
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoelectrochemical valorisation of organic waste for the cogeneration of solar fuels and value-added chemicals†
Fitri Nur Indah
Sari
a,
Ping-Chang
Chuang
b,
Shih-Ching
Huang
 a,
Chia-Yu
Lin
*ac and
Yi-Hsuan
Lai
*bc
a,
Chia-Yu
Lin
*ac and
Yi-Hsuan
Lai
*bc
aDepartment of Chemical Engineering, National Cheng Kung University, No. 1, University Road, Tainan City 701, Taiwan. E-mail: cyl44@mail.ncku.edu.tw
bDepartment of Materials Science and Engineering, National Cheng Kung University, No. 1, University Road, Tainan City 701, Taiwan. E-mail: yhlai@gs.ncku.edu.tw
cHierarchical Green-Energy Materials (Hi-GEM) Research Center, National Cheng Kung University, Tainan City 70101, Taiwan
First published on 21st July 2025
Abstract
Rapid industrialisation has resulted in severe greenhouse gas emissions through extensive fossil fuel extraction and an increasing volume of solid waste disposal. This perspective review examines the photoelectrochemical (PEC) valorisation of organic waste as a promising solution for addressing the energy crisis and environmental pollution. The current stage of PEC organic valorisation has yet to meet industrial requirements, hindered by its relatively low efficiency, limited robustness, and poor scalability compared to conventional technologies. A better understanding of the working principle of the PEC reaction mechanism, the properties of state-of-the-art photoelectrodes, and the organic waste pre-treatment process is required to pave the path toward practical implementation. In this perspective review, we demonstrate that the strategies employed in the design of photoanodes, including doping, heterojunction formation, co-catalyst modification, nanostructuring, and crystal facet engineering, have different effects on activity, selectivity, and stability. In addition to the hydrogen evolution reaction, selected organic reduction reactions for the synthesis of value-added chemicals in a PEC cell are also discussed, followed by recent progress on integrated PEC cells and their practical assessment for solar fuel and value-added chemical production. Since stability and scalability are also essential parameters beyond efficiency for assessing the techno-economic feasibility of a PEC organic valorisation system, we additionally addressed stability, scalability and the compatibility of photoelectrodes in organic waste valorisation. Finally, conclusions and future perspectives on feasible strategies for PEC valorisation are discussed. We hope this review will serve as a helpful guide in designing effective, robust, and scalable PEC organic waste valorisation systems, making it a viable technology for real-world applications.
 Fitri Nur Indah Sari | Fitri Nur Indah Sari received her PhD degree from the Department of Materials Science and Engineering at National Cheng Kung University in 2018. She is currently a postdoctoral fellow at the Department of Chemical Engineering, National Cheng Kung University. Her research interests focus on the design and synthesis of advanced organic and inorganic nanomaterials, including graphene and its composite, MoS2, ceramics, and high entropy material. These materials have been explored for functional applications, especially in energy conversion, energy storage, and environmental remediation. |
 Ping-Chang Chuang | Ping-Chang Chuang obtained a BS in Chemical and Materials Engineering from the National University of Kaohsiung in 2018, followed by an MS in Materials and Optoelectronic Science from National Sun Yat-Sen University in 2020. Since then, he has been working towards his PhD in the Department of Materials Science and Engineering at National Cheng-Kung University, mentored by Prof. Yi-Hsuan Lai. His current research focuses on electrocatalytic biomass valorisation and artificial photosynthesis. |
 Shih-Ching Huang | Shih-Ching Huang received her BSc, MSc, and PhD degrees in Chemical Engineering from National Cheng Kung University in 2024, under the supervision of Prof. Chia-Yu Lin. She is currently a postdoctoral research fellow at the National Synchrotron Radiation Research Centre, Taiwan. Her research focuses on (photo-)electrochemical energy conversion, electrosynthesis, environmental remediation, and the development of advanced catalytic materials for sustainable applications. |
 Chia-Yu Lin | Chia-Yu Lin received his BS degree in Chemical Engineering from National Cheng Kung University, Taiwan, in 2003. He received his MS and PhD degrees in Chemical Engineering from National Taiwan University, Taiwan in 2005 and 2010, respectively. After two-years of postdoctoral research in the Department of Chemistry, University of Cambridge, United Kingdom, he joined the Department of Chemical Engineering, National Cheng Kung University, Taiwan, as an Assistant Professor in 2013. Currently, he is a full Professor in the Department of Chemical Engineering, National Cheng Kung University, Taiwan. His research interests include the development of functional nanomaterials for chemical sensors, (photo-)electrocatalysis, and energy-conversion devices. |
 Yi-Hsuan Lai | Yi-Hsuan Lai received her PhD degree from the University of Cambridge in 2015. She started her independent career in 2016 as an assistant professor at National Sun Yat-sen University. In 2019, she moved to National Cheng Kung University as an assistant professor and was promoted to associate professor in 2024. Her research interests include photoelectrochemistry, photocatalysis, electrocatalysis, and sustainability, with a special focus on water splitting and organic waste valorisation. |
1. Introduction
Meeting the energy demand sustainably to alleviate greenhouse gas (GHG) emissions has reached a global consensus.1–3 Over the past few centuries, overdependence on fossil fuels has resulted in irreparable climate change due to the greenhouse effects. On the other hand, rapid technological development has led to a significant increase in the generation of solid waste, such as biomass and plastics, which also contribute to non-negligible GHG emissions.4,5 In addition, the predominant waste processing methodologies, landfilling and incineration, have caused severe environmental pollution and the loss of valuable resources. The exploration of technologies that align with the principles of a carbon circular economy for sustainable energy production and waste valorisation is therefore highly promising and urgently needed.6,7Photoelectrochemical (PEC) valorisation has been demonstrated to be one of the most promising technologies for addressing energy and environmental pollution issues. It supports carbon-neutral sustainability by converting organic waste into value-added chemicals using sustainable energy sources (Fig. 1).8–11 In particular, PEC reactions generally operate at room temperature under ambient conditions, thereby eliminating the substantial energy demands associated with the high temperatures and pressures typically required in conventional waste valorisation methods.12,13 In addition, PEC valorisation utilises solar energy, the most abundant renewable energy resource, to drive chemical reactions using organic waste as feedstock. Therefore, solar energy can be stored in the form of solar fuels in the co-production of value-added chemicals through PEC valorisation. Compared to water splitting, one of the most extensively studied reactions in PEC systems, organic waste valorisation offers an alternative pathway for generating more economically valuable products. For example, oxygen (O2) is the oxidation product in water splitting and has an economic value of only 0.04 USD per kg.8 In contrast, formic acid (FA), which can be generated from various organic oxidation processes, possesses an economic value approximately ten times higher than that of O2.14
 | ||
| Fig. 1 Overview of PEC organic waste valorisation to drive value-added chemical and solar fuels production in a circular economy. | ||
Decreasing the energy demand in PEC reactions and enhancing the production rate of products are additional advantages of replacing water oxidation with organic oxidation. Water splitting consists of an oxygen evolution reaction (OER) and a hydrogen evolution reaction (HER):15,16
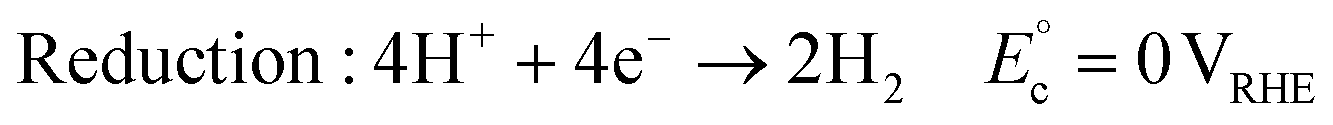 | (1) |
 | (2) |
| Overall reaction: 2H2O → O2 + 2H2 ΔG° = 237 kJ mol−1 | (3) |
 .17
.17
On the other hand, replacing the OER with organic oxidation, i.e., coupling HER with organic oxidation, results in a thermodynamically favourable or marginally endothermic reaction (Table 1). For example, the HER coupled with glycerol valorisation to dihydroxyacetone (DHA) requires a marginal endothermic ΔG° of only 10.3 kJ mol−1. The potential required to oxidise glycerol to DHA is approximately at 0.05 V vs. the reversible hydrogen electrode (VRHE). Another example is ethylene glycol (EG), a monomeric unit of polyethylene terephthalate (PET), whose valorisation to FA coupled with the HER constitutes a thermodynamically favourable reaction. The potential required to oxidise EG to FA is located at a −0.25 VRHE (Table 1).
 and standard electromotive force of the cell
and standard electromotive force of the cell 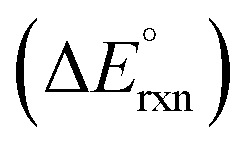 for the reaction of organics valorisation couple with H2 production
for the reaction of organics valorisation couple with H2 production
| Overall reaction | Cathodic half-cell reaction | Anodic half-cell reaction |
|---|---|---|
a Derived from ref. 6.
b Obtained by  c Obtained by
c Obtained by  , where m is the respective coefficient in the balanced chemical reaction and , where m is the respective coefficient in the balanced chemical reaction and 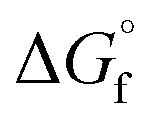 is the standard molar Gibbs free energy of formation (Table S1).
d is the standard molar Gibbs free energy of formation (Table S1).
d
 , where n is the number of electrons transferred (mol) and F is the Faraday constant of 96 , where n is the number of electrons transferred (mol) and F is the Faraday constant of 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 (C mol−1). HMF: 5-hydroxymethylfurfural; FDCA: 2,5-furandicarboxylic acid; FA: formate; EG: ethylene glycol; GLA: glycolic acid; DHA: dihydroxyacetone; GLAD: glyceraldehyde; GNA: gluconic acid; and GRA: glucaric acid. 485 (C mol−1). HMF: 5-hydroxymethylfurfural; FDCA: 2,5-furandicarboxylic acid; FA: formate; EG: ethylene glycol; GLA: glycolic acid; DHA: dihydroxyacetone; GLAD: glyceraldehyde; GNA: gluconic acid; and GRA: glucaric acid.
|
||
| C6H6O3 (HMF) + 2H2O ↔ C6H4O5 (FDCA) + 3H2 | 6H+ + 6e− ↔ 3H2 | C6H6O3 (HMF) + 2H2O → C6H4O5 (FDCA) + 6H+ + 6e− |
aΔG° = 173.7 kJ mol−1, b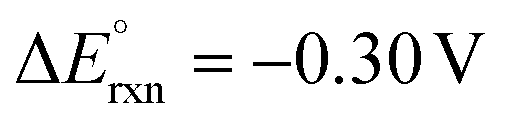 |

|
a

|
| C2H6O2 (EG) + 2OH− ↔ 2 HCOO−(FA) + 3H2 | 6H2O + 6e− ↔ 3H2 + 6OH− | C2H6O2 (EG) + 8OH− → 2 HCOO−(FA) + 6H2O + 6e− |
cΔG° = −147.5 kJ mol−1, d |

|
b

|
| C2H6O2 (EG) + H2O ↔ C2H4O3 (GLA) + 2H2 | 4H2O + 4e− ↔ 2H2 + 4OH− | C2H6O2 (EG) + 4OH− → C2H4O3 (GLA) + 3H2O + 4e− |
cΔG° = 105.2 kJ mol−1, d |

|
b
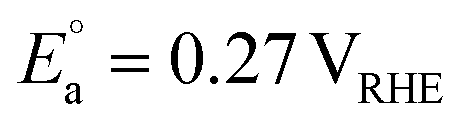
|
| C3H8O3 (glycerol) ↔ C3H6O3 (DHA) + H2 | 2H+ + 2e− ↔ H2 | C3H8O3 (glycerol) → C3H6O3 (DHA) + 2H+ + 2e− |
cΔG° = 10.3 kJ mol−1, d |

|
b
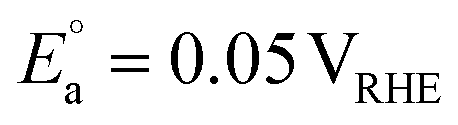
|
| C3H8O3 (glycerol) ↔ C3H6O3 (GLAD) + H2 | 2H+ + 2e− ↔ H2 | C3H8O3 (glycerol) → C3H6O3 (GLAD) + 2H+ + 2e− |
cΔG° = 37.3 kJ mol−1, d |

|
b
|
| C3H8O3 (glycerol) + 3OH− ↔ 3 HCOO− (FA) + 4H2 | 8H2O + 8e− ↔ 4H2 + 8OH− | C3H8O3 (glycerol) + 11 OH− → 3 HCOO− (FA) + 8H2O + 8e− |
cΔG° = 65.6 kJ mol−1, d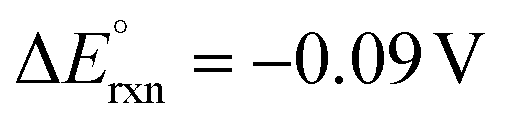 |

|
b

|
| C6H12O6 (glucose) + 6OH− ↔ 6 HCOO− (FA) + 6H2 | 12H2O + 12e− ↔ 6H2 + 12OH− | C6H12O6 (glucose) + 18OH− → 6 HCOO− (FA) + 12H2O + 12e− |
cΔG° = 47.9 kJ mol−1, d |

|
b

|
| C6H12O6 (glucose) + H2O ↔ C6H12O7 (GNA) + H2 | 2H+ + 2e− ↔ H2 | C6H12O6 (glucose) + H2O → C6H12O7 (GNA) + 2H+ + 2e− |
cΔG° = 70.9 kJ mol−1, d |

|
b
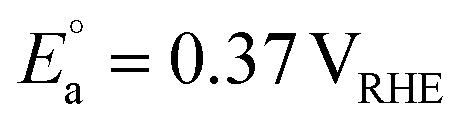
|
| C6H12O6 (glucose) + 2H2O ↔ C6H10O8 (GRA) + 3H2 | 6H+ + 6e− ↔ 3H2 | C6H12O6 (glucose) + 2H2O → C6H10O8 (GRA) + 6H+ + 6e− |
cΔG° = 179.1 kJ mol−1, d |

|
b

|
Therefore, using a PEC system to drive a PEC organic valorisation reaction coupled with H2 evolution generally results in a higher product formation rate than using the same PEC system to drive water splitting. Fig. 2a and b show that the photogenerated holes can provide greater kinetic energy, i.e., a higher overpotential (ηOX), to drive organic oxidation, as organic oxidation requires less thermodynamic energy than the OER. Severe surface charge recombination caused by the sluggish kinetics of the OER would result in the onset potential for OER (Eon,OER) deviating significantly from the flat band potential of the photoanode (EF,n), as shown in Fig. 2c. In contrast, surface charge recombination can generally be mitigated by replacing the OER with a suitable organic oxidation reaction, in which the onset potential of organic valorisation (Eon,VR) can be negatively shifted toward the EF,n of the photoanode. Taking α-Fe2O3 as an example, the onset potential shifted from 1.1 VRHE to 0.85 VRHE by replacing the OER with the EG oxidation reaction,9 while it shifted from 1.0 VRHE to 0.7 VRHE by replacing the OER with the glucose oxidation reaction.18 The deviation range between the onset potential and the EF,n of the photoanode is crucial in determining the overall PEC cell performance. If an integrated PEC system comprising a photocathode and a photoanode is employed, the operating photocurrent (JOP) is determined by the intersection of the photocurrents of the two half-reactions (oxidation and reduction) (Fig. 2c). The difference between Eon,OER and Eon,VR for the same photoanode resulted in the JOP of PEC water splitting (JOP1) being lower than that of PEC organic valorisation coupled with the same reduction reaction (JOP2).
 | ||
| Fig. 2 Tandem PEC cells for (a) water splitting and (b) organic waste valorisation. (c) Overlaid current (J) potential (E) curves measured in a three-electrode system with a photocathode (red) and a photoanode (blue) in water with (dash) or without (solid) organic substrates under light irradiation. The operating photocurrent of a two-electrode tandem PEC cell can be predicted by the intersection of the oxidation and reduction photocurrents. EF,n and EF,p represent the flat band edge of the n-type (photoanode) and p-type (photocathode) semiconductor, respectively, under light illumination. | ||
PEC organic waste valorisation may also provide an alternative approach to mitigate the instability of photoelectrodes in PEC water splitting. The thermodynamic stability of a photoelectrode depends on the relative thermodynamic potential of the semiconductor and the potential of the target redox reaction. Taking water oxidation as an example, if the thermodynamic oxidation potential (Eox) of a photoanode is more negative than the potential of the OER (1.23 VRHE), the photoanode would undergo photocorrosion by its photogenerated holes.19 Most non-oxide semiconductors, such as ZnS, CdS, ZnSe, etc., and some oxide semiconductors, such as ZnO, are susceptible to oxidation based on thermodynamic predictions.19 However, if these photoanodes are used for glycerol valorisation, for example, they are thermodynamically resistant to photocorrosion, as their Eox is more positive than the potential for glycerol valorisation to DHA (0.05 VRHE). Given these advantages, research on PEC organic waste valorisation has rapidly increased in recent years. This perspective review focuses on PEC organic waste valorisation. Organic waste substrates, including biomass, its derivatives, and plastics, are particularly highlighted in this review, since biomass derived from food and lignocellulosic sources accounts for the majority composition (∼60%) of solid waste. Although plastics represent a smaller fraction (12%) compared to plant-derived materials, their non-biodegradability poses a significantly detrimental effect on soil, water, marine life, and human health. Beyond the substrate used in PEC valorisation, variables such as solution pH, operation duration, and light intensity also influence PEC performance (Fig. 3). In particular, the properties of the photoelectrode represent the most crucial factor determining the upper limit of PEC valorisation system performance. Hence, in this perspective review, the working principle of a PEC reaction is introduced, followed by an overview of the materials and optical properties of the state-of-the-art photoelectrodes applied in PEC organic valorisation. Recent progress in designing efficient photoanode strategies for organic waste oxidation to generate value-added chemicals is subsequently discussed. Value-added chemical synthesis through reduction reactions using PEC cells is then covered, although the reported research in this area remains relatively limited compared to oxidation. Finally, recent developments in integrated PEC cells and their practical evaluation for solar fuel and value-added chemical production are reviewed, followed by a discussion of perspectives. Efficiency, stability, and scalability are the three key system requirements for the successful deployment of PEC systems.20 Long-term stability is an essential criterion for the practical implementation of PEC technologies. Without sustained performance over long-term operation, even highly efficient PEC devices cannot satisfy the economic, safety, and reliability standards required for commercialisation. Therefore, unlike most recent review papers, this work not only focuses on efficiencies but also addresses stability, scalability, and the compatibility of photoelectrodes and co-catalysts in integrated PEC cells. Finally, conclusions and future perspectives on feasible strategies for PEC valorisation are discussed. We hope this will guide readers in designing effective, robust, and scalable PEC systems for organic waste valorisation, particularly in the context of integrated PEC cells.
