
Topical BET PROTACs for locally restricted protein degradation in the lung†
Martin Hemmerling *a,
Jianming Liub,
Antonio Pirasc,
Rikard Pehrsond,
Ulf Hedströme,
Carlo Cassania,
Beatrice Ranieria,
Karolina Kwapieńa,
Karin Ribbinge,
Cecilia Forsse,
Oliwia Slettengrene,
Frederik Eiseleb,
Mei Dingb,
Pia Hanssonb,
Anna Novénb,
Markus Nordbergb,
Hyunsoo Parkb,
Annica Jarkef,
Lisa-Catherine Rosenbauma,
Jesper Malmberga,
Annika Bordec,
Lassina Badolod,
Madeleine Engsevie,
Johan Jirholte,
Perla Brecciag,
Stefan Schiessera,
Lena Ripaa and
Werngard Czechtizkya
*a,
Jianming Liub,
Antonio Pirasc,
Rikard Pehrsond,
Ulf Hedströme,
Carlo Cassania,
Beatrice Ranieria,
Karolina Kwapieńa,
Karin Ribbinge,
Cecilia Forsse,
Oliwia Slettengrene,
Frederik Eiseleb,
Mei Dingb,
Pia Hanssonb,
Anna Novénb,
Markus Nordbergb,
Hyunsoo Parkb,
Annica Jarkef,
Lisa-Catherine Rosenbauma,
Jesper Malmberga,
Annika Bordec,
Lassina Badolod,
Madeleine Engsevie,
Johan Jirholte,
Perla Brecciag,
Stefan Schiessera,
Lena Ripaa and
Werngard Czechtizkya
aMedicinal Chemistry, Research and Early Development, Respiratory and Immunology, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden. E-mail: martin.hemmerling@astrazeneca.com
bAssays, Profiling & Cell Science, Discovery Sciences, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
cBioscience In Vivo, Research and Early Development, Respiratory and Immunology, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
dDMPK, Research and Early Development, Respiratory and Immunology, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
eBioscience COPD/IPF, Research and Early Development, Respiratory and Immunology, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
fAdvanced Drug Delivery, Pharmaceutical Sciences, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
gMedicinal Chemistry, Neuroscience, BioPharmaceuticals R&D, AstraZeneca, Cambridge, UK
First published on 21st April 2025
Abstract
Proteolysis targeting chimeras (PROTACs) have been studied extensively to optimize their oral bioavailability. Based on Lipinski's and Veber's rules, molecular weight, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, logD, the number of hydrogen bond donors and acceptors, the polar surface area and the number of rotatable bonds play a critical role for the oral bioavailability of any given PROTAC. Multiple analyses of the published PROTAC chemical space show that the overall guidelines for obtaining orally available PROTACs are broadly in line with those of the bRo5 space established for oral drugs. In contrast to the significant knowledge that has been generated for the design of oral PROTACs, there is little expertise regarding inhaled and generally topically administered PROTACs. In this work we (1) introduce parameters influencing the inhaled route of administration of PROTACs and (2) describe the first examples of inhaled bromodomain and extra terminal domain (BET) PROTACs which were designed for inhaled delivery and characterized in vitro and in vivo.
P, logD, the number of hydrogen bond donors and acceptors, the polar surface area and the number of rotatable bonds play a critical role for the oral bioavailability of any given PROTAC. Multiple analyses of the published PROTAC chemical space show that the overall guidelines for obtaining orally available PROTACs are broadly in line with those of the bRo5 space established for oral drugs. In contrast to the significant knowledge that has been generated for the design of oral PROTACs, there is little expertise regarding inhaled and generally topically administered PROTACs. In this work we (1) introduce parameters influencing the inhaled route of administration of PROTACs and (2) describe the first examples of inhaled bromodomain and extra terminal domain (BET) PROTACs which were designed for inhaled delivery and characterized in vitro and in vivo.
Introduction
Our interest in inhaled PROTACs derives from three main characteristics PROTACs provide as compared to classical small molecule inhibitors. These include (1) their inherent propensity for tissue retention in lung and skin based on their compound properties, (2) their differentiated mode of action from inhibitors, and (3) the advantages they can offer regarding a reduced systemic toxicity.Inhaled synthetic molecules are predominantly retained in the lung either due to their lipophilicity/limited solubility and precipitation/dissolution characteristics, or due to basic properties causing prolonged interaction with lysosomes in the lung.1–3 PROTACs inherently carry a significant propensity for lung retention due to their structural properties; many being highly lipophilic with low overall solubility, and/or incorporating basic groups in either the target of interest binder and/or the linker.
PROTACs can be highly effective at low doses due to their mode of action, particularly when the target protein resynthesis rate is slow. Even with significant clearance from the tissue, a therapeutically relevant exposure for a short period is sufficient to maintain reduced protein expression for an extended time. PROTACs only require transient binding to the target of interest without the need for prolonged high-affinity binding to the target. PROTACs are recycled and can work catalytically, reducing the required overall drug exposure.4,5 This applies in contrast to small molecule inhibitors which require a long enough exposure to exert sufficient and durable target engagement over hours.
