
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence‘Vitrimer nanocomposites’ derived from graphene oxide and post-consumer recycled polypropylene†
Indranil
Dey‡
,
Debashrita
Kundu‡
 ,
Sayon
Ghosh
,
Samir
Mandal
,
Ketaki
Samanta
* and
Suryasarathi
Bose
*
,
Sayon
Ghosh
,
Samir
Mandal
,
Ketaki
Samanta
* and
Suryasarathi
Bose
*
Department of Materials Engineering, Indian Institute of Science, Bengaluru – 560012, India. E-mail: ketakisamanta123@gmail.com; sbose@iisc.ac.in
First published on 13th March 2025
Abstract
Post-consumer recycled polypropylene (PCR PP) is promising for sustainable applications, yet its limitations in electrical conductivity and mechanical properties require modifications. This study develops a vitrimer nanocomposite by modifying PCR PP via styrene-assisted maleic anhydride grafting and incorporating a molecule containing multiple epoxide groups facilitating effective crosslinking. Graphene oxide (GO) is added as a nanofiller, improving rheological, thermal, electrical and infrared thermal properties. Characterization techniques confirm structural enhancements, while tensile testing shows significant gains in strength and modulus. The vitrimer nanocomposite demonstrates recyclability and high performance, offering a sustainable path for advanced engineering applications within a circular economy framework.
1. Introduction
According to conservative predictions, the world's yearly plastic manufacturing will surpass 500 million tons by 2050.1 However, plastics do not currently have a recyclable lifespan. Only around 9% of plastic trash is recycled; the remainder is burned, dumped, or landfilled, which has a major and lasting negative influence on the environment.2–4 Globally, nations have implemented policies aimed at curbing the issue of plastic pollution.5 To reduce plastic waste pollution in the environment, two strategies have been explored: replacing commercial non-biodegradable plastic products with biodegradable plastic products and dumping, collecting, recycling, and reusing plastic trash.6–9 Short-term plastic pollution reduction is possible with the latter strategy, which is also a more effective and industrially viable approach. However, rather than directly recycling plastic waste into products of similar value, the current approach to recycling waste plastic primarily involves re-extrusion with virgin polymers, additives or fillers. This is necessary, otherwise the polymer degrades thermally and mechanically during the re-extrusion process, and it also degrades due to photo-oxidation during the service time.10–15 However, this is associated with a huge carbon footprint which sets a significant challenge to tackle in the coming decades. In order to reach net-zero carbon emissions, a closed-loop circular economy in the plastics industry is essential. However, gathering, classifying, and processing post-consumer recycled (PCR) plastics presents a significant obstacle to this circularity until a workable way is developed.16 In 2019, the global production of waste polypropylene (PP) exceeded 61 million tons, making up nearly 20% of all plastic trash.17 Consequently, it is essential for the sustainable development of polypropylene (PP) to either create new PP materials with enhanced properties or responsibly upcycle existing materials with reduced negative effects on the economy and environment.Incorporating dynamic crosslinkers into waste plastics presents a viable solution to this problem. This entails converting waste plastics into covalent adaptive networks (CANs), where the crosslinks can interchange dynamically in response to external triggers.18 The strong mechanical characteristics of conventional thermosets and the malleability or reprocessability of thermoplastics are combined in an ideal CAN.19–25 Bulk reprocessing using injection molding, compression molding, melt blowing, or twin-screw extrusion has been shown to be effective in CANs that are based on dynamic linkages such as acetals,26,27 vinylogous ureas/urethanes,28,29 Diels–Alder structures,30 hydrazines,31 dioxaborolanes,32 boroxines,33,34 olefins,35,36 silyl ether,37 disulfides,38,39 siloxanes,40 diketoenamines,41 and so forth. Even with these advancements, most CANs still rely on dynamic linkages that are typically absent from well-established commercial thermosets, necessitating the creation, synthesis, and development of novel materials. The integration of various polar structures through the use of CANs is anticipated to potentially expand the variety and value of PP's uses in addition to addressing the material's notable loss of mechanical qualities after repeated recycling.42 Yet, because of the strict synthetic requirements and the non-reactive PP backbones, the production of CANs using PP requires adherence to more specific concepts than when using many other polymers. To meet industrial demands, dynamic chemistry should be incorporated into polypropylene (PP) in a way that is scalable, features a universal structure, avoids costly additives, and remains resistant during post-processing.43 The potential to directly convert standard polymers into CANs without additional modification or optimization of current facilities makes the implantation of dynamic links via post polymerization functionalization attractive.25 Conversely, the effective synthesis of PP-CANs is contingent upon a restricted set of dynamic reactions because of their chemical and thermal stability within the matrix.44
Over the past ten years, transesterification has emerged as a widely renowned technique for creating CANs, thanks to the groundbreaking research conducted on vitrimers in 2011.45 Therefore, adding maleic anhydride (MA) side groups to PP, the most thoroughly researched commercial product among all post-functionalized PP materials, presents a workable platform for transesterification.46–48 To accomplish transesterification, such reliable dynamic reactions typically require the presence of external catalysts.49–52
We developed a straightforward, scalable method for upcycling PCR PP into vitrimers via transesterification using zinc acetate as an external catalyst. The process involved a two-step reaction: first, grafting maleic anhydride (MA) onto PCR PP with styrene as a co-agent and dicumyl peroxide (DCP) as an initiator, followed by crosslinking with TGDDM epoxy in a twin-screw extruder (Fig. 1). Post evaluating the vitrimer thoroughly, graphene oxide (GO) was incorporated to design vitrimer nanocomposites, allowing us to study the effects of GO on the nanocomposite's thermomechanical, rheological, electrical, and infrared thermal properties, both pre- and post-recycling. There are plenty of literature studies on polypropylene nanocomposites,53–58 however, currently, there is no literature exploring the impact of adding nanofillers to polyolefin vitrimer nanocomposites or recycling them. Therefore, understanding the role of nanofillers in polyolefin vitrimer systems is essential. This study investigates the thermomechanical, rheological, electrical, and infrared thermal properties of PCR PP vitrimer nanocomposites by varying the amount of graphene oxide (GO). Additionally, the performance of these properties was carefully evaluated after recycling to assess the nanocomposites' durability and potential for sustainable reuse.
