
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of triamine-functionalized rigid crosslinkers for materials science†
Niccolò
Braidi
 *a,
Aitor
Hernández
b,
Giulia
Scurani
a,
Francesca
Parenti
a,
Nezha
Badi
b and
Filip E.
Du Prez
*b
*a,
Aitor
Hernández
b,
Giulia
Scurani
a,
Francesca
Parenti
a,
Nezha
Badi
b and
Filip E.
Du Prez
*b
aDepartment of Chemical and Geological Sciences, University of Modena and Reggio Emilia, via Campi 103, 41125, Modena, Italy. E-mail: niccolo.braidi2@unibo.it
bPolymer Chemistry Research Group, Centre of Macromolecular Chemistry, Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281 (S4), 9000 Ghent, Belgium. E-mail: filip.duprez@ugent.be
First published on 25th February 2025
Abstract
In this study, a primary amine-terminated star-shaped polystyrene (PS) was synthesized using an Activators Regenerated by Electron Transfer Atom Transfer Radical Polymerization (ARGET ATRP) protocol, yielding products with low dispersity (<1.2) and molar masses in the range of 2 to 12 kDa. The influence of the trifunctional initiator's reactivity on the resulting polymer topology was investigated. The bromo-terminated PS was efficiently converted to its azide-terminated counterpart as confirmed by online ATR FT-IR and NMR spectroscopy. The targeted amine-terminated PS was then obtained by a Staudinger reduction of the azide groups using tributylphosphine. To assess the applicability of these novel amine-terminated PSs as well-defined trifunctional crosslinking agents, traditional epoxy thermoset networks and covalent adaptable networks (CANs) were synthesized using diepoxides or diacetoacetates, respectively. The resulting materials exhibited excellent thermal resistance, attributed to the high PS content. Moreover, by making use of the option of tuning the molar mass of such macromolecular crosslinkers, the network's crosslinking density could be tailored, enabling control over swelling degree, glass transition temperature, and, in the case of the obtained vinylogous urethane vitrimers, even reprocessability.
Introduction
The role of amines in materials science is prevalent, with applications spanning catalysis,1,2 epoxy thermoset materials,3 surface modification,4,5 and carbon capture,6,7 to name a few. Particularly in the field of polymer science, following the discovery of covalent adaptable networks (CANs),8–11 multifunctional amine-terminated (macro)molecules have become crucial in the development of recyclable crosslinked materials based on various exchange chemistries. These include vinylogous urethanes,12–14 dicarboxamide-imide,15,16 imine-based,17–19 and β-amino ester chemistries.20–22 As a result, there is considerable interest in creating novel multifunctional amines for using them as rigid crosslinkers in materials science and replacing for example the widely used tris(2-aminoethyl)amine (TREN) containing a tertiary amine moiety.Given the ability of Atom Transfer Radical Polymerization (ATRP) to precisely control the macromolecular architecture23,24 and, consequently, the mechanical properties of polymers, it is not surprising that the design of primary amine-terminated polymers derived from this technique has been widely investigated. This is particularly evident in the following examples, which summarize attempts to introduce amines at the polymer chain ends and highlight some of the challenges encountered in achieving these goals. Coessens and Matyjaszewski reported an unsuccessful attempt to obtain primary amine-terminated poly(methyl acrylate) by reacting a bromo-terminated precursor with ammonia.25 In contrast, an excess of diamines (e.g., hexamethylenediamine) reacted quantitatively, although this resulted in chain ends bearing both primary and secondary amine functionalities.26,27 Another approach involves Gabriel's synthesis, starting with nucleophilic substitution using potassium phthalimide, followed by cleavage via acid hydrolysis (or hydrazine in the Ing-Manske variant) to release the primary amine. Using this method, Monge et al. and subsequent researchers observed quantitative conversion of bromo-terminated polymers.28–33 However, Postma et al. reported that β-elimination competed with nucleophilic substitution, decreasing the chain-end functionality of the heterotelechelic polystyrene employed.34,35 Furthermore, Weimer et al. tested the Ing-Manske procedure on hyperbranched polystyrene but encountered crosslinking, attributed to nucleophilic displacement of residual halides by the newly formed primary amines.36 Another approach, offering arguably higher atom economy (depending on the reducing agent used), using less harmful reagents than the Ing-Manske variant, and with reduced susceptibility to β-elimination, involved the replacement of halogenated chain ends with azides (using sodium azide37–44 or trimethylsilyl azide38,45,46), followed by reduction to obtain primary amine chain ends. While the first step has been frequently reported in the literature due to its use in azide–alkyne Huisgen cycloaddition,47,48 fewer reports describe the reduction of azide-terminated polymers. Matyjaszewski et al. reported the quantitative reduction of both heterotelechelic and telechelic azide-terminated polystyrene using LiAlH4.45 However, they observed gelation of the reaction mixture during the reduction of telechelic polymers, necessitating a larger solvent volume to complete the reaction. Given the drawbacks of the aforementioned procedures, the Staudinger reduction of azide-terminated polymers with triphenylphosphine, initially proposed by Coessens et al., has been more widely applied.2,25,41,46 The primary amine-terminated polymers obtained through this method have been used in copolymerizations,49 (bio)conjugation,50 membrane fabrication for (nano)filtration,31 and catalyst production.2 However, to the best of our knowledge, they have not yet been tested as crosslinking agents.
Therefore, the objective of this study is twofold: first, the development of a synthetic pathway for rigid, star-shaped amino-terminated polystyrenes with variable molar mass (trisAminoPS); second, the evaluation of these products as crosslinkers in both conventional epoxy networks and CANs (Fig. 1).