The potential PK/PD disconnect for PROTACs can further facilitate addressing targets of the distal airways and the alveoli. These are highly perfused regions in the lung with little potential to retain enough of a small molecule inhibitor to have sufficient exposure. Small molecule inhibitors are usually very rapidly cleared without being able to maintain target engagement for targets in the periphery of the lung.6,7
Interestingly, Chen et al. have recently used a similar hypothesis for the development of topical Janus kinase (JAK) 1/2 PROTACs for skin indications.8 Topical formulations of tofacitinib (a JAK1/3 inhibitor) and delgocitinib (a pan-JAK inhibitor) have had limited efficacy as topical treatments for AD due to high threshold dose and low skin reservoir levels.9,10 Remarkably, with topical administration, JAK PROTACs exhibited superior potency as compared to e.g. ruxolitinib.8 The team demonstrated the effective degradation of JAK1/2 proteins in vitro and in vivo and proposed that their JAK1/2 PROTAC exhibited high efficacy while requiring lower dose and less frequent administration than any of the inhibitors. This could limit off-target effects seen with oral administration of inhibitors and dose restrictions caused by limited skin reservoirs of inhibitors upon topical administration.8
Bromodomain and extra terminal domain (BET) PROTACs
Modulation of BET protein activity, either by silencing with bromodomain-containing protein 4 (BRD4) siRNA or by the action of pan-BET inhibitors, was found to halt fibrotic processes in lung derived cells, fibroblasts and epithelial cells, as well as in different animal models for lung remodelling.11–13 Thus, targeting BRD4 could present a novel approach to treat interstitial lung diseases such as idiopathic pulmonary fibrosis (IPF). However, clinical trials with oral BET inhibitors in cancer patients revealed dose-limiting toxicities.14We planned to develop inhaled molecules with limited systemic exposure to reduce potential severe, systemic adverse effects such as platelet or gastrointestinal toxicity.14,15 Due to the necessity to treat the peripheral fibrotic IPF lung and to achieve a durable local effect in highly perfused tissue such as the alveoli, we selected PROTACs rather than inhibitors as a suitable modality. This based on the hypothesis that the degradation mode-of-action might provide a hit-and-run type of mechanism where the pharmacological effect can outlast the actual presence of the drug in the targeted tissue.4,5
Inhaled BET PROTACs for local treatment of the lung were not previously described. A review of published BET PROTACs pointed at cereblon (CRBN)-recruiting degraders as most common and potent degraders (Chart 1). Among those, QCA570,16 BETd-260,17 dBET23,18 or ZXH-3-2618 exploit the ZA channel of the BET protein with the option to place a linker between BET and CRBN binders, offering a rational design for a quick identification of highly potent BET PROTACs.
 | ||
| Chart 1 CRBN-recruiting BET-PROTACs. | ||
Results and discussion
Design of BET PROTACs for inhalation
We selected a suitable BET binding scaffold based on the following criteria: (1) low lipophilicity, anticipating an inevitable increase of molecular weight and lipophilicity upon addition of linker and CRBN binder; (2) similar affinity to both acetyl lysine binding domains to provide freedom for ternary complex formation engaging either binding domain; (3) a suitable handle to link to the CRBN binder outside of the BRD4 protein, with the trajectory along the ZA channel considered as most promising; (4) an intermediate binding affinity to enable dissecting inhibitory and degradation mode-of-action and to simplify understanding of the observed pharmacology. After a comprehensive literature review, we selected the GSK-derived benzimidazole 119–21 and the fragment 2 (Scheme 1 and Fig. 1) as starting points; superimposition of the ternary complex of dBET2318 with BRD4 and CRBN (Fig. 1) indicates that extension of the amide residue of benzimidazole 2 through the ZA channel should enable the recruitment of CRBN. | ||
| Scheme 1 Synthesis of PROTACs 3–6 with polar linkers (Table 1). Reagents and conditions: (a) HATU, DIPEA, DMF, rt. | ||
 | ||
| Fig. 1 Docking of benzimidazole 2 (magenta sticks) into the structure of the ternary complex of dBET23 (see Chart 1; yellow sticks) with BRD4-BD1 (light grey surface) and CRBN (light blue surface). ZA = loop connecting the αZ and αA helices; WPF = TrpProPhe motif; BD = binding domain. | ||
Synthesis of PROTACs
PROTACs 3–6 (Scheme 1, Table 1) are derivatives of the BET inhibitor 121 and were prepared by HATU-mediated amide couplings of the threonine-extended-(1H-benzo[d]imidazol-2-yl)pyridin-2-one 2420 and commercial primary amines of the generic structure 23 (Scheme 1).| Compound | logDa |
BRD2 bindingb,c pIC50 | BRD4 degradationb,d | ||
|---|---|---|---|---|---|
| BD1 | BD2 | pDC50 | Dmax (%) | ||
| a Distribution coefficient (logD7.4) between octanol and 10 mM sodium phosphate buffer with the solution adjusted to pH 7.4.b Values are means of at least two independent experiments with two replicates each, unless otherwise indicated. For standard error see Table S1.†c Compound binding to BRD2 BD1 or BD2 by displacement of a fluorescent Alexafluor 647-JQ-1-like probe in −log10(M). The 95% confidence interval of pIC50 values was ±0.24 for BD1 and ±0.23 for BD2.d NanoGlo degradation assay in a HEK293 cell line expressing human BRD4-HiBit fusion at endogenous expression levels in −log10(M). The 95% confidence interval of pDC50 values was ±0.14.e One test occasion. |
