
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphonium fullerides: isolable zwitterionic adducts of a phosphine with C60†
Maike B.
Röthel
,
Jonas H.
Franzen
,
Daniel
Leitner
,
Thomas S.
Hofer
 ,
Michael
Seidl
and
Fabian
Dielmann
*
,
Michael
Seidl
and
Fabian
Dielmann
*
Institute of General, Inorganic and Theoretical Chemistry, Universität Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: fabian.dielmann@uibk.ac.at
First published on 26th June 2025
Abstract
Although fullerene derivatization has been extensively studied for decades, zwitterionic adducts with neutral Lewis bases are rare, and those with tertiary phosphines remain elusive. This work presents a combined experimental and computational study on the first isolable zwitterionic phosphonium fullerides. The reaction of tris(tetramethylguanidinyl)phosphine ((tmg)3P) with C60 results in the formation of the zwitterionic adducts (tmg)3PC60 and (tmg)3PC60P(tmg)3 in quantitative yield. Stoichiometric studies demonstrate that up to two (tmg)3P molecules can reversibly bind to C60, forming bisphosphine adducts as regioisomeric mixtures with reduced P–C bond stability. Spectroscopic, crystallographic, and computational analyses reveal the presence of σ-type dative P–C bonds and significant charge redistribution within the fullerene cage. Furthermore, functionalizations of the phosphonium fullerides with electrophiles yield ionic derivatives, highlighting their reactivity and potential for further modification.
Introduction
With the development of a multigram preparation method for buckminsterfullerene (C60)1 and its ton-scale production,2,3 research into fullerene derivatization has intensified.3,4 A particular focus lies in the selective functionalization of the C60 cage, aiming to achieve unique structural and electronic properties with multifaceted application potential for high-tech, fullerene-based materials.5,6 Due to its pronounced electrophilicity,7 C60 reacts preferentially with nucleophilic reagents and accepts electrons from strong electron donors. In this context the reactivity with various neutral Lewis bases was investigated. A common reaction pathway of ambiphilic reagents is the cycloaddition at the C–C bond bisecting two six-membered rings of the C60 cage, as observed with carbenes8 and their heavier homologues9,10 (Fig. 1a). Primary and secondary amines undergo hydroamination,11,12 analogous to secondary phosphines, reacting to form 1,2-hydrophosphination products13,14 (Fig. 1b). While such 1,2-additions with neutral nucleophiles are widely documented, there are few examples where the nucleophile binds to a single carbon atom, forming a zwitterionic adduct. For example, the zwitterionic product A has been isolated from the reaction of the N-heterocyclic carbene (NHC) 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene with C60 (Fig. 1c).15–17 In the reaction between the non-nucleophilic base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and C60, initial electron transfer forms [DBU]+[C60]−, and spectroscopic data suggest subsequent formation of the zwitterionic adduct.18 Recent studies by Hobza and coworkers provide evidence for the formation of dative piperidine N–C60 bonds, which is enhanced cooperatively via hydrogen bonding interactions between the piperidine molecules.19 To date, however, isolable zwitterionic Lewis base adducts with tertiary amines or tertiary phosphines remain elusive.20,21 | ||
| Fig. 1 Selected fullerene derivatives obtained by reaction of buckminsterfullerene with neutral Lewis bases via (a) cycloadditions,10,22 (b) 1,2-hydroaminations12 or 1,2-hydrophosphinations,13 (c) formation of zwitterionic adducts15 including the herein presented phosphonium fulleride (Dipp = 2,6-diisopropylphenyl, tmg = tetramethylguanidinyl). | ||
Regarding tertiary phosphines, computational studies by Hobza and coworkers have shown that the substituents at the phosphorus atom significantly influence the stability of the resulting C60–P bond.23 Specifically, π-donating nitrogen substituents were found to enhance dative C60–P interaction, suggesting near thermoneutral C60 complexation when phosphines bear dimethylamino or 1-pyrrolidinyl substituents. Our group has further demonstrated that phosphines with strongly π-donating guanidine-type substituents exhibit superbasic character24 and act as strong nucleophiles towards various substrates, including carbon dioxide,25 chlorazolium salts,26 sulfur dioxide27 and sulfur hexafluoride.28 In this study we investigate the complexation reaction of tris(tetramethylguanidinyl)phosphine29 – a synthetically easily accessible phosphorus superbase – with buckminsterfullerene.