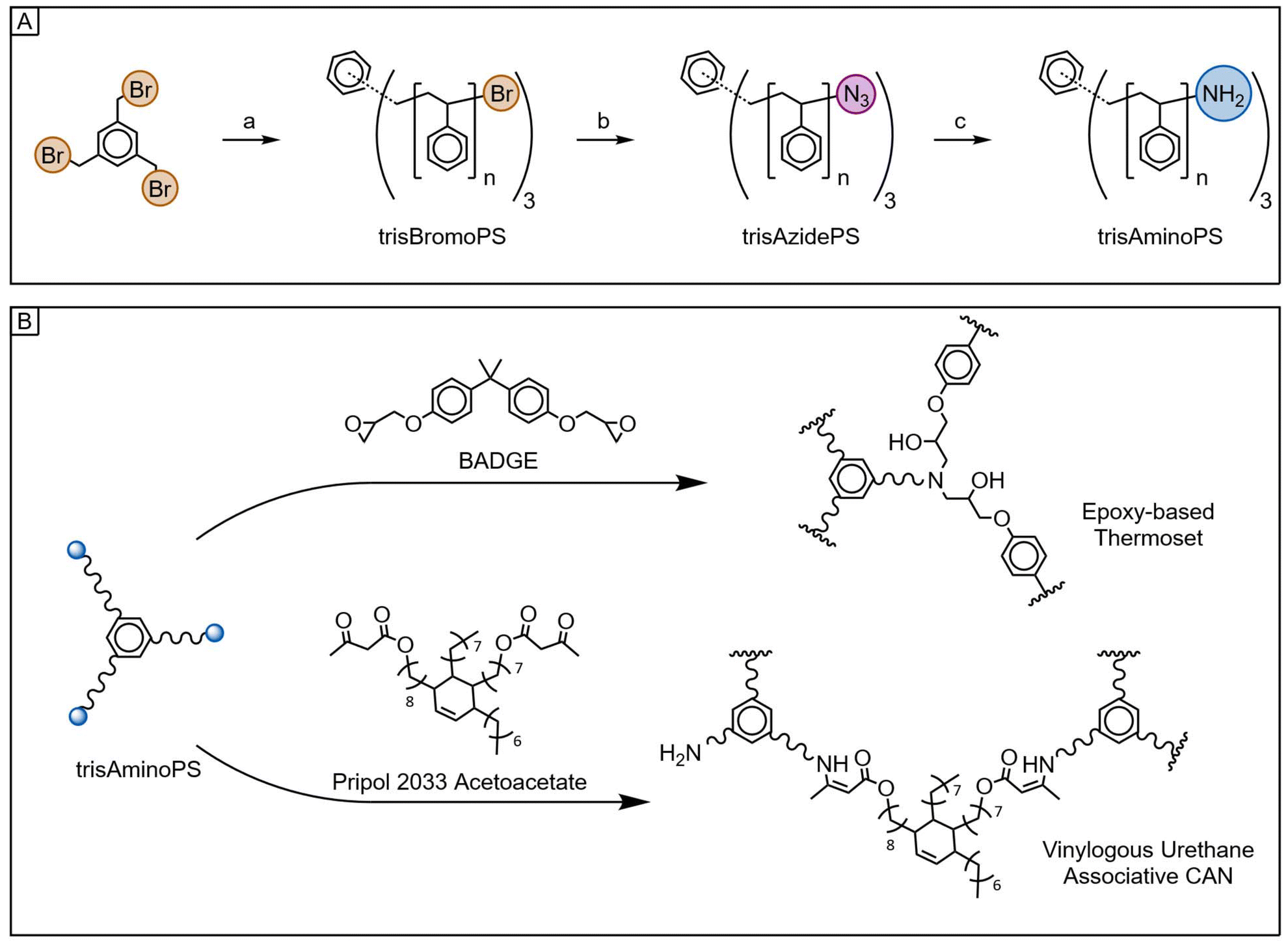 | ||
| Fig. 1 Overview of the reactions explored. (A) Synthetic pathway yielding trifunctional primary amine-terminated polystyrene: (a) ARGET ATRP of styrene initiated by 1,3,5-tris(bromomethyl)benzene, (b) nucleophilic substitution with sodium azide, (c) Staudinger reduction with tributylphosphine and water. (B) Crosslinking process using bisphenol A diglycidyl ether (BADGE) or Pripol 2033 acetoacetate to form a traditional epoxy-based network or a vinylogous urethane CAN, respectively. | ||
The proposed synthetic pathway begins with the Activators Regenerated by Electron Transfer (ARGET)51–53 ATRP of styrene using a trifunctional brominated initiator (1,3,5-tris(bromomethyl)benzene) for the design of trifunctional bromo-terminated polystyrenes (trisBromoPS) with inherently high glass transition temperature (Tg). This is followed by nucleophilic substitution with sodium azide to yield trisAzidePS and subsequent Staudinger reduction with tributylphosphine to obtain the trifunctional primary amine-terminated polystyrenes (trisAminoPS, Fig. 1A). The latter macromolecular structures are then reacted with a diepoxide or a diacetoacetate electrophilic compound to form the targeted networks (Fig. 1B).
Results and discussion
The choice of ARGET among the advanced ATRP techniques54–58 relies on the possibility of using mild organic bases such as 2,4,6-trimethylpyridine, in combination with a lipophilic derivative of ascorbic acid (5,6-isopropylidene-L-ascorbic acid). This allows precise tuning of the concentration of radicals, as discussed in a previous study,51 which is crucial to minimize radical–radical termination reactions that could lead to branched or even crosslinked products when using a trifunctional initiator.59–61 Since our previously reported reaction conditions were optimized for chlorinated initiators, some modifications were necessary to adapt them for brominated initiators, which yield more readily functionalizable polymers.62 Specifically, the solvent was changed to anisole (instead of an ethyl acetate/ethanol solution) to prevent nucleophilic substitution of the ethoxide anion—generated in small quantities by ethanol in a basic environment—on the more labile brominated chain-ends.52 Additionally, due to the altered polarity of the reaction mixture and thus to address solubility, a more lipophilic ligand, 4,4′-dinonyl-2,2′-dipyridyl, was employed instead of the previously used tris(2-pyridylmethyl)amine ligand.By varying the amount of initiator used, a series of polymers with different chain lengths were synthesized, by employing a volume of styrene (Vstyrene) of 20 mL. The number-average molar masses determined by gel permeation chromatography (MGPCn) were found to closely match the targeted theoretical values (Mthn), as reported in Table 1. Note that the linear fit of MGPCn has a slope slightly lower than unity, leading to significant discrepancies between the two values at higher molar masses (Fig. 2A).