 | ||
| Fig. 1 Schematic diagram for the preparation of the PCR PP vitrimer nanocomposite and reprocessing via dynamic exchange. | ||
2. Experimental section
2.1 Materials
The PCR PP granules were contributed by Manjushree Technopack Limited and subjected to a proper 5-hour vacuum drying process at 80 °C before undergoing reactive extrusion. GO with a thickness (z) of ∼0.8–2 nm and dimensions (x and y) of ∼5–10 μm was purchased from Adnano Technologies. Maleic anhydride (MA) (99%), dicumyl peroxide (DCP) (99%), 4,4′-methylenebis(N,N-diglycidylaniline) (TGDDM), Irganox 1010 (pentaerythritol tetrakis(3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionate)) (99%), zinc acetate dihydrate (99%), styrene (99%), xylene (99%), and acetone (99%) were procured from Sigma Aldrich (Merck).2.2 Reactive extrusion for the formulation of styrene assisted maleated PCR PP and PCR PP vitrimers and vitrimer nanocomposite batches
Every batch of styrene assisted maleated PCR PP (m′-PCR PP), PCR PP vitrimers and the corresponding nanocomposites with GO was prepared by employing reactive extrusion in a DSM Xplore twin-screw micro-compounder with 15 cm3 capacity. The device is equipped with co-rotating conical screws and a recirculation channel, enabling precise control over residence time. Initially, 10 wt% maleic anhydride (MA), 0.5 wt% DCP catalyst, and 0.5 wt% Irganox 1010 as an antioxidant were added to PCR PP along with styrene (St) which acts as a co-grafting agent to formulate m′-PCR PP (Table S1†) and mixed thoroughly at 180 °C temperature at 150 rpm screw speed for 2 min in a melt extruder. As widely elaborated and reported in the literature, the inclusion of styrene considerably diminishes PP chain scission while enhancing the grafting degree of MA on PP. This is achieved as styrene preferentially interacts with the PP macroradicals to form stable styryl macroradicals, which copolymerize with MA, generating branches that mitigate PP chain scission and elevate grafting efficiency.59 The molar ratio of maleic anhydride and styrene was maintained at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio according to earlier literature.59
:1 ratio according to earlier literature.59
In the following step, PCR PP vitrimer batches were fabricated by crosslinking the prepared m′-PCR PP batches with TGDDM in the presence of zinc acetate as a transesterification catalyst60 by subjecting them to extrusion at 180 °C and 150 rpm speed for 2 min residence time. PCR PP vitrimer batches were fabricated at different concentrations of TGDDM crosslinker (Table S2†). The nanocomposites of the most optimum crosslinked sample were formulated with the GO nanofiller at varying concentrations (Table S3†). The conditions for preparing the nanocomposites were kept consistent with those for the vitrimer samples. After extruding each batch, tensile samples were prepared through injection molding, with the barrel temperature set at 180 °C and the mold kept at ambient temperature. Four dog bone-shaped specimens were molded for each sample, using an injection pressure of 14 bar for mechanical testing.
3. Materials characterization
3.1 FT-IR
FT-IR spectroscopy of m′-PCR PP and PCR PP vitrimers was performed with a PerkinElmer Frontier spectrometer in ATR mode in the range of 650–4000 cm−1.3.2 Gel fraction studies
Initially, approximately about 250 mg of the PCR PP and crosslinked PCR PP samples were weighed and transferred to a 100 mL round-bottom flask (RB) accompanied by 50 mL of xylene in order to calculate the gel fraction of the respective PCR PP vitrimers and the corresponding nanocomposite with GO. Following the initial step, the RB was subjected to heating in an oil bath under reflux conditions at a temperature of 120 °C for 24 h. Following this, xylene was removed from the RB flask and the residual undissolved polymer was thoroughly rinsed with acetone followed by vacuum-drying for 24 hours at 80 °C, and the final weight of the dried residue was weighed. The gel fraction was estimated based on the mass ratio of the polymer both before and after heating in xylene. The matching equation is given below:| Gel fraction (%) = W1/W0 × 100 | (1) |
3.3 Tensile testing
Dumbbell-shaped specimens of PCR PP, vitrimers and vitrimer nanocomposites underwent uniaxial tensile testing at room temperature in accordance with the ASTM D638 standard, using Tinius Olsen 1ST universal testing apparatus equipped with a 5 kN load cell. The tests were conducted at a fixed crosshead speed of 50 mm min−1. Yield strength (YS) was determined, which is a material property that describes the amount of stress a material can withstand without permanently deforming. The elastic modulus was calculated as the gradient of the linear section in the elastic range of the stress–strain curve before yielding, while elongation at yield (EY) was determined as the percentage strain at the yield point.3.4 Differential scanning calorimetry (DSC)
The melting and crystallization transitions of neat PCR-PP, vitrimers, and vitrimer nanocomposites were analysed using a DSCQ2000 from TA Instruments. Prior to thermal analysis, 3–4 mg samples were vacuum-dried at 80 °C for 6 hours to remove moisture. A consistent temperature profile was maintained across all samples. The first temperature scan involved heating the samples to 200 °C and holding at this temperature for 2 minutes to achieve internal relaxation and eliminate any prior thermal history.The crystallization temperature (Tc) was determined from the exothermic peak observed during cooling to 40 °C. Subsequently, a second heating cycle was conducted to measure the melting temperature (Tm) from the endothermic peak. The heating and cooling rates were set at 10 °C min−1 under a nitrogen flow of 50 mL min−1.
The fraction of crystallinity (% Xc) of each vitrimer was determined by comparing its melting enthalpy (ΔHm) with the theoretical enthalpy of 100% crystalline isotactic polypropylene (i-PP), ΔHm0, which is 207.0 J g−1, where61
| % Xc = ΔHm/ΔHm0 × 100 | (2) |
3.5 Thermogravimetric analysis (TGA)