Results and discussion
Synthesis and characterization of tris(tetramethylguanidinyl)phosphonium fulleride (1)
Inspired by Hobza's prediction,23 we initially examined the reactivity of tris(dialkylamino)phosphines with C60. However, no reaction between tris(dimethylamino)phosphine or tris(diethylamino)phosphine and C60 was observed in the absence of molecular oxygen,21 even under irradiation with light at different wavelengths (ESI, Chapter 1.2†). By contrast tris(tetramethylguanidinyl)phosphine, (tmg)3P, reacts with C60 at room temperature in 1,2-dichlorobenzene (DCB) to form tris(tetramethylguanidinyl)phosphonium fulleride (1, (tmg)3P–C60) in quantitative yield (Fig. 2a). Compound 1 is isolated as a crystalline dark green solid after solvent removal. It is moderately soluble in THF, DCM and halogenated aromatics. In contrast to the air-sensitive free phosphine, the fullerene adduct 1 is stable in wet THF and can be briefly handled in air as a solid (see the ESI† for further details). The formation of the zwitterionic adduct is indicated by the pronounced highfield shift of the 31P resonance δ = −18.7 ppm (P(tmg)3: δ = 83.5 ppm). Phosphorus carbon coupling constants between the phosphonium center and the C60-cage (1JPC = 147 Hz and 2JPC = 9 Hz) are deduced from carbon satellites in the 31P{1H} NMR spectrum as well as from the corresponding resonances in the 13C{1H} NMR spectrum. | ||
| Fig. 2 (a) Reaction of (tmg)3P with C60 to give 1; (b) cyclic voltammogram of 1 in THF at a scan rate of 100 mV s−1 (Bu4NPF6, 0.1 M as electrolyte; Pt working electrode); (c) solid-state structure (positional disorder of C60 and THF solvent molecule as well as hydrogen atoms are omitted for clarity); thermal ellipsoids are set at 50% probability, selected bond lengths (Å) and angles (°): P–C1 1.873(2), P–N4 1.6021(16), P–N1 1.606(2), N1–C61 1.304(3), N4–C66 1.308(3), N4–P–N1 117.55(7), N4–P–N4 103.18(12). | ||
An X-ray diffraction study of single crystals obtained from a concentrated tetrahydrofuran solution confirmed that 1 is an adduct of the phosphine (tmg)3P and C60 connected via a single phosphorus–carbon bond (Fig. 2c). The bond length (1.873 Å) is in the range of a σ-single bond and in good agreement with the P–C bond length of the zwitterionic carbon dioxide complex (tmg)3P–CO2 (1.879 Å).29 The sum of N–P–N angles (338.3°) shows a planarization of the P moiety relative to the free phosphine (296.2°) which is slightly more pronounced than in the CO2 adduct (332.3°).
We next studied the electrochemical properties of 1 using cyclic voltammetry (Fig. 2b and S10–S14†). Four sequential reduction events were observed during the reductive sweep (Fig. 2b, 3–6), consistent with the stepwise reduction of the fulleride core of R3P–C60 (1), aligning with established fullerene electrochemistry.‡ These reduction processes remain consistent upon repeated cycling, reversing the scan direction, or using lower scan rates. Furthermore, the observed reduction potentials do not match those of buckminsterfullerene (Fig. S15†), indicating that the fullerene–phosphine adduct remains intact throughout the redox events during the cyclic voltammetry. Remarkably, the first reduction potential of 1 (Ered11/2 = −1.55 V) appears at significantly more negative potential than both that of the first and second reduction waves of pristine C60 (Ered11/2 = −0.82 V, Ered21/2 = −1.35 V). This observation is attributed to the zwitterionic nature of 1, consisting of a cationic phosphonium moiety bound to an anionic fulleride core. The addition of an electron to 1 during the first reduction therefore produces a radical dianionic fulleride moiety. By contrast, the dianionic fulleride C602− is generated at more positive potential due to the more efficient delocalization of the π electrons. The similarity in redox chemistry to the dianionic fulleride C602− can be explained using the orbital interaction diagram (Fig. S48†). This diagram shows that the formation of the P–C bond, arising from the orbital overlap between the HOMO of (tmg)3P and the LUMO of C60, results in a doubly occupied, high-energy, C60-centered HOMO. At the same time, the number and energies of the remaining frontier orbitals of C60 remain largely unchanged. Experimental evidence supporting the assignment of the reduction events was obtained by treating 1 with cobaltocene (E°(DCM) = −1.33 V vs. Fc)30 and decamethylcobaltocene (E°(DCM) = −1.94 V vs. Fc).30 While pristine C60 is already reduced by cobaltocene,31 no reaction was observed between 1 and cobaltocene. However, 1 is readily reduced by decamethylcobaltocene as the color of the solution changes from dark green to dark brown and the resonance of phosphine (tmg)3P appears in the 31P NMR spectrum of the reaction mixture. This observation shows that the dissociation of the radical anion 1− into free phosphine and C60− is energetically favored. However, cleavage of the P–C bond seems to be kinetically hindered, as even the polyanions 1n− (n = 1–4) were persistent during the cyclic voltammetry studies.
In addition to the reduction processes, the voltammogram of 1 exhibits two oxidation processes, labeled 1 and 2 (Fig. 2b). Based on the frontier orbital energies of 1 (Fig. 3 and S46†), the fulleride core undergoes the first oxidation (process 2), resulting in the formation of the corresponding cationic phosphonium fullerenyl radical. The second oxidation process is likely to involve the removal of another electron from the fullerene core since the guanidinyl groups are expected to be oxidized at significantly more positive potentials (for comparison: Emax[{tmg}4P+] = 0.80 V, Fig. S15†). The resulting phosphonium fullerenyl dication (R3P+–C60+) resembles organofullerenyl cations (R–C60+)32 which are important intermediates in fullerene derivatization and can be generated from the corresponding anions (RC60−) through successive oxidation using rather mild oxidants such as CuII ions or iodine.33,34 Notably, alkylfullerenyl cation intermediates are stabilized by adjacent donor atoms,33,35 a scenario that is also conceivable for the guanidinyl nitrogen atoms in 12+.