|||||
| dBET6 | 7.36 | 7.27 | 8.32 | 100 | |
| ZXH-3-26 | 7.63e | 7.51e | 8.15 | 100 | |
| 2 | 1.25 | 7.26 | 6.89 | <5.0e | NV |
| 3 | −0.02 | 7.18 | 6.82 | 6.53e | 100 |
| 4 | 0.29 | 7.19 | 6.74 | 6.42 | 98 |
| 5 | 0.30 | 7.26e | 6.93e | 6.76 | 78 |
| 6 | 0.19 | 7.19e | 6.82e | 7.29 | 100 |
The synthesis of PROTACs 7–14 (Scheme 2, Table 2) started with a Sonogashira coupling22 of 1-hydroxy-alkyl acetylene (31) with 4- or 5-bromo-thalidomide. The resulting primary alcohols 25 were transformed into the corresponding azides 27 in two steps via mesylation and subsequent azidation. An alternative route was used for the preparation of the azide 27a, featuring an ether linker, when the acetylene 32 was coupled to obtain the BOC-protected primary amine 28. After deprotection, FSO2N3-mediated diazotransfer23 to the resulting primary amine produced the azide 27a. The final PROTACs 7–14 (Table 2) were obtained by a copper-mediated triazole formation of the azides 27 and the acetylene 30 that was prepared by a Seyferth–Gilbert homologization of the aldehyde 29 using the Bestmann–Ohira reagent.19–21,24
 | ||
| Scheme 2 Synthesis of PROTACs 7–14 with lipophilic linker design. Reagents and conditions: (a) TEA, CuI, PdCl2(PPh3)2, DMF, 80 °C; (b) (MsO)2O, TEA, DMAP, DCM 0 °C to rt; (c) Me3SiN3, TBAF, DMF, 60 °C; (d) HCl (4 M in dioxane), rt; (e) FSO2N3, CuSO4, K2CO3, MeOH, rt; (f) L-ascorbic acid 2-phosphate trisodium salt, CuSO4, 1,4-dioxane/water (1:1), rt; (g) dimethyl (1-diazo-2-oxopropyl)phosphonate, K2CO3, MeOH, 0 °C to rt. | ||
| Compound | logDa |
BRD2 bindingb,c pIC50 | BRD4 degradationb,d | ||
|---|---|---|---|---|---|
| BD1 | BD2 | pDC50 | Dmax (%) | ||
| a Distribution coefficient (logD7.4) between octanol and 10 mM sodium phosphate buffer with the solution adjusted to pH 7.4.b Values are means of at least two independent experiments with two replicates each, unless otherwise indicated. For standard deviation see Table S1.†c Compound binding to BRD2 BD1 or BD2 by displacement of a fluorescent Alexafluor 647-JQ-1-like probe in −log10(M). The 95% confidence interval of pIC50 values was ±0.24 for BD1 and ±0.23 for BD2.d NanoGlo degradation assay in a HEK293 cell line expressing human BRD4-HiBit fusion at endogenous expression levels in −log10(M). The 95% confidence interval of pDC50 values was ±0.14.e nt = not tested. |
|||||
| 7 | 0.83 | 7.21 | 6.81 | 7.82 | 111 |
| 8 | 2.4 | nte | nte | 9.83 | 101 |
| 9 | 2.4 | 7.34 | 6.87 | 9.23 | 101 |
| 10 | 2.9 | 7.35 | 7.14 | 9.59 | 104 |
| 11 | 2.3 | 7.64 | 7.30 | 9.60 | 99 |
| 12 | 2.4 | 7.51 | 6.94 | 9.34 | 100 |
| 13 | 3.2 | 7.44 | 7.15 | 9.72 | 100 |
| 14 | >4.0 | 7.04 | 6.62 | 9.05 | 100 |
The PROTACs presented in Table 3 were derived from the dihydrouracils 33 and 34.25 A propylamine linker was attached to 33 by reductive amination to yield the primary amine 35. Subsequent HATU-mediated amide coupling with the acid 38 (Scheme 4) provided the amide 15 (Scheme 3). Alkylation of the dihydrouracils 33 and 34 with commercial alkyl tosylates featuring a terminal azide function provided the azides 36 and 37 that served as partners for the acetylene 30 in a final click-reaction to provide the triazole-linked PROTACs 16–20 (Table 3, Scheme 3).
| Compound | logDa |
BRD2 bindingb,c pIC50 | BRD4 degradationb,d | ||
|---|---|---|---|---|---|
| BD1 | BD2 | pDC50 | Dmax (%) | ||
| a Distribution coefficient (logD7.4) between octanol and 10 mM sodium phosphate buffer with the solution adjusted to pH 7.4.b Values are means of at least two independent experiments with two replicates each, unless otherwise indicated. For standard deviation see Table S1.†c Compound binding to BRD2 BD1 or BD2 by displacement of a fluorescent Alexa Fluor 647-JQ-1-like probe in −log10(M). The 95% confidence interval of pIC50 values was ±0.24 for BD1 and ±0.23 for BD2.d NanoGlo degradation assay in a HEK293 cell line expressing human BRD4-HiBit fusion at endogenous expression levels in −log10(M). The 95% confidence interval of pDC50 values was ±0.14.e One test occasion. |
|||||
| 15 | 0.18 | 7.37 | 7.43 | 7.17 | 99 |
| 16 | 0.97 | 7.83e | 7.54e | 9.02 | 97 |
| 17 | 0.93 | 7.46 | 7.27 | 8.79 | 100 |
| 18 | 0.79 | 7.62e | 7.42e | 9.15 | 100 |
| 19 | 1.5 | 7.64e | 7.34e | 9.61 | 100 |
| 20 | 1.3 | 7.60e | 7.36e | >9.46 | 99 |
| 21 | 1.7 | 7.38 | 7.00 | 10.1 | 101 |
| 22 | 2.1 | 7.56 | 7.13 | 10.1 | 101 |
 | ||
| Scheme 3 Synthesis of PROTACs 15–20 with basic linkers and dihydro uracil CRBN binder (Table 3). Reagents and conditions: (a) NaBH(OAc)3, DCM, rt; (b) HCl (4 M in dioxane), rt; (c) HATU, DIPEA, DMF, rt; (d) DIPEA, DMF, 90 °C; (e) sodium ascorbate, CuSO4, 1,4-dioxane/H2O, rt. | ||
The ester linked PROTACs 21 and 22 were prepared from the carboxylic acid 38 which was accessed from the corresponding aldehyde 29 using Pinnick oxidation conditions.26 Esterification of 38 was performed by condensation with primary alkanols featuring a terminal bromide to provide the bromides 39. The bromides were used for alkylation of the piperidine in the dihydrouracil 34 to produce the ester-linked PROTACs 21 and 22 (Table 3, Scheme 4).