The thermal stability of vitrimers was determined through thermogravimetric analysis using a thermogravimetric analyser Q5000 from TA Instruments. For this study, 10 mg sample of each vitrimer was heated in ceramic pans under a nitrogen atmosphere at a heating rate of 10 °C min−1, within the temperature range of 30 °C to 800 °C. The weight loss of the samples was observed and documented as the temperature increased.3.6 Rheological studies
A TA Instruments HR-3 DISCOVERY hybrid rheometer was used to investigate the viscoelastic properties of PCR PP, TGDDM crosslinked PCR PP vitrimer and GO infused vitrimer composite samples. The experiments were carried out under a N2 atmosphere utilizing a parallel plate geometry consisting of EHP steel (25 mm in diameter) and a sample thickness of ∼1 mm. Additionally, the entire experiment was conducted under a consistent 10–20 N axial force. The frequency sweep was performed over a range of 0.1 to 100 rad s−1 at a temperature of 180 °C maintaining a uniform strain amplitude of 0.5% throughout the experiment.3.7 SEM study
The fracture morphology of tensile-tested samples was analysed using a Zeiss EVO-10 at an accelerating voltage of 15 kV with 5000× magnification. By using this tool, the effect of the vitrimer network, GO, and recycling of PCRPP on fracture morphology was studied, and failure mechanisms were analysed.3.8 Conductivity study
The AC electrical conductivity of the samples was assessed using an impedance analyser (Alpha-A analyser, Novocontrol, Germany) across a wide frequency range of 101 to 107 Hz at room temperature.3.9 Infrared thermal analysis
Infrared thermal analysis of 15 TGDDM and the 0.5 GO vitrimer nanocomposite was conducted by measuring the surface temperature of the laser-exposed sample using an IR camera during laser exposure of the sample at 808 nm and a power density of 1.4 W cm−2 positioned 78 mm from the sample.4. Results and discussion
4.1 Spectroscopic evidence, the gel fraction and structural properties
FTIR analysis of m′-PCR PP and the corresponding PCR PP vitrimers was performed to check the dynamic network formation between functionalized PCR PP and the employed crosslinker molecule. The characteristic peak in the region of 1730–1735 cm−1 corroborates the successful formation of β-hydroxy ester bonds as a result of the reaction between reactive epoxy groups of TGDDM and maleic anhydride (MA) groups on the PCR PP backbone (Fig. 2a). These ester bonds can participate in transesterification exchange reactions at elevated temperatures. It is well reported that with a subsequent increase in concentration of TGDDM, the peak intensity at 1730–1735 cm−1 increases owing to consumption of more MA groups (which is characterized by the diminishing intensity of the peak at 1780–1785 cm−1 corresponding to symmetric stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of MA) due to the formation of more dynamic networks in the system. The presence of styrene (St) in the extruded m′-PCR PP and corresponding vitrimer samples is confirmed by its characteristic peak at 704 cm−1 owing to the asymmetric stretching vibration of the phenyl group in St.62
O of MA) due to the formation of more dynamic networks in the system. The presence of styrene (St) in the extruded m′-PCR PP and corresponding vitrimer samples is confirmed by its characteristic peak at 704 cm−1 owing to the asymmetric stretching vibration of the phenyl group in St.62
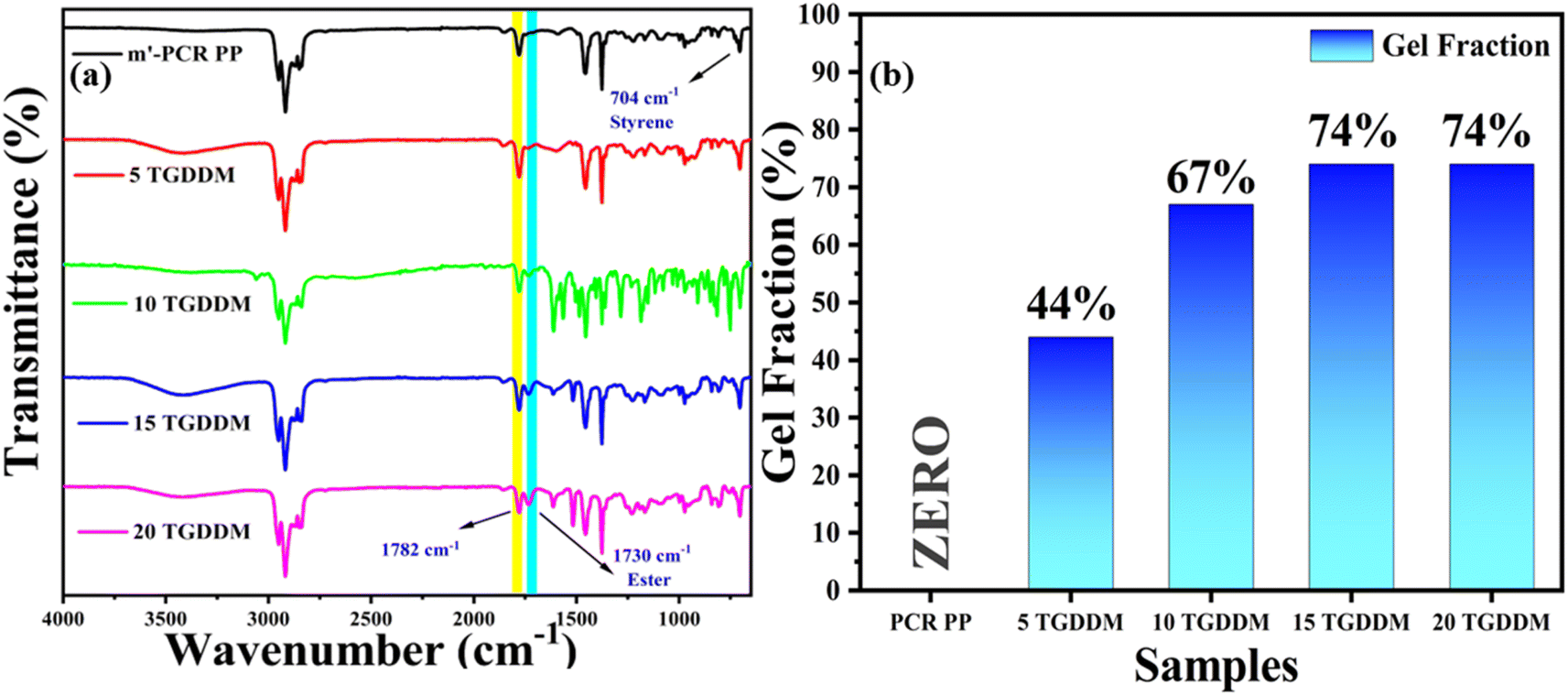 | ||
| Fig. 2 (a) FT-IR spectrum of m′-PCR PP and vitrimers with different concentrations of TGDDM. (b) Gel fraction of PCR PP and PCR PP vitrimers. | ||
Graphene oxide (GO) was specifically chosen as the nanofiller for the vitrimer nanocomposites due to its unique surface chemistry, which includes a high density of oxygen-containing functional groups such as hydroxyl, epoxy, and carboxyl groups. These functional groups facilitate strong interfacial interactions with the epoxide groups of TGDDM (the crosslinker used in our study) and maleic anhydride. FTIR spectra of 0.5 GO and 1 GO samples were analysed and compared with that of the 15 TGDDM sample (see Fig. S1†). It is evident that the intensity of the anhydride peak at 1782 cm−1 decreases from the 15 TGDDM to the 1 GO sample and the intensity of the ester peak at 1730 cm−1 increases. This confirms that GO reacted with maleic anhydride and formed an ester bond. This results in improved interfacial adhesion between GO and the vitrimer matrix.
The degree of crosslinking in PCR PP and the corresponding vitrimer samples was assessed through gel fraction studies by subjecting the samples to reflux conditions in xylene at 120 °C overnight. The analysis shows that the level of maleic anhydride groups grafted onto PCR PP is crucial for promoting crosslinking.16,43 Functionalized PCR PP (m′-PCR PP) systems present active functional sites capable of reacting with the TGDDM molecule, which promotes the crosslinking of the polymer and facilitates the formation of dynamic covalent adaptable networks. This leads to the conclusion that the degree of crosslinking is intrinsically linked to the maleation efficiency. As depicted in Fig. 2b, pristine PCR PP has a 0% gel fraction, implying its non-crosslinked network which accounts for its complete dissolution in hot xylene during the experiment. In contrast, the corresponding vitrimers of PCR PP with TGDDM did not dissolve, corroborating the formation of a crosslinked network that does not dissolve in xylene. The gel fraction progressively increased from 44% to 74% with TGDDM concentrations ranging from 5 wt% to 20 wt%, corroborating the enhancement in crosslinked networks in the system owing to availability of more TGDDM moieties to attach to the MA groups on the PCR PP backbone. Interestingly, the gel fraction saturated at 74% for both the samples 15 TGDDM and 20 TGDDM, suggesting that the crosslinking density of the PCR PP vitrimer approaches a saturation threshold at 15 wt% of TGDDM beyond which additional crosslinkers do not significantly enhance dynamic network formation and may be left dangling. Additional gel fraction analyses of GO incorporated vitrimer nanocomposites reveals that the addition of GO did not significantly alter the crosslink density (Fig. S2†). This result suggests that GO has minimal effect on the crosslinking of PCR PP.