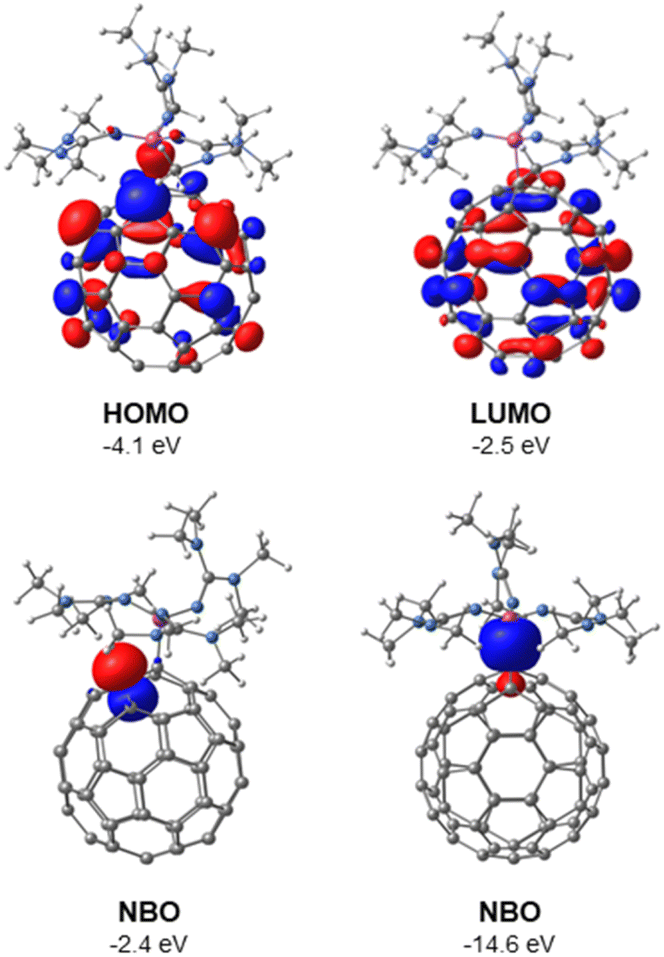 | ||
| Fig. 3 Molecular orbitals (HOMO and LUMO) and two selected natural bond orbitals (NBOs) of 1, determined by DFT using B3LYP(GD3BJ)/6-31G(d,p)/SMD(THF). | ||
A UV/Vis spectroscopic analysis of 1 in aromatic solvents (Fig. S15 and S16†) reveals a strong absorption at 330 nm, consistent with that observed for the neutral fullerene molecule.36 Additionally, a broad absorption band emerges around 650 nm which has been also observed for fulleride B.10
The molecular and natural bond orbitals of 1 were calculated using density functional theory (DFT) at the B3LYP(GD3BJ)/6-31G(d,p)/SMD(THF) level. In analogy to the zwitterionic phosphonium fulleride A,15 the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are both localized on the C60 cage. The HOMO (E = −4.1 eV) exhibits its highest orbital coefficients adjacent to the C–P bond, while the LUMO (E = −2.5 eV) is more centrally located on the C60 cage (Fig. 3 top). The natural bond orbitals (NBO) analysis indicates that the highest energy orbital corresponds to the lone pair on the carbon atom positioned adjacent to the quaternary carbon atom, both situated between two hexagonal rings (Fig. 3 bottom left, E = −2.4 eV, occupancy: 1.17 e−, 99.43% p orbital character).§ The NBO analysis furthermore confirms the σ-type nature of the C–P bond (Fig. 3 bottom right, Wiberg bond index = 0.72, bond length: 1.886 Å).
Reactivity of 1 with selenium and electrophiles
To evaluate the reversibility of the reaction between C60 and (tmg)3P, gray selenium was added to a solution of 1 in THF. No reaction was observed at ambient temperature. However, upon heating the mixture to 100 °C for 16 hours, complete conversion to the corresponding phosphine selenide was achieved, indicating dissociation of the P–C bond at elevated temperature.The potential frustrated Lewis pair (FLP) character of the zwitterionic fullerene–NHC adduct A was highlighted by Alcarazo and coworker.37 To investigate the possible ambiphilic behavior of 1, THF solutions of 1 were treated with carbon dioxide, dihydrogen, and diphenylacetylene, and the reaction mixtures were heated up to 100 °C. NMR analysis revealed no reaction with these substrates, which are known to undergo transformations with FLPs.38
The novel phosphine–fullerene adduct 1 was reacted with electrophiles (E) to investigate the feasibility of further functionalizing the anionic fulleride moiety. Treatment with proton and methyl cation sources resulted in the selective formation of a single regioisomer of the cation [(tmg)3P–C60–E]+ accompanied by a color change from green to brown¶ (Fig. 4a and Table 1). Specifically, the stochiometric reaction of 1 with [H(Et2O)2][B(C6F5)4] in dichloromethane quantitatively yielded [(tmg)3PC60H][B(C6F5)4] (2). Consistent with the NBO analysis, the protonation was found to occur at the carbon atom adjacent to the P–C bond, as evidenced by the characteristic 3JPH coupling constant of 32 Hz (Table 1). Additionally, the 1H–13C{1H} heteronuclear multiple bond correlation (HMBC) 2D NMR spectrum revealed long-range coupling between the newly attached proton and the quaternary carbon bonded to the phosphonium unit. It is noteworthy that 2,6-lutidinium triflate is also sufficiently acidic to protonate 1. However, attempts to remove 2,6-lutidine by heating to 90 °C under reduced pressure led to the formation of insoluble products. Treatment of 1 with methyl triflate in dichloromethane gave [(tmg)3PC60CH3][OTf] (3) in quantitative yield. The observation of a single set of signals in 1H, 13C and 31P NMR spectra suggests the selective methylation adjacent to the P–C bond, in agreement with the protonation reaction and NBO analysis. The ionic fullerene derivatives 2 and 3 exhibit good solubility in polar solvents such as dichloromethane, which is significantly better than that of the neutral adduct 1.