 | ||
Fig. 2 Characterization of trisBromoPSs obtained via the ARGET ATRP of styrene initiated by 1,3,5-tris(bromomethyl)benzene. Reaction conditions: [styrene]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CuBr2/(4,4′-dinonyl-2,2′-dipyridyl)2]0:[5,6-isopropyl-idene-L-ascorbic acid]0:[2,6-dimethylpyridine]0 = 100:0.0511:0.249:0.737 mol% (relative to styrene), with [1,3,5-tris(bromomethyl)benzene]0 varied from 2.76 to 0.447 mol% to target different Mthn, Vstyrene:Vanisole = 20:10 v/v, T = 100 °C, time = 14 h. (A) Plot of MGPCnversus Mthn (green triangles) with linear fitting, including the 95% confidence interval (solid line and shaded area, respectively), Mthnversus Mthn (dashed line); (B) plot of p versus Mthn (red circles, left ordinate) with the mean value (p = 0.49, red dashed line), plot of Đ versus Mthn (blue diamonds, right ordinate) with the mean value (Đ = 1.15, blue dashed line); (C) MWDs of three trisBromoPSs with Mn = 1.94, 4.03, and 11.51 kDa (green, blue, and magenta, respectively); (D) overlaid 1H-NMR spectra of trisBromoPSs, expanded in the δ = 4.25–4.6 ppm region, normalized to δ = 4.52 ppm to highlight the decrease in intensity of the broad shoulder assigned to the pendant PS-Ph(CH2Br)-PS, relative to the broad peak assigned to the chain-end PS-CH(Ph)Br, as MGPCn increases. :[CuBr2/(4,4′-dinonyl-2,2′-dipyridyl)2]0:[5,6-isopropyl-idene-L-ascorbic acid]0:[2,6-dimethylpyridine]0 = 100:0.0511:0.249:0.737 mol% (relative to styrene), with [1,3,5-tris(bromomethyl)benzene]0 varied from 2.76 to 0.447 mol% to target different Mthn, Vstyrene:Vanisole = 20:10 v/v, T = 100 °C, time = 14 h. (A) Plot of MGPCnversus Mthn (green triangles) with linear fitting, including the 95% confidence interval (solid line and shaded area, respectively), Mthnversus Mthn (dashed line); (B) plot of p versus Mthn (red circles, left ordinate) with the mean value (p = 0.49, red dashed line), plot of Đ versus Mthn (blue diamonds, right ordinate) with the mean value (Đ = 1.15, blue dashed line); (C) MWDs of three trisBromoPSs with Mn = 1.94, 4.03, and 11.51 kDa (green, blue, and magenta, respectively); (D) overlaid 1H-NMR spectra of trisBromoPSs, expanded in the δ = 4.25–4.6 ppm region, normalized to δ = 4.52 ppm to highlight the decrease in intensity of the broad shoulder assigned to the pendant PS-Ph(CH2Br)-PS, relative to the broad peak assigned to the chain-end PS-CH(Ph)Br, as MGPCn increases. | ||
| Entry | [Initiator]0 (mol%) | p |
M
thnc (kDa) |
M GPCn (kDa) | Đ | f | T g (°C) |
|---|---|---|---|---|---|---|---|
|
a Common conditions: [styrene]0:[CuBr2/(4,4′-dinonyl-2,2′-dipyridyl)2]0:[5,6-isopropyl-idene-L-ascorbic acid]0:[2,6-dimethylpyridine]0 = 100:0.0511:0.249:0.737 mol% with respect to styrene, Vstyrene:Vanisole = 20:10 mL, T = 100 °C, time = 14 h.
b The amounts of monomer, solvent, and reagents were increased fivefold.
c Calculated with eqn (S1).†
|
|||||||
| 1 | 2.76 | 0.416 | 1.93 | 1.94 | 1.21 | 2.84 | 64.2 |
| 2b | 0.902 | 0.225 | 2.95 | 3.09 | 1.17 | 2.79 | 76.5 |
| 3 | 1.54 | 0.420 | 3.20 | 3.30 | 1.16 | 2.95 | 76.8 |
| 4 | 1.01 | 0.409 | 4.57 | 4.03 | 1.16 | 2.82 | 80.2 |
| 5 | 0.832 | 0.489 | 6.48 | 5.91 | 1.13 | 2.89 | 83.3 |
| 6 | 0.671 | 0.517 | 8.38 | 7.54 | 1.12 | 2.76 | 84.6 |
| 7 | 0.487 | 0.554 | 12.20 | 10.75 | 1.13 | 2.85 | — |
| 8 | 0.447 | 0.547 | 13.10 | 11.51 | 1.13 | 2.75 | 90.2 |
This arises from the smaller radius of gyration of star polymers than that of the linear PS standards used for GPC calibration.63,64 Conversions (p) higher than 0.4 were achieved, yielding products on a ten-gram scale with dispersities (Đ) below 1.2. Similarly low Đ values were obtained when the reaction scale was increased fivefold, although this came at the expense of the yield (entry 2, Table 1). Moreover, targeting higher Mthn gratifyingly resulted in higher p and lower Đ values (Fig. 2B). Notably, the narrow and monomodal molecular weight distributions (MWD) recorded indicate minimal occurrence of radical–radical termination reactions, regardless of the targeted Mthn (Fig. 2C).
