torr total pressures, with varying O2 percentage. They observed significant enhancement in the NH2 decay at 1 torr O2 partial pressure and 570 torr total pressure (≈0.2% O2) compared to a similar experiment without oxygen. The authors explained that the increased decay rate was unlikely to be due to the reaction between NH2 and O2 but instead was due to their experimental setup. In the experiment, the NH2 radicals were generated via NH3 photolysis, yielding H radicals that can react with O2 to form HO2, which reacted irreversibly with NH2, resulting in the decay of NH2. This is in agreement with our results, which show that at such low O2 percentage, the formation of NH2O2 was insignificant (0.05%, ESI Table S4†). However, no increase in the NH2 decay rate was observed when further increasing the O2 pressure to 100 and 570 torr in the experiments (18% and 100% O2 at 570 torr total pressure). These observations are in contrast to our calculations, which suggest that the fraction of NH2O2 was 4% and 20% under these conditions, respectively (ESI Table S4†), which would affect the NH2 decay rate. The NH2 decay was saturated because significant NH2 decay rate was also observed upon increasing the N2 pressure from 100 torr to 570 torr without any O2 in the experiments.
Patrick and Golden21 conducted experiments in the temperature range 272–348 K where NH2 was generated via O3 photolysis forming atomic O, which reacted with NH3 to form NH2. The experiments with O2 were carried out at low total pressures of 230–240 torr and O2 partial pressures of only 0–14 torr (max. 6% O2). Our calculations show that the NH2O2 fraction is less than 3% at such low O2 pressures (ESI Table S4†). No significant reactivity between NH2 and O2 was observed in the experiments, in agreement with our results. Experiments with higher O2 partial pressures cannot be conducted in this setup because the reaction between O2 and atomic O would suppress the formation of NH2.
4 Conclusions
We have carried out high-level multireference calculations and kinetic modeling for studying the formation of the NH2O2 radical via the association reaction between NH2 and O2, which is a crucial step in the atmospheric oxidation of NH3. We find that the association reaction is much faster than the current IUPAC estimate and that NH2 + O2 is the dominant bimolecular reaction of NH2 in the atmosphere. Our calculations encompass a broad range of atmospherically relevant conditions, and we show that NH2O2 formation can play an important role in NH3 oxidation, especially under low-temperature and high-pressure conditions, where a substantial equilibrium fraction of NH2O2 is expected to be present. We encourage laboratory experiments to directly detect the NH2O2 radical and determine the rate of the NH2 + O2 reaction under relevant conditions.Data availability
Calculation output files related to the study are available in the ESI† and at http://doi.org/10.5281/zenodo.12657070.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Research Council of Finland Centre of Excellence: Virtual Laboratory for Molecular-Level Atmospheric Transformations (grant 346369), the Jane and Aatos Erkko (JAES) Foundation, and the Villum Fonden (VIL50443) for funding. We also thank the CSC – IT Center of Science (Finland) for providing the supercomputing platforms and computational resources.References
- L. Liu, W. Xu, X. Lu, B. Zhong, Y. Guo, X. Lu, Y. Zhao, W. He, S. Wang and X. Zhang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2121998119 CrossRef CAS PubMed.
- R. Zhu, J. Sun, Y. Liu, Z. Gong and L. Sun, Antarct. Sci., 2011, 23, 78–92 CrossRef.
- J. Lindaas, I. B. Pollack, J. J. Calahorrano, K. O'Dell, L. A. Garofalo, M. A. Pothier, D. K. Farmer, S. M. Kreidenweis, T. Campos and F. Flocke, et al. , J. Geophys. Res.: Atmos., 2021, 126, e2020JD033730 CrossRef CAS.
- S. Riddick, T. Blackall, U. Dragosits, F. Daunt, M. Newell, C. Braban, Y. Tang, J. Schmale, P. Hill and S. Wanless, et al. , Atmos. Environ., 2016, 134, 40–50 CrossRef CAS.
- J. Plautz, Science, 2018, 361, 1060–1063 CrossRef CAS PubMed.
- R. Pinder, A. Gilliland and R. Dennis, Geophys. Res. Lett., 2008, 35, L12808 CrossRef.