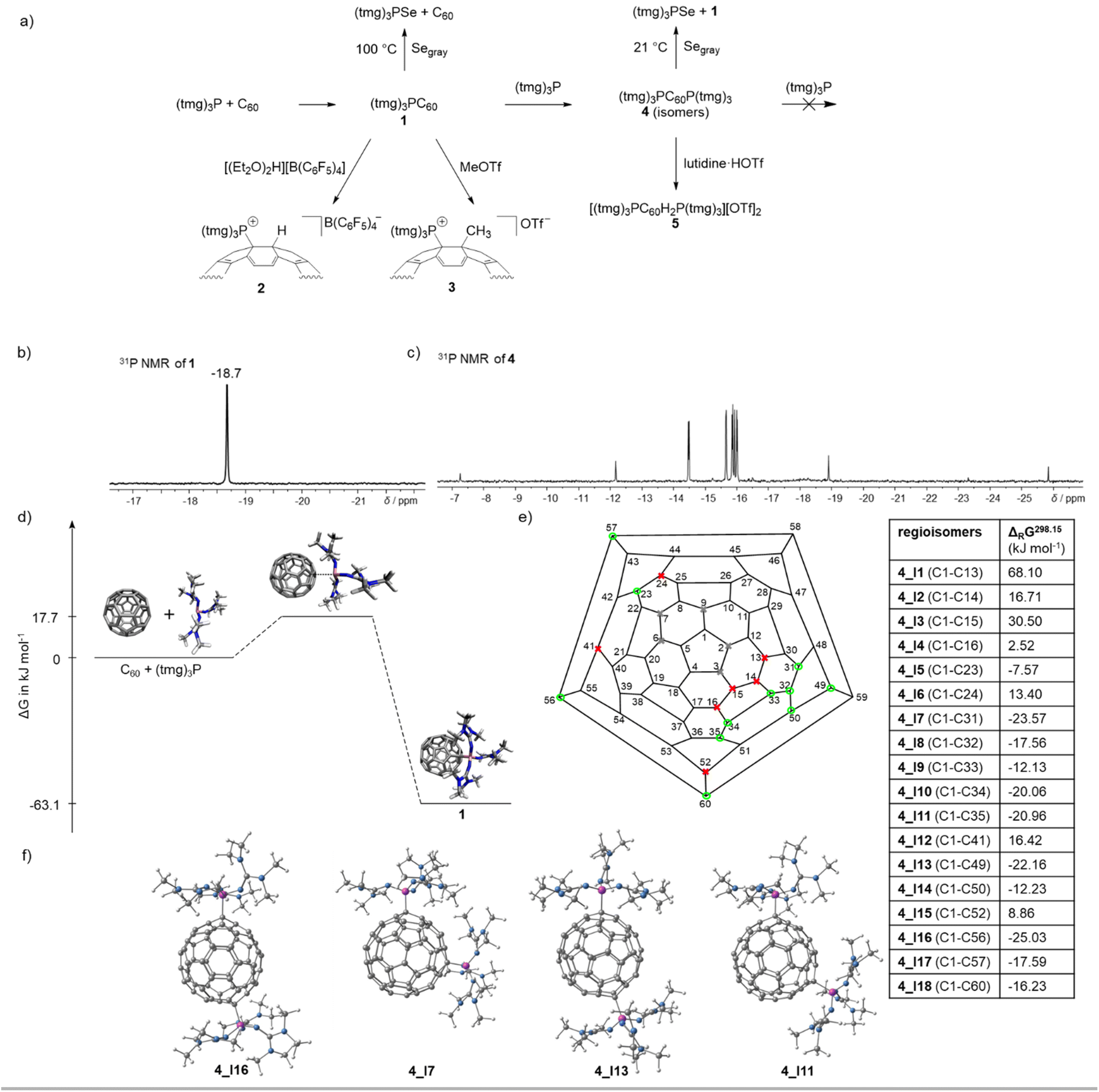 | ||
| Fig. 4 (a) Overview of the reactivity of 1 and 4; (b) 31P NMR spectrum of the monoadduct 1 in THF; (c) 31P NMR spectrum in THF of the bisadducts (tmg)3PC60P(tmg)3 (4) obtained as mixture of positional isomers; (d) reaction energy profile for the formation of 1 determined by DFT using B3LYP(GD3BJ)/6-31G(d,p)/SMD(THF); (e) calculated free energies for the formation of the positional isomers 4 according to: 1 + (tmg)3P → 4 determined by DFT using B3LYP(GD3BJ)/6-31G(d,p)/SMD(THF), Schlegel diagram of C60 with possible structural isomers39 if one phosphonium center is located at C1 (gray: positional isomers with less than three CC bond between the phosphonium centers that were not considered for steric reasons, red: endergonic isomers, green: exergonic isomers); (f) optimized geometries of the four most stable isomers of 4. | ||
| 1 (tmg)3PC60 | 2 [(tmg)3PC60H][B(C6F5)4] | 3 [(tmg)3PC60CH3][OTf] | |
|---|---|---|---|
| 31P of C–P | −18.7 | −20.7 (3JPH = 32 Hz) | −27.3 |
| 13C of C–P | 79.0 | 73.8 | 69.5 |
| 13C of C–E | — | 59.3 | 56.0 |
Synthesis and reactivity of fullerene adducts with two phosphines (4)
Monitoring the stoichiometric reaction of (tmg)3P with C60 by 31P NMR spectroscopy revealed the initial appearance of several new 31P resonances between −7 and −26 ppm. Over time, these resonances decreased in intensity, while the resonance of 1 at −18.7 ppm increased. This observation suggests the transient addition of more than one phosphine to C60, caused by the poor solubility of C60, which temporarily creates an excess of phosphine in solution. Indeed, varying the ratio of C60 to (tmg)3P reveals that up to two phosphines can attach to the fullerene core, forming a mixture of regioisomers (tmg)3PC60P(tmg)3 (4) (Table S1†). Adding more than two equivalents of phosphine does not lead to further association with the negatively charged C60 core. The bisphosphine fullerene adducts (4) exhibit 31P NMR resonances between −7 and −26 ppm (Fig. 4c), which is in a similar range to that of 1 (δ = −18.7 ppm). The most intensive resonances appear as two pairs of doublets at δ = −14.5 and −15.9 ppm and at δ = −15.7 and −16.0 ppm, with phosphorus–phosphorus coupling constants of JPP = 3.2 and 3.7 Hz, respectively. The two pairs of doublets correspond to two regioisomers as confirmed by 31P–31P COSY spectroscopy (Fig. S37†). Furthermore, five singlets appear at δ = −7.3, −12.2, −15.9 ppm, −18.9, and −25.8 ppm, which either originate from five individual isomers with isochronous phosphonium centers or may also represent regioisomers with different 31P chemical shifts where the JPP coupling is unresolved. Hence, among the 23 possible regioisomers of disubstituted fullerene RC60R39 (Fig. 4e), a total of five to seven isomers were detected via31P NMR spectroscopy. Notably, variable-temperature 31P and 31P–31P NOESY/EXSY NMR spectroscopy experiments at 70 °C reveal that the regioisomers of 4 undergo interconversion at elevated temperatures (Fig. S36 and S38†).The assignment of the 31P NMR resonances to different regioisomers 4 is further supported by the addition of 2,6-lutidinium triflate, acting as proton source, to the isomeric mixture 4. The resulting 31P NMR spectrum shows several new resonances with chemical shifts and 3JPH coupling constants similar to those of 2 (Fig. S41†). As expected, the number of regioisomers of the dications [((tmg)3P)2C60H2]2+ (5) increases, since the protons can attach to three different carbon sites adjacent to each phosphonium center.