Only a slight incidence of the expected β-elimination was observed, resulting in double bonds at some chain ends, as evidenced by the 1H-NMR spectra (Fig. S1–S8†).34,65,66 Consequently, across the explored Mthn range, the average PS functionality (f, calculated with eqn (S3)†) was found to be ![[f with combining macron]](https://www.rsc.org/images/entities/i_char_0066_0304.gif) NMR = 2.83 ± 0.06 instead of the ideal value of f = 3. These products do not possess perfect star-shaped structures because the bromides of 1,3,5-tris(bromomethyl)benzene are less reactive than those at the PS chain ends. This disparity arises from the fact that the bromides in the initiator are bound to primary carbons, whereas the chain-end bromides are attached to secondary carbons.67 As a result, the radicals generated on the initiator are less stable than those at the chain ends, leading to incomplete arm initiation. Evidence for this is provided by the 1H-NMR spectra of the trisBromoPS samples. When the spectra are overlaid and normalized at δ = 4.52 ppm, a decrease in the intensity of the broad signal attributed to the pendant PS-Ph(CH2Br)-PS, relative to the broad peak assigned to the chain-end PS-CH(Ph)Br, is apparent (Fig. 2D). This effect becomes more pronounced at lower MGPCn values, where the highest intensity of the PS-Ph(CH2Br)-PS signal was observed. In other words, the low MGPCn products are richer in telechelic structures with a pendant bromide in the middle compared to the desired three-arm star polymer. Accurate quantification of the three-arm star molar fraction (χstar) is challenging due to significant overlap with the PS-CH(Ph)Br signal. However, this limitation could be readily addressed by using a more reactive initiator.67
NMR = 2.83 ± 0.06 instead of the ideal value of f = 3. These products do not possess perfect star-shaped structures because the bromides of 1,3,5-tris(bromomethyl)benzene are less reactive than those at the PS chain ends. This disparity arises from the fact that the bromides in the initiator are bound to primary carbons, whereas the chain-end bromides are attached to secondary carbons.67 As a result, the radicals generated on the initiator are less stable than those at the chain ends, leading to incomplete arm initiation. Evidence for this is provided by the 1H-NMR spectra of the trisBromoPS samples. When the spectra are overlaid and normalized at δ = 4.52 ppm, a decrease in the intensity of the broad signal attributed to the pendant PS-Ph(CH2Br)-PS, relative to the broad peak assigned to the chain-end PS-CH(Ph)Br, is apparent (Fig. 2D). This effect becomes more pronounced at lower MGPCn values, where the highest intensity of the PS-Ph(CH2Br)-PS signal was observed. In other words, the low MGPCn products are richer in telechelic structures with a pendant bromide in the middle compared to the desired three-arm star polymer. Accurate quantification of the three-arm star molar fraction (χstar) is challenging due to significant overlap with the PS-CH(Ph)Br signal. However, this limitation could be readily addressed by using a more reactive initiator.67
Although the post-functionalization of bromo-terminated polystyrenes with sodium azide has been extensively reported, the procedures show a wide range of reaction times, spanning from a few38,68 to several2,39,41 hours. Therefore, to monitor the progress of the azidation reaction, online ATR FT-IR was employed, specifically tracking the emergence of the azide stretching peak at 2096 cm−1.2,39,46,69 After only two hours of reaction at 30 °C, the intensity of this peak approached an asymptotic value (Fig. S9†). Quantitative conversion from bromide to azide was confirmed by 1H-NMR analysis. As shown in previous studies,2,39,43,45,46 the chemical shift of the chain-end methines moved up-field from 4.2–4.9 ppm for PS-CH(Ph)Br to 3.8–4.1 ppm for PS-CH(Ph)N3 (Fig. S10–S16†). Notably, the peak attributed to PS-Ph(CH2N3)-PS (δ = 4.1–4.2 ppm) was sharper and significantly less overlapped with the PS-CH(Ph)N3 signal (δ = 3.8–4.1 ppm). This assignment was further confirmed by an HSQC spectrum (Fig. S17†). Unlike the brominated precursor, this enabled the assessment of χstar (calculated with eqn (S2), ESI†) which increased from 0.06 for the polymer with MGPCn = 2.22 kDa to χstar = 0.80 for the polymer with MGPCn = 11.20 kDa (Table S1†). The functionality value remained unchanged after the nucleophilic substitution with sodium azide (NMR = 2.84 ± 0.06). Additionally, the yields were consistently high for both low and high-molar mass polymers,  . Consequently, the MGPCn values of the azide-functionalized PS closely matched those of the brominated precursors, as reflected in the low mean percentage difference between the two,
. Consequently, the MGPCn values of the azide-functionalized PS closely matched those of the brominated precursors, as reflected in the low mean percentage difference between the two,  .
.
For the final step of the proposed synthetic pathway (i.e., the Staudinger reduction, step c in Fig. 1A), tributylphosphine was chosen because of its reportedly lower hazard profile, ease of byproduct removal, and superior atom economy compared to the more commonly used triphenylphosphine.2,41,46 Upon addition of the reagent to the trisAzidePS solution in THF, rapid nitrogen gas evolution was observed, corresponding to the formation of the iminophosphorane intermediate, which was subsequently cleaved upon the addition of water. As an example of this strategy, the reduction of a low-molar mass trisAzidePS (entry 1, Table S2†) demonstrated quantitative chain-end reduction, as confirmed by 1H-NMR analysis. The chemical-shift of the chain-end methines shifted up-field from δPS-CH(Ph)N3 = 3.8–4.1 ppm to δPS-CH(Ph)NH2 = 3.3–3.9 ppm (Fig. 3A). This result was consistently observed across the entire series of triamine-functionalized PS (Fig. S18–S24†).
 | ||
| Fig. 3 Characterization of the low-MW trisAminoPS (entry 1, Table S2†) obtained by the reduction of trisAzidePS with tributylphosphine and water. Reaction conditions: tributylphosphine (2 equiv. relative to the azide functionalities of the trisAzidePS), VTHF = 6 mL g−1 with respect to wtrisAzidePS, T = 60 °C, time = 0.5 h. Subsequently, VH2O = 0.6 mL g−1 was added, and the reaction mixture was stirred at 60 °C overnight. (A) Superimposed 1H-NMR spectra of trisBromoPS (orange), trisAzidePS (purple), and trisAminoPS (blue). The inset highlights the chemical shift changes of the proton(s) geminal to the respective functionalities. (B) MALDI-TOF MS spectrum of trisAminoPS measured using dithranol as the matrix and AgTFA as the ionizing salt. The main population appears at m/z = 2148 ± n·104 u (green dashed line), with progressively less intense populations observed at m/z = 2131 (orange), 2114 (magenta), and 2096 (red) ± n·104 u. Assignments are discussed in the text. (C) MWDs of the products. | ||
This result was further confirmed by MALDI-TOF MS analysis (Fig. 3B). The mass spectrum showed a main distribution with a peak at m/z = 2148 u, corresponding to the calculated mass for trisAminoPS (n = 18 leading to [C153H153(NH2)3, Ag]+ = 2148 u). A secondary distribution shifted by −17 u aligns with the expected polystyrene containing two amines and a dehydrohalogenated chain-end, [C153H152(NH2)2, Ag]+ = 2131 u. Only minor traces of products with one or no amine (from double or triple dehydrohalogenation) were detected, shifted by −34 and −51 u, respectively.