 | ||
| Scheme 4 Synthesis of PROTACs 21 and 22 with ester linker (Table 3). Reagents and conditions: (a) NaClO2, NaH2PO4, H2O2, MeCN/H2O (1:1); (b) EDC, DIPEA, DMAP, DCM, rt; (c) DIPEA, DMF, 90 °C. | ||
Structure activity relationship (SAR)
BET protein degradation was assessed in HEK293 cells with endogenous expression levels of HiBiT-tagged BRD4, allowing quantification of BRD4 protein in cell lysates. Isoform degradation was monitored in analogous HiBiT assays for BRD2 and BRD3 However, no relevant selectivity was found (see ESI,† Table S1). The JQ1-based PROTAC dBET618,27 was used as reference in the assay for full degradation. The most promising compounds were then profiled in normal human lung fibroblasts (NHLF) and mouse lung fibroblasts (MLF). The abundance of BRD4 was measured by immunofluorescence (IF). Functional activity was assessed in TGF-β1-stimulated primary human lung fibroblasts as inhibition of α-SMA (alpha smooth muscle actin) expression. Binding affinity to BET was tested in a Fluorescence Polarization assay measuring displacement of a fluorescently labelled BET ligand. The binding domains for acetyl-lysine are highly conserved in the different BET isoforms;28 here, BRD2-BD1 and -BD2 derived constructs were used to assess binding.Table 1 shows the first series of PROTACs with limited degradation potency in the range of pDC50 of 6–7 and a BRD4 degradation between 70 and 100%. The inhibitor 2 and the BRD4 PROTACs dBET6 and ZXH-3-2627 are included as references.
As a next step, we reduced the number of H-bond acceptors and donors in the linker unit, in concert with rigid attachment points to both head groups, with the intent to increase permeability for improved cytosolic and possibly nucleic delivery.29,30 In a radical attempt to eliminate all H-bonding capability, the amide and amino connecting groups at the BET binder unit were exchanged by a triazole group, conveniently accessed using click chemistry. In addition, an acetylene moiety was utilized to avoid hetero atoms at the other linker end. This design provided a quantum leap in degradation potency for all examples (Table 2); we observed a >100-fold improvement in degradation potency, surpassing our most potent references by one order of magnitude. The SAR in this subseries was flat with all examples reaching sub nanomolar potency with no apparent impact of the linker length. Notably, the introduction of a single PEG-unit in example 7 led to a significant drop in potency, underlining the unfavourable impact of H-bond acceptors on the potency.
Next, the hydrolysis-labile glutarimide moieties were exchanged with chemically stable indolyl-dihydro-uracils (DHUs) to explore options with potential for improved developability by replacing a chemical motif that has been demonstrated to be prone to isomerization (Table 3).31 The first examples directly provided similar satisfactory degradation potencies, again with little differentiation regarding linker length. In contrast, the variation of the intermittent heterocyclic motifs had significant impact: indole-based PROTACs (19 and 20) were more potent than azaindole derivatives (17 and 18). In Table 3, amide linked PROTAC 15 is included to emphasize the steep SAR with regards to H-donors in the linker region. In comparison to PROTACs with triazole units, this compound shows a more than one order of magnitude reduction in degradation potency. This SAR finding was confirmed by replacing amide and triazole linkers with ester functionalities (21 and 22) further increasing the degradation potency and finally introducing on top a soft spot for hydrolyzation and preventing the cleavable inhaled PROTACs from generating any significant systemic compound/degradation levels.
Selection of PROTACs for inhalation
To choose examples for further profiling, we selected PROTACs based on design principles for standard inhaled drugs, high potency and potential for prolonged lung retention. Maintaining compound exposure in lung tissue for extended time can be achieved via slow release of freely available drug from a reservoir of either non-dissolved or tissue-bound material.1 This requires exquisitely potent compounds as only small amounts of free drug will be available at any given timepoint. We selected the neutral examples 13 and 14 with low solubility (Table 4) to enable administration of a suspension, aiming at a slow dissolution. In contrast, examples 16, 18 and the ester linked PROTAC 22 feature a common piperidine motif that differs across these compounds in basicity, measured as pKa 5.9, 8.0 and 8.4, respectively. While, of these 3 PROTACs, 16 displayed a low solubility, 18 and 22 displayed a relatively high solubility and were considered for a solution formulation – where lung retention was anticipated to rather derive from basicity-driven tissue affinity.1| Compound | Sol pH 7.4a (μM) | hPPBb fu (%) | Plasma stabilityc (% at 18 h) | HLMd (μL min−1 mg−1) | mHEPe (μL min−1 1 × 106) | Caco2 ABf Papp (1 × 10−6 cm s−1) |
|---|---|---|---|---|---|---|
| a Aqueous solubility.b Unbound fraction in human plasma.c Intact compound remaining after 18 h incubation in plasma.d Intrinsic clearance in human liver microsomes.e Intrinsic clearance in mouse hepatocytes.f Intrinsic permeability.g NV = no value due to limited stability in assay. | ||||||
| 13 | <0.16 (n = 4) | 0.5 (n = 2) | 39 ± 5 (n = 2) | >300 (n = 3) | 148 ± 5 (n = 3) | 21 ± 7.7 (n = 4) |
| 14 | <0.16 (n = 2) | NVg (n = 1) | 2.3 (n = 1) | >300 (n = 1) | 48 ± 2 (n = 2) | <0.6 ± 0.1 (n = 2) |
| 16 | <0.16 (n = 3) | 66 ± 13 (n = 2) | 96 ± 24 (n = 2) | <3.0 (n = 4) | <1.0 (n = 2) | 0.4 ± 0.1 (n = 2) |
| 18 | 12 ± 3 (n = 2) | 58 ± 13 (n = 2) | 94 ± 10 (n = 2) | <3.0 (n = 3) | <1.0 (n = 2) | 0.1 ± 0.0 (n = 2) |
| 22 | 2.6 (n = 1) | NVg (n = 1) | 0.1 (n = 1) | 158 ± 9 (n = 2) | 87 ± 18 (n = 2) | 11 ± 3.2 (n = 2) |
In general, the α-SMA inhibitory potency in normal human lung fibroblasts (NHLF) correlated well with the HiBiT assay potency (Table 5). In mouse lung fibroblasts (MLF) however, basic examples 16, 18 and 22 were less active. This can be explained by a key residue difference in the CRBN binding pocket for these two species. For human CRBN there is an interaction between glutamic acid (Glu377) and piperidine, which potentially stabilizes ternary complex and subsequently increases the degradation potency (Fig. 2A). Mouse CRBN features instead a lipophilic residue (Val380) that is likely detrimental for accommodation of a polar amine, and as a result a drop-off in degradation potency is observed. There is a correlation between the potency drop-off and the measured pKa for subseries with basic linker. The higher the pKa, the stronger the interaction with glutamic acid (Glu377), therefore the highest drop-off is observed for 22 (pKa = 8.4). For neutral compounds (13 and 14), there is no basic group and the vector from the E3 warhead puts the linker far from the key residue (Fig. 2B), therefore there is no impact on potency.