- S. N. Behera, M. Sharma, V. P. Aneja and R. Balasubramanian, Environ. Sci. Pollut. Res., 2013, 20, 8092–8131 CrossRef CAS PubMed.
- N. Evangeliou, Y. Balkanski, S. Eckhardt, A. Cozic, M. Van Damme, P.-F. Coheur, L. Clarisse, M. W. Shephard, K. E. Cady-Pereira and D. Hauglustaine, Atmos. Chem. Phys., 2020, 2020, 1–41 Search PubMed.
- S. J. Pai, C. L. Heald and J. G. Murphy, ACS Earth Space Chem., 2021, 5, 1674–1685 CrossRef CAS.
- J. M. Roberts, A. O. Langford, P. D. Goldan and F. C. Fehsenfeld, J. Atmos. Chem., 1988, 7, 137–152 CrossRef CAS.
- R. D. Stephens, J. Phys. Chem., 1984, 88, 3308–3313 CrossRef CAS.
- C. P. Ennis, J. R. Lane, H. G. Kjaergaard and A. J. McKinley, J. Am. Chem. Soc., 2009, 131, 1358–1359 CrossRef CAS PubMed.
- M. Monge-Palacios and J. Espinosa-Garcia, J. Phys. Chem. A, 2010, 114, 4418–4426 CrossRef CAS PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2004, 4, 1461–1738 CrossRef CAS.
- G. Tyndall, J. Orlando, K. E. Nickerson, C. Cantrell and J. Calvert, J. Geophys. Res.: Atmos., 1991, 96, 20761–20768 CrossRef CAS.
- H. Gesser, J. Am. Chem. Soc., 1955, 77, 2626–2629 CrossRef CAS.
- R. Jayanty, R. Simonaitis and J. Heicklen, J. Phys. Chem., 1976, 80, 433–437 CrossRef CAS.
- S. Cheskis and O. Sarkisov, Chem. Phys. Lett., 1979, 62, 72–76 CrossRef CAS.
- P. B. Pagsberg, J. Eriksen and H. Christensen, J. Phys. Chem., 1979, 83, 582–590 CrossRef CAS.
- W. Hack, O. Horie and H. G. Wagner, J. Phys. Chem., 1982, 86, 765–771 CrossRef CAS.
- R. Patrick and D. M. Golden, J. Phys. Chem., 1984, 88, 491–495 CrossRef CAS.
- R. L. Caravan, M. A. H. Khan, J. Zádor, L. Sheps, I. O. Antonov, B. Rotavera, K. Ramasesha, K. Au, M.-W. Chen and D. Rösch, et al. , Nat. Commun., 2018, 9, 4343 CrossRef PubMed.
- M. B. Pushkarsky, S. J. Zalyubovsky and T. A. Miller, J. Chem. Phys., 2000, 112, 10695–10698 CrossRef CAS.
- C. Pouchan and M. Chaillet, Chem. Phys. Lett., 1982, 90, 310–316 CrossRef CAS.
- C. F. Melius and J. S. Binkley, ACS Symp. Ser., 1984, 103–115 CAS.
- J. W. Bozzelli and A. M. Dean, J. Phys. Chem., 1989, 93, 1058–1065 CrossRef CAS.
- R. Sumathi and S. D. Peyerimhoff, J. Chem. Phys., 1998, 108, 5510–5521 CrossRef CAS.
- S. J. Klippenstein, L. B. Harding, P. Glarborg and J. A. Miller, Combust. Flame, 2011, 158, 774–789 CrossRef CAS.
- J. L. Bao and D. G. Truhlar, Chem. Soc. Rev., 2017, 46, 7548–7596 RSC.
- R. A. Marcus, J. Chem. Phys., 1952, 20, 359–364 CrossRef CAS.
- J. W. Davies, N. J. Green and M. J. Pilling, Chem. Phys. Lett., 1986, 126, 373–379 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- B. O. Roos, P. R. Taylor and P. E. Sigbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- C. Kollmar, K. Sivalingam, B. Helmich-Paris, C. Angeli and F. Neese, J. Comput. Chem., 2019, 40, 1463–1470 CrossRef CAS PubMed.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS.
- Y. Guo, K. Sivalingam and F. Neese, J. Chem. Phys., 2021, 154, 214111 CrossRef CAS PubMed.