These findings support the quantitative conversion of azides to amines, suggesting, as previously reported,34,65,66 that the primary cause of the small functionality loss arises from the undesired dehydrohalogenation during the ARGET ATRP step. By comparing the peak intensities, the chain-end functionality determined by MALDI-ToF MS (fMALDI = 2.69) is consistent with the value obtained from 1H-NMR (fNMR = 2.75). As ARGET ATRP allows the synthesis of low-dispersity polymers with predetermined molar masses, it is crucial that the post-functionalization pathway does not compromise these properties. This requirement was fulfilled in the reduction step, where the low molar mass and dispersity of the starting trisBromoPS were maintained in the amine derivative (Fig. 3C). A slight shift toward higher molar masses was observed, likely due to the precipitation-based purification process, which removed lower molar mass chains that dissolve in methanol. This explanation is further supported by the increase in yields as the MGPCn of the precursor increased (Table S2†). While testing this synthetic step, a potential side reaction with high-molar mass azide-functionalized PSs was found. Specifically, a shoulder appeared at higher molar masses in the MWD, indicative of coupling reactions. This unwanted reaction was slightly more pronounced for longer precursors, as shown in the comparison between lower and higher MGPCn (Fig. 4A). Notably, decreasing the reaction time between tributylphosphine and water addition significantly suppressed these side reactions. Specifically, waiting 0.5 h instead of 3 h resulted in a less pronounced high-MW shoulder (Fig. 4B). A possible explanation for this phenomenon is the formation of isocyanate through the reaction of iminophosphorane complexes with CO2.70 Such isocyanates could then react further with the produced free-base amines, forming urea bridges. However, the significant impact of reaction conditions on this side reaction suggests that proper optimization could effectively mitigate it.
 | ||
| Fig. 4 MWDs of trisAminoPS (blue) compared to their respective precursors (purple). The entries (referencing Table S2†) differ in the molar mass of the precursor and the reaction time waited before the addition of water, while all other reaction conditions are kept constant. (A) Variation in the MGPCn of the precursor: increases going from the first to the second row. (B) Variation in the time waited between the addition of tributylphosphine and water: increases going from the first to the second column (0.5 to 3 h). | ||
To evaluate the sustainability of the proposed synthetic pathway, the E-factor has been calculated. According to the first principle of green chemistry, this value should be as close to zero as possible, indicating minimal waste generation.71,72 By definition, the E-factor accounts for solvent waste, which typically constitutes 80–90% of the reaction mixture's weight. In industrial settings, solvent management is significantly more optimized than at the laboratory scale. As a result, the true E-factor (i.e., the value achievable in an optimized industrial process) lies between a simplified E-factor (sEF), assuming 100% solvent recovery, and a complete E-factor (cEF), assuming total solvent waste. As a result of the additive nature of the E-factor, the overall sEF for the multistep synthesis of trisAminoPS was calculated as 1.85 (0.76 + 0.11 + 0.98 for the first, second, and third synthetic steps, respectively), placing it within the range for bulk chemicals. In contrast, the cEF-value is 156, with the primary contribution arising from solvents used in product recovery and purification via precipitation. When excluding the purification steps, the E-factor significantly decreases to 14, which highlights the main expected limitation of the proposed synthesis method, which is the waste generated during product recovery.
In the second part of this research, two examples have been selected as proof-of-concept to explore the potential applicability of trisAminoPSs as crosslinkers, namely for the synthesis of (1) traditional thermosets using bisphenol A diglycidyl ether (BADGE) with a 3:1 molar ratio relative to trisAminoPS and (2) vinylogous urethane associative CANs (vitrimers)12 using Pripol 2033 acetoacetate in a 3:2.1 molar ratio relative to trisAminoPS. Preliminary tests revealed that the latter reaction required demanding conditions to achieve significant conversion. Specifically, the reaction necessitated the use of a solvent (toluene), high reaction temperatures (100–140 °C), and extended reaction times (48 h). In contrast, while the BADGE-based thermoset also required elevated temperatures and prolonged reaction times, the synthesis could be done in bulk. These overall demanding conditions for the crosslinking were attributed to two factors: the relatively low concentration of reactive groups (compared to small polyamine molecules) and the positioning of these groups on less accessible sites, i.e. on a secondary carbon or on a primary carbon located in the middle of the polymeric chain. Despite these challenges, the relatively low soluble fractions of the resulting networks, with a mean value of 5.3% w/w for the epoxy-based thermoset and 16.0% w/w for the acetoacetate-based CAN, show that trisAminoPS can effectively be used as a crosslinker (Table 2). Furthermore, the results revealed that tuning the MGPCn of the trisAminoPS can effectively influence the properties of the resulting networks. First, it was observed that increasing the MGPCn of the precursor leads to a progressive increase in the swelling ratio of the networks as expected. Specifically, swelling increased from 