| Compound | BRD4 degradation (NHLF)a | BRD4 degradation (MLF)b | FMT (α-SMA inhibition) (NHLF)c | |||
|---|---|---|---|---|---|---|
| DC50 (SD) (nM) | pDC50 −log10(M) |
DC50 (SD) (nM) | pDC50 −log10(M) |
IC50 (SD) (nM) | pIC50 −log10(M) |
|
| a Based on n = 2 experiments in one NHLF donor for 22 and n = 3 NHLF donors for the other 5 compounds.b Based on n = 1 for 14 and 13, n = 2 for 18, n = 3 for 22, n = 4 for 16 and n = 5 for ZXH-3-26.c Based on n = 1 NHLF donor for 22 and n = 3 NHLF donors for the other 5 compounds; NHLF = normal human lung fibroblasts, MLF = mouse lung fibroblasts, FMT = fibroblast-to-myofibroblast transition, α-SMA = alpha smooth muscle actin. Standard deviations (SD) are shown in parentheses. | ||||||
| 13 | 0.29 (0.08) | 9.54 | 0.2 | 9.8 | 0.21 (0.08) | 9.69 |
| 14 | 0.30 (0.06) | 9.52 | 0.3 | 9.6 | 0.44 (0.1) | 9.36 |
| 16 | 4.3 (0.9) | 8.37 | 6.1 (4) | 8.22 | 3.6 (1) | 8.45 |
| 18 | 0.65 (0.08) | 9.18 | 7.2 (2) | 8.14 | 0.50 (0.2) | 9.30 |
| 22 | 0.013 (0.001) | 10.9 | 0.26 (0.1) | 9.59 | 0.002 | 12 |
| ZXH-3-26 | 17 (4) | 7.78 | 0.81 (0.3) | 9.09 | 13 (4) | 7.87 |
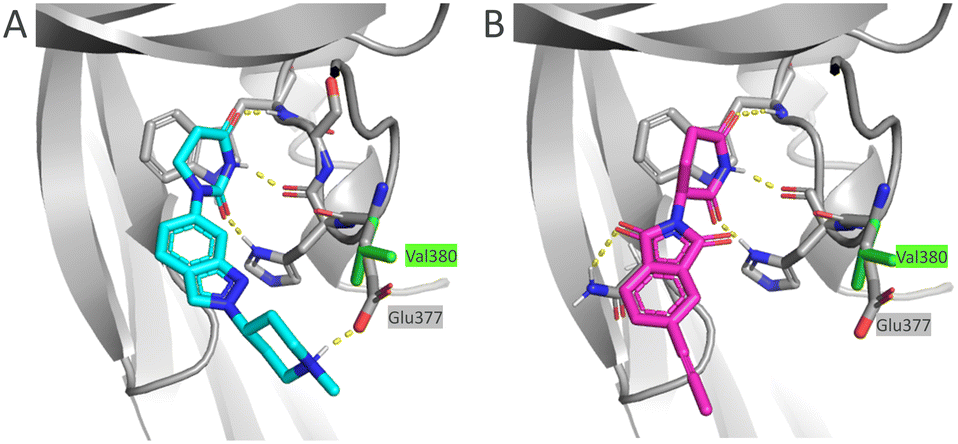 | ||
| Fig. 2 Docking of (A) 18 (light blue) and (B) 13 (magenta) E3 warheads into human CRBN (grey) and overlay with mouse CRBN (the different residue for mouse Val377 shown with green sticks). Polar interactions marked with yellow dashed lines. PDB codes for human and mouse CRBN are: 4TZ4 and 5YIZ, respectively. | ||
Diametral differences were found for physiochemical properties (Table 4). The lipophilic examples 13 and 14 exhibited low solubility, excessive unspecific protein binding, high intrinsic metabolism and, noteworthy, low serum plasma stability. The basic dihydro-uracil-derived PROTACs 16 and 18, in contrast, showed high metabolic and plasma stability. Further, these basic PROTACs exhibit low protein binding. For inhalation use, when seeking a local effect, the lipophilic examples 13 and 14, in contrary to 16 and 18, appeared to be prone for rapid elimination from circulation and were therefore preferred. The ester linked PROTAC 22 showed an almost complete disappearance in serum plasma in vitro after 18 h and otherwise resembled the profile of the neutral examples.