Further evidence for the composition of 4 was obtained through its reaction with gray selenium, which produced 1 and (tmg)3PSe in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Notably, this reaction occurs readily at ambient temperature, indicating that the P–C60 bond dissociation energy is significantly lower than that of 1. This observation can be rationalized by the increased negative charge imparted to the fullerene core with each successive phosphine complexation.
:1 ratio. Notably, this reaction occurs readily at ambient temperature, indicating that the P–C60 bond dissociation energy is significantly lower than that of 1. This observation can be rationalized by the increased negative charge imparted to the fullerene core with each successive phosphine complexation.
Collectively, these experiments demonstrate that up to two phosphines (tmg)3P can reversibly bind to C60. The formation of the first and second dative P–C bonds occurs with low energy barriers, as indicated by the rapid formation of 4 with excess phosphine. However, the P–C bonds in 4 are less stable than in 1, enabling selective conversion of 4 to 1 in the presence of free C60. The reactivity and trapping reactions with gray selenium indicate heterolytic dissociation of the dative P–C bond into neutral phosphine and fullerene fragments.
Mechanistic investigations
Mechanistic investigations using DFT at the B3LYP(GD3BJ)/6-31G(d,p)/SMD(THF) level of theory reveal that the formation of compound 1 from C60 and (tmg)3P follows a low-energy pathway with a transition state barrier of ΔG‡ = 17.7 kJ mol−1 and an exergonic free energy change of ΔG = −63.1 kJ mol−1 (Fig. 4d). This aligns with experimental observations of a reversible reaction at elevated temperatures. An alternative electron transfer (ET) mechanism was considered, but the generation of the radical ion pair [(tmg)3P+˙][C60−˙] was found to be significantly less favorable, with a minimum energy barrier of 41.8 kJ mol−1. This contrasts with the literature reports on the C60–DBU system,18 where a stepwise ET mechanism was proposed based on the decrease in EPR signal intensity of the C60˙− radical anion over time.To determine whether radical species are generated during the formation of the zwitterionic adduct 1, DCB solutions containing (tmg)3P and C60 in various stoichiometric ratios were analyzed by EPR spectroscopy. No radical species were detected with an excess of C60. However, in a 1:1 stoichiometric mixture or in the presence of excess phosphine, at least three paramagnetic species were observed. These species were assigned to the C60˙− radical anion and two phosphorus-containing radicals (ESI, Chapter 1.6.5†). Unlike the DBU–C60 system, the signal intensity of these species remained constant over time. The presence of paramagnetic species implies that ET processes or homolytic P–C bond cleavage may occur to some extent. However, the high selectivity of the trapping reactions, combined with the lack of reactivity between 1 and diphenylacetylene upon heating, indicates that these processes play a minor role. Instead, they are likely associated with ET from the electron rich bisadducts 4 (Fig. S44†).