210% to 560% for the epoxy-based network and from 620% to 2140% for the acetoacetate-based one. This trend is attributed to a dilution of the crosslinking points caused by the higher number of repeating polystyrene units. Second, it was observed that the glass transition temperature (Tg) of the networks decreases with increasing molar mass of the precursor. For the epoxy-based networks, Tg dropped from 109 to 74 °C, while for the acetoacetate-based networks, Tg decreased from 104 °C to 54 °C (Fig. S25† and Table 2).This was attributed to a progressive decrease in crosslink density, which appears to outweigh the inherent increase in the Tg of the precursor associated with higher MGPCn (Table S2†). For comparison, a previously reported Pripol-based network crosslinked with the common crosslinker TREN exhibited a low Tg of −24 °C (specifically, Pripol 2033 acetoacetate and TREN combined with Priamine 1074 at a ratio of 0.40:0.40:0.95 eq.), resulting in elastomeric materials.13 This shows that the PS-based crosslinkers significantly enhanced the rigidity of the networks, despite the use of an elastomeric acetoacetate. Third, the rheological properties of the vinylogous urethane network were evaluated. The material was compression molded at 160 °C for 60 min under a pressure of 4 metric tons using a steel mold, resulting in pressed samples that exhibited optical transparency (Fig. S26†). Discs of an appropriate diameter were subsequently punched out for testing. Amplitude-sweep experiments were conducted to determine the optimal conditions for stress-relaxation experiments, which were set at an amplitude of 2% and a frequency of 1 Hz (Fig. S27†). From these experiments, relaxation times were extrapolated across the temperature range of 160–195 °C, and Arrhenius plots were constructed (Fig. S28†). These allowed calculation of the following activation energies: 238.81 ± 18.66 kJ mol−1 (for the 4.69 kDa precursor) and 182.05 ± 6.12 kJ mol−1 (for the 7.94 kDa precursor). As previously observed when macromolecules are employed as precursors for CANs,73,74 these values are significantly higher than those reported for vinylogous urethane networks derived from small molecules.12 Moreover, a significant decrease in activation energy was observed with increasing MGPCn of the precursor. This trend was tentatively attributed to the synergic decrease in Tg and an increase in χstar, where the latter improves the accessibility of the amines as the MGPCn of the precursor increases. These effects counteract the decrease in concentration of active moieties (i.e., decreased dynamic crosslinking density), which has been previously reported to hinder the exchange dynamics.75 It is noteworthy that the modulus of relaxation in both samples increased slightly from one temperature to another, which is attributable to incomplete curing of the product, as evidenced by their soluble fractions (>10 wt%). Additionally, the exceptionally high temperatures required to obtain relaxation times are clearly imputable to the pronounced rigidity of the product. In fact, these last two observations prevent a reliable evaluation of the results in absolute terms. However, the comparison between the two samples highlights the significant influence of the PS-based precursor on the properties of the crosslinked product and thus its potential applicability in reprocessable materials with other dynamic chemistry platforms that make use of amine crosslinkers.18,21
| Entry | Bifunctional electrophile | M thn (kDa) | Swelling ratio (%) | Soluble fraction (%) | T g (°C) | w loss225 °C (%) | T onsetd,air (°C) |
|---|---|---|---|---|---|---|---|
| a Common conditions: trisAminoPS and the electrophile were added to a glass vial, alongside toluene in the case of Pripol 2033 acetoacetate. The vial was loosely sealed, heated overnight (100 °C), and the resulting networks were dried in an oven at 120 °C and under vacuum at 140 °C. b Extrapolated at wloss = 2.5%. c Extrapolated after two isotherms (160 °C for 1 h and 225 °C for 2 h). d Extrapolated after one isotherm (225 °C for 2 h). | |||||||
| 1 | BADGE | 2.58 | 207 ± 7 | 5.0 ± 0.3 | 109 | — | — |
| 2 | 4.69 | 300 ± 30 | 5.4 ± 5.3 | 71 | 1.46c | 217 | |
| 3 | 7.46 | 395 ± 13 | 5.3 ± 0.3 | 99 | — | — | |
| 4 | 7.94 | 511 ± 40 | 5.4 ± 2.3 | 75 | 3.94c | 277 | |
| 5 | 12.18 | 556 ± 7 | 5.5 ± 0.4 | 74 | — | — | |
| 6 | Pripol 2033 acetoacetate | 2.58 | 624 ± 95 | 10.3 ± 1.0 | 104 | — | — |
| 7 | 4.69 | 900 ± 50 | 13.7 ± 3.0 | 76 | 1.28d | 327 | |
| 8 | 7.46 | 1202 ± 130 | 19.9 ± 3.6 | 72 | — | — | |
| 9 | 7.94 | 1050 ± 60 | 12.7 ± 1.9 | 84 | 1.81d | 325 | |
| 10 | 12.18 | 2140 ± 197 | 23.3 ± 2.9 | 54 | — | — | |
Finally, thermogravimetric analysis (TGA) was conducted on selected samples (entries 2, 4, 7, and 9 in Table 2). First, we compared the thermal behavior of the samples under a heating ramp in an air atmosphere (10 °C min−1). As observed from the overlaid curves and the data in Table 2, the VU-based networks exhibit inherently higher thermal resistance (Fig. S29†). Additionally, the peak in the first derivative appears slightly shifted to lower temperatures in networks employing a lower molar mass precursor, which is a trend observed in both types of networks. Conversely, under isothermal degradation conditions, samples with a shorter precursor exhibit a lower overall weight loss (compare entry pairs 2 vs. 4 and 7 vs. 9, Table 2). Overall, these relatively low weight losses, combined with high degradation onset temperatures—particularly for the VU-based networks—indicate good thermal stability of the materials. This thermal behavior aligns well with the composition of the networks, which consist of over 90% PS in weight.