Due to their propensity for rapid systemic clearance, PROTACs 13, 14 and 22 were selected for efficacy testing in vivo. PROTACs 13 and 14 were short-lived in mice upon intravenous administration, with very similar residence time and clearance and only differing in distribution volumes. PROTAC 22 could not be detected, indicating a rapid elimination, likely accelerated by ester hydrolysis as intended by the softdrug design. To examine inhaled pharmacokinetics, the neutral PROTACs 13 and 14 were administered intratracheally to mice as a suspension of amorphous material.32 With this administration and formulation approach, lung retention could be extended to hours to provide a dissolution driven slow release of the agent that allowed a steady flow of the drug passing through the lung. The basic and more soluble ester PROTAC 22, given as solution, was not detected in lung tissue. The basic piperidine (measured pKa 8.4) would confer potential lung retention driven by accumulation in acidic compartments like lysosomes, but the low metabolic stability still limits this approach (Table 6).
| Compound | iva | itb | ||
|---|---|---|---|---|
| CLc (mL min−1 kg−1) | Vssd (L kg−1) | MRTe (h) | Lung T1/2f (h) | |
| a iv, intravenous.b it, intratracheally.c CL, clearance.d Vss, volume of distribution at steady state.e MRT, mean residence time.f Lung T1/2, half-life in lung after it administration of a suspension of amorphous material.g NE = no exposure; doses: for 22 iv 2.5 mg kg−1 and it 1 mg kg−1; for 13 and 14. iv 1 mg kg−1 and it 0.8 mg kg−1, respectively. | ||||
| 13 | 81 ± 2 (n = 2) | 0.8 ± 0.5 (n = 2) | 0.2 ± 0.1 (n = 2) | 3.4 (susp) (n = 6) |
| 14 | 57 ± 1 (n = 2) | 0.7 ± 0.3 (n = 2) | 0.2 ± 0.1 (n = 2) | 2.7 (susp) (n = 6) |
| 22 | NEg (n = 3) | NEg (n = 3) | NEg (n = 3) | NEg (n = 18) |
Based on these findings, the efficacy of both neutral PROTACs 13 and 14 to degrade BRD4 was evaluated in mice after intratracheal delivery at different timepoints (1 h, 7 h and 24 h, respectively) and intravenous delivery (24 h). Lungs were collected, fixed and BRD4 abundance was assessed by immunohistochemistry (IHC). The results for 14 showed a rather transient effect for all time points, while, for 13, a substantial reduction of BRD4 was detected at all time points (Fig. 3). For 13, no relapse to natural BRD4 levels was observed up to 24 hours. This enduring effect may be attributed to slow BRD4 resynthesis rate, as indicated by SILAC experiments across various primary cell types,33 or potentially also to slow dissolution of solid, deposited material in the lung providing a slow release of 13. Most interestingly, an intravenous administration of an equal dose resulted in no degradation. This finding indicates that the observed activity of locally administered PROTACs is not based on any significant systemic contribution.
 | ||
| Fig. 3 Time course study in C57BL/6 mice assessing the reduction of BRD4 positive cells by immunohistochemistry (IHC) in the lung after a single intratracheal or intravenous dose of PROTACs 13 and 14. Doses for 13 and 14. iv 1 mg kg−1 and it 0.8 mg kg−1, respectively. | ||
We finally progressed PROTAC 13 into an in vivo dose–response study to further evaluate its effects. In this study, both the lung and spleen were analysed to compare local effects with potential systemic spill-over. Following a single intratracheal administration, BRD4 levels were assessed after 7 hours in lung and spleen sections using immunohistochemistry (IHC).
In the analysis of BRD4 levels across the entire lung (Fig. 4A), a dose-dependent induction of BRD4 degradation by PROTAC 13 was observed. Notably, BRD4 reduction occurred only in the lung, with no significant changes detected in the spleen, thereby suggesting a local effect. Further analysis of lung segments demonstrated a degradation in BRD4 levels, in the central bronchial epithelium and the peripheral alveolar space (Fig. 4B). In particular, dose-dependent degradation of BRD4 was observed in the alveoli to demonstrate successful targeting of the peripheral lung tissue (Fig. 4C). Therefore, significant reduction in BRD4 levels was found throughout the entire lung following a single dose of PROTAC 13, sparing endogenous BRD4 levels in the spleen.
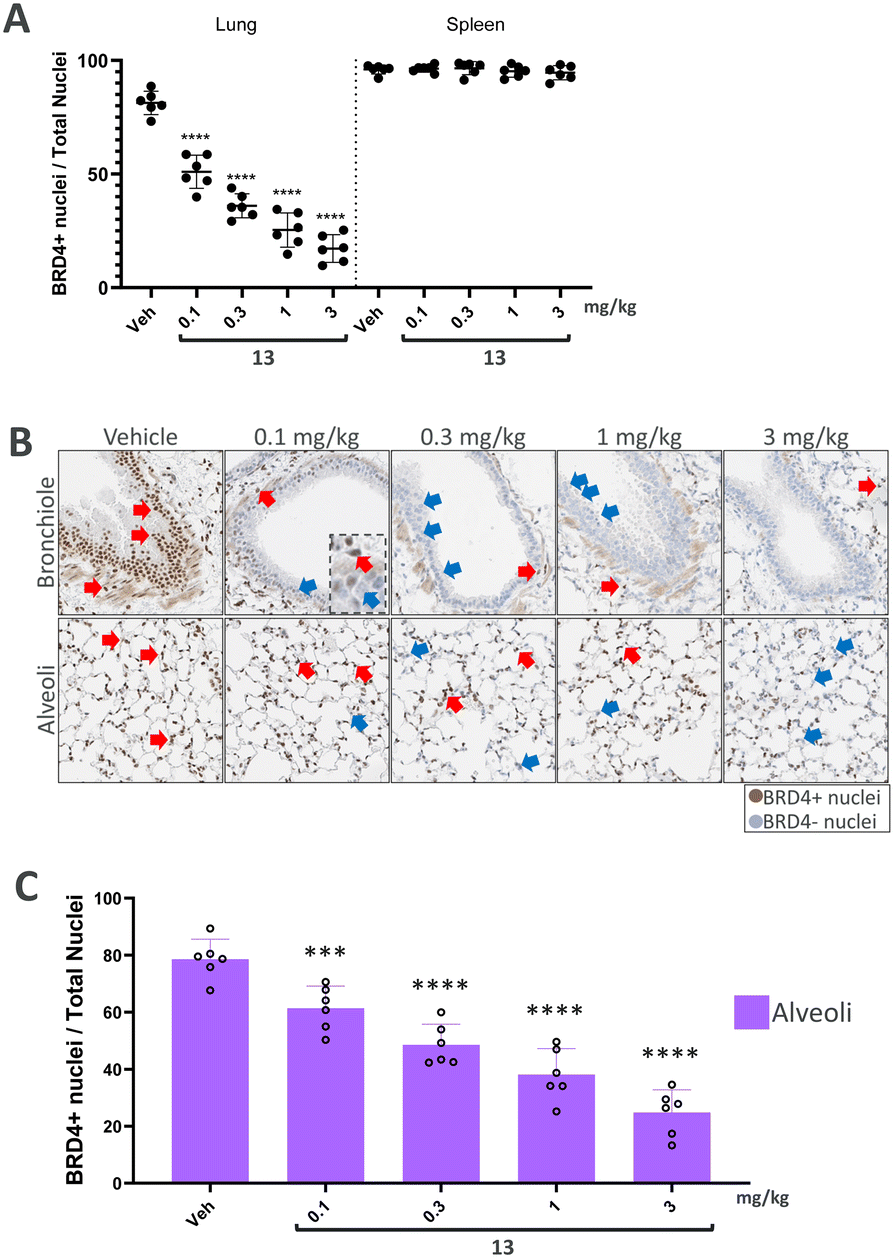 | ||
| Fig. 4 Dose–response study in C57BL/6 mice evaluating BRD4 positive cells using immunohistochemistry (IHC). (A) Dose related reduction of % BRD4 positive cells in lung but not spleen. (B) Representative IHC images showing BRD4 stained cells in the central lung (bronchiole) and the peripheral alveolar epithelium (alveoli). Red arrows point to BRD4 positive cells and blue arrows indicate BRD4 negative cells. (C) Dose-dependent reduction of % BRD4 positive cells in the peripheral lung (alveoli). Data are shown as bar graphs with mean ± SD (n = 6/group). Asterisks denote significant differences between vehicle and compound treated groups (***P < 0.001; ****P < 0.0001). | ||