DFT calculations were also performed to investigate the second addition of the phosphine to 1 and to assign the positional isomers of 4. Among the 23 possible positional isomers of RC60R (R = (tmg)3P),39 five were excluded due to the spatial proximity of the phosphonium centers (Fig. 4e). For the remaining 18 isomers (4_I1 to 4_I18), 31P NMR chemical shifts and the free energies of formation were calculated. The computed 31P NMR chemical shifts (δ = 2 to −23 ppm, Table S6†) fall within the observed range, but are not precise enough for definite isomer assignment. Free energy calculations (Fig. 4e) reveal that the distance between the phosphonium centers significantly influences stability. Isomers with three carbon–carbon bond separations (4_I1, Fig. S52† and 4_I3) are highly endergonic, while those with four CC bonds (4_I2, 4_I4 and 4_I6, Fig. S52†) are less endergonic. Starting at five CC bonds, isomers become exergonic (ΔG = −7 to −25 kJ mol−1). Exceptions, such as 4_I12 and 4_I15, demonstrate that the electronic structure also affects regioisomer stability. Overall, the DFT results suggest the existence of 11 stable isomers with varying stabilities. While steric factors significantly influence stability, electronic effects appear to play only a minor role. However, due to their similar stabilities, a definitive assignment of the isomers observed in the 31P NMR spectrum was not possible.
Conclusions
Herein, we report the preparation of the first adducts between buckminsterfullerene (C60) and a phosphine. The electron-rich phosphine (tmg)3P reacts selectively with C60via a concerted nucleophilic addition pathway to form the zwitterionic phosphonium fulleride 1, a stable crystalline adduct featuring a dative P–C bond. In the presence of excess phosphine, a second phosphine rapidly binds to the C60 core yielding bisphosphine fullerene adducts 4 as a mixture of regioisomers. Given the increased negative charge of the C60 core with each successive phosphine addition, this behavior highlights the high nucleophilicity of the superbasic phosphine (tmg)3P and has not been observed for other Lewis bases.15–18 The formation of stable adducts between (tmg)3P and C60 agrees with predictions from Hobza,23 and suggests that phosphines bearing the same or greater basicity should also form adducts with C60 of other fullerenes. Trapping experiments provide evidence for heterolytic P–C bond dissociation and demonstrate the reversibility of the adduct formation. Furthermore, functionalization of 1 with electrophiles provides access to ionic fullerene derivatives, illustrating the potential for further chemical modifications. These findings contribute to a deeper understanding of phosphine–fullerene interactions and pave the way for the development of new functional fullerene derivatives with tailored properties. Potential applications of these compounds may extend from semiconductor technology to molecular rotors and integration into optoelectronic devices.6,40Data availability
Further details of the experimental procedures, the computational studies, and the characterization data for the new compounds have been included as part of the ESI.† Crystallographic data for 1 has been deposited at the CCDC under 2411442 and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Author contributions
M. B. R. performed the experiments. DFT calculations were performed by J. H. F. and T. S. H. D. L. performed EPR measurements. SCXRD studies were performed by M. S. F. D. directed the investigation. The manuscript was written by M. B. R. and F. D. All authors have given approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. B. R. thanks the University of Innsbruck for the Early Stage Funding. J. H. F. thanks the Tiroler Wissenschaftsförderung (TWF, F.45075) for funding. The computational results presented here have been achieved (in part) using the LEO HPC infrastructure of the University of Innsbruck. We thank Christoph Kreutz for performing special NMR measurements of 4. We thank Samuel Ray Lawrence for scientific discussions.Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162 CrossRef CAS.
- W. Krätschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354 CrossRef.
- A. Hirsch and M. Brettreich, Fullerenes. Chemistry and Reactions, Wiley-VCH, Weinheim, 1st edn, 2005 Search PubMed.
- (a) F. Giacalone and N. Martín, Chem. Rev., 2006, 106, 5136 CrossRef CAS PubMed; (b) Y. Matsuo and E. Nakamura, Chem. Rev., 2008, 108, 3016 CrossRef CAS PubMed; (c) D. M. Guldi, B. M. Illescas, C. M. Atienza, M. Wielopolski and N. Martín, Chem. Soc. Rev., 2009, 38, 1587 RSC; (d) M. D. Tzirakis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262 CrossRef CAS PubMed; (e) S.-E. Zhu, F. Li and G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7535 RSC.