Conclusions
This study reported the development of a synthetic pathway for producing trifunctional amine-terminated polystyrenes and their potential use as crosslinking agents in both traditional and covalent adaptable networks. Specific attention was given to low molar mass products, demonstrating that the ARGET ATRP procedure produces bromide-bearing precursors in the 2–12 kDa range with low dispersities. The selection of the initiator was a critical factor as its reactivity was found to influence the topology of the resulting polymers. β-Elimination was identified as the primary source of functionality loss but in all cases an overall functionality above 2.8 was found. Post-functionalization with sodium azide was shown to be both highly effective, as confirmed by 1H-NMR analysis, and efficient as indicated by online ATR FT-IR. The quantitative reduction of trifunctional azide-terminated PS using tributylphosphine was demonstrated by 1H-NMR and MALDI-TOF MS. A potential side reaction was noticed, attributed to coupling due to isocyanate formation, and a strategy to minimize its occurrence by reducing the time waited before water addition was proposed. The scalability of the synthetic pathway was hinted by operating on a multi-gram scale, with one example involving a fivefold increase in the quantities of reagents at all steps. Then, the resulting amine-terminated star-shaped PSs, with varying molar masses and low dispersities, were successfully employed as crosslinkers to produce both traditional networks (using a diepoxide) and CANs (using a diacetoacetate molecule). This approach demonstrated how the precursor architecture (molar mass and topology) influences the properties of the resulting networks, including swelling degree, glass transition temperature, and, in the case of vinylogous urethane vitrimers, reprocessability. The final materials exhibited excellent thermal resistance, attributed to high PS content, further confirming the potential of this synthetic approach.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The NMR Expertise Centre (Ghent University) is acknowledged for providing support and access to NMR infrastructure funded by the Research Foundation Flanders (FWO I006920N) and the Bijzonder Onderzoeksfonds (BOF.BAS. 2022.0023.01). N. B. and F. D. P thank BOF-UGent for GOA-funding (BOF21/GOA/007). A. T.'s PhD thesis was funded by the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant Agreement No. 860911.References
- L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807–1821 RSC.
- S. Abraham, C.-S. Ha and I. Kim, Macromol. Rapid Commun., 2006, 27, 1386–1392 CrossRef CAS.
- F.-L. Jin, X. Li and S.-J. Park, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- N. Bouazizi, J. Vieillard, B. Samir and F. Le Derf, Polymers, 2022, 14, 378 CrossRef CAS PubMed.
- Y.-W. Jeong, S. Jung, J. J. Han, H.-J. Park, R. Y. Kim, B.-H. Kim and M.-S. Kook, Materials, 2019, 12, 2581 CrossRef CAS PubMed.
- J. Hack, N. Maeda and D. M. Meier, ACS Omega, 2022, 7, 39520–39530 CrossRef CAS PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- F. Van Lijsebetten, T. Debsharma, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202210405 CrossRef CAS PubMed.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS PubMed.
- F. Van Lijsebetten, K. De Bruycker, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202113872 CrossRef CAS PubMed.
- F. Van Lijsebetten, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 15834–15844 CrossRef CAS PubMed.
- H. Zhang, A. van Hertrooij, T. Schnitzer, Y. Chen, S. Majumdar, R. A. T. M. van Benthem, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2023, 56, 6452–6460 CrossRef CAS PubMed.
- S. K. Schoustra, V. Asadi, H. Zuilhof and M. M. J. Smulders, Eur. Polym. J., 2023, 195, 112209 CrossRef CAS.
- S. K. Schoustra, V. Asadi and M. M. J. Smulders, ACS Appl. Polym. Mater., 2024, 6, 79–89 CrossRef CAS PubMed.
- A. Liguori and M. Hakkarainen, Macromol. Rapid Commun., 2022, 43, 2100816 CrossRef CAS PubMed.
- L. T. Nguyen, C. Mertens and F. E. Du Prez, Macromolecules, 2024, 57, 4817–4825 CrossRef CAS.
- L. T. Nguyen, F. Portone and F. E. Du Prez, Polym. Chem., 2024, 15, 11–16 RSC.
- L. Stricker, C. Taplan and F. E. Du Prez, ACS Sustainable Chem. Eng., 2022, 10, 14045–14052 CrossRef CAS.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800616 CrossRef PubMed.
- K. Matyjaszewski, Chem. Mater., 2024, 36, 1775–1778 CrossRef CAS.
- V. Coessens and K. Matyjaszewski, J. Macromol. Sci., Part A:Pure Appl. Chem., 1999, 36, 811–826 CrossRef.
- J. Babin, D. Taton, M. Brinkmann and S. Lecommandoux, Macromolecules, 2008, 41, 1384–1392 CrossRef CAS.
- B. Lepoittevin, A. Elhiri, L. Bech, J. Belleney, J.-P. Baltaze, I. Capron, V. Planchot and P. Roger, Carbohydr. Polym., 2011, 83, 1174–1179 CrossRef CAS.
- S. Monge, O. Giani, E. Ruiz, M. Cavalier and J.-J. Robin, Macromol. Rapid Commun., 2007, 28, 2272–2276 CrossRef CAS.
- X. Zhang, O. Giani, S. Monge and J.-J. Robin, Eur. Polym. J., 2008, 44, 3676–3687 CrossRef CAS.
- R. P. Johnson, Y.-I. Jeong, E. Choi, C.-W. Chung, D. H. Kang, S.-O. Oh, H. Suh and I. Kim, Adv. Funct. Mater., 2012, 22, 1058–1068 CrossRef CAS.
- Q. Yang, H. H. Himstedt, M. Ulbricht, X. Qian and S. R. Wickramasinghe, J. Membr. Sci., 2013, 430, 70–78 CrossRef CAS.
- H. H. Himstedt, Q. Yang, L. P. Dasi, X. Qian, S. R. Wickramasinghe and M. Ulbricht, Langmuir, 2011, 27, 5574–5581 CrossRef CAS PubMed.
- T. Itoh, T. Iwai, E. Ihara and K. Inoue, Polym. J., 2007, 39, 853–860 CrossRef CAS.
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, React. Funct. Polym., 2006, 66, 137–147 CrossRef CAS.
- C. Shi, L. Lin, Y. Yang, W. Luo, M. Deng and Y. Wu, e-Polymers, 2020, 20, 317–327 CrossRef CAS.
- M. W. Weimer, J. M. J. Fréchet and I. Gitsov, J. Polym. Sci., Part A:Polym. Chem., 1998, 36, 955–970 CrossRef CAS.
- L. Li, C. Wang, Z. Long and S. Fu, J. Polym. Sci., Part A:Polym. Chem., 2000, 38, 4519–4523 CrossRef CAS.
- V. Coessens, Y. Nakagawa and K. Matyjaszewski, Polym. Bull., 1998, 40, 135–142 CrossRef CAS.