Conclusions
The optimization of a novel series of BET PROTACs towards inhaled route of delivery furnished PROTACs with a picomolar degradation potency and full BRD4 degradation. PROTAC 13 exhibited short half-life in circulation and low solubility allowing formulation as a suspension In target engagement studies, intratracheally administrated 13 induced BRD4-degradation leading to persistently reduced BRD4-levels in the lung after a single dose, lasting for 24 hours after dosing. Immunohistochemistry analysis of lung sections demonstrated a substantial BRD4 degradation across the entire lung tissue. A systemic contribution of 13 to the effect in lung could be excluded when intravenous administration of a dose marginally higher than the locally effective dose did not achieve target engagement, assessed as BRD4-degradation, in the lung. This finding was corroborated by a follow-up study where a dose–response relationship could be established across entire lung sections, including the alveolar space, while no spill-over effects were observed in the control organ spleen. This suggests that 13 is exclusively acting in the lung. To our knowledge, local action of a PROTAC in the lung periphery while sparing systemic effects has not been described previously, nor has it been achieved for a small molecule inhibitor. The concept of a lung-restricted degrader presented herein might inspire treatment options for lung diseases of distal airways where systemic treatment cannot be applied due to insufficient therapeutic index of either oral small molecule inhibitors or PROTACs.Experimental section
Synthesis of compounds 13 and 14
Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of AstraZeneca and approved by the local Ethical Committee for Laboratory Animals in Gothenburg.Data availability
Additional data supporting this article have been included as part of the ESI.† These include – synthesis of compounds 3–12 and 15–22, spectra and analyses, molecular docking protocol, in vitro, in vivo and drug metabolism and pharmacokinetics (DMPK) assays, including formulation descriptions.Author contributions
MH, WC, LR, SS and PB contributed to the conceptualization of the manuscript, MH and WC wrote the first draft with the assistance of KK and LR finalized the manuscript. MH and JL shared project administration, WC provided mentorship. CC, BR, LCR, JM performed synthesis and completed experimental write-ups. KK performed docking studies and contributed to analysis. PH and AN developed, completed and analysed binding assays. JL, FE, MD, MN and HP designed and completed method development and experimentation of the assessment of protein degradation by HiBiT methodology, including cell line generation. UH, KR, OS, CF and ME contributed to methodology, experiments and analysis and curation of results from fibroblasts. AP developed methodology for imaging in mouse lung sections and performed analysis and visualization. AP, RP and AB designed experiments in mice. RP supervised and contributed with analysis of DMPK related experiments. Formulations for in vivo dosing were prepared by AJ. WC, AB, LB and JJ provided supervision. All Authors contributed with writing, critical review & editing.Conflicts of interest
The authors declare the following competing financial interest(s): all authors that are or have been employees of AstraZeneca may own stock or stock options.Acknowledgements
We would like to thank the analytical and separation science team at AstraZeneca Gothenburg for analytical support and purification of final compounds. In particular, we thank Richard Lewis for the support in structure elucidation by NMR spectroscopy. We thank our colleagues at Pharmaron Beijing, in particular Wenzhen Yang and Ziyang Nan, for the synthesis support. Furthermore, we thank Christopher Kourra for supporting library synthesis, Ken Grime for constructive discussions on in vitro pharmacology and Jamie Scott for constructive feedback on the manuscript.Notes and references
- A. E. Cooper, D. Ferguson and K. Grime, Optimisation of DMPK by the inhaled route: Challenges and approaches, Curr. Drug Metab., 2012, 13, 457–473 CrossRef CAS PubMed.
- E. Pasqua, N. Hamblin, C. Edwards, C. Baker-Glenn and C. Hurley, Developing inhaled drugs for respiratory diseases: A medicinal chemistry perspective, Drug Discovery Today, 2022, 27, 134–150 CrossRef CAS PubMed.