- (a) R. C. Haddon, G. P. Kochanski, A. F. Hebard, A. T. Fiory and R. C. Morris, Science, 1992, 258, 1636 CrossRef CAS PubMed; (b) A. Y. Ganin, Y. Takabayashi, P. Jeglic, D. Arcon, A. Potocnik, P. J. Baker, Y. Ohishi, M. T. McDonald, M. D. Tzirakis, A. McLennan, G. R. Darling, M. Takata, M. J. Rosseinsky and K. Prassides, Nature, 2010, 466, 221 CrossRef CAS PubMed; (c) L. Hou, X. Cui, B. Guan, S. Wang, R. Li, Y. Liu, D. Zhu and J. Zheng, Nature, 2022, 606, 507 CrossRef CAS PubMed; (d) E. Meirzadeh, A. M. Evans, M. Rezaee, M. Milich, C. J. Dionne, T. P. Darlington, S. T. Bao, A. K. Bartholomew, T. Handa, D. J. Rizzo, R. A. Wiscons, M. Reza, A. Zangiabadi, N. Fardian-Melamed, A. C. Crowther, P. J. Schuck, D. N. Basov, X. Zhu, A. Giri, P. E. Hopkins, P. Kim, M. L. Steigerwald, J. Yang, C. Nuckolls and X. Roy, Nature, 2023, 613, 71 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CrossRef CAS.
- For selected works see: (a) P. J. Fagan, J. C. Calabrese and B. Malone, Science, 1991, 252, 1160 CrossRef CAS; (b) P. J. Fagan, J. C. Calabrese and B. Malone, Acc. Chem. Res., 1992, 25, 134 CrossRef CAS.
- (a) M. Yamada, T. Akasaka and S. Nagase, Chem. Rev., 2013, 113, 7209 CrossRef CAS PubMed; (b) H. Nikawa, T. Nakahodo, T. Tsuchiya, T. Wakahara, G. M. A. Rahman, T. Akasaka, Y. Maeda, M. T. H. Liu, A. Meguro, S. Kyushin, H. Matsumoto, N. Mizorogi and S. Nagase, Angew. Chem., Int. Ed., 2005, 44, 7567 CrossRef CAS.
- (a) T. Akasaka, W. Ando, K. Kobayashi and S. Nagase, J. Am. Chem. Soc., 1993, 115, 1605 CrossRef CAS; (b) T. Kusukawa and W. Ando, Angew Chem. Int. Ed. Engl., 1996, 35, 1315 CrossRef CAS; (c) M. Kako, D. Inaba, K. Minami, R. Iida, T. Nakahodo and T. Akasaka, Heteroat. Chem., 2014, 25, 584 CrossRef CAS.
- S. R. Lawrence, T. Rüffer, A. Stasch and R. Kretschmer, Chem. Commun., 2023, 59, 7923 RSC.
- (a) A. Hirsch, Q. Li and F. Wudl, Angew. Chem., Int. Ed., 1991, 30, 1309 CrossRef; (b) R. Seshadri, A. Govindaraj, R. Nagarajan, T. Pradeep and C. Rao, Tetrahedron Lett., 1992, 33, 2069 CrossRef CAS; (c) A. L. Balch, B. Cullison, W. R. Fawcett, A. S. Ginwalla, M. M. Olmstead and K. Winkler, J. Chem. Soc., Chem. Commun., 1995, 22, 2287 RSC; (d) K.-D. Kampe and N. Egger, Adv. Cycloaddit., 1995, 1995, 115 Search PubMed; (e) K.-D. Kampe, N. Egger and M. Vogel, Angew Chem. Int. Ed. Engl., 1993, 32, 1174 CrossRef; (f) S. N. Davey, D. A. Leigh, A. E. Moody, L. W. Tetler and F. A. Wade, J. Chem. Soc., Chem. Commun., 1994, 4, 397 RSC.
- G. Schick, K.-D. Kampe and A. Hirsch, J. Chem. Soc., Chem. Commun., 1995, 19, 2023 RSC.
- S. Yamago, M. Yanagawa, H. Mukai and E. Nakamura, Tetrahedron, 1996, 52, 5091 CrossRef CAS.
- V. A. Kuimov, E. A. Matveeva, S. F. Malysheva, D. O. Samultsev, L. V. Klyba, Y. V. Dem'yanov, N. K. Gusarova and B. A. Trofimov, Mendeleev Commun., 2017, 27, 198 CrossRef CAS.
- H. Li, C. Risko, J. H. Seo, C. Campbell, G. Wu, J.-L. Brédas and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 12410 CrossRef CAS PubMed ; additions of NHCs to metallofullerenes in the same manner have also been reported, see ref. 16 and 17..
- L. Bao, M. Chen, W. Shen, L. Yang, P. Jin and X. Lu, Inorg. Chem., 2017, 56, 14747 CrossRef CAS PubMed.
- W. Shen, L. Yang, Y. Wu, L. Bao, Y. Li, P. Jin, H. Fang, Y. Xie and X. Lu, J. Org. Chem., 2019, 84, 606 CrossRef CAS.
- A. Skiebe, A. Hirsch, H. Klos and B. Gotschy, Chem. Phys. Lett., 1994, 220, 138 CrossRef CAS.
- M. Lamanec, R. Lo, D. Nachtigallová, A. Bakandritsos, E. Mohammadi, M. Dračínský, R. Zbořil, P. Hobza and W. Wang, Angew. Chem., Int. Ed., 2021, 60, 1942 CrossRef CAS PubMed.