- W.-B. Zhang, Y. Tu, R. Ranjan, R. M. Van Horn, S. Leng, J. Wang, M. J. Polce, C. Wesdemiotis, R. P. Quirk, G. R. Newkome and S. Z. D. Cheng, Macromolecules, 2008, 41, 515–517 CrossRef CAS.
- H.-X. Wu, W.-M. Cao, R.-F. Cai, Y.-L. Song and L. Zhao, J. Mater. Sci., 2007, 42, 6515–6523 CrossRef CAS.
- L. Chen, X. Wang, R. Hou, H. Lu, Z. He, X. Zhou, W. Zhang and X. Wang, Polym. Chem., 2023, 14, 4659–4670 RSC.
- A. P. Vogt and B. S. Sumerlin, Macromolecules, 2006, 39, 5286–5292 CrossRef CAS.
- Q. Liu and Y. Chen, J. Polym. Sci., Part A:Polym. Chem., 2006, 44, 6103–6113 CrossRef CAS.
- S. Abraham, C.-S. Ha and I. Kim, J. Polym. Sci., Part A:Polym. Chem., 2006, 44, 2774–2783 CrossRef CAS.
- K. Matyjaszewski, Y. Nakagawa and S. G. Gaynor, Macromol. Rapid Commun., 1997, 18, 1057–1066 CrossRef CAS.
- E. Cloutet, J.-L. Fillaut, D. Astruc and Y. Gnanou, Macromolecules, 1999, 32, 1043–1054 CrossRef CAS.
- F.-A. Teng, Y. Guo, J. He, Y. Zhang, Z. Han and H. Li, Des. Monomers Polym., 2017, 20, 283–292 CrossRef CAS PubMed.
- H. Gao, G. Louche, B. S. Sumerlin, N. Jahed, P. Golas and K. Matyjaszewski, Macromolecules, 2005, 38, 8979–8982 CrossRef CAS.
- K. R. Brzezinska and T. J. Deming, Macromol. Biosci., 2004, 4, 566–569 CrossRef CAS PubMed.
- Y. Sasaki, N. Konishi, M. Kohri, T. Taniguchi, K. Kishikawa and T. Karatsu, Colloid Polym. Sci., 2022, 300, 319–331 CrossRef CAS.
- N. Braidi, F. Parenti, G. Scurani, F. Tassinari, M. Buffagni, L. Bonifaci, G. Cavalca, N. Pettenuzzo and F. Ghelfi, Polym. Chem., 2023, 14, 1567–1576 RSC.
- N. Braidi, M. Buffagni, F. Ghelfi, F. Parenti, A. Gennaro, A. A. Isse, E. Bedogni, L. Bonifaci, G. Cavalca, A. Ferrando, A. Longo and I. Morandini, J. Macromol. Sci., Part A:Pure Appl. Chem., 2021, 58, 376–386 CrossRef CAS.
- N. Braidi, N. Porcelli, F. Roncaglia, A. Mucci and F. Tassinari, Macromol. Rapid Commun., 2025, 46, 2400564 CrossRef CAS PubMed.
- M. Flejszar, P. Chmielarz, J. Smenda and K. Wolski, Polymer, 2021, 228, 123905 CrossRef CAS.
- G. Gazzola, S. Pasinato, M. Fantin, N. Braidi, C. Tubaro, C. Durante and A. A. Isse, Molecules, 2022, 27, 6312 CrossRef CAS PubMed.
- G. Damonte, I. Zaborniak, M. Klamut, D. Di Lisa, L. Pastorino, K. Awsiuk, K. Wolski, P. Chmielarz and O. Monticelli, Int. J. Biol. Macromol., 2024, 273, 132768 CrossRef CAS PubMed.
- J. Wang, L. Zhang, D. Chen, C. Wang, Z. Ren and Z. Wang, Macromolecules, 2024, 57, 6267–6274 CrossRef CAS.
- C. Wang, H. Deng and H. Zhao, Giant, 2022, 11, 100114 CrossRef CAS.
- S. Angot, K. S. Murthy, D. Taton and Y. Gnanou, Macromolecules, 1998, 31, 7218–7225 CrossRef CAS.
- K. Ohno, B. Wong and D. M. Haddleton, J. Polym. Sci., Part A:Polym. Chem., 2001, 39, 2206–2214 Search PubMed.
- Y. Gnanou and D. Taton, Macromol. Symp., 2001, 174, 333–341 CrossRef CAS.
- V. Coessens and K. Matyjaszewski, J. Macromol. Sci., Part A:Pure Appl. Chem., 1999, 36, 667–679 CrossRef.
- W. Radke, J. Gerber and G. Wittmann, Polymer, 2003, 44, 519–525 CrossRef CAS.
- J. Gerber, W. Radke and G. Wittmann, Int. J. Polym. Anal. Charact., 2004, 9, 39–52 CrossRef CAS.
- J.-F. Lutz and K. Matyjaszewski, J. Polym. Sci., Part A:Polym. Chem., 2005, 43, 897–910 CrossRef CAS.
- K. Matyjaszewski, K. Davis, T. E. Patten and M. Wei, Tetrahedron, 1997, 53, 15321–15329 CrossRef CAS.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed.
- H. Gao, D. J. Siegwart, N. Jahed, T. Sarbu and K. Matyjaszewski, Des. Monomers Polym., 2005, 8, 533–546 CrossRef CAS.
- D. Yang, L. Li and C. Wang, Mater. Chem. Phys., 2004, 87, 114–119 CrossRef CAS.
- D. Carnaroglio, K. Martina, G. Palmisano, A. Penoni, C. Domini and G. Cravotto, Beilstein J. Org. Chem., 2013, 9, 2378–2386 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2023, 25, 1704–1728 Search PubMed.
- T. V. T. Phan, C. Gallardo and J. Mane, Green Chem., 2015, 17, 2846–2852 RSC.
- J. Tellers, R. Pinalli, M. Soliman, J. Vachon and E. Dalcanale, Polym. Chem., 2019, 10, 5534–5542 RSC.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. Du Prez, Polym. Chem., 2020, 11, 5377–5385 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00098j |
| This journal is © The Royal Society of Chemistry 2025 |