The mechanical properties of post-consumer recycled polypropylene (PCR PP), along with vitrimers containing varying concentrations of TGDDM and GO-incorporated vitrimer-nanocomposites, were examined through tensile testing at room temperature. The mechanical properties and corresponding stress–strain curves are presented in Fig. 3a and S3† respectively and listed in Table S4.† All samples exhibited significantly higher yield compared to their thermoplastic precursor, PCR PP, prior to failure.
 | ||
| Fig. 3 (a) Mechanical properties of PCR PP vitrimers at room temperature and (b) the DSC thermogram illustrates the melting and crystallization behaviour of PCR PP vitrimers, both with and without nanofillers, emphasizing that the dynamic network has a trivial impact on the crystallization process of the vitrimers. | ||
A notable observation was the steady increase in Young's modulus (YM) and yield stress (YS) with increasing TGDDM concentrations, from 5% to 20%. This trend highlights the substantial impact of the degree of crosslinking and crystallinity (Xc) on the mechanical properties of these semi-crystalline polymers. The observed behaviour can be explained by the preferential reaction of styrene with macroradicals of PP, leading to the formation of more stable styryl macroradicals. These radicals copolymerize with MA in the modified PCR PP (m′-PCR PP), forming branches. As a result, the addition of styrene mitigates PP chain scission while simultaneously enhancing the grafting of MA, thus improving the overall mechanical performance of the vitrimer system.63,64 The crosslinking of maleic anhydride (MA) and TGDDM, resulting in the formation of a covalent adaptable network (CAN) within the PCR PP matrix results in this upcycling of PCR PP. Given that the transesterification reaction responsible for topological rearrangement of CANs is slow at room temperature, the PCR PP vitrimer samples behave similarly to non-dynamically crosslinked PCR PP. This creates a significant barrier to crystallite rearrangement, which explains the higher Young's modulus (YM) and yield stress (YS) compared to those of the precursor material. The CANs thus demonstrate their potential for upcycling PCR PP, as evidenced by the increase in YS from 28 MPa in PCR PP to 33 MPa for the 20% TGDDM system.65,66 All vitrimers and the vitrimer-nanocomposite showed a similar elongation at yield (EY) as that of PCR PP. At 20% TGDDM content, the vitrimer reached a saturation point in both Young's modulus (YM) and yield stress (YS), showing similar values to that of the 15% TGDDM system. This suggests that further increases in crosslinker concentration may lead to crosslinker aggregation, negatively affecting mechanical properties. As a result, the 15 TGDDM system was selected for further studies due to its optimum mechanical properties. To explain these observations, it is proposed that at lower gel content, the increased crosslink density enhanced the interactions between molecular chains, improving the mechanical properties of m′-PCR PP TGDDM vitrimers. However, at higher gel content, the denser crosslinked network restricted the mobility and elongation of the molecular chains, causing a decline in EB as the TGDDM content increased from 5% to 20%.67 The underlying reason lies in the fact that the mechanical strength of semi-crystalline polymers like polypropylene (PP) is largely derived from their crystalline morphology. When crosslinked with active agents, the mechanical strength initially increases as crosslinker concentration increases. However, at higher concentrations, the crystalline structure is disrupted, resulting in reduced elongation, toughness, and overall mechanical performance.68,69 Interestingly, the percentage crystallinity showed only a slight decrease from PCR PP to the 20% TGDDM vitrimer (which is further discussed in the later sections), indicating that TGDDM is primarily concentrated in the amorphous regions of PP and has minimal impact on the crystalline regions. Similarly, previous work by Kar et al. reported the use of a di-epoxy crosslinker, DGEBA, to upcycle recycled PP and PE, demonstrating a comparable approach to enhance the mechanical properties of recycled polymers.43 However, TGDDM, with its four epoxide functional groups, exhibits higher reactivity, potentially promoting twice as many crosslinking reactions during epoxy-anhydride curing compared to the former epoxy crosslinker. This enhanced reactivity occurs through interactions with the pendant maleic anhydride groups grafted onto the PCR PP chain via the styrene-maleic anhydride (SMA) charge transfer complex (CTC). As a result, the mechanical properties of PCR PP are significantly enhanced, facilitating more effective upcycling of the material post vitrimer formation.
In the case of graphene oxide (GO), especially in the 15% TGDDM vitrimer a significant increase in Young's modulus was observed while maintaining comparable tensile strength and elongation at yield as listed in Table S4.† This improvement can be attributed to the oxygen-containing functional groups on the GO surface, which likely formed covalent bonds with the epoxide groups of unreacted TGDDM. As a nanofiller, GO possesses a large surface area, facilitating strong interfacial adhesion with the matrix. Additionally, its surface chemistry fosters strong interfacial interactions with the epoxide groups, resulting in a marked enhancement of the composite's mechanical properties. The robust interfacial adhesion between GO and the vitrimer matrix translates to higher energy absorption at failure, as observed in the composites with as low as 0.5% GO in the vitrimer.
However, when the GO content was increased to 1.0%, the system exhibited a reduction in tensile strength, Young's modulus, and elongation at yield, leading to diminished overall toughness in the sample 1 GO. This decline is likely due to the short mixing time (2 minutes) during extrusion, which may have been insufficient to achieve proper dispersion of 1% GO within the vitrimer matrix. As a result, GO agglomerated creating stress concentration points that led to premature failure of the 1 GO vitrimer nanocomposite.70 Thus, the 0.5 GO vitrimer nanocomposite system was selected for further experimental work, as it demonstrated the most optimal mechanical performance.
4.2 Effect on crystallinity and flow properties
Crosslinking semi-crystalline polymers like polypropylene (PP) primarily affects the amorphous regions rather than the crystalline domains.71 Consequently, the thermal transition characteristics of the crosslinked polymers remain like those of their thermoplastic precursors. Table S5† lists the melting temperature (Tm), crystallization temperature (Tc), and crystallinity (% Xc) of the crosslinked vitrimers and their post-consumer recycled thermoplastic precursors, with the corresponding thermograms shown in Fig. S4.†Thermograms reveal a less intense peak at around 125 °C in heating scans and 114 °C in cooling scans of PCR PP, attributed to a minor concentration of polyethylene in the waste PCR PP stream, which potentially entered during collection, sorting, etc. As crosslinking density increased, Tc exhibited an upward trend, reflecting that less undercooling was needed for crystallization due to heterogeneous microdomains acting as nucleation sites in PCR PP. For the 15 TGDDM vitrimer with a gel fraction of 74%, Tc increased marginally as compared to neat PCR PP (Tc = 125 °C), and further addition of TGDDM did not elevate Tc beyond that of 15 TGDDM (Tc = 132 °C), indicating that non-bonded esters aggregated in the amorphous regions, as corroborated by a consistent gel fraction of 74% between 15% and 20% TGDDM.
Moreover, the peak of the crystallization exotherm decreased and broadened with increasing TGDDM concentration, signifying a rise in matrix heterogeneity. This trend was also observed for Tm, where reduced crystalline domains required less energy to melt. Crosslinking constraints significantly hindered the segmental alignment of polyolefin chains, creating a physical barrier to chain packing and reducing crystallinity. At lower TGDDM concentrations, nucleation and crosslinking were balanced, while higher TGDDM content led to dominant crosslinking as evidenced by the 20 TGDDM vitrimer, where crystallinity is lower compared to that of the 5 TGDDM system, although the crystallization temperature (Tc) is the highest as depicted in Table S5.†43 The incorporation of styrene during the melt mixing of PCR PP and maleic anhydride (MA) reduces PP chain scission. It activates MA double bonds via charge transfer complex (CTC) formation, enhancing grafting and reactivity with TGDDM.59,72 This results in increased gel content and decreased crystallinity. Despite the high TGDDM concentration, the vitrimer maintains a notable semi-crystalline structure with only a slight reduction in crystallinity compared to its thermoplastic precursor, PCR PP, thereby preserving its mechanical properties.