- C. Kollmar, K. Sivalingam, Y. Guo and F. Neese, J. Chem. Phys., 2021, 155, 234104 CrossRef CAS PubMed.
- K. Andersson, P. A. Malmqvist, B. O. Roos, A. J. Sadlej and K. Wolinski, J. Phys. Chem., 1990, 94, 5483–5488 CrossRef CAS.
- G. Ghigo, B. O. Roos and P.-Å. Malmqvist, Chem. Phys. Lett., 2004, 396, 142–149 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- S. Zhong, E. C. Barnes and G. A. Petersson, J. Chem. Phys., 2008, 129, 184116 CrossRef PubMed.
- F. Neese and E. F. Valeev, J. Chem. Theory Comput., 2011, 7, 33–43 CrossRef CAS PubMed.
- T. Helgaker, W. Klopper, H. Koch and J. Noga, J. Chem. Phys., 1997, 106, 9639–9646 CrossRef CAS.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- B. Chan and L. Radom, J. Chem. Theory Comput., 2015, 11, 2109–2119 CrossRef CAS PubMed.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, I. Miller, F. Thomas, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- M. Kállay, P. R. Nagy, D. Mester, Z. Rolik, G. Samu, J. Csontos, J. Csóka, P. B. Szabó, L. Gyevi-Nagy, B. Hégely, I. Ladjánszki, L. Szegedy, B. Ladóczki, K. Petrov, M. Farkas, P. D. Mezei and Á. Ganyecz, J. Chem. Phys., 2020, 152, 074107 CrossRef PubMed.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- R. L. Brown, J. Res. Natl. Bur. Stand., 1981, 86, 357–359 CrossRef CAS PubMed.
- D. R. Glowacki, C.-H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS PubMed.
- P. D. Lightfoot, R. Cox, J. Crowley, M. Destriau, G. Hayman, M. Jenkin, G. Moortgat and F. Zabel, Atmos. Environ., Part A, 1992, 26, 1805–1961 CrossRef.
- J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- A. Bossolasco, E. P. Faragó, C. Schoemaecker and C. Fittschen, Chem. Phys. Lett., 2014, 593, 7–13 CrossRef CAS.
- J. Chen, J. R. Lane, K. H. Bates and H. G. Kjaergaard, Environ. Sci. Technol., 2023, 57, 21168–21177 CrossRef CAS PubMed.
- B. M. Wong, D. M. Matheu and W. H. Green, J. Phys. Chem. A, 2003, 107, 6206–6211 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe and IUPAC Subcommittee, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- R. X. Fernandes, K. Luther and J. Troe, J. Phys. Chem. A, 2006, 110, 4442–4449 CrossRef CAS PubMed.
- J. B. Burkholder, S. P. Sander, J. Abbatt, J. R., C. Cappa, J. D. Crounse, T. S. Dibble, R. E. Huie, C. E. Kolb, M. J. Kurylo, V. L. Orkin, C. J. Percival, D. M. Wilmouth and P. H. Wine, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, JPL Publication, Jet Propulsion Laboratory, Pasadena, 19, 2019 Search PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, 2016 Search PubMed.
- Atmospheric Administration and United States Committee on Extension to the Standard Atmosphere, US Standard Atmosphere, National Oceanic and Atmospheric Administration, 1976 Search PubMed.
- B. Ruscic, R. E. Pinzon, M. L. Morton, G. von Laszevski, S. J. Bittner, S. G. Nijsure, K. A. Amin, M. Minkoff and A. F. Wagner, J. Chem. Phys. A, 2004, 108, 9979–9997 CrossRef CAS.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- P. Glarborg, H. Hashemi, S. Cheskis and A. W. Jasper, J. Phys. Chem. A, 2021, 125, 1505–1516 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further details and discussion of the CH3 + O2 → CH3O2 calculations, CVT calculations, Gibbs energy surface calculations, master equation simulations and sensitivity tests, NH2 + O2 → NH2O2 reaction potentials calculated with CASPT2 and CASPT2-IPEA methods, structural parameters and visualizations of CAS orbitals of relevant structures obtained at the NEVPT2 level of theory, and xyz-coordinates of the studied molecules. See DOI: https://doi.org/10.1039/d5ea00042d |
| This journal is © The Royal Society of Chemistry 2025 |