- J. S. Patton and P. R. Byron, Inhaling medicines: delivering drugs to the body through the lungs, Nat. Rev. Drug Discovery, 2007, 6, 67–74 CrossRef CAS PubMed.
- M. Békés, D. R. Langley and C. M. Crews, PROTAC targeted protein degraders: The past is prologue, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- K. Li and C. M. Crews, PROTACs: past, present and future, Chem. Soc. Rev., 2022, 51, 5214–5236 RSC.
- G. R. Hamm, et al., Revealing the regional localization and differential lung retention of inhaled compounds by mass spectrometry imaging, J. Aerosol Med. Pulm. Drug Delivery, 2020, 33, 43–53 CrossRef CAS PubMed.
- A. E. John, et al., Translational pharmacology of an inhaled small molecule αvβ6 integrin inhibitor for idiopathic pulmonary fibrosis, Nat. Commun., 2020, 11, 4659 CrossRef CAS PubMed.
- J. Wu, et al., JAK1/JAK2 degraders based on PROTAC for topical treatment of atopic dermatitis, Biomed. Pharmacother., 2024, 171, 116167 CrossRef CAS PubMed.
- T. Ju, A. Labib, A. V. Does and G. Yosipovitch, Topical Janus kinase-signal transducers and activators of transcription inhibitor tofacitinib is effective in reducing nonatopic dermatitis chronic itch: a case series, J. Am. Acad. Dermatol., 2022, 87, 400–403 CrossRef CAS PubMed.
- S. Dhillon, Delgocitinib: first approval, Drugs, 2020, 80, 609–615 CrossRef CAS PubMed.
- A. R. Brasier and J. Zhou, Validation of the epigenetic reader bromodomain-containing protein 4 (BRD4) as a therapeutic target for treatment of airway remodeling, Drug Discovery Today, 2020, 25, 126–132 CrossRef CAS PubMed.
- X. Tang, et al., BET bromodomain proteins mediate downstream signaling events following growth factor stimulation in human lung fibroblasts and are involved in bleomycin-induced pulmonary fibrosis, Mol. Pharmacol., 2013, 83, 283–293 CrossRef CAS PubMed.
- X. Tang, et al., Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis, Am. J. Pathol., 2013, 183, 470–479 CrossRef CAS PubMed.
- M. Ameratunga, et al., First-in-human Phase 1 open label study of the BET inhibitor ODM-207 in patients with selected solid tumours, Br. J. Cancer, 2020, 123, 1730–1736 CrossRef CAS PubMed.
- M. M. Mita and A. C. Mita, Bromodomain inhibitors a decade later: a promise unfulfilled?, Br. J. Cancer, 2020, 123, 1713–1714 CrossRef PubMed.
- C. Qin, et al., Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the Bromodomain and Extra-Terminal (BET) proteins capable of inducing complete and durable tumor regression, J. Med. Chem., 2018, 61, 6685–6704 CrossRef CAS PubMed.
- B. Zhou, et al., Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression, J. Med. Chem., 2017, 61, 462–481 CrossRef PubMed.
- R. P. Nowak, et al., Plasticity in binding confers selectivity in ligand induced protein degradation, Nat. Chem. Biol., 2018, 14, 706–714 CrossRef CAS PubMed.
- C. R. Wellaway, et al., Discovery of a bromodomain and extraterminal inhibitor with a low predicted human dose through synergistic use of encoded library technology and fragment screening, J. Med. Chem., 2020, 63, 714–746 CrossRef CAS PubMed.
- R. A. Bit, J. A. Brown, P. G. Humphreys and K. L. Jones, WO2016146738, 2016, (to GlaxoSmithKline Intellectual Property Development Limited).
- N. P. Henley, WO2018041946, 2018, (to GlaxoSmithKline Intellectual Property Development Limited).
- K. Sonogashira, Development of Pd–Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- G. Meng, et al., Modular click chemistry libraries for functional screens using a diazotizing reagent, Nature, 2019, 574, 86–89 CrossRef CAS PubMed.
- G. J. Roth, B. Liepold, S. G. Mueller and H. J. Bestmann, Further improvements of the synthesis of alkynes from aldehydes, Synthesis, 2004, 1, 59–62 Search PubMed.
- U. Borjesson, et al., Synthesis of heterocyclic substituted pyrimidine anticancer agents, WO2022069520, 2022.
- B. S. Bal, W. E. Childers and H. W. Pinnick, Oxidation of α,β-unsaturated aldehydes, Tetrahedron, 1981, 37, 2091–2096 CrossRef CAS.
- G. E. Winter, et al., BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment, Mol. Cell, 2017, 67, 5–18 CrossRef CAS PubMed.
- Y. Nakamura, et al., Crystal structure of the human BRD2 bromodomain: insights into dimerization and recognition of acetylated histone H4, J. Biol. Chem., 2007, 282, 4193–4201 CrossRef CAS PubMed.
- V. G. Klein, A. G. Bond, C. Craigon, R. S. Lokey and A. Ciulli, Amide-to-ester substitution as a strategy for optimizing PROTAC permeability and cellular activity, J. Med. Chem., 2021, 64, 18082–18101 CrossRef CAS PubMed.
- V. Poongavanam and J. Kihlberg, PROTAC cell permeability and oral bioavailability: a journey into uncharted territory, Future Med. Chem., 2022, 14, 123–126 CrossRef CAS PubMed.
- B. Knoche and G. Blaschke, Investigations on the in vitro racemization of thalidomide by high-performance liquid chromatography, J. Chromatogr. A, 1994, 666, 235–240 CrossRef CAS.
- The importance of formulation for lung retention was demonstrated in rats. Intratracheal administration in rats of 13 as a suspension had a similar lung half-life as in mice, whereas administration as a solution resulted in a negligible half life (Table SI2†).
- T. Mathieson, et al., Systematic analysis of protein turnover in primary cells, Nat. Commun., 2018, 9, 689 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00173k |
| This journal is © The Royal Society of Chemistry 2025 |