- (a) G. E. Lawson, A. Kitaygorodskiy, B. Ma, C. E. Bunker and Y.-P. Sun, J. Chem. Soc., Chem. Commun., 1995, 21, 2225 RSC; (b) G. P. Miller, C. R. Chim., 2006, 9, 952 CrossRef CAS; (c) S.-C. Chuang, K. C. Santhosh, C.-H. Lin, S.-L. Wang and C.-H. Cheng, J. Org. Chem., 1999, 64, 6664 CrossRef CAS PubMed.
- I. P. Romanova, V. F. Mironov, O. A. Larionova, V. I. Morozov, V. V. Zverev and O. G. Sinyashin, Russ. Chem. Bull., 2008, 57, 209 CrossRef CAS.
- D. Seyferth, Acc. Chem. Res., 1972, 5, 65 CrossRef CAS.
- R. Lo, D. Manna and P. Hobza, Chem. Commun., 2021, 57, 3363 RSC.
- M. A. Wünsche, P. Mehlmann, T. Witteler, F. Buß, P. Rathmann and F. Dielmann, Angew. Chem., Int. Ed., 2015, 54, 11857 CrossRef.
- (a) F. Buß, P. Mehlmann, C. Mück-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840 CrossRef PubMed; (b) P. Mehlmann, C. Mück-Lichtenfeld, T. T. Y. Tan and F. Dielmann, Chem.–Eur. J., 2017, 23, 5929 CrossRef CAS PubMed.
- M. D. Böhme, T. Eder, M. B. Röthel, P. D. Dutschke, L. F. B. Wilm, E. Hahn and F. Dielmann, Angew. Chem., Int. Ed., 2022, 61, e202202190 CrossRef.
- F. Buß, P. Rotering, C. Mück-Lichtenfeld and F. Dielmann, Dalton Trans., 2018, 47, 10420 RSC.
- (a) T. Eder, F. Buß, L. F. B. Wilm, M. Seidl, M. Podewitz and F. Dielmann, Angew. Chem., Int. Ed., 2022, 61, e202209067 CrossRef CAS PubMed; (b) M. B. Röthel, A. Schöler, F. Buß, P. Löwe and F. Dielmann, Chem.–Eur. J., 2024, e202402028 CrossRef PubMed; (c) F. Buß, C. Mück-Lichtenfeld, P. Mehlmann and F. Dielmann, Angew. Chem., Int. Ed., 2018, 130, 5045 CrossRef.
- F. Buß, M. B. Röthel, J. A. Werra, P. Rotering, L. F. B. Wilm, C. G. Daniliuc, P. Löwe and F. Dielmann, Chem.–Eur. J., 2022, 28, e202104021 CrossRef PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- J. Stinchcombe, A. Penicaud, P. Bhyrappa, P. D. W. Boyd and C. A. Reed, J. Am. Chem. Soc., 1993, 115, 5212 CrossRef CAS.
- H.-S. Lin and Y. Matsuo, Chem. Commun., 2018, 54, 11244 RSC.
- Y. Matsuo, Y. Yu, X.-Y. Yang, H. Ueno, H. Okada, H. Shibuya, Y. S. Choi and Y. W. Jin, J. Org. Chem., 2019, 84, 6270 CrossRef CAS PubMed.
- Y. Zhang, Y. Matsuo, C.-Z. Li, H. Tanaka and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 8086 CrossRef CAS PubMed.
- T. Kitagawa, H. Sakamoto and K. Takeuchi, J. Am. Chem. Soc., 1999, 121, 4298 CrossRef CAS.
- H. Ajie, M. M. Alvarez, S. J. Anz, R. D. Beck, F. Diederich, K. Fostiropoulos, D. R. Huffman, W. Kraetschmer and Y. Rubin, et al. , J. Phys. Chem., 1990, 94, 8630 CrossRef CAS.
- J. Iglesias-Sigüenza and M. Alcarazo, Angew. Chem., Int. Ed., 2012, 51, 1523 CrossRef PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed.
- K. Balasubramanian, J. Phys. Chem., 1993, 97, 6990 CrossRef CAS.
- (a) A. Lorbach, E. Maverick, A. Carreras, P. Alemany, G. Wu, M. A. Garcia-Garibay and G. C. Bazan, Phys. Chem. Chem. Phys., 2014, 16, 12980 RSC; (b) A. Carreras, A. Lorbach, G. C. Bazan, P. Alemany, G. Wu, M. A. Garcia-Garibay and E. F. Maverick, Helv. Chim. Acta, 2023, 106, e202200142 CrossRef CAS.
- Q. Xie, E. Perez-Cordero and L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 3978 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2411442. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00367a |
| ‡ Due to the limitations imposed by the solvent window, it remains unclear whether the fourth reduction of 1 is reversible, as would be expected for fullerenes. Notably, up to six reduction events have been observed for buckminsterfullerene, albeit at lower temperatures.41 |
| § The next NBO lower in energy represents an aromatic π bond of two carbon atoms of the C60 cage (E = −6.3 eV, occupancy: 1.62 e−), NBOs representing lone pairs at the nitrogen atoms of the phosphonium unit are even lower in energy (E = −8.3 eV to E = −6.9 eV). |
| ¶ This color change is also observed when water is added to 1, in which case the products could not be identified due to their insolubility. |
| This journal is © The Royal Society of Chemistry 2025 |