DOI:
10.1039/D5TC01598G
(Paper)
J. Mater. Chem. C, 2025,
13, 14044-14054
Low clearing asymmetric BTBT mesogens with perfect ambipolar charge transport†
Received
20th April 2025
, Accepted 24th May 2025
First published on 28th May 2025
Abstract
Two new asymmetrically substituted BTBT mesogens were successfully synthesised. These mesogens were designed to achieve low melting and clearing temperatures via the decoration with swallow-tailed chains containing peripheral siloxane units. The aromatic endgroups (phenyl and phthalonitrile) have a significant impact on the bilayer smectic self-assembly. For both materials the aromatic units intercalate completely into double layers, but the phenyl derivative generates a SmC phase and the phthalonitrile derivative a SmA phase. The latter is a weak donor–acceptor system showing outstanding charge transport properties in the mesophase. It exhibits perfectly balanced ambipolar charge carrier mobility in the range of 10−2 cm2 V−1 s−1 resulting in a material that is extremely promising for organic electronic applications.
Introduction
Organic semiconductors are very promising compounds for future flexible electronic materials used in organic photovoltaics (OPV),1–3 organic light-emitting diodes (OLEDs)4–6 or organic field-effect transistors (OFETs).7–10 They offer different advantages compared to their inorganic counterparts such as low cost, flexibility and transparency.11–15 However, organic materials often lack high charge carrier mobilities. A possibility of improving their performance is through the use of stimuli responsive materials such as thermotropic liquid crystals. Their fluid-like nature allows for a precise control of the required molecular orientation in a device. Often the material can be subsequently transformed to an almost defect-free crystalline structure templated by the liquid crystal (LC).14,16–19 Using this strategy, we propose to enhance the conducting properties significantly. One of the extremely promising organic semiconductors is [1]benzothieno[3,2-b]benzothiophene (BTBT), which shows extraordinarily high charge carrier mobilities.20–29 When combined with one or two alkyl chains BTBTs acquire thermotropic liquid crystalline properties.14,30,31
One of the best mesogenic BTBT compounds to date is Ph-BTBT-C10 (Fig. 1), reported by Iino et al. in 2015.14 They deployed this strategy and designed a calamitic theromotropic mesogen to obtain a highly ordered SmE mesophase in a thin film. The transformation by cooling from a well-aligned SmA phase guaranteed the formation of uniform crystalline thin films with a low number of defects. Using this method they were able to obtain extraordinarily high OFET-charge carrier mobilities of up to 13.9 cm2 V−1 s−1 for the non-symmetric Ph-BTBT-10 derivative.14 In contrast to Ph-BTBT-10, mesogen 10-BTBT-10 exhibits a less-ordered SmA phase and no SmE phase and therefore the charge carrier mobility drops to roughly 2 × 10−2 cm2 V−1 s−1 in the SmA phase.14
 |
| | Fig. 1 Overview of different BTBT materials and their hole mobilities, only for D(PhFCO)-BTBT the electron mobility is given. The molecules in the red box are reported to be liquid crystalline.14,20,21,25,26,32–35 Typically, the charge carrier mobilities are given for the crystalline phases. FI-TRMC, field-induced time-resolved microwave conductivity (intrinsic charge carrier mobility); OFET, charge carrier mobility derived from a transistor device; FP-TMRC, flash photolysis time-resolved microwave conductivity (intrinsic charge carrier mobility). | |
Mesogens with only one chain, such as Ph-BTBT-10 often exhibit high clearing temperatures >200 °C, which is unfavourable for cell preparation owing to decomposition of either the material or cell components. Therefore, we designed BTBT derivatives equipped on one side with a swallow-tailed chain with peripheral siloxane units, known to strongly reduce the transition temperatures.36,37 On the other side of the BTBT core, we coupled different aromatic units—a phenyl and a phthalonitrile unit—to investigate the impact on the charge carrier mobility (Fig. 2).
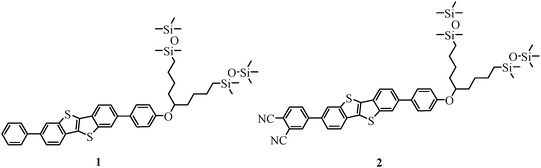 |
| | Fig. 2 Structure of target compounds 1 and 2. | |
Of special interest in this study was the influence of the phthalonitrile group in 2 on the material properties. Since phthalonitrile is a good acceptor and BTBT is a rather electron rich donor moiety promoting hole conductivity, we expected the formation of a donor–acceptor structure, which might evince ambipolar charge transport for this compound. The aim to achieve a balanced ambipolar transport is important for various organic electronic applications especially for photovoltaic devices, in which electrons and holes have to be extracted with the same velocity to prevent the generation of space charges.