The addition of 0.5% GO to the vitrimer results in a minor baseline shift in the DSC thermogram (Fig. 3b), indicating a transition towards a composite material. Given the low concentration of 0.5% GO, it does not significantly influence the crystallinity, since their number and size are insufficient compared to the crosslinking junctions, which are the primary nucleation sites. Consequently, the Tc and crystallization exotherm of the 0.5 GO vitrimer nanocomposite system (ΔHc = 49.6 J g−1) remain comparable to those of the 15 TGDDM vitrimer system (ΔHc = 50.19 J g−1). Similarly, the melting endotherm around Tm exhibits little change, reflecting the minimal impact on crystallinity and thermal transitions.
In contrast, the 1% GO system displays a pronounced baseline shift in the DSC thermogram, suggesting that it has evolved into a distinct vitrimer-nanocomposite system. Although 1% GO content is too low to act as major nucleation sites, the presence of GO domains impedes the smooth packing and folding of the PP matrix, reducing crystallinity (Xc) to 16% while keeping Tc relatively unchanged at 131 °C. This disruption leads to a lower crystallization exotherm of 35.81 J g−1. The diminished crystalline domains require less energy to melt and induce flow, resulting in a reduced melting endotherm of 33.87 J g−1, even though Tm remains intact. Thus, the presence of GO in the 1 GO sample creates barriers that hinder crystalline structure formation, affecting the thermal and mechanical properties of the vitrimer nanocomposite.
TGA thermograms of PCR PP, 15 TGDDM, and 0.5 GO are presented in Fig. S5† which illustrates the differences in thermal stability during the transition from thermoplastic to vitrimer to vitrimer-composite. Notably, the Td5 of both 15 TGDDM and 0.5 GO is observed at approximately 328 °C, which is lower than that of PCR PP (372 °C). However, the Td95 of 0.5 GO exceeds that of both PCR PP and 15 TGDDM, reaching 450 °C. The identical Td5 at 328 °C for both 15 TGDDM and 0.5 GO is presumably due to the volatiles originating from the onset of TGDDM decomposition. According to Lee,73 it was suggested that the thermal breakdown process of cured epoxy resins occurs through the decomposition of the glycidyl ether unit within the network. The TGA analysis demonstrated a significant change in slope at around 373 °C, indicating the decomposition of a substantial amount of TGDDM into glycidyl ether units.74 The higher Td95 of 15 TGDDM compared to PCR PP, along with a residual weight loss of 5.55% at 800 °C, can be attributed to the strong dynamic networks of the β-hydroxy ester in the vitrimer system undergoing thermally activated bond exchange even at elevated temperatures imparting enhanced thermal stability compared to neat PCR PP. Conversely, the Td95 of 0.5 GO, not even identified within the temperature scan range, with a residual weight loss of 7% at 800 °C, indicates that the decomposition temperature is elevated by the addition of GO nanofillers within the 15 TGDDM vitrimer matrix. The reason behind this originates from the phenomenon that the nanofiller GO shows spatial interference which enforces the development of the highly crosslinked molecular structure of 0.5 GO vitrimer-nanocomposites.75
Rheological experiments were conducted to better understand the flow behaviour and viscoelastic nature of vitrimers at increased temperatures, which has a direct impact on their processibility and end use. To investigate the viscoelastic properties of vitrimers, small amplitude oscillatory sweeps (SAOS) were used to assess flow characteristics at different frequencies or shear rates. The frequency sweeps at 180 °C showed that both the vitrimer and the vitrimer-composite exhibited power-law behaviour (G′ ∼ G′′ ∼ ωn), as shown in Fig. 4a. This common power-law behaviour has been regularly observed in various earlier vitrimer investigations.76,77 The classic behaviour of a viscoelastic polymer melt with a significant frequency dependency is evident in the case of PCR PP, where G′ and G′′ increase with an increase in frequency, probably resulting in difficulty in relaxation at higher frequencies. Interestingly, PCR PP exhibited no crossover points between its storage (G′) and loss (G′′) moduli. Furthermore, Fig. 4b shows that PCR PP shows more viscous character (G′′ > G′) than elastic across the full frequency range investigated. Dynamic crosslinking with TGDDM resulted in vitrimers showing elastic solid behaviour with a frequency-independent G′ at lower frequencies, virtually reaching a plateau, and a significantly lower G′′ than G′, as is typical of crosslinked materials.78 Furthermore, the G′ and G′′ trends of 15 TGDDM vitrimer samples in Fig. 4a demonstrated no crossover point in the examined frequency range.
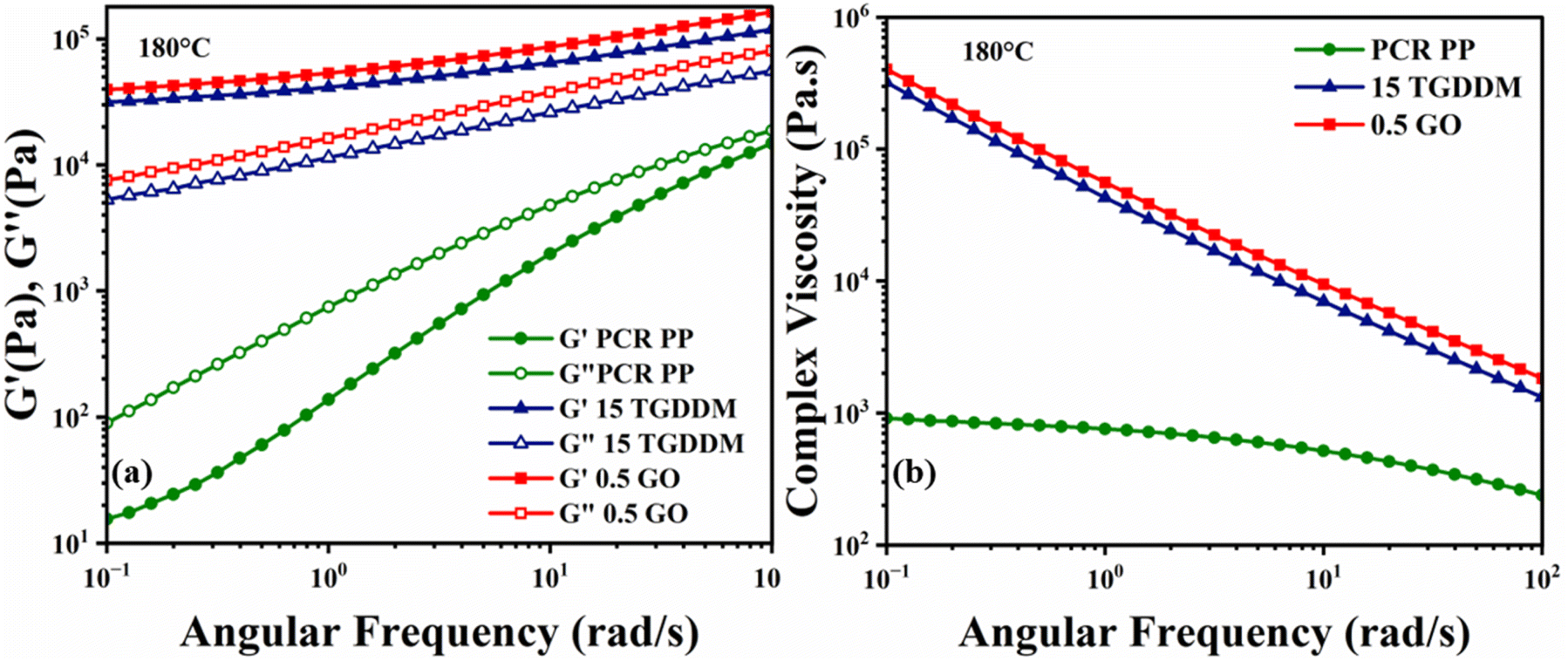 | ||
| Fig. 4 (a) Storage modulus (G′) and loss modulus (G′′) versus angular frequency and (b) complex viscosity versus angular frequency for PCR PP, vitrimer and the vitrimer nanocomposite at 180 °C. | ||
Interestingly, the addition of 0.5% GO in 15 TGDDM did not change its viscoelastic characteristics significantly, only enhancing both G′ and G′′ marginally. The minimal increase in G′ is presumably due to the reaction of GO with the unreacted TGDDM leading to the formation of covalent bonds, and therefore a better filler–matrix interface, and improved load transfer characteristics.70,79,80 The incorporated GO in the vitrimer matrix might bridge the separated polymer chains in the vitrimer-nanocomposite matrix reducing the slippage of the chains during viscous flow and therefore increasing the G′′.81
Fig. 4b shows that PCR PP attained zero-shear viscosity at roughly 103 Pa s, and that of 15 TGDDM continued to increase dramatically, nearly two orders of magnitude higher, with no evidence of zero-shear viscosity within the studied frequency range. This result represents a significant increase in melt strength due to the creation of a crosslinked network in the vitrimers. The 0.5 GO vitrimer-nanocomposite system behaves similarly to the 15 TGDDM vitrimer system in terms of shear thinning. The marginal increase in complex viscosity can be attributed to the formation of a GO nanoparticle network in the vitrimer system corroborated by the literature reported on graphite oxide suspensions in PDMS.81
The surface morphology of tensile fractured PCR PP is reported as shear bands and microfibres in our prior work.16 However, after transforming PCR PP into a vitrimer, the tensile fracture morphology remained unchanged and exhibited the same fibrous texture with no phase separation, as depicted in Fig. 5a. A similar fibrous morphology was observed following the addition of 0.5 wt% GO to the PCR PP vitrimer (Fig. 5b).