 | ||
| Fig. 3 Parameters affecting the performance of a PEC cell for organic waste valorisation. | ||
2. PEC valorisation
2.1 Working principle of PEC reactions
PEC devices directly employ the solid–liquid junction to drive desired reactions under light illumination. When the photoelectrode is immersed in the electrolyte, charge transfer occurs between the semiconductor and the electrolyte until equilibrium is reached between the Fermi level of the semiconductor and the redox potential of the electrolyte. The built-in electric field at the surface of an n-type semiconductor resulted in upward band bending, favouring photo-driven oxidation reactions. In contrast, the built-in electric field at the surface of a p-type semiconductor resulted in downward band bending, favouring photo-driven reduction reactions. Upon light irradiation, e−/h+ pairs are photo-generated within the photoelectrodes, as indicated by route (i) in Fig. 4. The photogenerated minority charge carriers, i.e., holes for n-type semiconductors and electrons for p-type semiconductors, are then transported to the electrode surface, driven by the built-in electric field at the semiconductor/liquid interface, and subsequently transferred into the electrolyte to carry out the redox reaction, as shown in routes (ii) and (iii). However, it should also be noted that to drive a desired oxidation reaction, the holes from the valence band (EVB) of a photoelectrode must possess sufficient driving force to overcome both the thermodynamic and kinetic barriers of the oxidation process. Conversely, the photogenerated electrons from the conduction band (ECB) must have adequate energy to drive the reduction reaction. | ||
| Fig. 4 Schematic of a photoanode performing a PEC reaction under light illumination. The process can be decoupled into three major steps: (i) light absorption to generate electron–hole pairs, (ii) transport of photogenerated holes to the photoanode surface, and (iii) interfacial charge transfer to oxidise Ared to Aoxi in the electrolyte. Unfavourable recombination paths, including (ii′) bulk recombination and (iii′) surface recombination, are also shown. | ||
The performance of a photoelectrode in terms of photocurrent density (JPEC) can be described by eqn (4):
| JPEC = Jmax × ηLHE × ηsep × ηint | (4) |
 | ||
| Fig. 5 (a) Maximum theoretical photocurrent of different bandgap semiconductors under one-sun illumination based on the air mass (AM) 1.5 G spectrum. (b) Band structure of representative photoelectrodes. | ||
On the other hand, the EVB of the semiconductor should be more positive than the potential of an oxidation reaction, such as water oxidation or organic oxidation. This ensures that the holes created by light absorption possess sufficient oxidative power to oxidise water or organic waste.29–32 In PEC water splitting, the state-of-the-art photoanode includes TiO2,33–35 ZnO,35–37 BiVO4,38,39 WO3,38,40–43 α-Fe2O3.44–46 Those photoanodes have an EVB located at ≥2.5 VRHE (Fig. 5b), providing significant overpotential for water oxidation. As previously mentioned, the redox potential of most organic oxidations in organic valorisation research is less positive than that of the OER. Most of the photoanodes used in PEC water oxidation can be utilised and have been investigated as effective photoanodes in PEC organic oxidation. However, the photo-driven holes have a sufficient driving force for both the oxidation of water and organics. The selectivity of organic valorisation might remain an issue, as the parasitic reaction of the OER could occur.
The replacement of water oxidation with less energy-demanding organic oxidation, at least, partially alleviates the thermodynamic and kinetic constraints in selecting photoanodes. For example, nanosheet-structured WO3 lost 50% of its initial photoactivity during water splitting in a neutral solution within a few minutes.53 However, the nanosheet-structured WO3 does not lose 50% of its initial photoactivity until 12 h of operation in the glycerol valorisation reaction.54 The stability difference between water oxidation and glycerol valorisation on WO3 possibly resulted from difference in their reaction kinetics. Peroxo-species are readily formed during sluggish water oxidation, which leads to the corrosion of WO3. In contrast, peroxo-species are possibly eliminated during glycerol valorisation. Similar results have also been reported for BiVO4.55 However, the mechanisms by which photocorrosion is alleviated when replacing water oxidation with organic valorisation might vary among different photoanodes. Eliminating surface charge recombination may be another contributing factor to why a photoanode, such as BiVO4, typically exhibits better stability for organic (e.g., EG, glycerol) valorisation than for water oxidation. To date, PEC organic valorisation in the half-cell has achieved a maximum operation time of up to 150 h,56 and 80 h has been reported for an integrated cell.18 This highlights that the current progress in stability remains significantly below the criteria required for practical implementation and warrants further effort. The long-term stability of PEC valorisation based on recently reported studies is further discussed in a later section.
TiO2 has been widely investigated in PEC water splitting; however, the application of simple TiO2 in PEC organic valorisation is relatively limited. TiO2 generally exhibits low selectivity toward selected value-added chemicals in PEC organic valorisation (Table 2). Surface modification with a secondary material is generally required to achieve high selectivity. Inspiring examples include Bi2O3 heterojunction with TiO2 for glycerol valorisation to DHA,56 ultrathin BiOx-covered TiO2 converting succinic acid into C2H4,63 and Bi2O3-modified TiO2 converting cellulose into FA.64
| Photoelectrode | Modification | Substrate | Spectrum and intensity of light | Supporting electrolyte | FE [%] of main product@E | J [mA cm−2]/φPEa@E | t 0.6 b [h]@E | Ref. |
|---|---|---|---|---|---|---|---|---|
| a φ PE is defined by the ratio of JPEC/Jmax, where j is the measured photocurrent and Jmax is the calculated theoretical photocurrent under 1 sun and AM 1.5 G, revealed in Fig. 5. b Defined as the time needed for the photocurrent to reach 60% of its respective initial photocurrent. c Operating at an external bias in a two-electrode system. d Operating at bias-free in a two-electrode system. | ||||||||
| TiO 2 -based photoelectrode | ||||||||
| TiO2 | N/A | Glycerol (0.1 M) | 385 nm, 10 mW cm−2 | Na2SO4 (0.5 M, pH 7) | FA: ≈33@1 VRHE | ≈2/N.A.@1.3 VRHE | N/A | 102 |
| TiO2 | N/A | Cellulose (0.15 M) | λ < 500 nm | NaOH (2 M) | Malonic acid: ≈45 at 1 VRHE | ≈5/N.A.@0.5 VRHE | >3@0 VRHE | 103 |
| TiO2 | Nanostructure | Glycerol (0.1 M) | AM 1.5 G, 100 mW cm−2 | Na2SO4 (1 M, pH 2) | GLAD: 32@1.23 VRHE | ≈0.9/0.84@0.5 VRHE | >10@1.23 VRHE | 104 |
| TiO2/Bi2O3 | Heterojunction | Cellulose | AM 1.5 G, 100 mW cm−2 | NaOH (0.1 M) | FA: 83.9 ± 4.9 at 0.5 VRHE | ≈0.36/0.34@0.5 VRHE | >6@0.5 VRHE | 64 |
| Bi2O3/TiO2 | Heterojunction/Co-catalyst | Glycerol (0.1 M) | AM 1.5 G, 100 mW cm−2 | Na2SO4 (0.5 M, pH 2) | DHA: 62.2@1.0 VRHE | ≈2.4/2.2@1.2 VRHE | N/A | 56 |
| Ag@LDH@TiO2 | Co-catalyst | Glycerol (0.1 M) | AM 1.5 G, 100 mW cm−2 | Na2SO4 (0.5 M, pH 7) | DHA: 55@1.2 VRHE | ≈2.3/2.1@1.2 VRHE | >4@1.2 VRHE | 105 |
| TiO2/nanoNiP | Co-catalyst | PET lysate | AM 1.5 G, 100 mW cm−2 | KOH (1 M, pH 14) | FA: 57.1 ± 1.7@c0.5 V | ≈0.6/0.57@1.2 VRHE | >2@0.5 VRHE | 106 |
|
||||||||
| WO 3 -based photoelectrode | ||||||||
| WO3 | N/A | HMF (5 mM) | 100 mW cm−2 | NaPi (0.1 M, pH 4) | N/A | ≈1.5/N.A@1.2 VRHE | N/A | 67 |
| WO3 | N/A | Cyclohexane (18 mL) | 100 mW cm−2 | tBuOH (12 mL) and HNO3 (2 mL) | KA oil: 76@c0.5 V | ≈2/N.A@1.2 VRHE | N/A | 65 |
| WO3 | N/A | Glycerol (0.1 M) | 350 mW cm−2 | Na2SO4 (0.5 M, pH 5.5) | GLAD: ≈30, DHA: ≈20@1.2 VRHE | ≈0.35/N.A@1.2 VRHE | >5@1.2 VRHE | 66 |
| WO3 | Nanostructure | Glucose (0.1 M) | AM 1.5 G, 100 mW cm−2 | NaCl (0.5 M, pH 4) | Gluconic acid: 20@1.23 VRHE | 6/1.5@1.2 VRHE | >20@1.23 VRHE | 107 |
| m-WO3 | Crystal phase | HMF (5 mM) | AM 1.5 G, 100 mW cm−2 | NaBi (0.1 M, pH 4) | N/A | ≈0.9/0.23@1.2 VRHE | ≈1@1.1 VRHE | 68 |
| WO3 (202) | Crystal facet | Glycerol (2 M) | AM 1.5 G, 100 mW cm−2 | Na2SO4 (0.1 M, pH 2) | GLAD: ≈75, DHA: ≈20@1.2 VRHE | ≈3.4/0.85@1.2 VRHE | >12@1.2 VRHE | 69 |
| WO3 | Crystal facet | Methane | 100 mW cm−2 | Na2SO4 (0.1 M, pH 2) | EG: 23.9@1.3 VRHE | ≈3/N.A@1.2 VRHE | >12@0.9 VRHE | 108 |
| WO3/TiO2 | Heterojunction | Glycerol (0.1 M) | AM 1.5 G, 100 mW cm−2 | Na2SO4 (0.5 M, pH 6) | GLAD&DHA: 70@1.2 VRHE | 2.89/0.72@1.23 VRHE | >1@1.23 VRHE | 109 |
| N-WO3 | Dopant | Glycerol (0.1 M) | 365 nm | Na2SO4 (0.5 M, pH 1) | CO: 40@0.6 VRHE | 1.5/N.A.@0.7 VRHE | c>30 V | 110 |
|
||||||||
| BiVO 4 -based photoelectrode | ||||||||
| BiVO4 | N/A | Glycerol (2 M) | AM 1.5 G, 100 mW cm−2 | Na2B4O7 (0.1 M, pH 9.4) | N/A | 1.3/0.17@1.2 VRHE | ≈7@0.7 VRHE | 76 |
| BiVO4 | N/A | Lignin (5 mg mL−1) | λ > 400 nm,100 mW cm−2 | KHCO3 buffer (10 mM, pH 8.2) | N/A | ≈0.53/N/A@0.6 VAg/AgCl | N/A | 71 |
| BiVO4 | N/A | HMF (5 mM) | AM 1.5 G, 100 mW cm−2 | Borate buffer (0.5 M, pH 9.2) containing 7.5 mM TEMPO | FDCA: 93–94@1.54 VRHE | N/A | N/A | 70 |
| Mo:BiVO4/NiCo-LDH | Dopant/Co-catalyst | EG (1 M) | 100 mW cm−2 | KOH (0.1 M) | FA: ≈100@1.2 VRHE | ≈2.3/N.A.@1.2 VRHE | >3@1.0 VRHE | 111 |
| Ta:BiVO4 | Dopant | Glycerol (0.1 M) | λ > 422 nm, AM 1.5 G, 100 mW cm−2 | H2SO4 (25 mM) | DHA: 61@1.0 VRHE | 3.07/0.4@1.23 VRHE | >2@1.0 VRHE | 112 |
| NiOx (OH)y/W:BiVO4 | Co-catalyst | Glycerol (0.1 M) | AM 1.5 G, 100 mW cm−2 | KBi (0.5 M, pH 9.3) | FA: ≈30, DHA: ≈19@1.2 VRHE | 3.5/0.49@1.2 VRHE | >24@0.8 VRHE | 55 |
| BiVO4 | Structural | Glycerol (0.1 M) | AM 1.5 G, 100 mW cm−2 | Na2SO4 (0.5 M, pH 2) | DHA: 29.1@1.23 VRHE | 6/0.8@1.23 VRHE | >4@1.23 VRHE | 74 |
| BiVO4 | Nanostructure | Glycerol (0.1 M) | AM 1.5 G, 100 mW cm−2 | Na2SO4 (0.5 M, pH 2) | DHA: ≈30@0.6 VRHE | 3.7/0.49@1.2 VRHE | >5@1.0 VRHE | 75 |
| Bi-rich BiVO4−x | Surface atom engineered | Glycerol (0.1 M) | AM 1.5 G, 100 mW cm−2 | Na2SO4 (0.5 M, pH 2) | DHA: ≈45@1.23 VRHE | 4.26/0.57@1.2 VRHE | >5@1.23 VRHE | 113 |
| BiVO4(010) | Crystal facet | Glycerol (0.1 M) | AM 1.5 G, 100 mW cm−2 | Na2B4O7 (0.1 M, pH 2) | N/A | ≈1.2/0.16@1.2 VRHE | >2@1.1 VRHE | 114 |
| Mo:BiVO4(001) | Crystal facet/dopant/nanostructure | Glycerol (0.5 M) | AM 1.5 G, 100 mW cm−2 | Na2SO4 (0.1 M, pH 2) | DHA: 23@1.23 VRHE | 7.45/0.99@1.23 VRHE | >100@0.8 VRHE | 115 |
|
||||||||
| Fe 2 O 3 -based photoelectrode | ||||||||
| Fe2O3 | N/A | Cyclohexene (10 mM) | AM 1.5 G, 100 mW cm−2 | CH3CN (20% H2O) containing 0.1 M tetrabutylammonium tetrafluoroborate | Cyclohexene oxide: 35.2 ± 1.6@0.8 VAg/AgCl | ≈0.20/0.017@1.2 VAg/AgCl | >2@0.8 VAg/AgCl | 116 |
| Fe2O3 | N/A | Glycerol (2 M) | AM 1.5 G, 100 mW cm−2 | NaOH (1 M) | FA: ≈ 46@1.1 VRHE | 2.01/0.17@1.23 VRHE | >36@1.23 VRHE | 117 |
| Ni-Pi/α-Fe2O3 | Co-catalyst | PET hydrolysate | AM 1.5 G, 100 mW cm−2 | NaOH (1 M, pH 13.6) | FA: ≈ 87@1.1 VRHE | ≈2/0.17@1.2 VRHE | >12@1.1 VRHE | 9 |
| NiOOH/α-Fe2O3 | Co-catalyst | Sawdust-derived sugar | AM 1.5 G, 100 mW cm−2 | KOH (1 M) | FA: ≈90@1 VRHE | ≈2/0.17@1.1 VRHE | >100@1.0 VRHE | 18 |
| Fe2O3/Ni(OH)x | Co-catalyst | PET hydrolysate (50 mM) | N/A | KOH (1 M) | FA: ≈100@1.2 VRHE | ≈4/0.34@1.2 VRHE | N/A | 118 |
| Fe2O3|m-CuO | Co-catalyst/heterojunction | Glucose (20 mM) | AM 1.5 G, 100 mW cm−2 | NaOH (0.1 M) | FA: 97.3 ± 2.8@1.0 VRHE | ≈0.8/0.07@1.2 VRHE | >2@1.0 VRHE | 119 |
| Fe2O3 | N/A | Methanol (95% vol in water) | 365 nm, 100 mW cm−2 | NaOH (0.1 M) | Formaldehyde: ≈100%@1.1 VRHE | ≈0.7/N.A.@0.55 VAg/AgCl | N/A | 99 |
| Ti-doped Fe2O3 | Dopant | Polyimide (≈60 mg mL−1) | AM 1.5 G, 100 mW cm−2 | KOH (1 M) | N/A | ≈1.5/0.13@1.2 VRHE | >4@1.2 VRHE | 120 |
|
||||||||
| Other photoelectrodes | ||||||||
| Bi2WO6 | N/A | Glycerol (10% v/v) | λ > 350 nm, 120 mW cm−2 | K2SO4 (0.1 M, pH 6) | N/A | 0.69/0.22@1.3 VRHE | N/A | 121 |
| CuWO4 | Nanostructure | Glycerol (1 M) | AM 1.5 G, 100 mW cm−2 | Pi (0.1 M, pH 7) | GLAD: 60 ± 6, DHA: 30 ± 1@1.23 VRHE | ≈0.6/0.07@1.23 VRHE | >38 h@1.23 VRHE | 54 |
| CoNiFe-LDH/Ta3N5 | Nanostructure/Co-catalyst | Glycerol | AM 1.5 G, 100 mW cm−2 | NaOH (1.0 M) | FA: 60@d0 V | 3.59/0.29@1.23 VRHE | >0.5 h@d0 V | 122 |
Owing to its widespread application in the OER, the WO3 photoanode has also been intensively studied in PEC organic valorisation in recent years (Table 2). Examples include the conversion of cyclohexane to KA oil (a mixture of cyclohexanol and cyclohexanone),65 glycerol into value-added C3 products,66 and HMF to furandicarboxaldehyde (DFF) and FDCA.67 The crystal structure and predominantly exposed facets of WO3 also significantly affect the selectivity of PEC organic valorisation,68,69 which are also discussed in a later section.
Like TiO2 and WO3, BiVO4 has been intensively investigated for PEC organic valorisation. Pioneering work on BiVO4 for PEC biomass valorisation was reported by K.-S. Choi's group.70 In the presence of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), a faradaic efficiency (FE) of over 90% toward FDCA from HMF is achievable using BiVO4 as the photoanode.70 Beyond the valorisation of HMF, BiVO4 has also been investigated for the PEC valorisation of lignin71,72 and methanol,73 and has been particularly well explored for glycerol valorisation (Table 2).55,74–76
α-Fe2O3 has also shown great potential and has attracted considerable attention in PEC organic valorisation. α-Fe2O3 has been investigated in glycerol, glucose, cyclohexene, methanol, and EG valorisation (Table 2). In addition, α-Fe2O3 is particularly suitable for polymer-waste valorisation. Because depolymerisation of plastics by alkaline hydrolysis is generally required prior to PEC valorisation, the high stability of α-Fe2O3 in strongly alkaline solutions makes it the most frequently applied photoanode for plastic valorisation.
As mentioned in the previous section, most work on PEC organic valorisation has focused on earth-abundant photoanodes such as TiO2, WO3, and α-Fe2O3. Given its extensive investigation in PEC reactions, BiVO4 is also reviewed here, though Bi and V are not highly earth-abundant elements. Other earth-abundant photoelectrodes beyond those mentioned above, including CuWO4,77–79 Ta3N5,80,81n-Si,82,83 ZnO,84 Cu2O,49,84 and CuBi2O4,85–87 have also been intensively studied for PEC water splitting; nevertheless, they have received significantly less, or no, attention in PEC valorisation (Table 2). For instance, CuWO4, an n-type semiconductor with a bandgap of 2.2 eV, is capable of utilising visible light up to approximately 550 nm to drive PEC oxidation reactions and exhibits chemical stability from acidic to slightly alkaline conditions.88,89 Only recently has CuWO4 been demonstrated as a versatile photoelectrode for converting glucose, fructose, and glycerol into several value-added chemicals, including FA, glycolate (the conjugate base of GLA), DHA, and GLAD.54
2.2 System and half-cell figures of merit
Solar-to-fuel energy conversion efficiency (ηSTF) is a critical performance indicator for systems used in solar water splitting and CO2 reduction reactions, as it is broadly applicable to various designs of artificial photosynthesis systems. ηSTF is determined by the ratio of the output energy of the solar fuel to the incident solar energy, as described in the following equation. Measurements are generally conducted under one-sun solar irradiation (1000 W m−2) with a solar spectrum of Air Mass 1.5 Global (AM 1.5 G): | (5) |
However, in PEC organic waste valorisation, ηSTF values are generally low, regardless of Rproduct, due to the typically small ΔG. In particular, some organic waste reactions have a negative ΔG, which limits the applicability of ηSTF. Given the substantial variation in ΔG across different organic waste valorisation reactions, relying on ηSTF would result in inequitable comparisons among solar-driven valorisation systems. As an alternative, areal activity (Rareal) can serve as another indicator, providing a unified quantification of product generation per unit of illuminated area and reaction time.1
 | (6) |
To recognize the economic value of each type of organic waste valorisation process, the solar-to-value (STV) creation rate (rSTV) proposed by E. Reisner's group has been introduced:1
 | (7) |
However, the cost of PEC cells should also be considered and is not negligible, particularly when noble metals are used in the composition of the photoelectrodes or when the synthesis of photoelectrodes involves arduous procedures. Therefore, rSTV is further modified as follows:
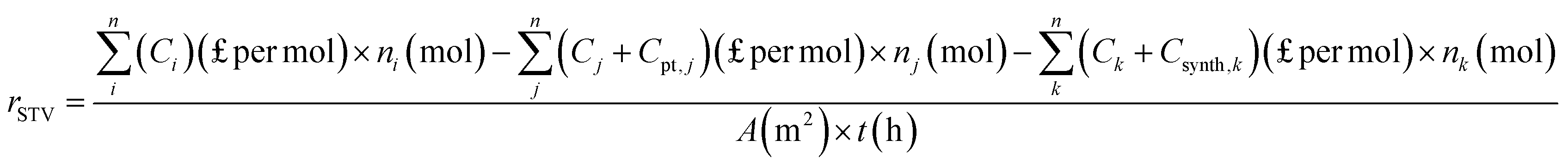 | (8) |
Although the ηSTF, Rareal, and rSTV parameters can be used to estimate the overall performance of an integrated PEC cell, additional indicators are necessary to evaluate half-cell performance. Since half-cell performance is predominantly governed by the intrinsic properties of the photoelectrodes, the inherent performance of a photoelectrode (φPE) using the following equation is suggested for the assessment of the half-cell performance of a PEC cell:
 | (9) |
The φPE allows for the assessment of the intrinsic performance of a photoelectrode by neglecting reaction substrates in a PEC system. It could be a promising indicator for comparing the performance of a PEC cell using the same photoelectrode for different organic valorisation reactions. The ηint generally deviates from unity during water oxidation due to its sluggish kinetics. However, ηint can be assumed to be unity, as most photoelectrodes provide significantly greater driving force than that required for water oxidation. Table 2 summarises the key parameters of reported PEC organic valorisation systems and categories of photoelectrodes based on their composition and modification methods. The φPE in Table 2 is calculated using eqn (9) and assuming ηint is unity.
The FE and power-saved metrics are advantageous for comparing the performance of different photoelectrodes under the same PEC organic valorising reaction. FE, defined as the percentage of passed charge used in the desired reaction, is an essential metric for evaluating selectivity in both EC and PEC systems. FE can be described by the following reaction:
 | (10) |
485 is the Faraday constant, ni is the number of moles of the generated specific product (i), and Q is the amount of charge passed during the PEC reaction.
Some literature presents selectivity using the ratio of the amount of a desired product to the total amount of product or the ratio of the amount of a desired product to the amount of substrate. If there is an unknown product, it will lead to a deviation using the former method, while the selectivity can be enhanced within a specific reaction time by lowering the initial amount of substrate using the latter method. Therefore, these two types of selective presentations may not be sufficiently objective, and FE will be used primarily in the discussion about selectivity.
Power-saved metrics enable the evaluation of the effects of input solar illumination on a half-cell.90 In a three-electrode system, the power stored at a specific current density I (Psave(I), W m−2) is derived by the product of the current density I and the potential difference of driving a half-reaction at current density I between a selected working electrode in the dark and a photoelectrode in the light:
| Psaved(I) = I(A m−2) × (Edark(I) − Elight(I)(V)) | (11) |
The Edark(I) and Elight(I) are the potential of driving a half-reaction at current density I of the dark electrode and photoelectrode, respectively.
2.3 Comparison of electrochemical (EC) and PEC organic valorisation
In addition to PEC organic valorisation, EC organic valorisation has also been demonstrated to convert organic waste into fuel and value-added chemicals. In EC systems, the energy required to drive the reaction relies on the external bias, and the catalytic current increases monotonically with the applied overpotential until it becomes limited by mass transfer. In contrast, in PEC systems, the photocurrent reaches a saturation plateau at a certain potential when the photogenerated electrons are fully extracted. Thus, EC systems typically achieve higher current densities at high overpotentials compared to PEC systems, particularly if PEC systems are operated under low photon flux conditions, such as one-sun illumination. For example, the EC system of nickel-modified cobalt phosphide on nickel foam (CoNi0.25P/NF) has been demonstrated to exhibit an electrocatalytic current for EG oxidation with an onset potential of approximately 1.2 VRHE, and its current significantly increases with the applied potential, reaching approximately 180 mA cm−2 at 1.35 VRHE (Fig. 6a).91 On the other hand, a BiVO4/NiCo-LDH photoanode exhibits a much lower saturation photocurrent of approximately 2 mA cm−2 for EG oxidation under one-sun illumination, i.e. 100 mW cm−2 (Fig. 6b).92 However, the external bias required in the PEC system is typically smaller than that of the EC system due to the presence of a light-induced photovoltage, resulting lower energy consumption in the PEC system at low overpotential regions. In the case of BiVO4/NiCo-LDH, the photocurrent for EG valorisation starts from 0.2 VRHE, which is much lower than the onset potential (1.2 VRHE) of the CoNi0.25P/NF EC system. The amount of energy saved by using a photoelectrode can be readily evaluated using eqn (11). In addition, the drawback of the low saturation photocurrent density of a photoelectrode can potentially be mitigated by using concentrated solar light as the light source93 or by incorporating the thermoelectric effects.93,94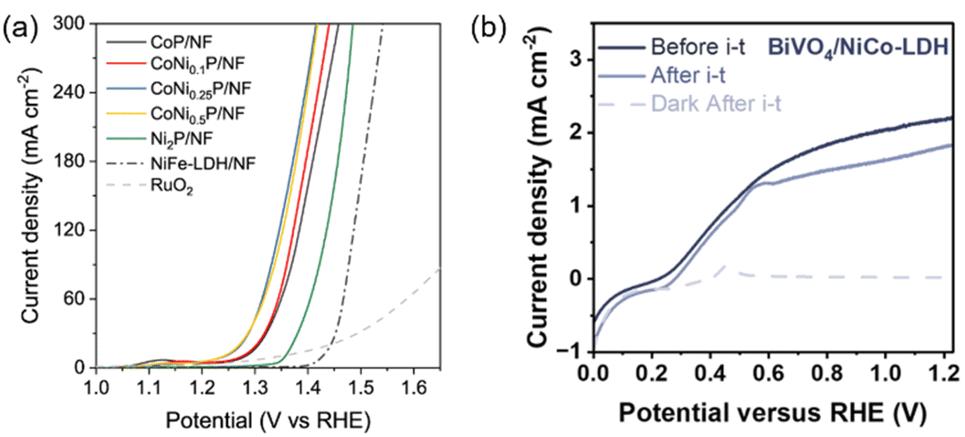 | ||
| Fig. 6 (a) Linear sweep voltammetry (LSV) curves for EC oxidation using CoNi0.25P/NF and other electrocatalysts. Reproduced with permission under the terms of a CC BY 4.0 license.91 Copyright 2021, The Author(s), published by Springer Nature. (b) Photocurrent (solid lines) of BiVO4/NiCo-LDH before and after i–t measurements in an EG containing KOH solution. Reproduced with permission.92 Copyright 2024, John Wiley and Sons. | ||
The advantages of using a PEC system over an EC system can be highlighted by the work on glycerol valorisation in a membrane-separated continuous-flow PEC cell reported in 2024.95 In this work, direct PEC oxidation of glycerol paired with the dark H2 evolution or CO2 reduction was achieved using a Si photoanode coupled with a silver nanoparticle-coated carbon cloth cathode. On the other hand, in the EC system, a Ni sheet was used as the anode. Intense sunlight of up to 10 suns was applied to the PEC cell, and a photocurrent exceeding 110 mA cm−2 was achieved for glycerol oxidation (Fig. 7a). Compared to the EC cell, the PEC cell required less energy input from the external voltage source to attain the same current density (Fig. 7b). Approximately 1 V less cell voltage was needed for the PEC cell compared to the EC cell to generate a current density of 100 mA cm−2 (Fig. 7b). Notably, this study also demonstrated that the product distribution of glycerol oxidation in the PEC cell differs from that of the EC cell at the same current density (Fig. 7c). The FE for value-added products was higher in the PEC cell than in the EC cell (≈82% vs. ≈63%). Oxygen quantification experiments confirmed that the PEC system can effectively suppress the parasitic OER. This difference may arise from the inherent discrepancy in potential dependence between PEC and EC systems. The oxidative force in the EC system depends on the applied potential, whereas in the PEC system, it is determined and fixed by the EVB position and is independent of the applied potential.
 | ||
| Fig. 7 (a) LSV of a Ni/Si photoanode under chopped light illumination and a Ni sheet-based electrode in the dark for glycerol oxidation. (b) Cell voltage difference between the Ni/Si photoanode and the Ni sheet-based anode at a current density of 100 mA cm−2 for glycerol oxidation. (c) FE of glycerol oxidation products after 30 min at a current density of 100 mA cm−2. Reproduced with permission under the terms of a CC BY 4.0 license.95 Copyright 2024, The Author(s), published by Springer Nature. | ||
Nonetheless, the PEC system undergoes complex charge transfer processes, beginning with the extraction of photogenerated charges, followed by hole transfer to the semiconductor surface, and subsequently to the organic reactant. Therefore, an effective photoelectrode design that facilitates rapid charge transfer is necessary for PEC organic valorisation.
2.4 Kinetics of PEC reactions
PEC reactions that involve multiple steps of charge transfer are typically kinetically challenging, and the reaction kinetics play a critical role in determining both efficiency and selectivity. For example, OER in water splitting is generally considered the bottleneck, as it involves a four-electron transfer process, and its sluggish kinetics constrain the overall efficiency of water splitting. Many organic valorisation reactions listed in Table 1 also involve multiple charge transfer steps, and various products may be formed during the process. Research into PEC reaction kinetics is therefore essential for advancing the development of PEC organic valorisation.The rate law analysis on α-Fe2O3 for PEC water oxidation was first demonstrated by Le Formal et al.96 Photoinduced absorption (PIA) spectroscopy combined with transient photocurrent (TPC) measurements was employed to elucidate the quantitative relationship between the reaction rate and the accumulation of photogenerated holes on the α-Fe2O3 surface.96 PIA enables monitoring of long-lived photogenerated charge carriers, i.e., surface hole density for the photoanode, while TPC monitors the net flux of holes transferred to the electrolyte. The reaction kinetics under steady-state PEC oxidation conditions was then estimated by rate law analysis (eqn (12)):96–98
| J = kr[hole]β | (12) |
It was found that a transition occurs from a first-order reaction at low densities of accumulated surface holes (slow reaction) to a third-order reaction at high densities of accumulated surface holes (fast reaction), indicating that the accumulation of multiple holes is required to drive water oxidation efficiently over α-Fe2O3.96
In addition to PIA, the rate law analysis can also be conducted using EC impedance spectroscopy (EIS) measurement. Zhang et al. determined the surface hole density on the α-Fe2O3 photoanode using this EIS measurement and subsequently correlated it with the photocurrent density to calculate the reaction order of water oxidation.97 They found that the reaction order was approximately 1 and 2 for water oxidation in near-neutral and alkaline conditions, respectively, highlighting the dependency of the water oxidation reaction order on the solution pH.
Shifting the focus toward PEC organic valorisation, PIA coupled with TPC measurements has also been applied to derive the kinetic parameters and elucidate the mechanism of PEC alcohol oxidation.99,100 Mesa et al. found that methanol, when PEC oxidised to formaldehyde on α-Fe2O3 photoanodes, followed a second-order reaction with rate orders of 1.88 and 1.89 at applied potentials of 0 V vs. Ag/AgCl (VAg/AgCl) and 0.55 VAg/AgCl, respectively (Fig. 8a).99 They confirmed that the rate order on α-Fe2O3 is determined solely by the density of holes accumulated at the surface of the photoelectrode and is independent of the applied potential. This observation corresponds to the earlier discussion comparing EC and PEC systems. In contrast to EC systems (Section 2.3), in which the oxidative force varies with the applied potential, the energy, i.e., the oxidative force of the holes, in a PEC system is determined by the EVB of photoanodes.
 | ||
| Fig. 8 (a) Rate law analyses showing the reaction order of PEC methanol oxidation on α-Fe2O3 at 0.55 VAg/AgCl (dark red) and 0.00 VAg/AgCl (light red), and on TiO2 at −0.80 VAg/AgCl (blue). Reproduced with permission under the terms of a CC BY 4.0 license.99 Copyright 2017, American Chemical Society. (b) Rate law analyses showing the reaction order of PEC water oxidation at 1.3 VRHE (blue), ethanol oxidation at 1.2 VRHE (black), and acetaldehyde oxidation at 1.1 VRHE (red) on α-Fe2O3. Reproduced with permission.100 Copyright 2022, John Wiley and Sons. | ||
It has been found that α-Fe2O3 exhibits highly selective aldehyde production through PEC alcohol oxidation.99,100 The underlying reason was also elucidated by operando spectroelectrochemical PIA coupled with TPC, in parallel with the Arrhenius analyses.100 Rate law analyses suggest that α-Fe2O3 exhibits a second-order reaction for ethanol oxidation, while the reaction order of PEC acetaldehyde oxidation on α-Fe2O3 is only 0.5 (Fig. 8b). Temperature-dependent rate law analyses further demonstrated that activation energy for ethanol oxidation by photogenerated holes in α-Fe2O3 is much lower than that for acetaldehyde oxidation (195 vs. 398 meV). These results indicate that the high selectivity for aldehyde production on α-Fe2O3 results from its much more facile kinetics toward ethanol oxidation compared to acetaldehyde oxidation, which suppresses the overoxidation of acetaldehyde. Compared to α-Fe2O3, TiO2 exhibits only a modest selectivity toward acetaldehyde production, and overoxidation of acetaldehyde was observed. The disparate selectivity toward acetaldehyde production from ethanol over different metal oxide photoelectrodes is correlated with their varying EVB.101 The measured acetaldehyde oxidation activation energies are 45 and 427 meV, respectively, for photogenerated holes in TiO2 and α-Fe2O3. The study highlights the significance of band potential in determining the selectivity of PEC organic valorisation.
Understanding reaction kinetics in PEC systems can guide the design of photoanodes for PEC organic valorisation, emphasising the importance of achieving a high density of accumulated holes and ensuring the suitability of the EVB for a photoanode. This can be achieved by designing photoelectrodes with a suitable electronic band structure, increasing the intensity of illumination using concentrated solar light, tuning hole densities through doping, modifying co-catalysts, nanostructuring, and creating heterojunctions. Strategies for enhancing the performance of photoanodes for organic valorisation is further discussed in Section 4.
3. Polymeric organic waste and pre-treatment process
Biomass and plastic are the major organic substrates of interest in PEC valorisation. Glycerol, glucose, and HMF are among the most widely studied biomass-derived platform molecules in PEC organic valorisation, while EG is the most extensively investigated plastic-derived molecule. However, some of these molecules originate from the depolymerisation of natural and synthetic polymeric waste. A pre-treatment process of reducing crystallinity or depolymerising polymeric organic waste is generally required to enhance reforming efficiency.3.1 Natural polymer waste
Biomass is a naturally synthesised polymer that primarily contains lignocellulose, proteins, and lipids, with lignocellulose being its main constituent and an earth-abundant resource.123,124 Lignocellulose is the most abundant biomass and is readily found in wood stems, nut shells, corn cobs, grasses, and rice husks,125,126 consisting of three major components: cellulose, hemicellulose, and lignin. Cellulose and hemicellulose are composed of long chains of sugars. The general chemical formula for cellulose is (C6H12O5)n, in which glucose-based monomers are linked via β-1,4-glycosidic bonds.127 Hemicellulose is a copolymer composed of five sugar monomers: D-xylose, L-arabinose, D-galactose, D-glucose, and D-mannose.128 In contrast, lignin is composed of three hydrophobic units: p-coumaryl, coniferyl alcohol, and sinapyl alcohol.129 The polar nature and high hydroxyl content of cellulose and hemicellulose enable their application in reforming reactions following a depolymerisation pre-treatment, whereas lignin presents a challenge due to its recalcitrant structure. On the other hand, proteins such as amino acids and lipids such as glyceride and fatty acid are found in algae and wood wastes.1243.2 Synthetic polymer waste
Affordability, lightweight nature, and versatility make plastics indispensable in human life. However, the high stability and durability of plastics result in prolonged degradation times. Significant “white pollution” is caused by the widespread use of plastics without a proper circular economy. Polyethylene (PE), PET, polystyrene (PS), polypropylene (PP), poly (vinyl chloride) (PVC), polylactic acid (PLA), and polyurethane (PU) are the primary types of plastics employed in daily life.30,130 These plastics are classified into two groups based on the type of bond in the main chain and the synthetic methods used.30,130(a) The first group, PE, PS, PP, and PVC, is composed of C–C bonds in the main chain and is synthesised via addition polymerisation. The high stability of these hydrocarbon chains limits their applicability in reforming reactions.
(b) The second group, comprising PET, PLA, and PU, features C–O or C–N bonds in the backbone chain and is synthesised via condensation polymerisation. Oxygenated plastics account for approximately 15% of global primary plastic production.131 Owing to their polarity and ester bonds, they are typically the target substrates in solar-driven plastic reforming.7,132
3.3 Pre-treatment of polymeric organic waste
The hydrogen bonding or crosslinking features within polymeric waste restricts their solubility in aqueous solution. A pre-treatment process that reduces crystallinity or depolymerises polymeric organic waste is generally required to enhance reforming efficiency. An effective pre-treatment method must be scalable, cost-effective, and capable of yielding a high amount of monomer components. Various pre-treatment technologies, ranging from conventional to green approaches, are used to solubilise organic waste. However, the scope of this section is to summarise the primary pre-treatment technologies used in solar-driven valorisation and to discuss their respective advantages and challenges. The pre-treatment processes are categorised into three major types.4. PEC oxidation for organic electrosynthesis: strategies to enhance the performance of photoanodes
Optimising the design of photoelectrode is essential not only for enhancing the conversion rate of solar energy into chemical energy but also for achieving efficient, selective, and robust PEC valorisation of organic waste. Several photoelectrodes are constrained by limited light absorption and high recombination of photo-generated electrons and holes. The limitation in light absorption is due to the wide bandgap. Photo-generated charge recombination primarily results from the intrinsic low conductivity and short minority carrier diffusion length of the semiconducting materials. Sluggish interfacial transfer of photo-generated holes further contributes to surface recombination.142,143 Several key strategies have been demonstrated to mitigate recombination without compromising the light-harvesting capability of the photoelectrodes. These strategies include doping, heterojunction, and nanostructuring. On the other hand, co-catalyst modification and crystal facet engineering effectively enhance the selectivity of the target products in organic waste valorisation. The aforementioned strategies used in PEC oxidation for organic electrosynthesis are summarised and discussed in the following sections.4.1 Doping
Doping offers a viable method for effectively modulating the electronic structure and properties of photoelectrodes.144 Doping may regulate the bandgap structure by altering the positions of EVB and ECB, thereby extending light absorption into the visible range. Additionally, electronic states are introduced, including donor levels below the ECB and/or acceptor levels above the EVB.145 In particular, doping is an effective strategy to eliminate significant charge recombination by enhancing carrier mobility and density or internal electric field in a photoelectrode.The suppression of charge recombination by enhancing the internal electric field has been recently demonstrated to boost the performance of a photoelectrode in organic valorisation. Kong et al. introduced nitrogen into WO3 (N-WO3) to enhance the PEC valorisation of polyol biomass into CO.110 Density functional theory (DFT) calculation revealed that nitrogen doping regulates the electronic structure polarity by creating an asymmetric distribution of charge (Fig. 9a). The asymmetric distribution of charge enhances the internal electric field, effectively preventing the recombination of charge carriers. As a result, N-WO3 exhibited a higher photocurrent density in PEC glycerol valorisation than pristine WO3 (Fig. 9b). N-WO3 presented a higher CO evolution rate than WO3 at a similar CO selectivity throughout the voltage range of 0.4–0.9 VRHE (Fig. 9c and d). N-WO3 also exhibited promising long-term stability in an acidic solution (0.5 M Na2SO4, pH 1), with around 80% selectivity towards CO, 40% CO FE, and 10% carbon yield after 50 h (Fig. 9e). The excellent stability was attributed to the absence of any phase or structural changes during the PEC process. Similarly, Kim et al. demonstrated an enhancement in PET microplastic valorisation over α-Fe2O3 by Zr doping (Zr:α-Fe2O3).146 PET microplastic was obtained by grinding commercial PET into microplastic (≤5 mm), followed by immersion in 5 M NaOH for 3 days at 80 °C to obtain EG. Zr doping served as an electron donor, accelerating electron transport kinetics and promoting more upward band bending. Zr:α-Fe2O3 exhibited a faster production rate of FA and acetate compared to α-Fe2O3. Moreover, Zr:α-Fe2O3 demonstrated excellent long-term stability over seven cycles, each lasting 12 h. Notably, there was no change in phase or surface oxidation state after the cycling test, and no metal leaching was observed, indicating excellent structural and chemical stability.
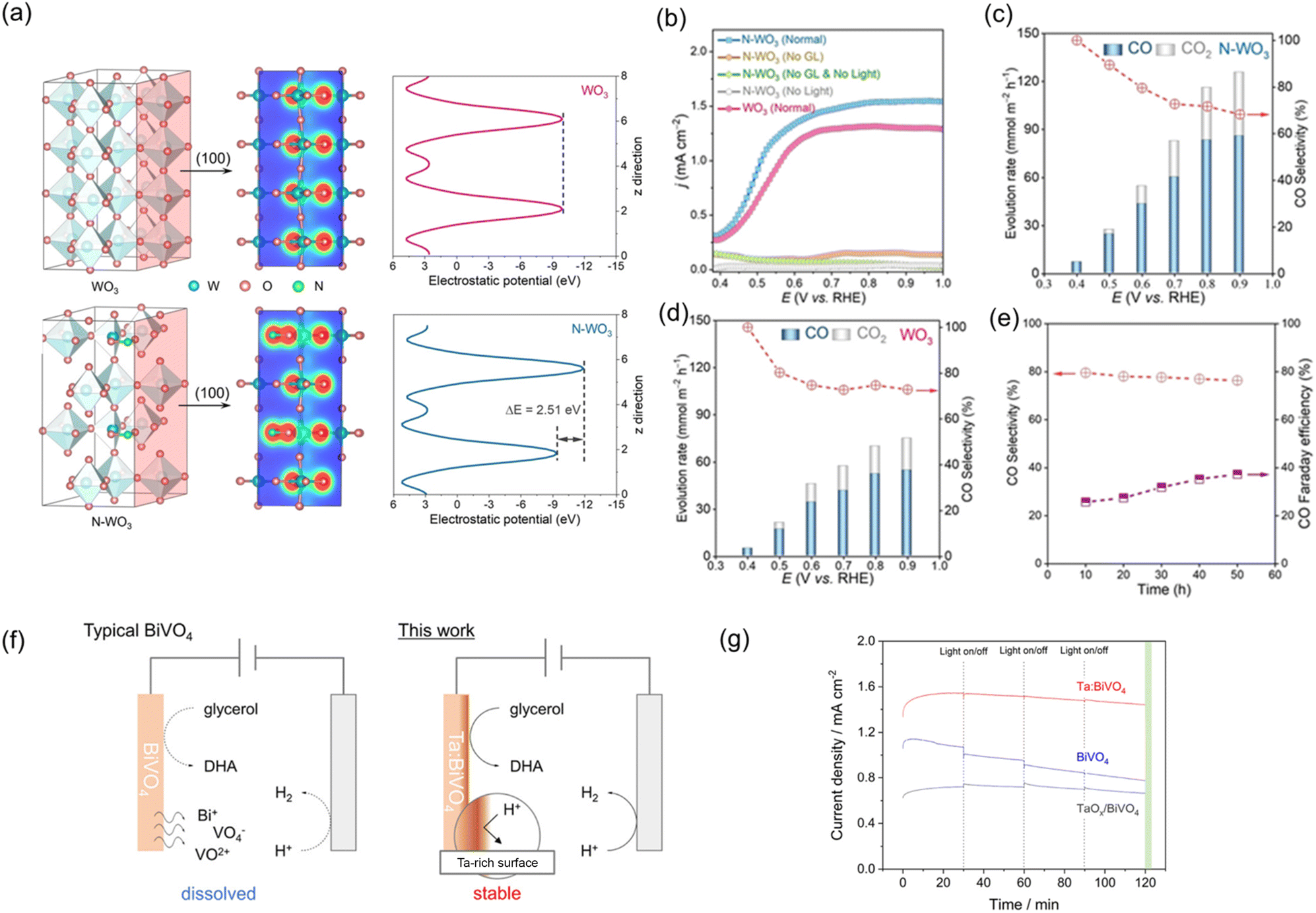 | ||
| Fig. 9 (a) Calculated isosurface of electron density and electrostatic potential of WO3 and N-WO3. (b) LSV curves of N-WO3 in the absence and presence of glycerol under dark or light irradiation. Evolution rate and CO selectivity of (c) N-WO3 and (d) WO3. (e) Long-term stability of PEC glycerol oxidation toward CO generation at 0.6 VRHE. Reproduced with permission.110 Copyright 2022, John Wiley and Sons. (f) Schematic of PEC glycerol oxidation over BiVO4 and Ta:BiVO4 in acidic medium. (g) Long-term stability of PEC glycerol oxidation over BiVO4, Ta:BiVO4, and TaOx/BiVO4 at 1.0 VRHE. Reproduced with permission.112 Copyright 2022, American Chemical Society. | ||
Beyond regulating the photocurrent of the pristine photoelectrode, increasing the selectivity for the target product and enhancing stability can also be realised by incorporating a suitable dopant. The enhancement of photocurrent, selectivity of DHA in PEC glycerol oxidation, and immunity to the dissolution in an acidic medium were achieved through Ta doping of BiVO4 (Ta:BiVO4), as shown in Fig. 9f and g.112 Doping with Ta in BiVO4 was found to effectively enhance the photocurrent density and FE of DHA from 1.70 to 3.07 mA cm−2 at 1.23 VRHE and from 41% to 61%, respectively. In terms of stability, BiVO4 retained only approximately 70% of the photocurrent in the acidic solution after 2 h of operation during glycerol oxidation. In contrast, both Ta:BiVO4 and TaOx-coated BiVO4 (TaOx/BiVO4) remained stable in photocurrent during 2 h PEC glycerol valorisation. These results confirm that the presence of Ta or TaOx on the surface of BiVO4 protects BiVO4 from dissolution in an acidic medium, thus providing high stability.
4.2 Heterojunction
The concept of a semiconductor–semiconductor heterojunction configuration is employed to prevent the recombination of photo-generated charges and accelerate the charge transfer driven by a built-in electric field.147,148 In some cases, the guest semiconductor is selected to have a narrower bandgap than the host semiconductor to extend the utilisation of photons in the solar light spectrum. The selectivity might also be tuned by altering the catalytic kinetics, oxidation potentials, or reaction routes through the appropriate selection of the heterojunction layer. Heterojunctions can be categorised into three conventional types according to their band alignment between the two semiconductors: straddling-gap (type-I), staggered-gap (type-II), and broken-gap heterojunctions (type-III).149 However, only type-II heterojunction can promote spatial charge separation, thereby eliminating charge recombination.150 The charge separation in a type-II heterojunction is realised by the photo-generated electrons in semiconductor B transporting to semiconductor A, while the photo-generated holes follow the reverse route. The junctions can be further divided into n–n junctions, p–p junctions, or p–n junctions based on the conductivity features of the semiconductors.PEC organic waste valorisation is typically performed in water, and the OER inevitably occurs as a side reaction if the photo-generated holes possess sufficient oxidative force toward OER. As previously mentioned, organic oxidation in common valorisation studies generally requires less oxidative force than OER. Selecting a guest semiconductor with insufficient EVB to oxidise water can eliminate the OER side reaction during PEC organic compound valorisation. Chuang et al. constructed a type-II p–n heterojunction of nanoFe2O3 and CuO as a nanoFe2O3|CuO photoanode for PEC glucose oxidation into FA.119 With regard to the EVB position of nanoFe2O3, there is competition between OER and glucose oxidation. The incorporation of CuO effectively inhibits the OER side reaction because of the insufficient driving force of holes on its EVB. Additionally, CuO is an efficient catalyst for glucose valorisation to FA and also acts as a co-catalyst in the nanoFe2O3|CuO heterojunction photoelectrode, promoting FA formation (Fig. 10a). The nanoFe2O3|CuO outperformed nanoFe2O3 by exhibiting a stable photocurrent density, while nanoFe2O3 exhibited a 33% decay photocurrent density after 2 h of operation (Fig. 10b). Furthermore, nanoFe2O3|CuO also exhibited nearly 100% of FE for FA production at 1.0 VRHE (Fig. 10c). Another analogous case was presented by Luo et al., showing that the addition of a p-type semiconductor of Bi2O3 to n-type TiO2 (Bi2O3/TiO2) significantly enhanced charge separation, transfer, and light absorption, thereby promoting larger photocurrent density in the PEC glycerol oxidation.56 The Bi2O3/TiO2 exhibited a higher DHA selectivity than TiO2 alone (65.7% vs. 8.2%). In particular, DFT calculation also revealed that Bi2O3 tends to adsorb the middle hydroxyl of glycerol, selectively generating DHA rather than formic acid (Fig. 10d). Meanwhile, the Bi2O3/TiO2 maintained a high conversion rate (>200 mmol m−2 h−1) and FE of DHA for 150 h (Fig. 10e) due to its excellent structural and chemical stability.
 | ||
| Fig. 10 (a) Band diagram of the nanoFe2O3|m-CuO. (b) CPE measurements of nanoFe2O3 and nanoFe2O3|m-CuO, and (c) their corresponding FE and production rate of FA in 0.1 M NaOH with 20 mM glucose under solar light irradiation (AM 1.5 G, 100 mW cm−2). Reproduced with permission.119 Copyright 2022, Royal Society of Chemistry. (d) Schematic of PEC glycerol oxidation over TiO2 and Bi2O3/TiO2 heterojunction and (e) long-term stability test of Bi2O3/TiO2 photoanode for PEC glycerol oxidation. Reproduced with permission.56 Copyright 2022, American Chemical Society. | ||
n–n heterojunctions have also been employed to promote the reaction rate for PEC organic valorisation. The formation of a type-II heterojunction between defective WO3 (m-H-WO3) and TiO2, coupled with interfacial and defect engineering, effectively accelerates charge separation and mass transfer.109 A photocurrent of 2.89 mA cm−2 was achieved at 1.23 VRHE for the heterojunction photoelectrode, which is more than 1.5 times higher than that of WO3. Likewise, the improved photocurrent stability of m-H-WO3/TiO2, 3 times higher than that of m-WO3, was demonstrated over 60 min. m-WO3 exhibited poor photocurrent stability, with a 60% decay occurring within only 35 min. The superior photocurrent stability in m-H-WO3/TiO2 was attributed to its massive oxygen vacancies, which acted as electron mediators. The production rate of GLYAD and DHA reached 353 mmol m−2 h−1 at 1.2 VRHE for the heterojunction photoelectrode, whereas WO3 achieved only 133 mmol m−2 h−1.
4.3 Co-catalyst modification
Oxidation of organic waste in the absence of a soluble redox mediator generally involves an inner-sphere electron transfer process. For this process, the reaction kinetics is significantly influenced by the interaction between the electrode material and the reactant/product.151,152 In this circumstance, the nature of electrode surface is crucial. In other words, to improve the selectivity of the desired products during organic waste conversion, the surface modification of the photoelectrodes with suitable co-catalysts is often necessary. The charge separation and overall activity of a photoelectrode are also possibly enhanced by accelerating charge transfer from the photoelectrode to the co-catalyst.29,153 For example, Huang et al. successfully transformed an inactive BiVO4 photoanode into a highly efficient photoanode toward FA production from methanol by nanoFe:Ni-Bi co-catalyst modification (Fig. 11a–c).154 nanoFe:Ni-Bi was synthesized by pulse-current electrodeposition and was found to be an active catalyst for methanol oxidation, in which the catalytic activity is controlled by the amount of iron species. Cooperating nanoFe:Ni–Bi with BiVO4 resulted in remarkable FA production with an FE of 94.6% ± 12.3% through PEC methanol oxidation at 0.55 VRHE at neutral pH, while negligible FA formation was observed in pristine BiVO4. In addition, BiVO4∣nanoFe:Ni-Bi exhibited a stable photocurrent density over 2 h. A co-catalyst modification strategy has also been revealed for glycerol valorisation using tungsten-doped BiVO4 (W:BiVO4).55 The co-catalyst of NiOx(OH)y not only enhanced the production rate but also altered the product distribution. The most valuable product, DHA, was not observed during glycerol valorisation using W:BiVO4 without the presence of NiOx(OH)y in a mild alkaline solution (Fig. 11d–f). Upon addition of NiOx (OH)y co-catalyst, W:BiVO4 generated DHA with an FE of approximately 20% at 1.2 VRHE. However, continuous decay in photocurrent due to the gradual dissolution of the NiOx(OH)y co-catalyst in alkaline conditions remains a challenge requiring further investigation.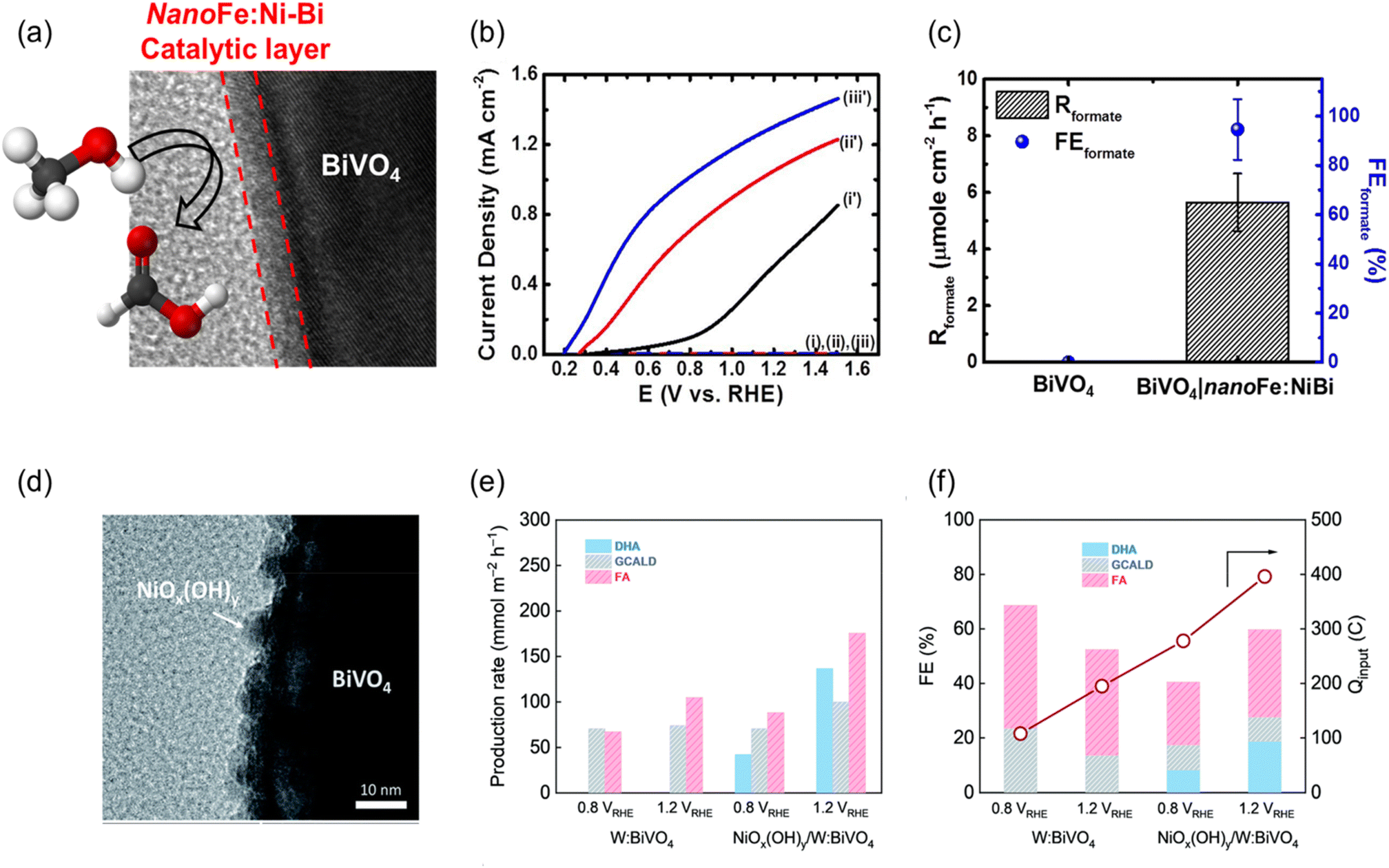 | ||
| Fig. 11 (a) High-resolution transition electron microscopy (HRTEM) image shows the surface modification of the BiVO4 photoanode with nanoFe:Ni-Bi. (b) LSV curves at a scan rate of 10 mV s−1 of (i and i′) the pristine BiVO4, (ii and ii′) BiVO4|nanoNi-Bi, and (iii and iii′) BiVO4|nanoFe:Ni-Bi photoanodes in near-neutral pH containing 0.1 M methanol in the dark (i′–iii′) and under light illumination (i–iii). (c) Production rate of formate (Rformate) and FEformate from PEC methanol oxidation using the pristine BiVO4 and BiVO4|nanoFe:Ni-Bi photoanodes. Reproduced with permission.154 Copyright 2020, Elsevier. (d) HRTEM image illustrating NiOx(OH)y co-catalyst incorporation on the surface of the W:BiVO4 electrode. (e) Production rate and (f) FE with total charge passed (Qinput) obtained in 0.5 M KBi. Reproduced with permission under the terms of a CC BY-NC 3.0 license.55 Copyright 2021, Royal Society of Chemistry. | ||
Co-catalyst modification has also been widely adopted for α-Fe2O3 in the valorisation of organic waste. For instance, CuO acts both as a heterojunction and co-catalyst in nanoFe2O3|CuO for PEC glucose valorisation, as discussed in the heterojunction section (Fig. 10a–c). Taking advantage of the corrosion-resistant properties of α-Fe2O3 in alkaline solutions, this material is of particular interest in polymer waste valorisation. However, the FE of some target products, such as FA, is often limited. Li et al. demonstrated that surface modification of Fe2O3 photoanode with Ni(OH)x co-catalyst significantly enhanced the FE of FA from PET valorisation, increasing it from 10% to nearly 100% (Fig. 12a–c).118 In this study, Ni(OH)x co-catalyst was loaded on the surface of α-Fe2O3 using a hydrothermal method under a mild temperature. Ni2+ in Ni(OH)x is proposed to be first oxidized to the higher valence of Ni3+ by the photo-generated holes from Fe2O3 and then reduced back to Ni2+ by oxidising EG, a monomer of PET, to FA. A similar result was observed with a Ni phosphate (Ni-Pi) co-catalyst modified α-Fe2O3 for EG and PET valorisation.9 Monomer EG was also obtained by grinding waste PET bottles into micro-sized powder, followed by alkaline hydrolysis in 1 M NaOH at 90 °C for 24 h. Ni phosphate (Ni-Pi) co-catalyst was deposited on α-Fe2O3 using a simple photo-deposition method (Fig. 12d–f), and the Ni-Pi-modified α-Fe2O3 (Ni-Pi/α-Fe2O3) photoanode demonstrated an enhancement of FE (87%) toward FA production from EG valorisation compared to unmodified α-Fe2O3 (40%). The experimental results confirmed that the enhanced PEC performance accounted for the enhancement in charge separation and charge transfer kinetics by Ni-Pi. This study also compared the performance of Ni-Pi-modified α-Fe2O3 with α-Fe2O3 modified by other co-catalysts. Ni-Pi demonstrated the highest activity and FE on α-Fe2O3 compared to CoOOH, NiOOH, and Co-Pi. DFT also revealed that Ni-Pi exhibits a stronger absorption ability toward the EG and promotes efficient desorption of the product of FA compared to NiOOH. Ni-Pi/α-Fe2O3 photoanode also successfully oxidised waste PET plastic bottle, achieving a high FA FE (82%) and functioned stably for 12 h (6 cycles) due to its structural and chemical stability.
 | ||
| Fig. 12 (a) Schematic of the surface modification of the Fe2O3 photoanode with Ni(OH)x for selective oxidation of EG into FA. Enhancement of (b) photocurrent and (c) the yield and FE of FA with the charge passed using Fe2O3/Ni(OH)x photoanodes modified with different concentrations of Ni(OH)x. Reproduced with permission.118 Copyright 2022, American Chemical Society. (d) TEM image of the Ni-Pi/α-Fe2O3 photoanode. (e) LSV curves and (f) FE for α-Fe2O3 with different co-catalysts. FE was derived at 1.1 V for 2 h. Reproduced with permission.9 Copyright 2023, Elsevier. | ||
4.4 Nanostructuring
As revealed in eqn (4), the activity in terms of photocurrent in a PEC reaction is determined by three factors, ηLHE, ηsep, and ηint, for a photoelectrode. Among them, ηLHE and ηsep have contradictory requirements regarding film thickness for a planar photoelectrode. Nanostructuring the photoelectrode can resolve the contradictory issues, thereby enhancing the photocurrent by decoupling the light-harvesting and charge-transport directions. Therefore, it is essential for photoelectrode materials with short charge carrier diffusion lengths, such as α-Fe2O3. In addition, the surface area and number of active sites of a photoelectrode can also be enhanced by nanostructuring. Nanostructuring has, therefore, also been demonstrated as a practical approach to boost the reaction rates in PEC organic valorisation.V2O5 has been demonstrated as an example of the effect of nanostructuring on the performance of methanol oxidation over V2O5 (Fig. 13a–d).10 Exfoliation of V2O5 into nanosheet bundles can be readily achieved by direct top-down exfoliation of sub-micron V2O5 plates using formamide as the exfoliation agent. The photocurrent for methanol oxidation doubled at 1.3 VRHE if the V2O5 microplate was exfoliated. An impedance study confirms that exfoliating V2O5 reduces charge transport resistance, owing to a shorter travel distance for photo-generated holes. In addition, the oxygen vacancy (VO) increases upon exfoliation, thereby enhancing the conductivity and active sites of V2O5.
 | ||
| Fig. 13 SEM images of (a) microV2O5 and (b) exfoliated V2O5. (c) TEM image of the exfoliated V2O5. (d) Photocurrent density recorded at 1.3 VRHE of (i) microV2O5 and (ii) exfoliated V2O5 in 0.1 M Na2SO4 electrolyte containing 0.1 M methanol. Reproduced with permission under the terms of a CC BY 4.0 license.10 Copyright 2022, The Author(s), published by Elsevier. (e) SEM image of nanosheet structured CuWO4 (f) FE derived from PEC valorisation of glucose (0.8 M), fructose (0.8 M) and glycerol (1.0 M) at 1.23 VRHE at the variation of pH. Reproduced with permission.54 Copyright 2024, John Wiley and Sons. (g) Schematic of the effect of microscale fluid in the PEC glycerol valorisation. (h) LSV curves of the different pore size of TiO2 nanotube in Na2SO4 (0.5 M, pH = 2) with and without the addition of glycerol. (i) FE of the main products formed from glycerol oxidation using TiO2 nanotube photoanodes with different pore sizes. Reproduced with permission.104 Copyright 2024, American Chemical Society. | ||
As mentioned in the previous section, the majority of work in PEC organic valorisation focuses on state-of-the-art photoanodes such as TiO2, WO3, Fe2O3, and BiVO4. However, except for TiO2, the application of these photoelectrodes is limited to specific reactions due to the narrow operational range in which they can function stably. Most recently, nanosheet-structured CuWO4 (nanoCuWO4) has been investigated across a wide range of PEC valorisation systems, including glucose, fructose, and glycerol, owing to its broad operational stability (Fig. 13e and f).54 FE of 76% ± 5% for FA and 61% ± 8% for GLA were achieved at pH 10.2 over CuWO4 from PEC glucose and fructose valorisation, respectively. Notably, the FEs of the primary products, GLAD and DHA, were over 85% at neutral and slightly acidic pH using CuWO4 for PEC glycerol valorisation. NanoCuWO4 exhibited a significantly higher photocurrent at 1.23 VRHE than a planar CuWO4 for PEC water oxidation and glycerol valorisation, demonstrating that nanoCuWO4 provides more active sites in PEC reactions. Notably, the nanoCuWO4 maintained a high total FE of value-added chemicals (100%) over 72 h of photoelectrolysis and retained 60% of its initial photocurrent density after prolonged operation of more than 38 h. This excellent stability was attributed to the preservation of phase properties following long-term operation.
Tuning product selectivity has also been reported to be achieved through nanoconfinement. Lu et al. introduced a nanoconfined environment into the glycerol valorisation process using one-dimensional TiO2 nanotubes (Fig. 13g–i).104 Nanotubes with three pore sizes, 50 nm (TNT-50), 75 nm (TNT-75) and 100 nm (TNT-100), were investigated, among which TNT-50 exhibited the highest photocurrent for PEC glycerol oxidation. The nanoconfined environment accelerated the mass transfer process by the microscale fluid effects. The diffusion rate of glycerol on TiO2 nanotubes increased almost fivefold when the tube diameter was reduced from 100 to 50 nm. The faster diffusion rate inhibited the overoxidation of glycerol and enhanced the selectivity of C3 products, such as GLAD. The TiO2 nanotube also demonstrated promising stability, with no significant photocurrent degradation observed during 10 h of operation.
4.5 Crystal facet and crystal phase engineering
Beyond doping, co-catalyst addition, or heterojunction formation, crystal facet and crystal phase engineering have also been demonstrated to enhance the performance of photoelectrodes for organic valorisation. These crystalline and phase properties influence surface reactivity by promoting favourable adsorption or facilitating the activation of specific reactants. These strategies have been applied on both WO3 and BiVO4 to improve the activity and selectivity for organic valorisation. Ma et al. optimised the reactivity of hydroxyl radicals (˙OH) by tuning the crystal facet of WO3 to achieve efficient EG production from CH4 (Fig. 14a–c).108 The photo-generated ˙OH radicals played a significant role in CH4 activation and subsequent EG formation. Their DFT calculations revealed that (010) facet exhibited the highest presence of ˙OH on the surface compared to the (100) and (001) facets. This was attributed to the spatial proximity of adsorbed ˙OH radicals on the (100) and (001) surfaces, which led to their disappearance via the formation of surface-bound O and H2O. As a result, WO3 nanobar (WO3 NB), which had the highest proportion of the (010) facet, showed superior EG production rate and selectivity compared to WO3 nanoplate (WO3NP) and WO3 nanoflake (WO3NF), both of had lower (010) facet ratios. This trend was corroborated by electron paramagnetic resonance (EPR) measurements, which showed that WO3 NB exhibited the strongest signal of DMPO-˙OH among the three morphologies. The WO3 NB also exhibited stable photocurrent density during 12 h of photoelectrolysis, confirming its promising stability for CH4 conversion. | ||
| Fig. 14 (a) Representation of the atomic structures of the lowest-energy ˙OH adsorption on twinning W atoms of different facets, in which, from left to right, the sequence is facets of (010), (100), and (001). The corresponding adsorption energy is presented at the bottom. (b) EPR analyses of ˙OH in the presence of WO3 NB, WO3 NP or WO3 NF after light illumination (c) PEC CH4-to-EG conversion selectivity at various potential using the photoanode of WO3 NB, WO3 NP or WO3 NF. Reproduced with permission.108 Copyright 2021, John Wiley and Sons. (d) Energy profiles derived from DFT calculation of glycerol oxidation on WO3 at the facets of (200) and (202). (e) The comparison of WO3 in OCP change between facets of (200) and (202) in a supporting electrolyte with subsequent injection of 0.1 M glycerol. (f) FT-IR absorbance spectra of the C–O bond vibration peaks of WO3 photoanodes with predominant facets of (200) and (202) in a Na2SO4 solution with or without glycerol. Reproduced with permission.69 Copyright 2022, American Chemical Society. (g) Schematic representation of the process of synthesising textured and untextured MBVO. (h) The ηsep and ηint (ηtrans) of textured and untextured MBVO. (i) The PEC stability of textured MBVO under glycerol oxidation and OER (inset) conditions. Reproduced with permission.115 Copyright 2024, John Wiley and Sons. | ||
Engineering the WO3 facet for optimising the generation of GLAD has also been reported (Fig. 14d–f).69 The production of GLAD originates from the two-electron oxidation of the primary hydroxyl group of glycerol. A WO3 photoanode with predominant (202) facets, denoted as WO3 (202), exhibited a higher production rate and selectivity for GLAD than WO3 with predominated (200) facets (WO3 (200)). This was attributed to the superior light absorption properties and higher carrier concentration of WO3 with predominated (202) facet. In addition, the DFT calculations revealed that the crystal facet of (202) showed stronger glycerol adsorption and facilitated its activation more effectively than the (200) facet. The (202) facet also exhibited easier desorption of GLAD. The stronger glycerol absorption tendency on WO3 (202) was evidenced by a more pronounced cathodic shift in open-circuit potential (OCP) after the addition of glycerol. Furthermore, Fourier-transform infrared (FT-IR) spectra indicated that, compared to WO3 (200), the C–O bond vibration peaks of the primary and secondary hydroxyl groups shifted to higher and lower wavenumbers, respectively, on WO3 (202). Both OCP and FT-IR studies experimentally demonstrated that WO3 (202) had a higher tendency for PEC GLAD production from glycerol. Notably, WO3 (202) also exhibited excellent photocurrent stability without any decay for 12 h, while the photocurrent density of WO3 (200) remained at approximately 70% of its initial photocurrent density at 1.2 VRHE.
In addition to the crystal facet effect in WO3, Wu et al. investigated the effect of the crystal phase of WO3, including monoclinic (m-WO3) and hexagonal (h-WO3), on HMF oxidation to DFF.68 m-WO3 outperformed h-WO3, attributed to enhanced light absorption, improved charge separation, optimised reactant adsorption and a higher oxidation capacity of the photo-generated holes. Consequently, m-WO3 exhibited a lower onset potential at 0.6 VRHE and a significant increase in photocurrent density, reaching 1.1 mA cm−2 at 1.3 VRHE (a sixfold enhancement over h-WO3). Under constant irradiation at 1.1 VRHE, m-WO3 retained 50% of its initial photocurrent.
Benefiting from the enhancement of reaction rate or selectivity by tuning the preferential crystal orientation has also been demonstrated in the case of BiVO4. Wu et al. synthesized textured Mo-doped BiVO4 (MBVO) with a (001) crystal orientation via a rapid-ramping annealing method and applied it for glycerol oxidation reaction (GOR) (Fig. 14g–i).115 The (001) crystal orientation in MBVO-textured provided a substantially enhanced ηsep of 90% at 1.23 VRHE, exceeding that of randomly oriented untextured MBVO (74%). Accordingly, the textured MBVO exhibited a photocurrent density of 7.45 ± 0.19 mA cm−2 at 1.23 VRHE, corresponding to 99% of the Jmax of BiVO4 (Table 2). Notably, the textured MBVO also showed promising stability of photocurrent density up to 100 h at 0.8 VRHE in GOR, originating from its high ηsep from (001) crystal orientation coupled with optimisation of photo-generated hole utilisation in glycerol oxidation. In another study, the difference in activity and selectivity toward DHA production from PEC glycerol valorisation between facets of (010) and (121) has been explored.114 Their FT-IR results indicated that (010) had a much stronger absorption signal for the C–O bond vibration peak of the secondary hydroxyl group than that of the facet (121). Although the DFT calculation aligned with the FT-IR result, there was only a subtle difference in the adsorption energy between facets of (010) and (121). This might result from the DFT used being the simplest one, without considering the solvent effect.
The above discussion on strategies related to the effect of achieving different levels of improvement on photoelectrode activity, target product selectivity, and PEC stability are summarised in Table 3.
| Activity | Selectivity | Stability | |
|---|---|---|---|
| Doping | ●●○ | ●○○ | ●○○ |
| Heterojunction | ●●○ | ●●○ | ●○○ |
| Co-catalyst modification | ●●○ | ●●● | ●○○ |
| Nanostructuring | ●●● | ●○○ | ○○○ |
| Crystal facet and phase engineering | ●●○ | ●●● | ●○○ |
5. PEC reduction for organic synthesis
PEC organic valorisation typically takes the thermodynamic benefits of organic oxidation to replace the sluggish OER. Therefore, most PEC organic valorisation studies focus on the oxidation process. Some studies still focus on organic reduction, although a cathode, rather than a photocathode, is normally employed in a PEC system. The production of BHMF from HMF using a PEC system consisting of an Ag cathode and a BiVO4 photoanode serves as a representative example.28 BHMF is an essential starting agent in the polymerisation and etherification industries. BHMF is produced under conditions of high pressure (>28 bar) and temperature (>403 K) using H2 as the hydrogen source. On the other hand, the EC reduction of HMF to BHMF can be operated under ambient conditions and utilise water as a hydrogen source, bypassing the need for H2. The authors systematically investigated HMF reduction using a dendritic fractal structured Ag cathode synthesised by galvanic displacement, denoted as Aggd. The Aggd exhibited an FE close to 100% toward BHMF. To reduce energy consumption, a PEC cell containing a BiVO4 photoanode and an Ag cathode was then constructed. Photo-generated holes from BiVO4 were used for OER, while the photoexcited electron was transferred to the Ag cathode to reduce HMF into BHMF (Fig. 15a). This PEC cell not only exhibited a comparable FE (94%) with an EC cell, but it also reduced the energy consumption required from the applied voltage. | ||
| Fig. 15 Schematic of (a) the PEC cell consisting of a BiVO4 photoanode and Ag cathode for HMF reduction into BHMF. Reproduced with permission.28 Copyright 2016, American Chemical Society. (b) Photo-electro-biochemical reactor with a TiO2 photoanode harvesting sunlight and a Co–N/CNT cathode to generate H2O2, which is provided to the biocatalyst for lignin valorisation. Reproduced with permission under the terms of a CC BY 4.0 license.155 Copyright 2019, The Author(s), published by Springer Nature. (c) Inorganic-biological hybrid PEC system with a Si/C photocathode for light harvesting and a LPMO catalysing α-chitin valorisation. (d) Long-term stability of the Si/C photocathode. Reproduced with permission.156 Copyright 2020, Elsevier. | ||
A PEC cell integrated with an enzyme, a biocatalyst, has been recently investigated for the valorisation of lignin and α-chitin (Fig. 15b). Myohwa Ko et al. demonstrated a PEC system consisting of a TiO2 photoanode for photovoltage generation and a Co–N/carbon nanotube (Co–N/CNT) cathode for oxygen reduction reaction.155 The cathodic product H2O2 acted as an electron acceptor for lignin peroxidase isozyme H8, which has high activity toward selective β-O-4 bonds in lignin cleavage. To prevent damage to the enzyme from high concentrations of H2O2, an additional cellulose membrane was used to separate the cathode and enzyme. The enzyme-assisted PEC catalyzed the lignin dimer depolymerisation with 93.7% conversion and 98.7% selectivity for 3,4-dimethoxybenzaldehyde, a vanillin derivative.
Generation of chitin oligosaccharides from α-chitin over a Si/C integrated with lytic polysaccharide monooxygenases enzyme mediator (LPMO) in the presence of a redox mediator is a rare example of using a photocathode for PEC reduction valorisation (Fig. 15c and d).156 The mediator, such as 2,6-dimethyl-1,4-benzoquinone (DMBQ), was first photoelectrochemically reduced by the Si/C photocathode. The reduced DMBQ subsequently provided the reductive power to LPMO, assisted by O2 activation, to convert α-chitin to oligosaccharide. The PEC system can be realised under benign environmental conditions for the sustainable valorisation of α-chitin. However, this system exhibited a photocurrent density decay of more than 60% after 12 h. Improving the stability of this system remains a subject for further investigation.
6. Integrated PEC cell for cogeneration of solar fuel and value-added chemical
This section discusses the integrated PEC systems for simultaneous solar fuel and value-added chemical production. There are many configurations of PEC systems. However, a tandem PEC cell consisting of a cathode and an anode, in which at least one of the electrodes is a photoelectrode, is focused on, as this type of PEC cell has the most concise design and is easily scalable. In addition, a review of a photovoltaic (PV) integrated with a two-electrode PEC system is also included, as this type of design is widely applied to PEC cells that require an external bias to achieve a sufficient production rate.6.1 Photoanode paired with a cathode
The pioneering work of an integrated PEC cell for the cogeneration of value-added chemicals and H2 was demonstrated in 2015.70 The PEC cell consists of a BiVO4 photoanode connected to a Pt cathode in a tandem configuration and was used for FDCA and H2 production. Additionally, TEMPO in the electrolyte is employed as a mediator and catalyst. Under light illumination, photo-generated holes from BiVO4 oxidise TEMPO to TEMPO+, further catalyses HMF conversion into FDCA. Meanwhile, the photoexcited electrons were transferred to the Pt cathode for H2 evolution. The authors also compared the TEMPO oxidation activity at the BiVO4 photoanode under light illumination with that at an Au electrode. Compared to the Au electrode, the bare BiVO4 photoanode exhibited a reduction of 0.7 V in the potential necessary for HMF oxidation due to its sufficient EVB to drive the TEMPO oxidation under light illumination. TEMPO-mediated PEC HMF oxidation resulted in 93% FE of FDCA. A PEC cell consisting of a photoanode and a cathode was also employed for PET valorisation and H2 co-generation. Lin et al. demonstrated an integrated PEC cell consisting of nickel phosphide nanoparticle-modified TiO2 nanorods photoanode (nanoTiO2/nanoNiP) and carbon nanotubes/nickel phosphide nanoparticles nanocomposite (CNT/nanoNiP) cathode for the valorisation of PET lysate (EG) into FA and H2 (Fig. 16a and b), respectively.106 PET bottles were pre-treated by cutting into flakes, followed by alkaline hydrolysis in 2 M KOH at 80 °C for 24 h to get EG. CV activation was beneficial in the surface reconstruction of nanoNiP into β-NiOOH active site of EG oxidation. Furthermore, P in nanoNiP was critical in suppressing further transformation of β-NiOOH into γ-NiOOH. Meanwhile, the pristine nanoNiP was found to be active as an HER catalyst. In the 4-h photoelectrolysis, the PEC cell containing nanoTiO2/nanoNiP photoanode and CNT/nanoNiP cathode exhibited an FE of 57.1% ± 1.7% toward FA and an FE of 76.8% ± 7.8% toward H2 at an external bias of 0.5 V. | ||
| Fig. 16 (a) Schematic of an integrated PEC cell comprising a nanoTiO2/nanoNiP photoanode paired with a CNT/nanoNiP cathode, and the chronoamperometric curve under light illumination of the PEC cell. (b) LSVs of (i) bare nanoTiO2, (ii) nanoTiO2|nanoNi-Pop(CV), (iii) Pt foil, and (iv) CNT/nanoNi-Pop. A solid line was recorded under light illumination in the deaerated PET lysate, whereas a dashed line was obtained in the dark in the deaerated KOH solution. Reproduced with permission under the terms of a CC BY 4.0 license.106 Copyright 2021, The Author(s), published by Elsevier. (c) Schematic representation of a photovoltaic device (Si solar cell) connected to the nanoCuWO4-based PEC cell for simultaneous glycerol valorisation and H2 production. (d) Overlaid J–V curves of the Si solar cell and nanoCuWO4-based PEC cell either for glycerol oxidation (green) or water oxidation (red). Solid and dash lines represent the J–V curves measured in the light and dark, respectively. (e) Amount of H2 generated by the nanoCuWO4-based PV-PEC cell from glycerol valorisation and water splitting. Reproduced with permission.54 Copyright 2024, John Wiley and Sons. | ||
As revealed in the last example, a PEC system might still need an external bias supply, although the required external bias is generally much less than in an EC system. A PV cell is typically further connected and stacked with a PEC cell to form an integrated, standalone PV-PEC system. For instance, a PV-PEC system has been applied to water splitting and glycerol valorisation over a CuWO4 photoanode (Fig. 16c–e).54 The PV-PEC cell exhibited a significantly higher operating photocurrent for glycerol valorisation than for water splitting. Product analyses also confirmed that the production rate of H2 in glycerol valorisation was approximately double that of water splitting. This corresponding finding supports that PEC valorisation can generate more valuable chemicals than water splitting and accelerate the H2 production rate.
6.2 Photocathode paired with an anode
The majority of cathodic reactions, including HER, CO2RR, and NH3 production, have been demonstrated to couple with organic oxidation in the configuration of a photocathode paired with an anode. Bhattacharjee et al. demonstrated a PEC cell to valorise organic waste into valuable chemicals and co-generate H2.157 The PEC cell consisted of a Pt-decorated lead-halide perovskite (perovskite|Pt) photocathode for PEC HER and Cu30Pd70 alloy micro-flowers for oxidation of organic waste such as PET and cellulose (Fig. 17a–e). The PET bottle was first cut and ground into a smaller pieces, followed by alkaline hydrolysis in 1 M KOH at 80 °C for 120 h to obtain monomer EG. Under 10 h bias-free photoelectrolysis, Cu30Pd70‖perovskite|Pt PEC cell, both in two-compartment and “artificial leaf” configurations, exhibited photocurrent density of 4–9 mA cm−2 from valorisation of plastic, biomass, and glycerol. Product selectivity from the oxidation of organic wastes ranged from 60% to 90%, while H2 was co-generated with FEs of exceeding 90% during the valorisation of different organic waste. | ||
| Fig. 17 Illustration of the Cu30Pd70|perovskite|Pt PEC system in (a) the two-compartment and (b) artificial leaf. PEC analyses using the two-compartment system or the artificial leaf in a solution containing pre-treated polymeric and real-world substrates in (c) chopped scans and (d) bias-free chronoamperometry. (e) Amount of oxidation product along with the corresponding selectivity generated from different substrates after 10 h of bias-free PEC reactions using the two-compartment system or the artificial leaf. Reproduced with permission under the terms of a CC BY 4.0 license.157 Copyright 2021, The Author(s), published by John Wiley and Sons. (f) Chopped scan and (g) bias-free chronoamperometry of the Ru@TiNS/Ni/perovskite photocathode in a two-electrode integrated system. (h) SAP, the amount of reduction product produced from under AM 1.5 G simulated one-sun condition per hour over an area of 1 cm2, of NH3 and NO2−, and (i) FE of the oxidation products from glycerol using the two-electrode system under bias-free condition. Ru@TiNS/Ni/perovskite and Pt@TiNS electrocatalysts were used as the photocathode and the anode in the two-electrode system, respectively. Reproduced with permission.159 Copyright 2024, Springer Nature. | ||
CO2RR, coupled with organic waste valorisation, was further demonstrated by the same group using a similar perovskite photocathode-based PEC system. A zero-bias PEC cell consisting of a CO2 reduction catalyst (CO2Rcat)-modified lead-halide perovskite (PVK) photocathode and a Ni foam|Cu27Pd73 microflower anode was used for PEC CO2RR and plastic valorisation.158 Three different CO2Rcat, including cobalt porphyrin (CoPL), Cu91In9 alloy, and tungsten-formate dehydrogenase (FDH) biocatalyst, were incorporated onto the perovskite surface, and their CO2RR performance was investigated. PEC reduction products at the photocathode part were CO (FE of 90%), syngas (CO/H2 = 1/1, FE of 43% (CO) and 49% (H2)), and FA (95%) using PVK|COPL, PVK|Cu91In9, and PVK|FDH photocathodes, respectively. On the other hand, the Cu27Pd73-based anode exhibited high selectivity with an FE exceeding 90% toward GLA from PET plastics. In addition, the PEC cells consisting of Ni foam|Cu27Pd73 anode and either a PVK|Cu91In9 or PVK|FDH photocathode also demonstrated stable photocurrent densities for up to 10 h. Meanwhile, a slight decay in the photocurrent density was observed when PVK|COPL was employed as the photocathode. This system is significant for developing high-value chemicals from the CO2RR platform, as conventional PEC CO2RR is limited by the high thermodynamic barrier associated with the OER.
PEC nitrate reduction (NO3RR) to form ammonia (NH3) has recently attracted considerable attention, as it generates green NH3 from nitrate-contaminated solutions. Furthermore, NH3 is a promising H2 carrier due to its safer and more efficient transportation. Tayyebi et al. recently demonstrated a bias-free PEC cell for co-generating NH3 and glyceric acid from NO3− and glycerol, respectively.159 The PEC cell was composed of a Ru-loaded titanate nanosheet loaded on a Ni-modified Pb-perovskite (Ru@TiNS/Ni/perovskite) photocathode and a Pt-decorated TiNS electrocatalyst, and could be operated bias-free. The bias-free PEC full cell achieved a stable photocurrent density of 21.2 ± 0.7 mA cm−2 over 24 h, which was attributed to the effective Ni coating that acted as a protecting layer over the perovskite photoelectrode, along with strong bonding between the Pt and TiNS at the anode (Fig. 17f and g). The reduction product was NH3, with a high yield of 1744.9 μg NH3 cm−2 h−1, corresponding to an FE of 99.5% ± 0.8%. Meanwhile, the major oxidation products were glyceric acid and lactic acid, which has a combined FE of 98.1% ± 2.4% (Fig. 17h and i).
6.3 Photocathode paired with a photoanode
There is relatively limited research on using two photoelectrodes in a PEC cell. Recently, PEC CO2RR coupled with PEC biomass oxidation for concurrent FA production has been demonstrated by Pan et al. A dual-photoelectrode PEC cell consisting of Bi/GaN/Si and NiOOH/α-Fe2O3 wafers was used for CO2RR and biomass valorisation, respectively (Fig. 18a–c).18 For the single photoelectrode performance in a three-electrode system, the NiOOH/α-Fe2O3 photoanode outperformed other α-Fe2O3-based photoanodes, including α-Fe2O3, CoOOH/α-Fe2O3, and FeOOH/α-Fe2O3, with the FEFA of approximate 97% and stable performance for 10 h. This was due to NiOOH enhancing the hole separation and charge transfer kinetics of α-Fe2O3, as evident from its lowest charge transfer resistance and the smallest calculated Gibbs free energy barrier for converting glucose into FA. The NiOOH/α-Fe2O3 photoanode was also able to oxidise real-world biomass to FA with an FE of over 90% and remain stable for 100 h. Shifting the focus to the cathodic side, the Bi/GaN/Si photocathode exhibited an FE of 85.2% toward FA at −0.2 V and stable for 10 h. The Bi/GaN/Si and NiOOH/α-Fe2O3 were subsequently integrated into a two-electrode tandem configuration. The top electrode of NiOOH/α-Fe2O3 first absorbed the simulated solar light, and the attenuated light was absorbed by the bottom electrode of Bi/GaN/Si. The energy consumption from external voltage was significantly reduced by over 300 mV in the biomass oxidation-CO2RR system compared to the conventional OER-CO2RR system. By integrating commercial crystalline Si solar cells with the tandem PEC cell, the system produced FA at 1044 μmol cm−2 and 817 μmol cm−2 at the photoanode and photocathode, respectively, and worked stably for 80 h. | ||
| Fig. 18 (a) Schematic representation of the two-photoelectrode tandem PEC cell consisting of NiOOH/α-Fe2O3 paired with Bi/GaN/Si for FA production from biomass and CO2. (b) LSV curves of the tandem PEC cell with (BOR) and without (OER) the addition of biomass. (c) Chronoamperometric curve and corresponding productivity and FEs of FA from biomass and CO2 using the tandem PEC cell integrated with a Si solar cell under AM 1.5 G one-sun illumination. Reproduced with permission under the terms of a CC BY 4.0 license.18 Copyright 2023, The Author(s), published by Springer Nature. (d) Schematic of the two-photoelectrode tandem PEC cell for EGOR coupled with HER. PEC cell consists of a BiVO4-based photoanode (Mo:BiVO4/NiCo-LDH) and a Cu2O-based photocathode (Au/Cu2O/Ga2O3/TiO2/RuOx) and can be operated without bias. (e) LSV curves and (f) unbiased stability investigation along with the corresponding FE of FA and H2 of the BiVO4-based photoanode and the Cu2O-based photocathode placed behind the BiVO4-based photoanode under simulated AM 1.5 G illumination in buffer solution containing EG at pH 9.0. Reproduced with permission.111 Copyright 2024, John Wiley and Sons. | ||
A Cu2O photocathode paired with a BiVO4 photoanode in a two-electrode tandem configuration (Cu2O/BiVO4) has been demonstrated as a state-of-the-art PEC system for solar water splitting.160 The two-electrode Cu2O/BiVO4 tandem cell, with the assistance of a co-catalyst, generated a photocurrent of 1 mA cm−2 under one-sun illumination. However, the Cu2O/BiVO4 tandem cell exhibited significant instability for water splitting, losing the majority of its photocurrent within 1 min. Most recently, Kang et al. synthesized and demonstrated BiVO4/NiCo-LDH photoanodes for EG oxidation reaction (EGOR), subsequently coupled with Cu2O photocathode for H2 production (Fig. 18d–f).111 In the photoanode, NiCo-LDH significantly improved the photostability of BiVO4 by accepting the photogenerated holes from BiVO4 and thereby facilitating the formation of Ni3+/Co3+ active sites for EGOR. Furthermore, they also found that Ni(OH)x in NiCo-LDH controlled the selectivity toward EG oxidation into FA, whereas Co(OH)x enhanced the photostability by creating a protective layer and passivating surface defects to promote hole transfer to the electrolyte. The synergistic effect between BiVO4 and NiCo-LDH resulted in a high photocurrent density of 2.3 mA cm−2 at 1.23 VRHE, stable operation up to 200 min, and FEFA above 85% in 0.1 M KOH, which outperformed BiVO4, BiVO4/Ni(OH)x, and BiVO4/Co(OH)x. Additionally, an unassisted PEC device for EGOR and HER by coupling the Mo:BiVO4/NiCo-LDH photoanode with a Cu2O photocathode resulted in a maximum photocurrent density of 2.3 mA cm−2 and stable operation up to 2 h with FEFA of ∼60% and FEH2 of ∼100%. However, degradation began at 4 h, with photocurrent density decreasing to 1.7 mA cm−2 (74% of the initial photocurrent density), reducing FEFA by ∼40%, and finally stabilising at ∼30% after 8.3 h. This decline might be attributed to unfavourable product generation from the peroxidation of EG into CO2.
7. Conclusions and perspectives
PEC organic waste valorisation is an advantageous technique that simultaneously produces value-added chemicals and solar fuels, thereby supporting the goals of a circular economy and sustainable energy systems. This work provides a comprehensive review of the PEC working principle, the properties of state-of-the-art earth-abundant photoelectrodes, the organic waste pre-treatment process, and strategies for enhancing PEC performance. By taking advantage of its intrinsic photovoltage and fixed hole-oxidising ability (determined by the EVB of a photoanode) or electron reduction ability (determined by the ECB of a photocathode), a PEC system potentially offers greater energy savings with improved selectivity control compared to an EC system. Significant progress has been made in tuning the photocurrent and selectivity of photoelectrodes through strategies such as doping, heterojunction formation, co-catalyst modification, nanostructuring, and crystal facet engineering. Each strategy offers specific advantages for improving PEC performance. However, significant challenges remain before PEC organic waste valorisation can become commercially viable. These challenges include, but are not limited to, the need for cost-effective pre-treatment processes, integrated and rational photoelectrode design, the development of selective and compatible co-catalysts, the exploration of novel, earth-abundant photoelectrodes, the investigation of PEC organic reduction pathways, and the development and design of large-scale flow systems. These aspects are discussed in detail in the subsequent text.7.1 Cost-effective pre-treatment process
An alkaline pre-treatment process is commonly reported for PEC valorisation of polymeric waste such as cellulose and plastics. However, alkaline pre-treatment processes are unlikely to be suitable for system scale-up due to their adverse impact on both cost and the environment. Previous analyses indicate that the pre-treatment process dominates the economic viability of a solar-driven organic waste valorisation plant on a pilot scale.132 The alkaline pre-treatment process also limits the selection of photoelectrodes to mostly TiO2 and Fe2O3 for polymeric waste valorisation (Table 2). Biological pre-treatment using enzymes is an alternative strategy to depolymerise polymeric waste under benign conditions. However, the accessibility and scalability of some enzymes remain a challenge. Developing catalysts made of earth-abundant elements that can depolymerise polymeric waste in neutral or near-neutral conditions is a possible strategy for alleviating current limitations in the selection of photoelectrodes for PEC polymeric waste valorisation.7.2 Integrated rational design of photoelectrodes
Optimising the photocurrent and stability of photoelectrodes is necessary to realise high-performance PEC organic waste conversion. The indicator of φPE is suggested to evaluate the intrinsic performance of a photoelectrode, as it only considers the factors of light harvesting and the charge separation ability of a photoelectrode. However, as shown in Table 2, many studies still have a φPE much below 1, especially for Fe2O3, Bi2WO6, TaON, CuWO4, etc. Integrating multiple strategies, such as doping, heterojunction formation, nanostructuring, and crystal facet modulation, might further enhance the φPE of these photoelectrodes. Mo:BiVO4(001) serves as an inspiring example of integrating multiple strategies of doping, nanostructuring, and crystal facet modulation, which achieves a φPE of close to 1.115 Persistent fundamental investigation of the material properties and electronic structure is also particularly essential for those photoelectrode with low φPE. For example, the kinetic study using operando spectroelectrochemical PIA coupled with TPC provides a comprehensive understanding of why α-Fe2O3 exhibits selective aldehyde production from PEC alcohol oxidation. However, such relative studies remain limited in PEC organic valorisation using other photoelectrodes and substrates. In addition to thermodynamic research, kinetic studies are also undeniably necessary for the development of efficient and selective photoelectrodes for organic waste valorisation.On the other hand, stability is a crucial parameter for assessing the performance and practicality of a PEC system. Nonetheless, stability continues to be neglected in PEC organic waste valorisation, although 100 h operation has been demonstrated in the studies of Mo:BiVO4(001) for PEC glycerol valorisation115 and NiOOH/α-Fe2O3 for sawdust-derived sugar solution.18 Strategies such as doping and heterojunction formation, widely applied in the current PEC valorisation studies, offer only modest enhancements to stability (Table 3). Other strategies to improve the stability of PEC systems, such as surface modification with a stable passivation layer, are suggested to be emphasised in future research. However, several requirements must be met to design a stable and efficient passivation layer for a photoelectrode. For example, the passivation layer should be thin enough to avoid charge transfer inhibition and parasitic light absorption,161 and the passivation process must be compatible with the photoelectrode.
7.3 Selective and compatible co-catalyst development
Most PEC organic waste valorisation involves an inner-sphere electron transfer process, and the interaction between the electrode interface and the reactant/product determines the reaction kinetics and selectivity. As reviewed in this work, co-catalyst modification is a widely used approach to enhance the selectivity of the target product. However, for PEC polymeric waste valorisation, the co-catalyst was mainly limited to enhancing the FE of the C1 product, i.e., FA. As an illustration, Ni-Pi in Ni-Pi/α-Fe2O3 enhances the FE of FA to 87% for PEC valorisation of PET hydrolysate,9 and NiOOH in NiOOH/α-Fe2O3 enhances the FE of FA to 90% for PEC valorisation of sawdust-derived sugar.18 On the other hand, the co-catalysts for enhancing the FE of C2+ are significantly lacking, which restricts the development of PEC organic valorisation from an economic value perspective. It is known that many C2+ products have a higher economic value than FA. For example, GLA has a market value five times higher than that of FA.9 Although research on the production of C2+ compounds from EC organic waste valorisation is abundant,162,163 most of the electrocatalyst synthetic processes are not readily deposited on or compatible with photoelectrode deposition. Research into electrocatalysts for C2+ products from organic waste, which can be readily applied to a photoelectrode, would help alleviate the constraints imposed by the limited selection of target products in PEC polymeric waste valorisation.7.4 Novel earth-abundant photoelectrodes exploration
The reported research on PEC organic waste valorisation is mainly limited to using state-of-the-art photoanodes (TiO2, WO3, BiVO4, and Fe2O3). These photoanodes have an EVB at >2.5 eV, providing sufficient driving force even for water oxidation. However, the strong oxidative power of the photo-generated holes from these photoanodes may result in parasitic OER and CO2 formation during the PEC oxidation of organics. A wide bandgap also limits the utilisation of higher wavelength photons in the solar spectrum. Since organic oxidation requires much less oxidative force than water oxidation, a narrower bandgap helps maximise the utilisation of solar light. Exploring other novel earth-abundant semiconductors with narrower bandgaps would significantly impact the progress of PEC organic waste valorisation. Recently, lead halide-based perovskites have attracted immense attention in this area due to their narrow and tunable bandgaps and high light absorption coefficients. Nonetheless, research into robust and lead-free perovskite remains a fundamental challenge in using perovskite-based photoelectrodes for PEC reactions. On the other hand, CuWO4 has recently been demonstrated to utilise wavelengths up to 550 nm to perform selective and stable PEC biomass valorisation.54 However, exploring and investigating other earth-abundant and robust photoelectrodes that can utilise broader visible light or near-infrared light to perform organic valorisation still requires considerable future effort.7.5 PEC organic reduction investigation
Fabricating an integrated PEC cell that generates value-added chemicals or fuels at both half-cells is crucial for fully leveraging incident solar energy. Several breakthroughs have been achieved by pairing organic oxidation with a reduction reaction of HER, CO2RR, or NO3RR,111,157–159 in which HER is the most prevalent paired reduction reaction. Unfortunately, many state-of-the-art photocathodes, such as Cu2O, undergo severe photocorrosion during HER. Albeit passivating a protective layer partially alleviates the instability issue, the passivation layer often requires arduous or non-scalable procedures, such as atomic layer deposition. An alternative is to replace HER with another reduction reaction with a more positive redox potential than (E(H+/H2)) and Ered of the photocathode. In particular, given the greater energy savings, some electroreduction reactions of organics, such as the hydrogenation of biomass derivatives, are more suitable for pairing with organic oxidation reactions.164,165 Although paired organic reduction and oxidation have been recently highlighted in EC systems,166–168 the relative research on a PEC system is still absent. Research into PEC organic reduction may offer a sought-after solution for photocathodes that easily undergo photocorrosion during HER, with the potential for greater energy savings.7.6 Large-scale flow system development and design
Large-scale development is critical for PEC devices to harvest solar light over a wide area due to the decentralised property of solar light.169 Therefore, investigating large-scale PEC or PC cells has drawn increasing attention to water splitting.170–172 An illuminated area of a minimum of 50 cm2 is typically considered the first phase of large-scale applications.173,174 However, most of the PEC organic waste valorisation research is still limited to laboratory-scale investigations using small areas of photoelectrodes. Limitations at the laboratory scale are generally attributed to arduous synthetic procedures, manufacturing equipment, and the electronic resistance of electrode substrates. Integrating module units of smaller dimensions to achieve large-scale production is an alternative approach. However, regardless of the design, the need for photoelectrode scale-up must be considered in material selection, engineering, and manufacturing.In contrast to water splitting, which has an endless supply of substrate, i.e., water, the amount of substrate is limited by the solubility of organic waste in water for organic valorisation reaction. The effects of reactant consumption and mass transfer on product selectivity in organics valorisation must be carefully considered. Current research mainly performs the PEC valorisation reaction in a batch system. However, continuous reactant consumption and product accumulation in a batch system result in the unfavourable overoxidation of the target products. Examples can be easily found in the reported research.6,54 In addition, product accumulation might lead to altered solution pH and, thereby, affect product selectivity. Shifting the batch design to a flow reactor powered by a peristaltic pump, which can provide a continuous reactant supply, is necessary to the maintain stable production of the target product. Alternatively, the microfluidic effect could be applied to the reactor design to enhance mass transfer and, thereby, improve selectivity. As shown in Fig. 3, other system parameters beyond photoelectrodes, such as substrate concentration, solution pH, temperature, applied potential, and substrate absorption model, also contribute to the performance of a PEC organic valorisation reaction. Research into the optimisation and investigation of its mechanism is also essential to maximise the efficiency of a PEC system. Additionally, a techno-economic analysis helps assess the technical feasibility of PEC organic waste valorisation.
In conclusion, PEC valorisation of organic waste could be a promising route for not only waste mitigation but also the cogeneration of value-added chemicals and solar fuels. However, research into cost-effective pretreatment processes and the maximisation of the operational efficiency of robust PEC systems in a manner that allows for scalability is required for a commercially viable technology. To meet this demand, research directions are suggested to focus on investigating cost-effective catalysts for polymer waste depolymerisation in neutral or near-neutral conditions, integrating rational design strategies for the existing state-of-the-art photoelectrodes, developing compatible co-catalysts that can target C2+ products, and exploring novel photoelectrodes that meet high PEC performance and stability requirements. Additionally, integrating PEC oxidation with PEC reduction or cathodic reaction for value-added chemical production and advanced PEC reactor design could facilitate a circular carbon economy while addressing the energy crisis and environmental pollution.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Fitri Nur Indah Sari: conceptualisation, investigation, writing – original draft (lead), review and editing. Ping-Chang Chuang: writing – original draft (supporting). Shih-Ching Huang: writing – original draft (supporting). Chia-Yu Lin: supervision and funding acquisition. Yi-Hsuan Lai: conceptualisation, investigation, writing – original draft (lead), review and editing, supervision and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Science and Technology Council of Taiwan (NSTC 111-2221-E-006-019-MY3 and NSTC 113-2221-E-006-085-MY3) is gratefully acknowledged. The authors also acknowledge the support from the Hierarchical Green-Energy Materials (Hi-GEM) Research Center at National Cheng Kung University by the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education in Taiwan.References
- S. Bhattacharjee, S. Linley and E. Reisner, Nat. Rev. Chem., 2024, 8, 87–105 CrossRef PubMed.
- K. de Kleijne, M. A. J. Huijbregts, F. Knobloch, R. van Zelm, J. P. Hilbers, H. de Coninck and S. V. Hanssen, Nat. Energy, 2024, 9, 1139–1152 CrossRef CAS.
- H. Song, S. Luo, H. Huang, B. Deng and J. Ye, ACS Energy Lett., 2022, 7, 1043–1065 CrossRef CAS.
- P. Stegmann, V. Daioglou, M. Londo, D. P. van Vuuren and M. Junginger, Nature, 2022, 612, 272–276 CrossRef CAS PubMed.
- S. Kaza, L. Yao, P. Bhada-Tata and F. Van Woerden, What a Waste 2.0: A Global Snapshot of Solid Waste Management to 2050, World Bank Publications, 2018 Search PubMed.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- Y.-H. Lai, P.-W. Yeh, M.-J. Jhong and P.-C. Chuang, Chem. Eng. J., 2023, 475, 146413 CrossRef CAS.
- T.-K. Liu, G. Y. Jang, S. Kim, K. Zhang, X. Zheng and J. H. Park, Small Methods, 2024, 8, 2300315 CrossRef CAS.
- B. Zhang, H. Zhang, Y. Pan, J. Shao, X. Wang, Y. Jiang, X. Xu and S. Chu, Chem. Eng. J., 2023, 462, 142247 CrossRef CAS.
- T.-R. Ko, C.-Y. Lin and Y.-H. Lai, Chem. Eng. J., 2022, 433, 133607 CrossRef CAS.
- Q. Shi and H. Duan, Chem Catal., 2022, 2, 3471–3496 CAS.
- Y. Liu, C. W. S. Yeung and E. Reisner, Energy Environ. Sci., 2025 10.1039/D5EE00689A.
- D. Sajwan, A. Sharma, M. Sharma and V. Krishnan, ACS Catal., 2024, 14, 4865–4926 CrossRef CAS.
- Alibaba product search: formic acid 85%, category: organic acid, min order: 1 metric ton, https://www.alibaba.com/products/formic_acid_85%2525.html, accessed April, 2025.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- Y. Pan, H. Zhang, B. Zhang, F. Gong, J. Feng, H. Huang, S. Vanka, R. Fan, Q. Cao, M. Shen, Z. Li, Z. Zou, R. Xiao and S. Chu, Nat. Commun., 2023, 14, 1013 CrossRef CAS.
- S. Chen and L.-W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- M. Zhang, J. Wang, H. Xue, J. Zhang, S. Peng, X. Han, Y. Deng and W. Hu, Angew Chem. Int. Ed. Engl., 2020, 59, 18463–18467 CrossRef CAS PubMed.
- L. Pan, J. H. Kim, M. T. Mayer, M.-K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412–420 CrossRef CAS.
- L. Pan, Y. Liu, L. Yao, R. Dan, K. Sivula, M. Grätzel and A. Hagfeldt, Nat. Commun., 2020, 11, 318 CrossRef CAS PubMed.
- S. P. Berglund, F. F. Abdi, P. Bogdanoff, A. Chemseddine, D. Friedrich and R. van de Krol, Chem. Mater., 2016, 28, 4231–4242 CrossRef CAS.
- L. Qu, R. Tan, A. Sivanantham, M. J. Kang, Y. J. Jeong, D. H. Seo, S. Kim and I. S. Cho, J. Energy Chem., 2022, 71, 201–209 CrossRef CAS.
- C. Li, J. He, Y. Xiao, Y. Li and J.-J. Delaunay, Energy Environ. Sci., 2020, 13, 3269–3306 RSC.
- Y. J. Jang, Y. B. Park, H. E. Kim, Y. H. Choi, S. H. Choi and J. S. Lee, Chem. Mater., 2016, 28, 6054–6061 CrossRef CAS.
- J. J. Roylance, T. W. Kim and K.-S. Choi, ACS Catal., 2016, 6, 1840–1847 CrossRef CAS.
- C. Y. Toe, C. Tsounis, J. Zhang, H. Masood, D. Gunawan, J. Scott and R. Amal, Energy Environ. Sci., 2021, 14, 1140–1175 RSC.
- M. Ashraf, N. Ullah, I. Khan, W. Tremel, S. Ahmad and M. N. Tahir, Chem. Rev., 2023, 123, 4443–4509 CrossRef CAS.
- M. Han, S. Zhu, C. Xia and B. Yang, Appl. Catal., B, 2022, 316, 121662 CrossRef CAS.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS.
- X. Yan, K. Sakai and H. Ozawa, ACS Catal., 2023, 13, 13456–13465 CrossRef CAS.
- T. Zhou, J. Wang, S. Chen, J. Bai, J. Li, Y. Zhang, L. Li, L. Xia, M. Rahim, Q. Xu and B. Zhou, Appl. Catal., B, 2020, 267, 118599 CrossRef CAS.
- L. Li, H. Shi, H. Yu, X. Tan, Y. Wang, S. Ge, A. Wang, K. Cui, L. Zhang and J. Yu, Appl. Catal., B, 2021, 292, 120184 CrossRef CAS.
- S. Cao, X. Yan, Z. Kang, Q. Liang, X. Liao and Y. Zhang, Nano Energy, 2016, 24, 25–31 CrossRef CAS.
- Y. Pihosh, I. Turkevych, K. Mawatari, T. Asai, T. Hisatomi, J. Uemura, M. Tosa, K. Shimamura, J. Kubota, K. Domen and T. Kitamori, Small, 2014, 10, 3692–3699 CrossRef CAS PubMed.
- J. Liu, J. Li, Y. Li, J. Guo, S.-M. Xu, R. Zhang and M. Shao, Appl. Catal., B, 2020, 278, 119268 CrossRef CAS.
- Y. Li, Q. Mei, Z. Liu, X. Hu, Z. Zhou, J. Huang, B. Bai, H. Liu, F. Ding and Q. Wang, Appl. Catal., B, 2022, 304, 120995 CrossRef CAS.
- M. Ma, K. Zhang, P. Li, M. S. Jung, M. J. Jeong and J. H. Park, Angew Chem. Int. Ed. Engl., 2016, 55, 11819–11823 CrossRef CAS PubMed.
- M. B. Costa, M. A. de Araújo, M. V. d. L. Tinoco, J. F. d. Brito and L. H. Mascaro, J. Energy Chem., 2022, 73, 88–113 CrossRef CAS.
- C. Loka, D. Gelija, S. V. P. Vattikuti and K.-S. Lee, ACS Sustain. Chem. Eng., 2023, 11, 11978–11990 CrossRef CAS.
- Y. Li, Q. Wu, Y. Chen, R. Zhang, C. Li, K. Zhang, M. Li, Y. Lin, D. Wang, X. Zou and T. Xie, Appl. Catal., B, 2021, 290, 120058 CrossRef CAS.
- Z.-Y. Wang, H.-M. Li, S.-S. Yi, M.-Z. You, H.-J. Jing, X.-Z. Yue, Z.-T. Zhang and D.-L. Chen, Appl. Catal., B, 2021, 297, 120406 CrossRef CAS.
- L. Liccardo, E. Lushaj, L. Dal Compare, E. Moretti and A. Vomiero, Small Sci., 2022, 2, 2100104 CrossRef CAS.
- J. Schneidewind, Adv. Energy Mater., 2022, 12, 2200342 CrossRef CAS.
- M. E. Aguirre, R. Zhou, A. J. Eugene, M. I. Guzman and M. A. Grela, Appl. Catal., B, 2017, 217, 485–493 CrossRef CAS.
- C.-Y. Lin, Y.-H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482–3487 RSC.
- D. S. Kim, S. Y. Oh, H. H. Lee and H. K. Cho, Adv. Energy Mater., 2024, 14, 2304239 CrossRef CAS.
- Y. Liu, H. Tan, Y. Wei, M. Liu, J. Hong, W. Gao, S. Zhao, S. Zhang and S. Guo, ACS Nano, 2023, 17, 5994–6001 CrossRef CAS PubMed.
- R. S. Lillard, G. S. Kanner and D. P. Butt, J. Electrochem. Soc., 1998, 145, 2718 CrossRef CAS.
- Y.-H. Lai, Y.-J. Lai, C.-Y. Yen and P.-C. Chuang, Sustainable Energy Fuels, 2020, 4, 5005–5008 RSC.
- P.-C. Chuang, C.-Y. Lin, S.-T. Ye and Y.-H. Lai, Small, 2024, 20, 2404478 CrossRef CAS PubMed.
- Y.-H. Wu, D. A. Kuznetsov, N. C. Pflug, A. Fedorov and C. R. Müller, J. Mater. Chem. A, 2021, 9, 6252–6260 RSC.
- L. Luo, W. Chen, S.-M. Xu, J. Yang, M. Li, H. Zhou, M. Xu, M. Shao, X. Kong, Z. Li and H. Duan, J. Am. Chem. Soc., 2022, 144, 7720–7730 CrossRef CAS PubMed.
- W. Septina and S. D. Tilley, Curr. Opin. Electrochem., 2017, 2, 120–127 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS PubMed.
- K. M. P. Wheelhouse, R. L. Webster and G. L. Beutner, Org. Process Res. Dev., 2023, 27, 1157–1159 CrossRef CAS.
- R. M. Bullock, Science, 2013, 342, 1054–1055 CrossRef CAS PubMed.
- G. Gunn, in Critical Metals Handbook, 2014, pp. 284–311, DOI:10.1002/9781118755341.ch12.
- Q. Shi, J. Li, Y. Liu, K. Kong, A.-Z. Li and H. Duan, ACS Catal., 2024, 14, 10728–10736 CrossRef CAS.
- N. Le Duy, P.-C. Chuang, C.-Y. Lin and Y.-H. Lai, J. Photochem. Photobiol., A, 2025, 458, 115932 CrossRef CAS.
- H. Tateno, S. Iguchi, Y. Miseki and K. Sayama, Angew. Chem., Int. Ed., 2018, 57, 11238–11241 CrossRef CAS PubMed.
- J. Yu, J. González-Cobos, F. Dappozze, F. J. López-Tenllado, J. Hidalgo-Carrillo, A. Marinas, P. Vernoux, A. Caravaca and C. Guillard, Appl. Catal., B, 2022, 318, 121843 CrossRef CAS.
- C. R. Lhermitte, N. Plainpan, P. Canjura, F. Boudoire and K. Sivula, RSC Adv., 2021, 11, 198–202 RSC.
- C.-C. Wu, T.-G. Vo, M. B. Sullivan, K. P. Ong, H. Jin, A. Chuang, M.-T. Huynh Pham and C.-Y. Chiang, Inorg. Chem., 2025, 64, 1579–1586 CrossRef CAS.
- J. Ouyang, X. Liu, B.-H. Wang, J.-B. Pan, S. Shen, L. Chen, C.-T. Au and S.-F. Yin, ACS Appl. Mater. Interfaces, 2022, 14, 23536–23545 CrossRef CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- D. Wang, S. H. Lee, S. Han, J. Kim, N. V. T. Trang, K. Kim, E.-G. Choi, P. Boonmongkolras, Y. W. Lee, B. Shin, Y. H. Kim and C. B. Park, Green Chem., 2020, 22, 5151–5160 RSC.
- T. Li, J. Y. Mo, D. M. Weekes, K. E. Dettelbach, R. P. Jansonius, G. M. Sammis and C. P. Berlinguette, ChemSusChem, 2020, 13, 3622–3626 CrossRef CAS PubMed.
- S. Li, F. Chen, T. Ma and H. Huang, Chem. Eng. J., 2023, 467, 143421 CrossRef CAS.
- C. Lin, C. Dong, S. Kim, Y. Lu, Y. Wang, Z. Yu, Y. Gu, Z. Gu, D. K. Lee, K. Zhang and J. H. Park, Adv. Mater., 2023, 35, 2209955 CrossRef CAS.
- D. Liu, J.-C. Liu, W. Cai, J. Ma, H. B. Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1779 CrossRef.
- L.-W. Huang, T.-G. Vo and C.-Y. Chiang, Electrochim. Acta, 2019, 322, 134725 CrossRef CAS.
- I. Grigioni, A. Polo, C. Nomellini, L. Vigni, A. Poma, M. V. Dozzi and E. Selli, ACS Appl. Energy Mater., 2023, 6, 10020–10029 CrossRef CAS.
- C. M. Tian, M. Jiang, D. Tang, L. Qiao, H. Y. Xiao, F. E. Oropeza, J. P. Hofmann, E. J. M. Hensen, A. Tadich, W. Li, D. C. Qi and K. H. L. Zhang, J. Mater. Chem. A, 2019, 7, 11895–11907 RSC.
- C. R. Lhermitte and B. M. Bartlett, Acc. Chem. Res., 2016, 49, 1121–1129 CrossRef CAS PubMed.
- L. Wang, X. Zhou, N. T. Nguyen, I. Hwang and P. Schmuki, Adv. Mater., 2016, 28, 2432–2438 CrossRef CAS PubMed.
- Y. Li, T. Takata, D. Cha, K. Takanabe, T. Minegishi, J. Kubota and K. Domen, Adv. Mater., 2013, 25, 125–131 CrossRef CAS PubMed.
- C. Feng, Z. Liu, H. Ju, A. Mavrič, M. Valant, J. Fu, B. Zhang and Y. Li, Nat. Commun., 2024, 15, 6436 CrossRef CAS.
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS PubMed.
- J. Kegel, I. M. Povey and M. E. Pemble, Nano Energy, 2018, 54, 409–428 CrossRef CAS.
- Y.-H. Lai, K.-C. Lin, C.-Y. Yen and B.-J. Jiang, Faraday Discuss., 2019, 215, 297–312 RSC.
- C.-Y. Lin, S.-Y. Lin, M.-C. Tsai and C.-H. Wu, Sustainable Energy Fuels, 2020, 4, 625–632 RSC.
- O. C. Olatunde, W. Lei, H. Ferjani and D. C. Onwudiwe, Mater. Today Sustain., 2024, 26, 100747 Search PubMed.
- J. E. Yourey and B. M. Bartlett, J. Mater. Chem. A, 2011, 21, 7651–7660 RSC.
- J. E. Yourey, K. J. Pyper, J. B. Kurtz and B. M. Bartlett, J. Phys. Chem. C, 2013, 117, 8708–8718 CrossRef CAS.
- R. H. Coridan, A. C. Nielander, S. A. Francis, M. T. McDowell, V. Dix, S. M. Chatman and N. S. Lewis, Energy Environ. Sci., 2015, 8, 2886–2901 RSC.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- F. Kang, Q. Wang, D. Du, L. Wu, D. W. F. Cheung and J. Luo, Angew. Chem., Int. Ed., 2025, 64, e202417648 CrossRef CAS.
- I. Holmes-Gentle, F. E. Bedoya-Lora, L. Aimone and S. Haussener, J. Mater. Chem. A, 2023, 11, 23895–23908 RSC.
- C. Pornrungroj, V. Andrei and E. Reisner, J. Am. Chem. Soc., 2023, 145, 13709–13714 CrossRef CAS PubMed.
- Á. Balog, E. Kecsenovity, G. F. Samu, J. He, D. Fekete and C. Janáky, Nat. Catal., 2024, 7, 522–535 CrossRef.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2015, 137, 6629–6637 CrossRef CAS PubMed.
- Y. Zhang, H. Zhang, A. Liu, C. Chen, W. Song and J. Zhao, J. Am. Chem. Soc., 2018, 140, 3264–3269 CrossRef CAS PubMed.
- C. A. Mesa, L. Francàs, K. R. Yang, P. Garrido-Barros, E. Pastor, Y. Ma, A. Kafizas, T. E. Rosser, M. T. Mayer, E. Reisner, M. Grätzel, V. S. Batista and J. R. Durrant, Nat. Chem., 2020, 12, 82–89 CrossRef CAS.
- C. A. Mesa, A. Kafizas, L. Francàs, S. R. Pendlebury, E. Pastor, Y. Ma, F. Le Formal, M. T. Mayer, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 11537–11543 CrossRef CAS PubMed.
- J. Yuan, Y. Yuan, J. Zhang, H. Xu, Z. Mao and Y. Ma, ChemSusChem, 2022, 15, e202102313 CrossRef CAS PubMed.
- Z. Ma, X. Guo, Y. Yuan, Y. Wu, Y. Hai, J. Wu, W. Wu and Y. Ma, J. Catal., 2024, 437, 115661 CrossRef CAS.
- M. S. Martí Molera, C. Fàbrega and T. Andreu, J. Electrochem. Soc., 2024, 171, 086503 CrossRef.
- Y. Kageshima, H. Wada, K. Teshima and H. Nishikiori, Appl. Catal., B, 2023, 327, 122431 CrossRef CAS.
- Y. Lu, T.-K. Liu, C. Lin, K. H. Kim, E. Kim, Y. Yang, X. Fan, K. Zhang and J. H. Park, Nano Lett., 2024, 24, 4633–4640 CrossRef CAS PubMed.
- Y. Liu, M. Wang, B. Zhang, D. Yan and X. Xiang, ACS Catal., 2022, 12, 6946–6957 CrossRef CAS.
- C.-Y. Lin, S.-C. Huang, Y.-G. Lin, L.-C. Hsu and C.-T. Yi, Appl. Catal., B, 2021, 296, 120351 CrossRef CAS.
- K. Jakubow-Piotrowska, B. Witkowski and J. Augustynski, Commun. Chem., 2022, 5, 125 CrossRef CAS PubMed.
- J. Ma, K. Mao, J. Low, Z. Wang, D. Xi, W. Zhang, H. Ju, Z. Qi, R. Long, X. Wu, L. Song and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 9357–9361 CrossRef CAS PubMed.
- Z. Gu, X. An, R. Liu, L. Xiong, J. Tang, C. Hu, H. Liu and J. Qu, Appl. Catal., B, 2021, 282, 119541 CrossRef CAS.
- F. Kong, H. Zhou, Z. Chen, Z. Dou and M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202210745 CrossRef CAS PubMed.
- F. Kang, Q. Wang, D. Du, L. Wu, D. W. F. Cheung and J. Luo, Angew Chem. Int. Ed. Engl., 2025, 64, e202417648 CrossRef CAS PubMed.
- H. Tateno, S.-Y. Chen, Y. Miseki, T. Nakajima, T. Mochizuki and K. Sayama, ACS Sustain. Chem. Eng., 2022, 10, 7586–7594 CrossRef CAS.
- Y. Lu, B. G. Lee, C. Lin, T.-K. Liu, Z. Wang, J. Miao, S. H. Oh, K. C. Kim, K. Zhang and J. H. Park, Nat. Commun., 2024, 15, 5475 CrossRef CAS PubMed.
- T.-G. Vo, C.-C. Kao, J.-L. Kuo, C.-c. Chiu and C.-Y. Chiang, Appl. Catal., B, 2020, 278, 119303 CrossRef CAS.
- Z. Wu, K. Ren, J. Zhou, Y. Zhang and L. Qi, Adv. Funct. Mater., 2025, 35, 2418294 CrossRef CAS.
- M. Tayebi, Z. Masoumi, A. Tayyebi, J.-H. Kim, H. Lee, B. Seo, C.-S. Lim and H.-G. Kim, ACS Appl. Mater. Interfaces, 2023, 15, 20053–20063 CrossRef CAS.
- S. Kim, D. Oh and J.-W. Jang, Nano Lett., 2024, 24, 5146–5153 CrossRef CAS.
- X. Li, J. Wang, T. Zhang, T. Wang and Y. Zhao, ACS Sustain. Chem. Eng., 2022, 10, 9546–9552 CrossRef CAS.
- P.-C. Chuang and Y.-H. Lai, Catal. Sci. Technol., 2022, 12, 6375–6383 RSC.
- H. Zhao, X. Zhao, J. Zhang, S. Anandita, W. Liu, S. W. Koh, S. Yu, C. Li, Z. Chen, R. Xu, Z. Zou, W. Tu and H. Li, Adv. Energy Mater., 2024, 14, 2400037 CrossRef CAS.
- L. L. Nascimento, J. Z. Marinho, A. L. R. dos Santos, A. M. de Faria, R. A. C. Souza, C. Wang and A. O. T. Patrocinio, Appl. Catal., A, 2022, 646, 118867 CrossRef CAS.
- Q. Wang, X. Ma, P. Wu, B. Li, L. Zhang and J. Shi, Nano Energy, 2021, 89, 106326 CrossRef CAS.
- F. W. S. Lucas, R. G. Grim, S. A. Tacey, C. A. Downes, J. Hasse, A. M. Roman, C. A. Farberow, J. A. Schaidle and A. Holewinski, ACS Energy Lett., 2021, 6, 1205–1270 CrossRef CAS.
- E. J. Cho, L. T. P. Trinh, Y. Song, Y. G. Lee and H.-J. Bae, Bioresour. Technol., 2020, 298, 122386 CrossRef CAS PubMed.
- A. Yousuf, Waste Manage., 2012, 32, 2061–2067 CrossRef CAS PubMed.
- S. Paul and A. Dutta, Resour., Conserv. Recycl., 2018, 130, 164–174 CrossRef.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- S.-J. Jung, S.-H. Kim and I.-M. Chung, Biomass Bioenergy, 2015, 83, 322–327 CrossRef CAS.
- O. Lanzalunga and M. Bietti, J. Photochem. Photobiol., B, 2000, 56, 85–108 CrossRef CAS PubMed.
- https://plasticseurope.org/knowledge-hub/plastics-the-facts-2022-2/, 2022.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- T. Uekert, C. M. Pichler, T. Schubert and E. Reisner, Nat. Sustain., 2021, 4, 383–391 CrossRef.
- A. W. Bhutto, K. Qureshi, K. Harijan, R. Abro, T. Abbas, A. A. Bazmi, S. Karim and G. Yu, Energy, 2017, 122, 724–745 CrossRef CAS.
- Y. You, S. Chen, J. Zhao, J. Lin, D. Wen, P. Sha, L. Li, D. Bu and S. Huang, Adv. Mater., 2024, 36, 2307962 CrossRef CAS PubMed.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- M. Du, Y. Zhang, S. Kang, X. Guo, Y. Ma, M. Xing, Y. Zhu, Y. Chai and B. Qiu, ACS Catal., 2022, 12, 12823–12832 CrossRef CAS.
- M. Li and S. Zhang, ACS Catal., 2024, 14, 2949–2958 CrossRef CAS.
- E. Lam and E. Reisner, Angew Chem. Int. Ed. Engl., 2021, 60, 23306–23312 CrossRef CAS.
- S. Linley and E. Reisner, Adv. Sci., 2023, 10, 2207314 CrossRef CAS PubMed.
- E. Lam, M. Miller, S. Linley, R. R. Manuel, I. A. C. Pereira and E. Reisner, Angew Chem. Int. Ed. Engl., 2023, 62, e202215894 CrossRef CAS PubMed.
- S. Bhattacharjee, C. Guo, E. Lam, J. M. Holstein, M. Rangel Pereira, C. M. Pichler, C. Pornrungroj, M. Rahaman, T. Uekert, F. Hollfelder and E. Reisner, J. Am. Chem. Soc., 2023, 145, 20355–20364 CrossRef CAS.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS PubMed.
- M. Z. Rahman, T. Edvinsson and J. Gascon, Nat. Rev. Chem., 2022, 6, 243–258 CrossRef PubMed.
- J. Wang, X. Li, T. Zhang, X. Qian, T. Wang and Y. Zhao, Appl. Catal., B, 2023, 332, 122744 CrossRef CAS.
- F. Amano, M. Nakata, A. Yamamoto and T. Tanaka, J. Phys. Chem. C, 2016, 120, 6467–6474 CrossRef CAS.
- J. Kim, J. Jang, T. Hilberath, F. Hollmann and C. B. Park, Nat. Synth., 2022, 1, 776–786 CrossRef CAS.
- H. Zhao, C.-F. Li, L.-Y. Liu, B. Palma, Z.-Y. Hu, S. Renneckar, S. Larter, Y. Li, M. G. Kibria, J. Hu and B.-L. Su, J. Colloid Interface Sci., 2021, 585, 694–704 CrossRef CAS PubMed.
- S. Guo, Y. Huang, D. Li, Z. Xie, Y. Jia, X. Wu, D. Xu and W. Shi, Chem. Commun., 2023, 59, 7791–7794 RSC.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef.
- S. Li, W. Xu, L. Meng, W. Tian and L. Li, Small Sci., 2022, 2, 2100112 CrossRef CAS PubMed.
- H. Zhou, Z. Li, L. Ma and H. Duan, Chem. Commun., 2022, 58, 897–907 RSC.
- A. J. Bard, J. Am. Chem. Soc., 2010, 132, 7559–7567 CrossRef CAS PubMed.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- S.-C. Huang, C.-C. Cheng, Y.-H. Lai and C.-Y. Lin, Chem. Eng. J., 2020, 395, 125176 CrossRef CAS.
- M. Ko, L. T. M. Pham, Y. J. Sa, J. Woo, T. V. T. Nguyen, J. H. Kim, D. Oh, P. Sharma, J. Ryu, T. J. Shin, S. H. Joo, Y. H. Kim and J.-W. Jang, Nat. Commun., 2019, 10, 5123 CrossRef PubMed.
- H. Zhang, Y. Zhao, H. Zhang, H. Zhou, H. Wang, X. Zong, H. Yin and C. Li, Appl. Catal., B, 2020, 265, 118558 CrossRef CAS.
- S. Bhattacharjee, V. Andrei, C. Pornrungroj, M. Rahaman, C. M. Pichler and E. Reisner, Adv. Funct. Mater., 2022, 32, 2109313 CrossRef CAS.
- S. Bhattacharjee, M. Rahaman, V. Andrei, M. Miller, S. Rodríguez-Jiménez, E. Lam, C. Pornrungroj and E. Reisner, Nat. Synth., 2023, 2, 182–192 CrossRef CAS.
- A. Tayyebi, R. Mehrotra, M. A. Mubarok, J. Kim, M. Zafari, M. Tayebi, D. Oh, S.-h. Lee, J. E. Matthews, S.-W. Lee, T. J. Shin, G. Lee, T. F. Jaramillo, S.-Y. Jang and J.-W. Jang, Nat. Catal., 2024, 7, 510–521 CrossRef CAS.
- P. Bornoz, F. F. Abdi, S. D. Tilley, B. Dam, R. van de Krol, M. Graetzel and K. Sivula, J. Phys. Chem. C, 2014, 118, 16959–16966 CrossRef CAS.
- R. Liu, Z. Zheng, J. Spurgeon and X. Yang, Energy Environ. Sci., 2014, 7, 2504–2517 RSC.
- D. Si, B. Xiong, L. Chen and J. Shi, Chem Catal., 2021, 1, 941–955 CAS.
- X. Liu, X. He, D. Xiong, G. Wang, Z. Tu, D. Wu, J. Wang, J. Gu and Z. Chen, ACS Catal., 2024, 14, 5366–5376 CrossRef CAS.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, C. Tian, A. Wu, Y. Wang and H. Fu, Nat. Commun., 2022, 13, 3125 CrossRef CAS PubMed.
- S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS PubMed.
- M. Zhang, Z. Zheng, X. Zhang, Z. Jiang, X. Yong, K. Li, X. Tu and K. Yan, JACS Au, 2025, 5, 937–947 CrossRef CAS PubMed.
- L. Xiao, W. Dai, S. Mou, X. Wang, Q. Cheng and F. Dong, Energy Environ. Sci., 2023, 16, 2696–2704 RSC.
- A. Chaturvedi, S. Gaber, S. Kaur, K. C. Ranjeesh, T. C. Nagaiah and D. Shetty, ACS Energy Lett., 2024, 9, 2484–2491 CrossRef CAS.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- A. Vilanova, P. Dias, T. Lopes and A. Mendes, Chem. Soc. Rev., 2024, 53, 2388–2434 RSC.
- V. Andrei, Y.-H. Chiang, M. Rahaman, M. Anaya, T. Kang, E. Ruggeri, S. D. Stranks and E. Reisner, Energy Environ. Sci., 2025, 18, 3623–3632 RSC.
- T. Hisatomi, Q. Wang, F. Zhang, S. Ardo, E. Reisner, H. Nishiyama, A. Kudo, T. Yamada and K. Domen, Front. Sci.Energy Storage Mater., 2024, 2, 1411644 Search PubMed.
- I. Y. Ahmet, Y. Ma, J.-W. Jang, T. Henschel, B. Stannowski, T. Lopes, A. Vilanova, A. Mendes, F. F. Abdi and R. van de Krol, Sustainable Energy Fuels, 2019, 3, 2366–2379 RSC.
- A. Vilanova, T. Lopes, C. Spenke, M. Wullenkord and A. Mendes, Energy Storage Mater., 2018, 13, 175–188 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03146j |
| This journal is © The Royal Society of Chemistry 2025 |