Synthesis
The synthesis of BTBT target compounds 1 and 2 follows a convergent route, with known iodocyclisation and photocyclization key steps to produce the BTBT core,38 and is outlined in Scheme 1. The boronic acid substituted head group 9 (HG-B(OH)2) is prepared starting with the synthesis of the swallow tailed chains 7via a Grignard reaction.39–41 The subsequent Mitsunobu reaction42,43 attached the branched chain to the aromatic core. This bromo-substituted aromatic unit 8 was eventually transformed into the boronic acid 944 in a very good overall yield of 79% after three steps. The acceptor component 5 was synthesised in a Sandmeyer-like reaction followed by a Miyaura borylation giving 5 in a 66% yield after two steps. The BTBT precursor 12 was synthesised via an SNAr reaction of 11 with NaSMe followed by Sonogashira coupling with TIPS acetylene. At this point it is important to introduce the head group with the solubilising chains, since the BTBT derivatives become increasingly insoluble in organic solvents.20,45 Consequently, the HG boronic acid 9 was reacted with 12 in a Suzuki reaction to obtain the TIPS protected compound 13. In this reaction and all subsequent Pd-catalysed reactions, it is important to use a Pd(0) catalyst, as otherwise isomerisation of the terminal double bonds occurs.46 After deprotection of the TIPS group, followed by a Sonogashira coupling with the iodophenyl derivative 11, the iodine catalysed cyclisation and subsequent photocyclization gave the BTBT derivative 18 in a total yield of 45% after seven steps starting from compound 10. Depending on the desired substitution pattern of the phenyl group, either compound 5 or the commercially available phenyl boronic acid is used for the Suzuki-coupling with the BTBT derivative 18 to isolate either intermediate 19 or 20. After hydrosilylation of these compounds, the corresponding target molecules 2 and 1 could be isolated. Note that the hydrosilylation of the swallow tails leads always to some isomerisation of the terminal double bonds affording internal double bonds which are confirmed by 1H NMR signals between 5.30 and 5.40 ppm. However, it was possible to separate the isomerised impurities using tedious gel permeation chromatography (GPC) due to the distinct hydrodynamic radii of the different species. The overall yield of 2 was 11% over 14 steps, which corresponds to an average yield of 86% per step, which is appreciably high for such an elaborated synthesis. The overall yield of 1 was 2.6% over 12 steps corresponding to a good average yield of 74% per individual step.
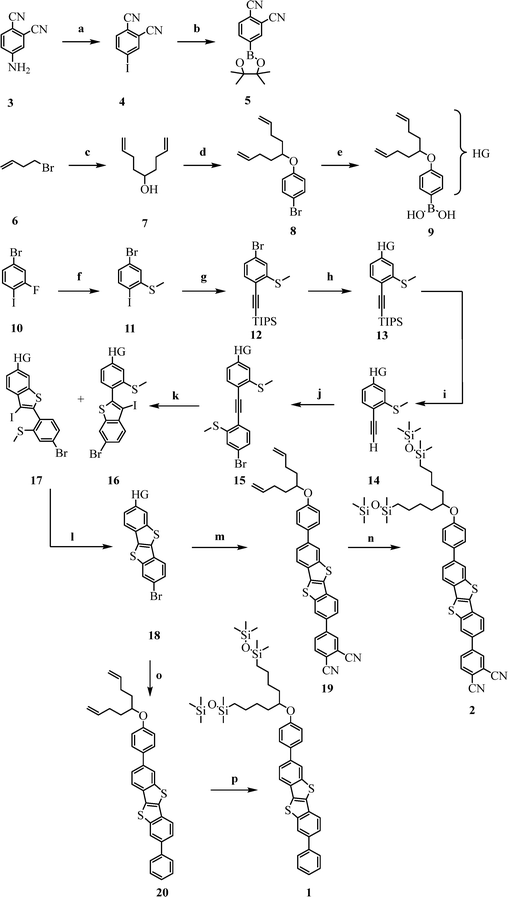 |
| | Scheme 1 (a) (1) H2SO4, NaNO2, 0 °C, (2) KI, 0 °C; 78% (b) B2pin2, Pd(PPh3)4, NaOAc, toluene, 100 °C, 84%; (c) (1) Mg, I2, THF, (2) NH4Cl, H2O, 99%; (d) PPh3, DIAD, NEt3, THF, RT, 81%; (e) (1) n-BuLi, THF, −78 °C, (2) B(OMe)3, −78 °C, 99%; (f) NaSMe, DMF, 0 °C, 99%; (g) Pd(dppf)Cl2, CuI, DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NEt3 1:1 (v/v), 0 °C, 97%; (h) 9, Pd(PPh3)4, K2CO3, THF, H2O, reflux, 85%; (i) TBAF·3H2O, THF, RT, 93%; (j) 11, Pd(PPh3)4, CuI, DCM:NEt3 1:1 (v/v), 0 °C, 85%; (k) I2, PhI(OAc)2, DCM, 0 °C, 73%; (l) hν, DCM, RT, 95%; (m) 5, Pd(PPh3)4, K2CO3, THF, H2O, reflux, 70%; (n) Karstedt-catalyst, Toluol, 80 °C, 68%; (o) PhB(OH)2, Pd(PPh3)4, K2CO3, THF, H2O, reflux, 27%; (p) Karstedt-catalyst, Toluol, 80 °C, 27%; pin = pinacol, Ac = acetate, DIAD = diisopropyl azodicarboxylate, dppf = 1,1′-bis(diphenyl-phosphino)ferrocene, TBAF = tetra-n-butylammonium fluoride. :NEt3 1:1 (v/v), 0 °C, 97%; (h) 9, Pd(PPh3)4, K2CO3, THF, H2O, reflux, 85%; (i) TBAF·3H2O, THF, RT, 93%; (j) 11, Pd(PPh3)4, CuI, DCM:NEt3 1:1 (v/v), 0 °C, 85%; (k) I2, PhI(OAc)2, DCM, 0 °C, 73%; (l) hν, DCM, RT, 95%; (m) 5, Pd(PPh3)4, K2CO3, THF, H2O, reflux, 70%; (n) Karstedt-catalyst, Toluol, 80 °C, 68%; (o) PhB(OH)2, Pd(PPh3)4, K2CO3, THF, H2O, reflux, 27%; (p) Karstedt-catalyst, Toluol, 80 °C, 27%; pin = pinacol, Ac = acetate, DIAD = diisopropyl azodicarboxylate, dppf = 1,1′-bis(diphenyl-phosphino)ferrocene, TBAF = tetra-n-butylammonium fluoride. | |