 | ||
| Fig. 5 SEM micrograph of tensile fractured surfaces of (a) the PCR PP vitrimer (15 TGDDM) and (b) its nanocomposites (0.5 GO). | ||
4.3 Infrared thermal properties, electrical conductivity and recyclability studies
A series of thermal maps were recorded with an IR camera at 30 second intervals during the heating phase and throughout the cooling phase after the laser was turned off, until reaching room temperature. The resulting data, illustrated in Fig. S6† and 6, present the temperature changes over time, highlighting the notable heat dissipation capabilities of the 15 TGDDM vitrimer and 0.5 GO vitrimer nanocomposite, respectively. This indicates its potential as an effective energy-harvesting material, capable of converting laser energy into thermal energy.82,83 For the 15 TGDDM sample, the temperature increase was minimal (∼74 °C) after 300 seconds of laser exposure (see Fig. S6†). In contrast, the 0.5 GO sample exhibited a sharp temperature increase, reaching up to 108 °C at the same exposure duration, followed by a rapid decline to 32 °C within 120 seconds, as shown in Fig. 6. This temperature increase is attributed to the uniform dispersion of GO as conductive nanofillers within the crosslinked PCR PP matrix, as evidenced by the SEM analysis presented in Fig. 5b. With the ability to generate heat upon laser exposure, this vitrimer nanocomposite could be applied in thermal management systems for electronic devices. For instance, it could be incorporated into systems requiring controlled dissipation of heat generated by components such as chips and processors. The strong photothermal conversion also makes it suitable for solar energy applications, particularly in solar-thermal devices that convert sunlight into heat for water heating, power generation, or other energy applications. | ||
| Fig. 6 (a–d) Infrared thermal image of the PCR PP vitrimer nanocomposite (0.5 GO) subjected to laser heating and (e) temperature vs. time graph upon heating and cooling for the PCR PP vitrimer (15 TGDDM) and its nanocomposite (0.5 GO). | ||
The electrical conductivity of PCR PP and its vitrimer 15 TGDDM is very low due to the insulating nature of the polymer. However, the incorporation of 0.5 wt% GO into the vitrimer increased AC conductivity to 10−10 S cm−1 (Fig. 7). This enhancement is likely due to the formation of a network structure within the polymer matrix, which facilitates charge transport. Chammingkwan et al. observed a similar range of AC conductivity by the addition of 1 wt% GO into PP.84 Therefore, lightweight conducting composites derived from PCR PP and GO can potentially serve as antistatic packaging for electronic components, automotive parts for static dissipation, and EMI shielding materials for sensitive electronic equipment.85
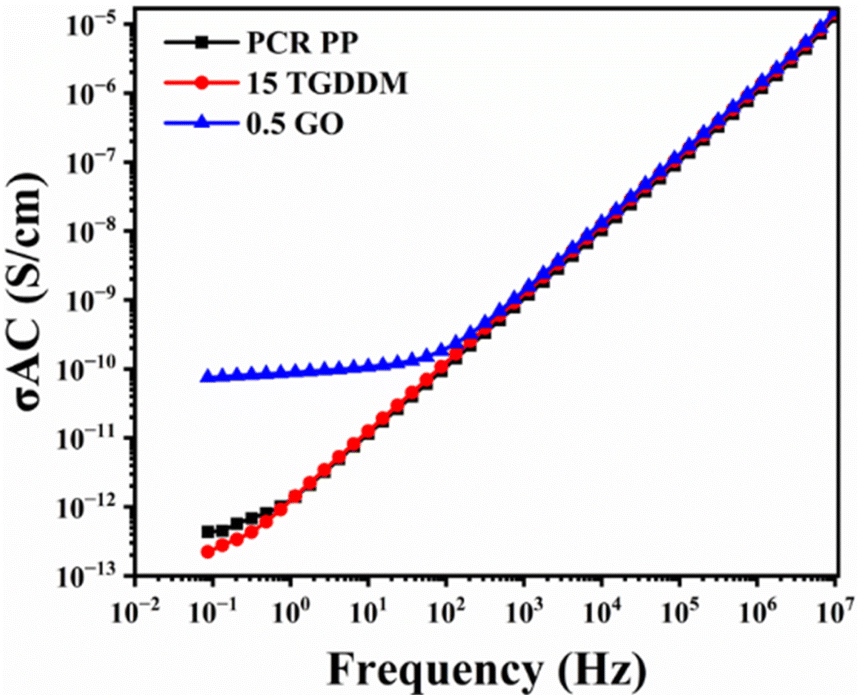 | ||
| Fig. 7 AC electrical conductivity of PCR PP, the PCR PP vitrimer (15 TGDDM), and its nanocomposite (0.5 GO). | ||
The thermally activated bond exchange induced by transesterification in the vitrimer enables the vitrimer-nanocomposite to be reprocessable. The initially extruded strands of 0.5 GO nanocomposite samples were cut into small pieces, re-extruded, and then injection molded under the same processing conditions to produce thermo-mechanically reprocessed vitrimer-nanocomposite samples (0.5 GO-r). The TGA thermogram (Fig. S7†) of the recycled 0.5 GO-r closely matches with that of the original 0.5 GO vitrimer nanocomposite sample showing that no change in the weight loss pattern has occurred, proving that no early degradation occurred due to recycling. Mechanical data (Table S6†) showed that the yield strength (YS), elongation at yield (EY), and Young's modulus (YM) remained almost unchanged after recycling, as evidenced in the stress–strain graph (Fig. S8†) for the 0.5 GO samples. This is further supported by the recovery rates of 100% for both TS and EY including 90% for YM, as depicted in the recovery rate (RR) bar plot (Fig. 8b). The fracture morphology of 0.5 GO-r was analyzed after tensile testing which revealed that the fracture surface exhibited characteristics similar to those of the parent material, including shear bands and microfibers aligned with the load direction, with no apparent phase separation (Fig. S9†). However, during the recycling of 0.5 GO, the AC conductivity slightly decreased to 10−11 S cm−1 due to a minor breakdown of the network during extrusion (Fig. 8c).86 Despite this reduction being minimal, it can be concluded that the AC conductivity is largely preserved after recycling. Furthermore, in the infrared thermal study of 0.5 GO-r, the temperature increase was up to 104 °C at 300 s of laser exposure time and there was an instantaneous fall in temperature (up to 32 °C) within 120 s of turning off the laser beam (Fig. 8d). This suggests the excellent heat dissipation capability of the prepared 0.5 GO vitrimer nanocomposite due to the high thermal conductivity of GO even after vigorous recycling. Thus, a thermally conductive vitrimer-nanocomposite produced from PCR PP helped to synthesise a recyclable nanocomposite for reprocessable CANs that fully recover their crosslink densities, associated thermomechanical properties and AC conductivity.