Photophysical studies
The UV-Vis spectrum of 1, visualised in Fig. 3A, shows a maximum in chloroform at λmax = 340 nm (ε = 5.18 × 104 L mol−1 cm−1) and a shoulder at λ = 357 nm (ε = 4.52 × 104 L mol−1 cm−1) while the maxima of 2 are bathochromically shifted to λmax = 351 nm (ε = 3.67 × 104 L mol−1 cm−1) and λmax = 381 nm (ε = 4.03 × 104 L mol−1 cm−1). In comparison, compound 2 possesses a much broader absorption band and the long wavelength absorption maximum shifts bathochromically by 24 nm, which also explains the different colours of the solid materials (Fig. 3C). We assumed that the bathochromic shift can be attributed to the donor–acceptor nature of 2. This would also imply a larger transition dipole compared to 1 and should manifest in a larger integral over the absorption band. However, the integrals over the absorption bands of 1 and 2 are surprisingly similar. An even larger difference does exhibit the emission spectra of both materials. Compound 2 exhibits one broad signal of high intensity with a maximum at 487 nm and a band shift of 105.0 nm relative to the absorption maximum, while 1 shows vibration progression with two peaks at λmax = 386 nm and λmax = 405 nm and a shoulder at λmax = 430 nm with a much smaller band shift of only 46 nm. Again, these observations can be rationalised by the donor–acceptor nature of 2, which should lead to an efficient intramolecular charge transfer transition. Eventually, excitation fluorescence spectra of both compounds were recorded (ESI,† Fig. S1) confirming the presence of only one emitting species.
 |
| | Fig. 3 Photophysical studies of 1 and 2; (A) absorption spectra in CHCl3 at a concentration of 3.09 × 10−5 mol L−1 (1) and 5.93 × 10−6 mol L−1 (2); (B) concentration- and absorption coefficient-corrected fluorescence spectra in CHCl3 at a concentration of 1.08 × 10−6 mol L−1 (1) and 1.13 × 10−6 mol L−1 (2) with an excitation wavelength of 380 nm (2) and 340 nm (1), respectively. (C) Shows the appearance of the bulk material of 1 (colourless/left) and 2 (bright yellow/right). | |
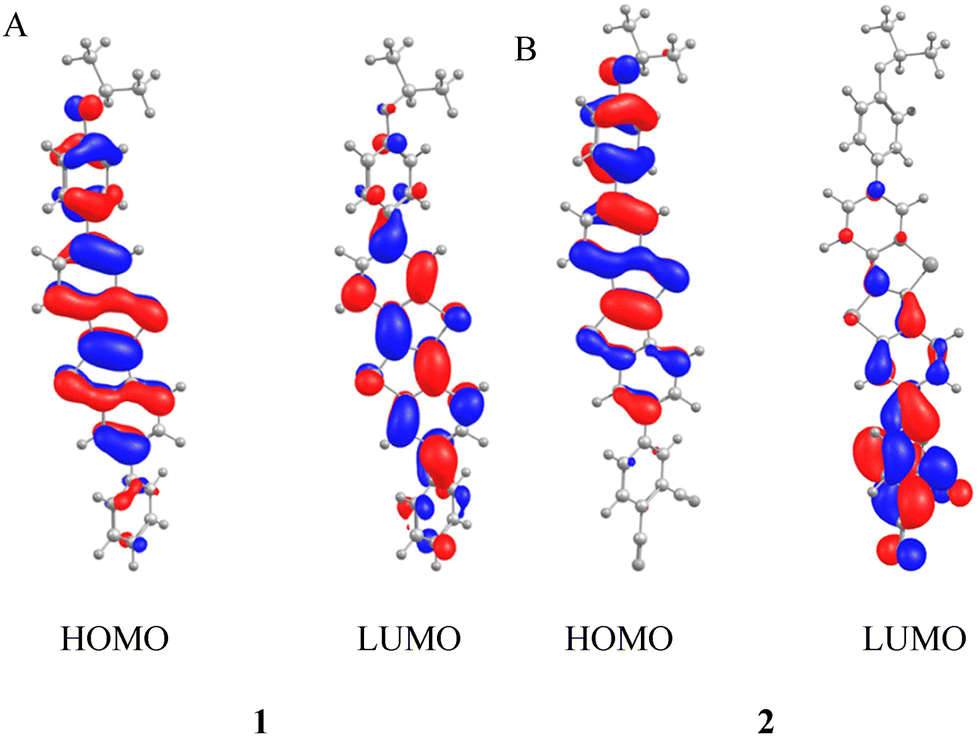
The DFT calculations were performed using the ωB97X-D3 functional47 with def2-TZVP48 as the basis set. The results show for 1 that both HOMO and LUMO are delocalised over the entire molecule (Fig. 4A). In contrast, the HOMO of 2 is strongly located on the side of the alkoxy substituted aryl unit of BTBT while the LUMO is mainly positioned at the phthalonitrile acceptor (Fig. 4B). These results support the expected donor–acceptor structure of 2. The calculated transition dipole moments of the two target compounds are 8.50 D (1) and 9.24 D (2) and are only 0.74 D apart. This small theoretical difference was confirmed by the calculation of the square of the integrals of the experimental absorption bands, which are proportional to the transition dipoles (see the ESI† for calculation details). The ratio between the theoretical transition dipole moments (1.12) is almost equal to the experimental ratio of the absorption band integrals (1.09). Consequently, the presence of the acceptor moiety only induces a weak donor–acceptor interaction. This can be rationalised by the molecular structure in which the phenyl groups realise a torsional angle of 43.5° with the BTBT core and prevent a strong conjugation.
 |
| | Fig. 4 Results of the DFT calculations showing the HOMO and LUMO of 1 (A) and 2 (B), respectively. | |
Thermotropic properties
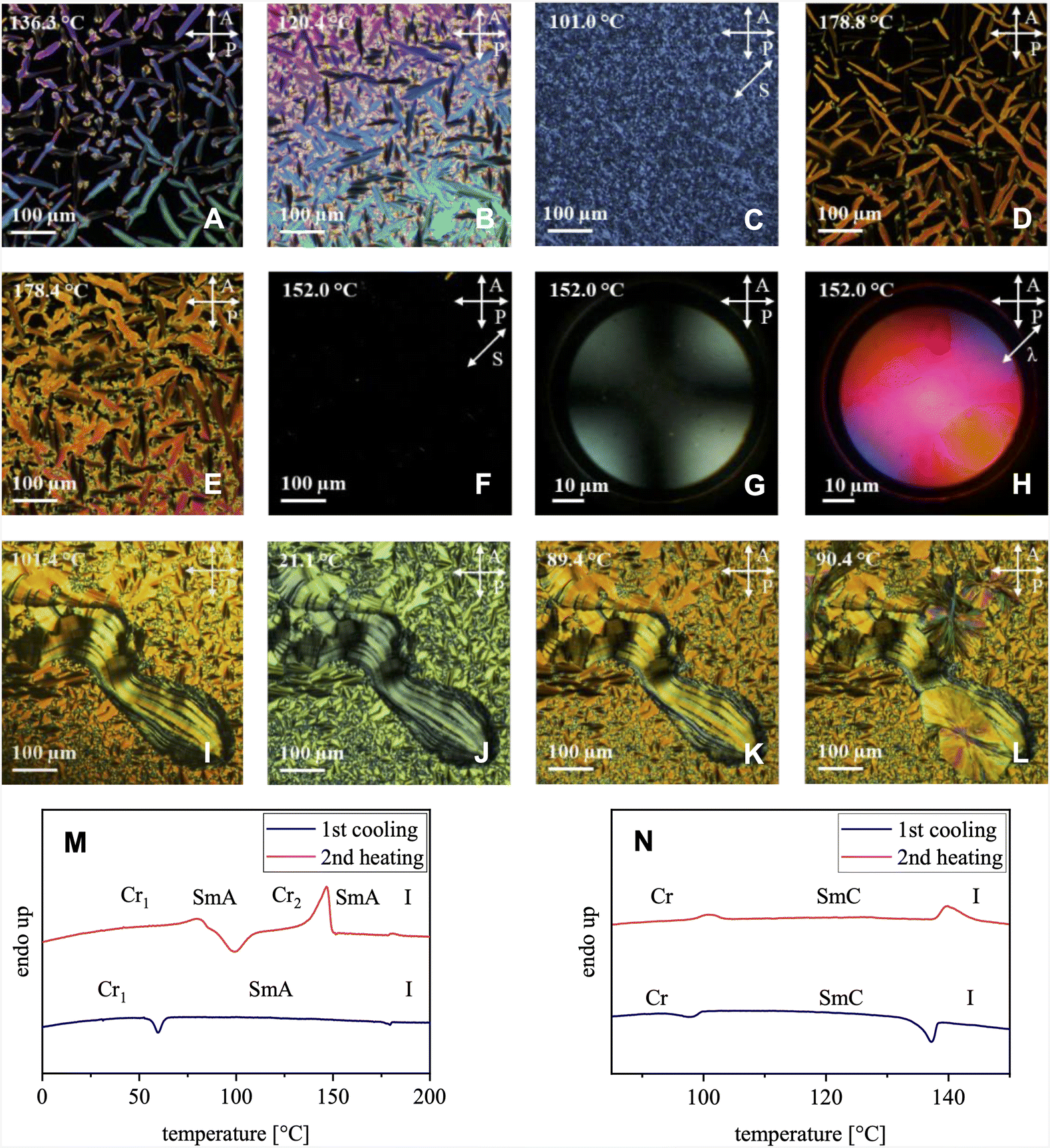
The thermotropic properties of both compounds were studied by DSC and POM (Fig. 5 and Table 1). Upon cooling 1 from the isotropic phase, bâtonnet structures (Fig. 5A) appeared at 136.3 °C indicating either a SmA or a SmC phase. The batônnets eventually turned into a focal conic texture at 120.4 °C (Fig. 5B). After shearing the sample at 101 °C, the birefringence was still observed (Fig. 5C), indicating the presence of a SmC phase. This interpretation is further strengthened by the DSC measurements (Fig. 5M), which show only a small transition enthalpy from the crystalline to liquid crystalline state of 0.389 kJ mol−1 at 96.5 °C and the rather large transition enthalpy of 1.24 kJ mol−1 at the clearing temperature of 140.5 °C.
 |
| | Fig. 5 POM images of 1 (A)–(C) and 2 (D)–(L) at different temperatures and DSC studies of 1 (M) and 2 (N). | |
Table 1 Onset temperature and transition enthalpy values of compounds 1 and 2 for heating and cooling cycles at a heating rate of 10 K min−1
| Compound |
Onset-temperature [°C]/transition enthalpy [kJ mol−1] |
|
1
|
Heating |
Cr 96.5/0.389 SmC 140.5/1.24 I |
| Cooling |
I 138.5/−1.02 SmC 99.7/−0.319 Cr |
|
|
|
2
|
Heating |
Cr1 81.3/6.54 (SmA 91.3/−19.87) Cr2 140.5/31.07 SmA 177.7/0.67 I |
| Cooling |
I 178.1/−0.57 SmA 62.8/−5.48 Cr1 |
The donor–acceptor compound 2 also exhibits a batônnet structure at 183.3 °C upon cooling from the isotropic phase (Fig. 5D). The batônnets slowly transformed into a focal conical texture at 178.4 °C (Fig. 5E). The mesophase possesses a rather low viscosity. Shearing the sample at 152.0 °C generated a homeotropically aligned phase (Fig. 5F), which is a typical behaviour for a SmA phase. Its uniaxial nature is confirmed by the conoscopic cross (Fig. 5G). Introducing the λ-retardation plate leads to the colours of the quadrants verifying the expected positive optical anisotropy of the rod-shaped mesogens (Fig. 5H). Further cooling results in the transition to a soft crystal phase Cr1 at 62.8 °C. The texture remained essentially the same as in the SmA phase (Fig. 5I and J) but turned out to be highly viscous and non-shearable. Subsequent heating to 89.4 °C generated again the SmA liquid crystal phase (Fig. 5K) and annealing the sample at 90.4 °C eventually resulted in the crystallisation (Cr2) of the material (Fig. 5L). DSC studies confirm the POM observations (Fig. 5N). The first cooling cycle shows a very small transition enthalpy of < 0.6 kJ mol−1 at the clearing transition (178.1 °C) with no hysteresis compared to the second heating cycle. This points to a rather low order of the mesophase. Further cooling resulted in a transition at 62.8 °C to the soft Cr1 phase with a still smaller transition enthalpy. Reheating indeed generates first the SmA phase before the sample shows a cold crystallisation and melts at 140.5 °C with a large transition enthalpy of 31.1 kJ mol−1 to the SmA phase.
X-ray scattering studies
Aligned fibres of compounds 1 and 2 were produced by extrusion and investigated using wide-angle X-ray scattering (WAXS). The fibres of 1 were extruded from the mesophase at 120 °C and were cooled rapidly to room temperature during this process. Interestingly, the diffractogram at 25 °C (Fig. 6A) shows an aligned crystalline phase Cr1 which is identical to the phase obtained by slow cooling (Fig. 6D). After heating to the mesophase at 126 °C (Fig. 6B), the X-ray pattern revealed an intense 001 reflection corresponding to a layer spacing of 38.3 Å with a correlation length of 526 Å (13.7 units) and a 002 reflection of low intensity corresponding to 19.2 Å. In addition, a halo can be identified corresponding to a superposition of average siloxane distances of 6.27 Å and the aliphatic chain distances of 4.85 Å. The position of the maximum intensity of this halo reveals that it is displaced by 22.2° from the fibre extrusion direction. This implies that the molecules are tilted versus the layer normal indicating a SmC structure. The molecular length derived from the DFT calculations comprises 30.8 Å. Since this distance is much shorter than the layer spacing, the mesogens can be assumed to form dimers self-assembling in a double layer. It is reasonable that the flat aromatic cores of the dimer are intercalated because the siloxane chains are rather voluminous. A complete intercalation leads to a total length of 44.9 Å and rationalises a tilt angle of 28.4° (see the ESI,† Fig. S30 for details). Since the aliphatic chains need to fill the gap between the aromatic interdigitated layer and voluminous siloxanes, a folding of these chains is reasonable and accounts for the further reduction of the dimer length by 1.5 Å, which is necessary to explain the tilt angle of 22.2°. When the temperature is reduced to 103 °C (Fig. 8C), the 002 reflection gains intensity and even the intensity of the 003 reflection increases indicating an expected increasing degree of order with decreasing temperature.
 |
| | Fig. 6 WAXS diffraction patterns of extruded fibres of 1 in the heating cycle at 25 °C (A) and 126 °C (B) and in the cooling cycle at 102 °C (C) and 25 °C (D). The insets at 126 °C and 102 °C highlight the small angle scattering at a different maximum intensity level to show the alignment. | |
Similar to compound 1, DA-mesogen 2 produces an aligned crystalline phase Cr1 at 25 °C upon extrusion of a fibre (Fig. 7A). With a slower first cooling history Cr1 is formed but without any special orientation (Fig. 7C and D). In the first heating cycle the diffraction pattern at 151 °C reveals an orientated smectic liquid crystal with an intense 001 and a very modest 002 reflection (Fig. 7B and inset). The principal reflection corresponds to a layer distance of 45.9 Å with a correlation length of 442 Å (9.63 units). The length of one single molecule of 2 is 31.6 Å. A complete intercalation of the aromatic units gives the length of a model dimer of 44.0 Å self-assembling in a double layer. This is in a rather good agreement with the experimental results. The halo consists of a broad signal typical for hydrosilylated aliphatic chains corresponding to an average distance of 6.31 Å and a small correlation length of 8.70 Å (1.38 units), which has its maximum exactly at the meridian of the XRS pattern. Thus, the mesogens are not tilted. All these results confirm the SmA structure of the mesophase of compound 2.
 |
| | Fig. 7 WAXS diffraction patterns of extruded fibres of 2 in the heating cycle at (A) 25 °C and (B) 151 °C and in the cooling cycle at (C) 25 °C. The inset at 151 °C highlights the small angle scattering at a different maximum intensity level to show the alignment; (D) plots of diffractograms of a powder sample of 2 at different temperatures. | |
 |
| | Fig. 8 Model of the mesophase of 2 (A), magnification of a single layer (B), schematic illustration of the HOMO–HOMO and LUMO–LUMO overlap of 1 when perpendicular and with a 22° tilt to the layer (C) and (D), and schematic illustration of the HOMO–HOMO (E) and LUMO–LUMO (F) overlap of 2. | |
A temperature-dependent study of a powder sample of 2 revealed that upon heating and annealing at 91 °C the formation of Cr2 is observed, which is clearly distinguishable from the powder pattern of Cr1 (Fig. 9D). Cooling from the isotropic phase to 73 °C uncovered the slow transition to the Cr1 phase, which was completed at 33 °C. These observations are in good agreement with POM and DSC studies. To gain a deeper insight into the self-assembly, the smectic A phase of compound 2 were modelled using the program Materials Studio (see the ESI† for details). For this reason, the density was measured using the buoyancy method and amounted to ρ = 0.972 g cm−3. This allowed us to determine the number of 36 mesogens in a section of 40.2 × 40.2 Å2 of the double layer. Two double layers were placed on top of each other and the structure was geometry optimised (Fig. 8A and B).
 |
| | Fig. 9 Electrons (red dots) and holes (black squares) mobilities depending on the temperature in the cooling cycle of 1 (A) and 2 (B). The mobilities were measured using the time-of-flight (TOF) method. The phase transition temperatures are indicated by dashed lines. | |
The model reveals that the intercalation in the SmA double layer is maintained and that the siloxane chains are nanosegregated from the aromatic units. In this model the molecules are not tilted, which is in agreement with the SmA nature of the phase. The aromatic layer seems to be rather disordered, a typical feature of a highly fluidic SmA phase. While at the centre of the layer the donor BTBT units are always in contact, only half of the acceptor units are organised at the two peripheries of the aromatic double layer.
The possible impact of these structures on the charge carrier mobility can be estimated by considering the HOMO and LUMO splitting within the molecular stacks. According to V. Lemaur an optimised overlap of HOMOs or LUMOs causes a splitting of the frontier orbital energies.49 The larger the splitting, the easier will be the charge transfer within the frame of the Marcus theory. A large HOMO splitting assists a fast hole transport, while a large LUMO splitting fosters a fast electron transport. In the structural model of double layers, we consider here exemplarily only the more favourable overlap of parallel mesogens (Fig. 8C–F). In this assembly the HOMO–HOMO (Fig. 8C) and LUMO–LUMO (Fig. 8D) overlap of 1 will be largest, when the mesogens are parallel with the layer normal. However, when the mesogens are tilted by an angle of 22° the overlap of HOMOs, which are orientated perpendicular to the molecular long axis, is reduced. The orbitals are then positioned above a node of the closest neighbours, and they will interact not only constructively with orbitals of the same sign, but also destructively with orbitals of the opposite sign. In contrast, the LUMOs are aligned parallel to the long axis of the molecule and are alternatively distributed at the front and the back sites. Even after tilting the molecules, a destructive overlap of the LUMOs is not possible although the interaction will be also reduced. These considerations hint that compound 1 should be a better electron than a hole conductor. In the case of compound 2 the SmA structure of the mesophase implies that molecules are orientated parallel to the layer normal, and this should maximise the HOMO and LUMO splitting (Fig. 8E and F) at a comparable level of disorder in the lamellar phase. This suggests that an improved charge transport and possibly an ambipolar charge transport can be expected.
Charge carrier mobility
The charge carrier mobilities for holes and electrons were determined via the time-of-flight transient photocurrent technique using Nd:YAG laser of wavelength 355 nm. The results of these temperature-dependent measurements in the cooling cycle are depicted in Fig. 9. A typical behaviour for a disordered smectic LC phase is observed for the hole and electron mobility of compound 1. The mobilities are different and slightly higher for the electrons (1.09 × 10−4 cm2 V−1 s−1vs. 7.88 × 10−4 cm2 V−1 s−1 at 110 °C), which can be rationalised by the discussed HOMO–HOMO and LUMO–LUMO overlap in the tilted smectic C phase.49 The mobility increases in the crystalline phase to almost the same value for both charge carriers (1.04 × 10−3 cm2 V−1 s−1 for electrons, 1.23 × 10−3 cm2 V−1 s−1 for holes) at 80 °C. Surprisingly, the DA mesogen 2 with the lower order phase exhibits a completely balanced hole and electron mobility, which is at least one order of magnitude higher than for compound 1. It shows the highest electron mobility of 1.09 × 10−2 cm2 V−1 s−1 and hole mobility of 1.03 × 10−2 cm2 V−1 s−1 at 150 °C. With decreasing temperature, the charge carrier mobility remains almost constant down to 120 °C. Subsequently, hole and electron mobilities drop to <7.00 × 10−4 cm2 V−1 s−1 at 30 °C. The latter can be explained by the formation of Cr2 during the slow cooling process. These mobilities are comparable to the one reported by Hanna et al. for 10-BTBT-10 in the SmA, which were also found to be in the range of 10−2 cm2 V−1 s−1.14 Another BTBT derivative Ph-BTBT-10 exhibited significantly higher mobilities of up to 13.9 cm2 V−1 s−1 in the SmE phase, while it was not measured in the SmA mesophase at higher temperatures.14 In that work, the SmE phase was correctly aligned through the presence of the SmA phase and the charge carrier mobility was determined using the OFET method.14 This measurement differs from the TOF experiments used for compound 2 and might overestimate the mobilities, which was demonstrated by others.50
Conclusions
The synthesis of two asymmetric BTBT derivatives 1 and 2, equipped with a swallow-tailed chain with peripheral siloxane units, has been successfully demonstrated. They differ only in the attached aromatic groups—a phenyl and a phthalonitrile unit. The latter turns material 2 into a weak donor–acceptor compound, which is demonstrated by the photophysical studies, the bathochromic shifts of the absorption and emission spectra and theoretical calculations (DFT) of the molecular orbitals HOMO and LUMO. The molecular design has been chosen to lower the clearing temperature below 200 °C to prevent decomposition during sample preparation. Both materials 1 (177.1 °C) and 2 (140.5 °C) show the transition from a smectic to the isotropic phase significantly below the anticipated temperature limit. The incommensurate size of the siloxane peripheries causes the mesogens to pack in LC double layers with completely intercalated aromatic units. However, the different substitution at the phenyl group results in distinct self-assemblies. The phenyl derivative 1 arranges in SmC layers and transforms to a soft crystal. The introduction of a strong dipole by the cyano groups in the phthalonitrile derivative 2 increases the clearing temperature but also changes the smectic structure to the more fluid SmA phase, which generates a monotropic soft crystalline phase below 60 °C but also a stable crystalline phase upon reheating the sample. The molecular tilt and the electronic structure have a significant impact on the charge transport properties. The weak donor acceptor structure in the SmA phase reveals a surprisingly high TOF charge carrier mobility for such a disordered mesophase of up to 1.08 × 10−2 cm2 V−1 s−1. Moreover, material 2 is outstanding with regard to the extremely balanced mobility for holes and electrons, i.e. compound 2 is a perfect ambipolar charge transport LC material. The charge mobility is indeed comparable to previously reported hole mobilities of SmA BTBTs measured in OFETs.14 The extremely balanced charge carrier mobility of moderate high value is especially interesting for photovoltaic applications, where both charges, holes and electrons, have to be extracted without generating space charges remaining in the material.
Author contributions
L. F.: conceptualization (equal); formal analysis (lead); funding acquisition (support); investigation (lead); methodology (equal); project administration (lead); supervision (support); visualization (lead); writing – original draft (lead); writing – review & editing (equal). L. M.: investigation of TOF (equal); resources (supporting). D. P. S.: TOF investigation (equal); formal analysis (supporting); resources (supporting); visualization (support); writing – review & editing (supporting). M. L.: conceptualization (equal); funding acquisition (lead); methodology (equal); project administration (support); resources (lead); supervision (lead); writing – original draft (support); writing – review & editing (equal).
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to Xincheng Miao for the DFT-calculations of the mesogens, and the Studienstiftung des deutschen Volkes, the Elitenetzwerk Bayern and the German Science Foundation DFG via LE1571/11-1 for funding. D. P. S. is thankful to the ULCO for BQR funding.
References
- K. Leo, Nat. Rev. Mater., 2016, 1, 16056 CrossRef CAS.
- G. Zhang, F. R. Lin, F. Qi, T. Heumuller, A. Distler, H. J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Jen and H. L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- A. Distler, C. J. Brabec and H. J. Egelhaaf, Prog. Photovolt.: Res. Appl., 2020, 29, 24–31 CrossRef.
- A. Salehi, X. Fu, D. H. Shin and F. So, Adv. Funct. Mater., 2019, 29, 1808803 CrossRef.
- J. Bauri, R. B. Choudhary and G. Mandal, J. Mater. Sci., 2021, 56, 18837–18866 CrossRef CAS.
- S. Banerjee, P. Singh, P. Purkayastha and S. Kumar Ghosh, Chem. – Asian J., 2025, 20, e202401291 CrossRef CAS PubMed.
- I. Suzuki, S. Koizumi, Y. Kito, J.-I. Hanna and H. Iino, Phys. Status Solidi A, 2023, 220, 2300281 CrossRef CAS.
- Z. Chen, S. Duan, X. Zhang and W. Hu, Mater. Chem. Front., 2024, 8, 2227–2272 RSC.
- S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS PubMed.
- J. D. Myers and J. Xue, Polym. Rev., 2012, 52, 1–37 CrossRef CAS.
- S. Ahmad, J. Polym. Eng., 2014, 34, 279–338 Search PubMed.
- H. Iino, T. Usui and J. Hanna, Nat. Commun., 2015, 6, 6828 CrossRef CAS PubMed.
- G. Schweicher, G. Garbay, R. Jouclas, F. Vibert, F. Devaux and Y. H. Geerts, Adv. Mater., 2020, 32, e1905909 CrossRef PubMed.
- R. K. Gupta and A. A. Sudhakar, Langmuir, 2019, 35, 2455–2479 CrossRef CAS PubMed.
- M. O'Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566–584 CrossRef PubMed.
- Y. C. Lin, G. S. Li, P. J. Yu, E. Ercan and W. C. Chen, J. Chin. Chem. Soc., 2022, 69, 1289–1304 Search PubMed.
- D. A. Ivanov, R. I. Gearba, D. Anokhin, S. Magonov and M. Lehmann, PMSE Preprints, 2006, 94, 655–656 CAS.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- W. Y. Wei, C. Yang, J. Y. Mai, Y. F. Gong, L. S. Yan, K. Zhao, H. L. Ning, S. J. Wu, J. W. Gao, X. S. Gao, G. F. Zhou, X. B. Lu and J. M. Liu, J. Mater. Chem. C, 2017, 5, 10652–10659 RSC.
- P. Xie, T. Liu, J. Sun, J. Jiang, Y. Yuan, Y. Gao, J. Zhou and J. Yang, Sci. Bull., 2020, 65, 791–795 CrossRef CAS PubMed.
- A. Tamayo, S. Hofer, T. Salzillo, C. Ruzie, G. Schweicher, R. Resel and M. Mas-Torrent, J. Mater. Chem. C, 2021, 9, 7186–7193 RSC.
- I. Suzuki, J.-I. Hanna and H. Iino, Appl. Phys. Express, 2024, 17, 051007 CrossRef CAS.
- R. Akai, K. Oka, S. Dekura, K. Yoshimi, H. Mori, R. Nishikubo, A. Saeki and N. Tohnai, J. Phys. Chem. Lett., 2023, 14, 3461–3467 Search PubMed.
- H. Usta, D. Kim, R. Ozdemir, Y. Zorlu, S. Kim, M. C. Ruiz Delgado, A. Harbuzaru, S. Kim, G. Demirel, J. Hong, Y.-G. Ha, K. Cho, A. Facchetti and M.-G. Kim, Chem. Mater., 2019, 31, 5254–5263 CrossRef CAS.
- Z. Ma, H. Geng, D. Wang and Z. Shuai, J. Mater. Chem. C, 2016, 4, 4546–4555 RSC.
- M. Alkan and I. Yavuz, Phys. Chem. Chem. Phys., 2018, 20, 15970–15979 RSC.
- H. Monobe, L. An, P. Hu, B.-Q. Wang, K.-Q. Zhao and Y. Shimizu, Mol. Cryst. Liq. Cryst., 2017, 647, 119–126 Search PubMed.
- T. Oka, N. Shioya, R. Abe, S. Maruyama and T. Hasegawa, J. Phys. Chem. C, 2025, 129, 4806–4813 Search PubMed.
- E. Ferrari, L. Pandolfi, G. Schweicher, Y. Geerts, T. Salzillo, M. Masino and E. Venuti, J. Phys. Chem. C, 2024, 128, 4258–4264 CrossRef CAS.
- H. Iino and J.-I. Hanna, J. Appl. Phys., 2011, 109, 074505 CrossRef.
- K. Tsujita, S. Maruyama, Y. Shibata, T. Koganezawa, K. Kaminaga, H. Fujikake and Y. Matsumoto, Cryst. Eng. Commun., 2023, 25, 64–71 RSC.
- P. Pandey, L. Fijahi, N. McIntosh, N. Turetta, M. Bardini, S. Giannini, C. Ruzié, G. Schweicher, D. Beljonne, J. Cornil, P. Samorì, M. Mas-Torrent, Y. H. Geerts, E. Modena and L. Maini, J. Mater. Chem. C, 2023, 11, 7345–7355 RSC.
- Y. Tsutsui, G. Schweicher, B. Chattopadhyay, T. Sakurai, J. B. Arlin, C. Ruzie, A. Aliev, A. Ciesielski, S. Colella, A. R. Kennedy, V. Lemaur, Y. Olivier, R. Hadji, L. Sanguinet, F. Castet, S. Osella, D. Dudenko, D. Beljonne, J. Cornil, P. Samori, S. Seki and Y. H. Geerts, Adv. Mater., 2016, 28, 7106–7114 CrossRef CAS PubMed.
- M. Funahashi and A. Sonoda, J. Mater. Chem., 2012, 22, 25190–25197 RSC.
- M. Lehmann, Front. Soft Matter, 2023, 3, 1089726 CrossRef.
- T. Kitamura, K. Morita, H. Nakamori and J. Oyamada, J. Org. Chem., 2019, 84, 4191–4199 CrossRef CAS PubMed.
- L. Steemers, M. J. Wanner, B. R. C. van Leeuwen, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2018, 874–878 CrossRef CAS.
- G. Lemiere, V. Gandon, K. Cariou, A. Hours, T. Fukuyama, A. L. Dhimane, L. Fensterbank and M. Malacria, J. Am. Chem. Soc., 2009, 131, 2993–3006 CrossRef CAS PubMed.
- K. J. Arrington, C. B. Murray, E. C. Smith, H. Marand and J. B. Matson, Macromolecules, 2016, 49, 3655–3662 CrossRef CAS.
- O. Mitsunobu and M. Yamada, Bull. Chem. Soc. Jpn., 1967, 40, 2380–2382 CrossRef CAS.
- S. Fletcher, Org. Chem. Front., 2015, 2, 739–752 RSC.
- D. Sleveland and H.-R. Bjørsvik, Org. Process Res. Dev., 2012, 16, 1121–1130 CrossRef CAS.
- R. Ozdemir, K. Ahn, İ. Deneme, Y. Zorlu, D. Kim, M.-G. Kim and H. Usta, J. Mater. Chem. C, 2020, 8, 15253–15267 RSC.
- A. Sen and T. W. Lai, Inorg. Chem., 1984, 23, 3257–3258 CrossRef CAS.
- Y. S. Lin, G. D. Li, S. P. Mao and J. D. Chai, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS.
- A. Hellweg and D. Rappoport, Phys. Chem. Chem. Phys., 2015, 17, 1010–1017 RSC.
- V. Lemaur, D. A. da Silva Filho, V. Coropceanu, M. Lehmann, Y. Geerts, J. Piris, M. G. Debije, A. M. van de Craats, K. Senthilkumar, L. D. Siebbeles, J. M. Warman, J. L. Bredas and J. Cornil, J. Am. Chem. Soc., 2004, 126, 3271–3279 CrossRef CAS PubMed.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, e1801079, DOI:10.1002/adma.201801079.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c and
Matthias
Lehmann
*c and
Matthias
Lehmann