 | ||
| Fig. 8 Recyclability study of the PCR PP vitrimer nanocomposite. (a) Recycling scheme, (b) recovery rate of mechanical characteristics, (c) AC conductivity, and (d) temperature vs. time graph upon heating and cooling along with infrared thermal imaging for the vitrimer nanocomposite (0.5 GO-r) post recycling. | ||
5. Conclusions
In conclusion, this study demonstrates the successful development of vitrimer nanocomposites from post-consumer recycled polypropylene (PCR PP), modified with styrene-assisted maleic anhydride grafting, TGDDM epoxy, and graphene oxide (GO) as a nanofiller. The maleic anhydride-grafted PCR PP promotes strong interfacial adhesion, enhancing the composite's mechanical properties. TGDDM epoxy contributes a dynamic covalent network, adding reprocessability and durability. GO further reinforces the material, enhancing thermomechanical and rheological performance while ensuring uniform stress distribution. Together, these features position the vitrimer nanocomposites as robust, recyclable materials suitable for diverse industrial applications where longevity, sustainability, and mechanical integrity are paramount. Future work should focus on formulation optimization to expand these nanocomposites' applicability and performance.Data availability
The authors declare that all the data in this manuscript are available upon request.Author contributions
ID and DK contributed equally to this work. SG and SM performed FT-IR, gel fraction, SEM and conductivity analyses. KS performed photothermal analysis. SB and KS conceptualized the work and the manuscript was thoroughly reviewed/edited by SB and KS.Conflicts of interest
The authors declare no competing interest.Acknowledgements
Among the authors, SB is grateful to ANRF (SP/ANRF-24-0018) for the generous funding. ID is grateful to MHRD for a PhD fellowship and KS acknowledges IISc for a postdoctoral fellowship.References
-
A. Gurría, Improving Markets for Recycled Plastics: Trends, Prospects and Policy Responses, OECD Publishing, 2018, pp. 1–160 Search PubMed
.
- H. Sardon and A. P. Dove, Science, 2018, 360, 380–381 CrossRef CAS PubMed
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed
- T. R. Walker and L. Fequet, TrAC, Trends Anal. Chem., 2023, 160, 116984 CrossRef CAS
- L. Jia, S. Evans and S. van der Linden, Nat. Commun., 2019, 10, 4582 CrossRef PubMed
- E. Schmaltz, E. C. Melvin, Z. Diana, E. F. Gunady, D. Rittschof, J. A. Somarelli, J. Virdin and M. M. Dunphy-Daly, Environ. Int., 2020, 144, 106067 CrossRef CAS PubMed
- M. Shen, B. Song, G. Zeng, Y. Zhang, W. Huang, X. Wen and W. Tang, Environ. Pollut., 2020, 263, 114469 CrossRef CAS PubMed
- T. Narancic, S. Verstichel, S. Reddy Chaganti, L. Morales-Gamez, S. T. Kenny, B. De Wilde, R. Babu Padamati and K. E. O'Connor, Environ. Sci. Technol., 2018, 52, 10441–10452 CrossRef CAS PubMed
- L. Cabernard, S. Pfister, C. Oberschelp and S. Hellweg, Nature Sustainability, 2021, 5, 139–148 CrossRef
- S. M. Al-Salem, M. S. El-Eskandarani and A. Constantinou, Waste Manage. Res., 2021, 39, 910–913 CrossRef CAS PubMed
- S. Okamoto, Nat. Mater., 2020, 19, 929–930 CrossRef CAS PubMed
- B. Luijsterburg and H. Goossens, Resour., Conserv. Recycl., 2014, 85, 88–97 CrossRef
- F. Gu, J. Guo, W. Zhang, P. A. Summers and P. Hall, Sci. Total Environ., 2017, 601–602, 1192–1207 CrossRef CAS PubMed
- M. K. Eriksen, J. D. Christiansen, A. E. Daugaard and T. F. Astrup, Waste Manage., 2019, 96, 75–85 CrossRef CAS PubMed
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed
- I. Dey, A. Mohammed N, S. S. Rege, S. S. Islam, A. Misra, K. Samanta, K. Manna and S. Bose, ACS Appl. Mater. Interfaces, 2023, 15(45), 53003–53016 CAS
-
M. Cormann, Global Plastics Outlook: Economic drivers, environmental impacts and policy options, OECD Publishing, 2022, pp. 1–199 Search PubMed
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS
- Y. Yang, Y. Xu, Y. Ji and Y. Wei, Prog. Mater. Sci., 2021, 120, 100710 CrossRef CAS
- F. Van Lijsebetten, J. O. Holloway, J. M. Winne and F. E. Du Prez, Chem. Soc. Rev., 2020, 49, 8425–8438 RSC
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29(14), 1606100 CrossRef PubMed
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustain. Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS
- Q. Li, S. Ma, S. Wang, W. Yuan, X. Xu, B. Wang, K. Huang and J. Zhu, J. Mater. Chem. A, 2019, 7, 18039–18049 RSC
- Q. Li, S. Ma, S. Wang, Y. Liu, M. A. Taher, B. Wang, K. Huang, X. Xu, Y. Han and J. Zhu, Macromolecules, 2020, 53, 1474–1485 CrossRef CAS
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2018, 140, 13272–13284 CrossRef CAS PubMed
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed
- X. Xu, S. Ma, S. Wang, J. Wu, Q. Li, N. Lu, Y. Liu, J. Yang, J. Feng and J. Zhu, J. Mater. Chem. A, 2020, 8, 11261–11274 RSC
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed
- W. A. Ogden and Z. Guan, J. Am. Chem. Soc., 2018, 140, 6217–6220 CrossRef CAS PubMed
- Y.-X. Lu, F. Tournilhac, L. Leibler and Z. Guan, J. Am. Chem. Soc., 2012, 134, 8424–8427 CrossRef CAS PubMed
- Y.-X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed
- C. A. Tretbar, J. A. Neal and Z. Guan, J. Am. Chem. Soc., 2019, 141, 16595–16599 CrossRef CAS PubMed
- W. Gu, F. Li, T. Liu, S. Gong, Q. Gao, J. Li and Z. Fang, Adv. Sci., 2022, 9(11), 2103623 CrossRef CAS PubMed
- Y. Li, M. Zhou, R. Wang, H. Han, Z. Huang and J. Wang, Eur. Polym. J., 2024, 214, 113159 CAS
- N. Zheng, J. Hou, H. Zhao, J. Wu, Y. Luo, H. Bai, J. A. Rogers, Q. Zhao and T. Xie, Adv. Mater., 2019, 31(11), 1807326 CrossRef PubMed
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS
- G. P. Kar, M. O. Saed and E. M. Terentjev, J. Mater. Chem. A, 2020, 8, 24137–24147 RSC
- M. Ahmadi, A. Hanifpour, S. Ghiassinejad and E. van Ruymbeke, Chem. Mater., 2022, 34, 10249–10271 CrossRef CAS
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed
- K. E. Russell and E. C. Kelusky, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 2273–2280 CrossRef CAS
- G. Moad, Prog. Polym. Sci., 1999, 24, 81–142 CrossRef CAS
- J. Cha and J. L. White, Polym. Eng. Sci., 2001, 41, 1227–1237 CrossRef CAS
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read de Alaniz and C. M. Bates, ACS Macro Lett., 2018, 7, 817–821 CrossRef CAS PubMed
- J.-H. Chen, J.-H. Lu, X.-L. Pu, L. Chen and Y.-Z. Wang, Chemosphere, 2022, 294, 133778 CrossRef CAS PubMed
- M. C. De Castro, M. C. C. Dos Santos, D. C. Bastos, J. Trota Filho and L. S. De Moraes, Revista Contemporânea, 2023, 3, 3475–3490 CrossRef
- Y. Huang, Y. Qin, Y. Zhou, H. Niu, Z.-Z. Yu and J.-Y. Dong, Chem. Mater., 2010, 22, 4096–4102 CrossRef CAS
- B. Yuan, C. Bao, L. Song, N. Hong, K. M. Liew and Y. Hu, Chem. Eng. J., 2014, 237, 411–420 CrossRef CAS
- C. Poornima, U. S. Mallik, A. G. Shivasiddaramaiah, N. Pushpalakshmi and B. S. Puneeth, Mater. Today: Proc., 2021, 46, 2477–2482 CAS
- M. Monti, M. Zaccone, A. Frache, L. Torre and I. Armentano, Nanomaterials, 2021, 11, 550 CrossRef CAS PubMed
- S. H. Yetgin, J. Mater. Res. Technol., 2019, 8, 4725–4735 CrossRef CAS
- Y. Li, X.-M. Xie and B.-H. Guo, Polymers, 2001, 42, 3419–3425 CrossRef CAS
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2020, 42(3), 2000415 Search PubMed
- C. Ruiz-Orta, J. P. Fernandez-Blazquez, A. M. Anderson-Wile, G. W. Coates and R. G. Alamo, Macromolecules, 2011, 44, 3436–3451 CAS
- J. Du, Y. Wang, X. Xie, M. Xu and Y. Song, Polymers, 2017, 9, 623 Search PubMed
- C. Ladavière, T. Delair, A. Domard, C. Pichot and B. Mandrand, Polym. Degrad. Stab., 1999, 65, 231–241 Search PubMed
- G. Schoukens and P. Samyn, Thermochim. Acta, 2014, 580, 28–37 CAS
- M. Maaz, A. Riba-Bremerch, C. Guibert, N. J. Van Zee and R. Nicolaÿ, Macromolecules, 2021, 54, 2213–2225 CAS
- Y. Gao and H. Niu, Polym. Chem., 2024, 15, 884–895 CAS
- Y. Zhao, H. Mao, T. Zhang, Z. Guo, D. Bai, H. Bai, Q. Zhang, H. Xiu and Q. Fu, Ind. Eng. Chem. Res., 2022, 61, 13126–13135 CAS
- M. O. Saed, X. Lin and E. M. Terentjev, ACS Appl. Mater. Interfaces, 2021, 13, 42044–42051 CAS
- S. Wang, S. Ma, J. Qiu, A. Tian, Q. Li, X. Xu, B. Wang, N. Lu, Y. Liu and J. Zhu, Green Chem., 2021, 23, 2931–2937 CAS
- H. Yang, C. Shan, F. Li, Q. Zhang, D. Han and L. Niu, J. Mater. Chem., 2009, 19, 8856 CAS
- Y. Zhou, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2017, 50, 6742–6751 CAS
- S. H. P. Bettini and A. C. Ruvolo Filho, J. Appl. Polym. Sci., 2008, 107, 1430–1438 CrossRef CAS
- L. Lee, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 859–882 CrossRef CAS
- M. F. Mustafa, W. D. Cook, T. L. Schiller and H. M. Siddiqi, Thermochim. Acta, 2014, 575, 21–28 CrossRef CAS
- B. S. Bohra, P. Singh, A. Rana, H. Sharma, T. Arya, M. Pathak, A. Chaurasia, S. Rana and N. G. Sahoo, Compos. Sci. Technol., 2023, 241, 110143 CrossRef CAS
- C. R. López-Barrón, J. Lu, J. A. Throckmorton, H. Passino and M. Gopinadhan, Macromolecules, 2024, 57, 2729–2745 CrossRef
- J. Qiu, S. Ma, S. Wang, Z. Tang, Q. Li, A. Tian, X. Xu, B. Wang, N. Lu and J. Zhu, Macromolecules, 2021, 54, 703–712 CrossRef CAS
- H. H. Winter and F. Chambon, J. Rheol., 1986, 30, 367–382 CrossRef CAS
- G. B. Olowojoba, S. Kopsidas, S. Eslava, E. S. Gutierrez, A. J. Kinloch, C. Mattevi, V. G. Rocha and A. C. Taylor, J. Mater. Sci., 2017, 52, 7323–7344 CrossRef CAS PubMed
- B. Krishnakumar, R. V. S. Prasanna Sanka, W. H. Binder, C. Park, J. Jung, V. Parthasarthy, S. Rana and G. J. Yun, Composites, Part B, 2020, 184, 107647 CrossRef CAS
- M. Martin-Gallego, M. M. Bernal, M. Hernandez, R. Verdejo and M. A. Lopez-Manchado, Eur. Polym. J., 2013, 49, 1347–1353 CrossRef CAS
- X. Cui, Q. Ruan, X. Zhuo, X. Xia, J. Hu, R. Fu, Y. Li, J. Wang and H. Xu, Chem. Rev., 2023, 123, 6891–6952 CrossRef CAS PubMed
- Z. Wang, M. Wang, X. Wang, Z. Hao, S. Han, T. Wang and H. Zhang, Biosens. Bioelectron., 2023, 220, 114883 CrossRef CAS PubMed
- P. Chammingkwan, K. Matsushita, T. Taniike and M. Terano, Materials, 2016, 9, 240 CrossRef PubMed
- S. K. H. Gulrez, M. E. Ali Mohsin, H. Shaikh, A. Anis, A. M. Pulose, M. K. Yadav, E. H. P. Qua and S. M. Al-Zahrani, Polym. Compos., 2014, 35, 900–914 CrossRef CAS
- I. Lorero, A. Mujica, M. Campo and S. G. Prolongo, Polym. Compos., 2024, 45, 6041–6058 CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00904e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |