
Cell-penetrating poly(disulfide)-based star polymers for simultaneous intracellular delivery of miRNAs and small molecule drugs†
Wenhua
Yang
a,
Changmin
Yu
 *a,
Chunxian
Wu
a,
Shao Q.
Yao
b and
Shuizhu
Wu
*a
*a,
Chunxian
Wu
a,
Shao Q.
Yao
b and
Shuizhu
Wu
*a
aState Key Laboratory of Luminescent Materials & Devices, College of Materials Science & Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: yucm@scut.edu.cn; shzhwu@scut.edu.cn
bDepartment of Chemistry, National University of Singapore, Singapore 117543
First published on 19th June 2017
Abstract
MicroRNAs (miRNAs) are small regulatory noncoding RNAs that control a variety of biological processes. The regulation of many endogenous miRNAs is tightly associated with various diseases. Thus, the design of miRNA delivery systems with minimal endolysosomal trapping, efficient delivery and controlled miRNA release is urgently needed. Herein, we have developed a new star polymer which consists of a β-cyclodextrin (βCD) core and multiple cell-penetrating poly(disulfide) (CPD) arms. By subsequently loading the system with miRNAs and small molecule drugs, we have successfully proven that this novel drug delivery platform could efficiently enter mammalian cells (<2 h) without apparent endolysosomal trapping. The global Pearson's R value was calculated to be 0.31 between our complex and endolysosome, which is far below the threshold of >0.5 required for correlation. In addition, the GSH-triggered degradation of CPD arms and the subsequent intracellular release of miR-203 and CPT, as well as the combination therapeutic effects have been successfully demonstrated. In this way, we show that this novel platform could be used in future to minimize potential cytotoxicity encountered by many existing cationic branched polymer systems in miRNA delivery. Our results provide important starting points for using CPD-based polymers to design personalized delivery platforms.
1. Introduction
MicroRNAs (miRNAs) are a class of endogenous non-coding RNAs of 21–23 nucleotides that control at least 30% of human genes and a variety of biological processes by acting post-transcriptionally to target mRNAs for translational cleavage, repression, and destabilization.1,2 miRNAs have been shown to play important regulatory roles in spatio-temporal patterning of the vertebrate body, as well as adult physiology.3 Accordingly, aberrant miRNA expression has been tightly linked to various diseases including cancer, cardiovascular diseases, viral infection, and neurological disorders.4Recently, hyperbranched or star polymers have emerged as promising candidates for gene delivery due to their unique characteristics, including a three-dimensional dendritic architecture, good solubility and low viscosity, and a large number of terminal functional groups.5 Particularly, cationic branched polymers, which possess the ability to complex with nucleic acids and deliver them into cells, have been extensively developed as a viable route for potential therapeutic intervention.5–8 However, these polymers are still prone to various degrees of cytotoxicity, poor blood circulation and particularly endosomal entrapment, thus limiting their in vivo therapeutic efficacy. The development of novel branched polymer-based gene delivery systems to solve these obstacles is therefore needed.
Recently, a kind of polyarginine cell-penetrating-peptide (CPP) mimic, named cell-penetrating poly(disulfide)s (CPDs), in which the polypeptide backbone of CPP was replaced with poly(disulfide)s, has been reported.9 Importantly, Matile et al. showed in several elegant studies that, CPDs could rapidly enter mammalian cells via thiol-mediated pathways, thus minimizing the major issue of endosomal trapping commonly associated with CPP and other means of delivery systems.10–13 In addition, CPDs are also characterized by minimal cytotoxicity and efficient cytosolic degradation/cargo release triggered by glutathione (GSH), a metabolite endogenously expressed in mini-molar concentrations by most mammalian cells. We and others further showed that CPD-conjugated large cargos such as proteins and nanoparticles could be rapidly delivered to mammalian cells with no apparent endolysosomal trapping.14–17
Studies have shown that co-delivery of anticancer drugs and therapeutic genes could enhance chemotherapy efficacy. Until now, various nanocarriers including cationic lipids, polymer micelles and inorganic nanoparticles have been reported as co-delivery candidates.18 As a result of possessing a hydrophobic interior and a hydrophilic exterior, as well as water solubility and low cytotoxicity, β-cyclodextrin (βCD; Scheme 1) has been widely used to improve the solubility and stability of many small molecules including hydrophobic drugs.19–23 In addition, βCD-based hyperbranched or star polymers have been designed as small-molecule hydrophobic drugs and gene carriers.24 For instance, Li et al. reported a system based on cationic CD-oligoethylenimine star-shaped polymers for co-delivery of hydrophobic anticancer drug PTX and cancer-therapeutic gene p53, which subsequently entered into cells via FR-mediated endocytosis.25 Zhang and co-workers reported star-shaped polymers for the drug/gene co-delivery by conjugating βCD with a positively charged PAMAM dendrimer.26 However, some obstacles remain for current CD-based hyperbranched or star polymers to be used as an ideal delivery system, such as cytotoxicity, low transfection efficacy or endosomal trapping.24 In particular, extensive investigations on the inclusion complexes formed between camptothecin (CPT) and βCD, as well as its derivatives, have been reported.27–29 CPT is an anticancer drug which shows powerful anticancer activity by binding to its cellular target, DNA topoisomerase I (TOP I), and subsequently inhibiting TOP I to relax supercoiled DNA prior to transcription through the formation of a single strand break and relegation.30 Therefore, a combination of CPT and RNA interference (RNAi) (such as miRNAs) has the potential to achieve a synergistic therapeutic effect in disease treatment.
 | ||
| Scheme 1 (a) Synthetic scheme of βCD-CPD. Reaction conditions: (a) β-Cyclodextrin, triphenylphosphane, iodine, DMF, 15 h, 80 °C, 88%; (b) thiourea, NaOH, KHSO4, DMF, 20 h, 70 °C, 85%. (b) Overall strategy for the βCD-CPD-based system for simultaneous delivery and controlled release of miRNAs and small molecule drugs in live cells. Other chemicals are shown in the ESI.† In this scheme, the number of the polymers does not reflect the actual number and is for illustration only. | ||
Herein, we have reported a new star-like delivery system based on the βCD core and multiple CPD arms, which is capable of rapid and simultaneous delivery of miRNAs/small molecule drugs (CPT) to mammalian cells, followed by subsequent release of cargos triggered by endogenous GSH. As shown in Scheme 1, after the synthesis of βCD-CPD, small molecule hydrophobic drug CPT was first encapsulated into the βCD core of our system to obtain CPT@βCD-CPD and the system was subsequently complexed with miRNAs via charge interaction to form the co-delivery system named CPT@βCD-CPD-miRNAs. Upon incubation with live cells (step I), the system successfully crossed the cell membrane efficiently via thiol-mediated pathways and subsequently entered the cytosol (step II). Next, the degradation of CPD arms triggered by endogenous GSH led to cytosolic release of drugs/miRNAs (step III). Thus, this novel drug delivery system could provide the following key features: (1) it is the first reported CPD-based star-like system that could rapidly and efficiently deliver cargos to mammalian cells via endocytosis-independent pathways; (2) by simultaneously delivering both miRNAs and the hydrophobic CPT to live cancer cells, the system is able to achieve a synergistic therapeutic effect in disease treatment with two mechanistically different anticancer agents; (3) intracellular controlled-release of miRNAs is possible, triggered by endogenous GSH-promoted rapid degradation of CPDs, thus minimizing the cytotoxicity of the overall system.
2. Materials and methods
2.1. Materials and measurements
β-Cyclodextrin (βCD), triphenylphosphane, thiourea, iodoacetamide and camptothecin (CPT) were purchased from Sigma-Aldrich and used without further purification. Iodine, sodium methoxide, potassium bisulfate (KHSO4), sodium hydroxide (NaOH) and triethanolamine (TEOA) were purchased from Aladdin Reagents and used without further purification. Antibodies were purchased from the following vendors: mouse anti-TUBB1 antibody (D198906, Shanghai Sangon Biotech), rabbit anti-survivin antibody (ab76424, Abcam), HRP-conjugated secondary antibodies: goat anti-mouse IgG (D110087, Shanghai Sangon Biotech), goat anti-rabbit IgG (AS014, ABclonal Technology). Dichloromethane (DCM) and N,N-dimethylformamide (DMF) were dried with CaH2 and distilled under a nitrogen atmosphere. Water used in all experiments was prepared by using a Milli-Q water purification system and further treated by ion exchange columns. All other reagents used were of analytical grade and used without further purification. 1H NMR spectra were recorded on a Bruker Avance 600 MHz NMR spectrometer. Mass spectra were obtained by using a Bruker Esquire HCT Plus mass spectrometer. Absorption spectra were recorded on a Hitachi U-3010 UV-vis spectrophotometer. Laser confocal microscopy imaging experiments were performed on a Zeiss LSM 710 Confocal Microscope equipped with a 40× water immersion objective. Flow cytometry was performed on a BD Accuri™ C6 cell analyzer. The particle size and distribution was determined on a Malvern Nano-ZS90 particle size analyzer based on the dynamic light scattering (DLS) mechanism. Transmission electronic microscopy (TEM) experiments were carried out on a JEOL JEM-1400Plus transmission electron microscope. The gel retardation assay and western blotting (WB) were visualized and photographed with a Bio-Rad Gel Doc XR+ Imaging System. Oligonucleotides were purchased from Sangon Biotech (Shanghai). The sequences are as follows:Cy5-labeled miR-203 (miR-203Cy5):
Cy5-5′-GUGAAAUGUUUAGGACCACUAG-3′
MiR-203: 5′-GUGAAAUGUUUAGGACCACUAG-3′
Poly21A DNA (21A): 5′-AAAAAAAAAAAAAAAAAAAA-3′
2.2. Synthesis of per-6-thio-β-cyclodextrin (βCD-SH)
The βCD-SH initiator was synthesized and characterized based on a published procedure.31 Briefly, triphenylphosphane (5.25 g, 20 mmol), β-CD (1.13 g, 1 mmol) and iodine (5.05 g, 20 mmol) were dissolved in DMF (20 mL), and the solution was stirred at 80 °C for 15 h. Upon concentration in vacuo to ∼half the volume, the pH of the solution was adjusted to 9–10 with sodium methoxide (3 M in methanol) and then stored for 30 min at 25 °C, before being poured into 200 mL of methanol to form a precipitate. The precipitate was washed three times and extracted with methanol for 24 h by using a Soxhlet extractor. After rigorous drying, per-6-iodo-β-cyclodextrin was obtained as a white powder (1.67 g, 88%). Next, per-6-iodo-β-cyclodextrin (0.951 g, 0.5 mmol) and thiourea (0.304 g, 4 mmol) were dissolved in DMF (5 mL), and the resulting mixture was heated to 70 °C under a nitrogen atmosphere for 20 h. Upon DMF removal under reduced pressure, the resulting yellow oil was next dissolved in NaOH solution (0.13 M) and heated to reflux under a nitrogen atmosphere for 1 h. The resulting suspension was acidified with aqueous KHSO4 and filtered, the precipitate was collected, washed thoroughly with water, and dried in vacuo to yield per-6-thio-β-cyclodextrin (βCD-SH) as an off-white powder (0.536 g, 85%). 1H NMR (600 MHz, DMSO-d6) δ 5.87 (s, 14 H, OH-2, OH-3), 4.93 (d, J = 3.4 Hz, 7 H), 3.71–3.65 (m, 7 H, H-3), 3.61 (t, J = 9.2 Hz, 7 H, H-5), 3.35 (dd, J = 9.5, 3.9 Hz, 14 H, H-2, H-4), 3.22–3.16 (m, 7 H, H-6a), 2.75 (dt, J = 14.6, 7.8 Hz, 7 H, H-6b), 2.13 (t, J = 8.3 Hz, 7 H, SH). MS m/z 1269.1 for [M + Na]+, calcd for (C42H70O28S7) 1247.1.2.3. General procedure for βCD-CPD synthesis
The βCD-CPD polymers were synthesized and characterized according to a previous report with modifocations.16 Briefly, the monomer (M in Scheme 1, 2.0 M in DMF), initiator (βCD-SH, 50 mM in DMF), terminator (iodoacetamide, 0.5 M in H2O) and TEOA buffer solution (1.0 M, pH = 7.0) were freshly prepared. The polymerization mixture was generated by mixing 10 μL of the monomer stock solution, 80 μL of TEOA buffer and 10 μL of the initiator βCD-SH stock solution. After 30 min of vigorous agitation at 25 °C, the polymerization reaction was quenched by addition of 1.9 mL of the terminator stock solution. The polymers were purified by a dialysis method. Briefly, the solution was transferred into a dialysis bag (MWCO 1 kDa) and dialyzed for 24 h to remove any small molecules. Finally, the solution was lyophilized for the following tests after purification with a NAP™-5 desalting column (GE Healthcare) against H2O by following the protocols provided by the vendor.2.4. Preparation of CPT@βCD-CPD inclusion complex
The encapsulation of CPT molecules into βCD was prepared according to previous literature reports.27–29 CPT was first suspended in water containing βCD-CPD and further stirred for 24 h in the dark (the overall solution was kept acidic in order to avoid formation of the carboxylate form of CPT). Excessive un-complexed CPT was subsequently removed by filtration with a 0.22 μm filter and the resulting filtrate was freeze-dried to obtain the CPT@βCD-CPD inclusion complex. For the determination of drug loading (DL), CPT@βCD-CPD was diluted and analyzed by UV-vis absorption spectrometry. DL was calculated according to the following formula based on a calibration curve obtained with CPT in DMSO of known CPT concentrations: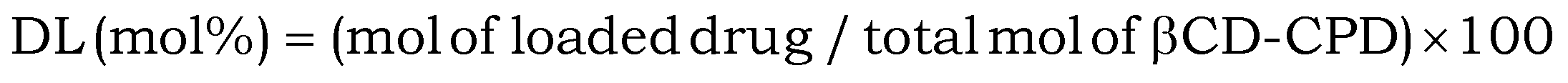
2.5. Preparation of CPT@βCD-CPD-miR-203 complex system
CPT@βCD-CPD-miR-203 nanoparticles were prepared by adding miR-203 (dissolved in DEPC-treated water) into a CPT@βCD-CPD solution (dissolved in HEPES buffer, 10 mM, pH 7.4) at varying volumes to give the complex system. After vigorous agitation with a vortex mixer, the resultant solution was incubated for 30 min at room temperature before use.2.6. Binding capacity and GSH-triggered release analysis
After the formation of complexes of βCD-CPD with miR-203 at different ratios, the binding capacity was analysed on a 3% agarose gel (100 V, 10 min). For the triggered release of miR-203, after the addition of GSH (10 mM) or HeLa cell lysates (1 mg per mL of proteins) for 1 h at 37 °C, respectively, aliquots were taken from the suspension and determined via running the supernatant on a 3% agarose gel. The release of CPT from the CPT@βCD-CPD-miR-203 complex was performed by using the dialysis technique. 15 mg of CPT@βCD-CPD-miR-203 was dispersed in 5 mL HEPES buffer, which was charged into a dialysis tube (molecular weight cut-off size: 3.5 kDa). Then the tube was immersed in 30 mL of dialysis media and stirred at 37 °C in the presence and absence of GSH (10 mM). At certain time intervals, 0.5 mL aliquot of the dialysis medium was taken out and the released amount of CPT was calculated via a UV-vis spectrum.2.7. Cell culture
Cell culture experiments were performed by using HeLa cell lines cultured in Dulbecco's Modified Eagle's Medium (DMEM; Gibco) supplemented with 10% Fetal Bovine Serum (FBS) and 2% penicillin/streptomycin (Gibco) and maintained at 37 °C under a 5% CO2 atmosphere.2.8. CLSM imaging of polymer complex
To visualize the complex, miR-203 was labeled with Cy5 (miR-203Cy5). Cells were seeded in a 20 mm glass-bottom dish and grown until 60–70% confluency. After medium removal, the HeLa cells were incubated in fresh medium with different polymer complexes of βCD-CPD-miR-203Cy5 (10 μM) and CPT@βCDs-CPD-miR-203Cy5 (10 μM) for 0.5 h, 2 h and 12 h, respectively. The culture medium was removed and the cells on the glass-bottom dish were washed three times with PBS buffer. Then the cell nuclei were stained with Hoechst for 10 min, followed by imaging analysis on a Zeiss LSM 710 Confocal Microscope System at different detection channels (λex = 405 nm, 643 nm; λem = 440–470 nm for Hoechst, 665–740 nm for Cy5, respectively). For the 3D CLSM imaging, the HeLa cells were seeded in a 30 mm dish and incubated for 12 h. After medium removal, the cells were incubated in fresh medium with βCD-CPD-miR-203Cy5 (10 μM) for 2 h, then stained with Hoechst 33342 and LysoTracker™ for 15 min, followed by analysis with the Zeiss LSM 710 Confocal Microscope equipped with a 40× water immersion objective, and with a step size of 0.163 μm (λex = 405 nm, 488 nm and 643 nm; λem = 385–470 nm for Hoechst, 500–540 nm for LysoTracker™ Green and 665–740 nm for Cy5). The acquired images were processed with ZEN software.2.9. MTT cell viability assay and flow cytometry (FACS) experiments
The cytotoxicity of βCD-CPD and CPT@βCD-CPD-miR-203 was studied by using MTT assays in HeLa cells. HeLa cells were seeded in 96-well plates at a density of 5000–7000 cells in each well. After 12 h of incubation, the medium was replaced with 100 μL of fresh DMEM medium containing different formulations of varied concentrations. Untreated cells were run concurrently as negative controls. The same loading amounts of free CPT (0.7 μM, 3.5 μM, 7 μM and 14 μM were dissolved in DMEM containing 1% DMSO respectively) and miR-203 (13.3 nM, 26.5 nM, 66.5 nM and 133 nM respectively) treated cells were used as positive controls. The miR-203 was transfected with Lipofectamine 3000 (Lipo 3000). After 24 h incubation, the plates were washed with PBS buffer and treated with fresh medium containing 0.5 mg mL−1 MTT for another 4 h. Upon careful removal of the medium, the resulting formazan crystals were dissolved in 150 μL of DMSO and the absorbance was recorded at 570 nm with a Thermo MK3 ELISA reader. All experiments were conducted in triplicate, in which the statistical mean and standard deviation were used to estimate cell viability. For FACS experiments, HeLa cells were seeded in 6-well plates and cultured until the cell density reached 70–80% of confluence. After medium removal, the cells were washed with PBS buffer, and then incubated in fresh medium with different formulations of varied concentrations. After 24 h incubation, the cells were harvested with trypsin and washed with cold PBS buffer. Untreated cells were run concurrently as negative controls. PI and annexin V-FITC staining were performed with a commercially available FITC annexin V Apoptosis Detection Kit by following the manufacturer's guidelines. Briefly, cells were resuspended in 1× binding buffer at a concentration of 1 × 106 cells per mL. Then 100 μL cells were transferred to a 1.5 mL culture tube, followed by sequential addition of 5 μL of FITC annexin V, 10 μL PI and 400 μL 1× binding buffer. The cells were subsequently analyzed on a BD Accuri™ C6 cell analyzer.To quantify the cellular uptake, HeLa cells seeded in a 6-well plate were incubated with βCD-CPD-miR-203Cy5 or free miR-203Cy5 for 2 h. The cells were digested and the suspensions were centrifuged, pelleted in eppendorf tubes, washed twice with cold PBS buffer and re-suspended in 500 μL PBS. The cells were subsequently analyzed on a BD Accuri™ C6 cell analyzer.
2.10. Western blotting (WB) of survivin protein
HeLa cells were seeded in 6-well plates and cultured until the cell density reached 70–80% of confluence. Upon medium removal, the cells were incubated in fresh medium with different formulations. Untreated cells were set as controls. After 12 h incubation, the cells were washed with PBS buffer three times and collected. The cell pellets were lysed in the Laemmli buffer (62.5 mM Tris-HCl, pH 6.8, 20% glycerol, 2% SDS, 2 mM DTT, phosphatase inhibitor and proteinase inhibitor cocktail) and boiled at 100 °C for 10 min. Protein concentrations were determined by using Bradford Protein Assay (Bio-Rad, #162-0177). Subsequently, WB analysis was carried out by using the appropriate primary and secondary antibodies.2.11. Cell migration assay
HeLa cells were seeded in a 30 mm dish and incubated for 12 h. After incubation, the cells on the upper side of the chamber were removed by using a needle. And then the cells were incubated in fresh medium with different formulations and different periods of time. The number of invading cells was enumerated under a light microscope equipped with a 10× objective lens.3. Results and discussion
3.1. Synthesis and characterization of βCD-CPD
The βCD-CPD-containing multi-arm star polymers were synthesized according to our previous literature with suitable modifications.16,17 As shown in Scheme 1, βCD-CPD is synthesized from a thiol-modified βCD (βCD-SH, as initiators/drug carriers), a guanidinium-propagating monomer and a terminator, grown directly on βCD substrates in solution by ring-opening disulfide-exchange polymerization. The synthesized βCD-CPD was successfully confirmed by 1H NMR (Fig. S5†). In addition, the number of guanidinium cation units on each βCD core was determined to be 33.1 according to the 1H NMR spectroscopy of βCD-CPD. The number and weight-averaged molecular weights (Mn and Mw) and polydispersity index (PDI = Mw/Mn) of 1.32 were determined by gel permeation chromatography (GPC) (Fig. S6†).3.2. Formation of inclusion complex and GSH-triggered release of miR-203 and CPT from the CPT@βCD-CPD-miR-203 complex
Subsequently, CPT molecules were encapsulated into the hydrophobic β-CD cores by dissolving βCD-CPD into a CPT suspension solution in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio and stirring for 24 h in the dark. The DL of encapsulated CPT was determined to be 69.1% (mol% per βCD) by UV-vis measurement (Fig. S7†). In our study, miR-203 was chosen as a model miRNA. Previous studies showed that miR-203 functions as a key tumor suppressor in various cancers by inhibiting the endogenous expression of its cellular protein targets, including survivin and Bmi-1.32 Correspondingly, its over-expression significantly reduces tumor growth and metastasis to the bone in nude mice.33,34 The CPT@βCD-CPD-miR-203 complex was prepared by adding different ratios of miR-203 in a CPT@βCD-CPD solution. Gel retardation assay was first performed to investigate the miR-203 binding capabilities of βCD-CPD. As shown in Fig. 1a, βCD-CPD could effectively bind with miR-203 and completely complex with miR-203 at a βCD-CPD/miRNA molar ratio of 40 (N/P, μmol/μmol). The diameters of the newly synthesized inclusion complexes in the presence of miR-203 (N/P = 40) were ∼60 nm according to TEM and DLS studies (Fig. 1b & c). Furthermore, the in vitro controlled release of miR-203 was successfully observed by gel electrophoresis, in the presence of either GSH or a HeLa cell lysate (which contains mini-molar concentrations of GSH). The in vitro release profiles of CPT from the CPT@βCD-CPD-miR-203 complex were assessed using the dialysis technique in the presence and absence of GSH. As shown in Fig. 1c, the addition of GSH could promote the CPT release behaviour, which is probably due to the GSH-induced dissociation of the complex. In addition, GSH-triggered degradation of βCD-CPD resulting in changes in particle size was also confirmed via DLS measurements (Fig. 1d).
:1 molar ratio and stirring for 24 h in the dark. The DL of encapsulated CPT was determined to be 69.1% (mol% per βCD) by UV-vis measurement (Fig. S7†). In our study, miR-203 was chosen as a model miRNA. Previous studies showed that miR-203 functions as a key tumor suppressor in various cancers by inhibiting the endogenous expression of its cellular protein targets, including survivin and Bmi-1.32 Correspondingly, its over-expression significantly reduces tumor growth and metastasis to the bone in nude mice.33,34 The CPT@βCD-CPD-miR-203 complex was prepared by adding different ratios of miR-203 in a CPT@βCD-CPD solution. Gel retardation assay was first performed to investigate the miR-203 binding capabilities of βCD-CPD. As shown in Fig. 1a, βCD-CPD could effectively bind with miR-203 and completely complex with miR-203 at a βCD-CPD/miRNA molar ratio of 40 (N/P, μmol/μmol). The diameters of the newly synthesized inclusion complexes in the presence of miR-203 (N/P = 40) were ∼60 nm according to TEM and DLS studies (Fig. 1b & c). Furthermore, the in vitro controlled release of miR-203 was successfully observed by gel electrophoresis, in the presence of either GSH or a HeLa cell lysate (which contains mini-molar concentrations of GSH). The in vitro release profiles of CPT from the CPT@βCD-CPD-miR-203 complex were assessed using the dialysis technique in the presence and absence of GSH. As shown in Fig. 1c, the addition of GSH could promote the CPT release behaviour, which is probably due to the GSH-induced dissociation of the complex. In addition, GSH-triggered degradation of βCD-CPD resulting in changes in particle size was also confirmed via DLS measurements (Fig. 1d).
 | ||
| Fig. 1 (a) Gel retardation assay of complexes of βCD-CPD with miR-203 at different ratios and treated with GSH (10 mM) or HeLa cell lysates (1 mg per mL of proteins) for 1 h at 37 °C, respectively; (b) DLS determination of βCD-CPD-miR-203 (N/P = 40) and the TEM image (inset); (c) in vitro CPT release profiles from the CPT@βCD-CPD-miR-203 complex in the presence and absence of GSH (10 mM) at 37 °C; (d) size changes of the βCD-CPD-miR-203 complex triggered by GSH (5 mM) for different periods of time. | ||
3.3. Cellular uptake of βCD-CPD-miR-203 complex
In order to demonstrate the cellular uptake of our complexes, and their subcellular localization, we first used Cy5-labelled miR-203 to form the complex (without CPT loading) and confocal laser scanning microscopy (CLSM) was performed for convenient assessment in live cells. Real-time imaging experiments were initially carried out. As shown in Fig. 2a, in the first 30 min, most βCD-CPD-miR-203Cy5 had accumulated around the cell membrane, and as incubation continued, the nanoparticles continuously crossed the cell membrane and were eventually distributed throughout the cytosol (excluding nucleus). However, naked miR-203Cy5 without the assistance of βCD-CPD did not enter cells even after prolonged incubation in live cells (12 h, Fig. 2b), which is consistent with previous reports.17,35 These results demonstrated that βCD-CPD could significantly improve the cellular uptake of miRNAs. A similar conclusion was made from flow cytometric analysis (Fig. 2c). We also noted the slight slower cellular uptake of βCD-CPD-miR-203Cy5 when compared to the uptake of previously reported CPD-conjugated proteins (which took <30 min for cell uptake16) – this is presumably due to significantly larger sizes of our complexes.17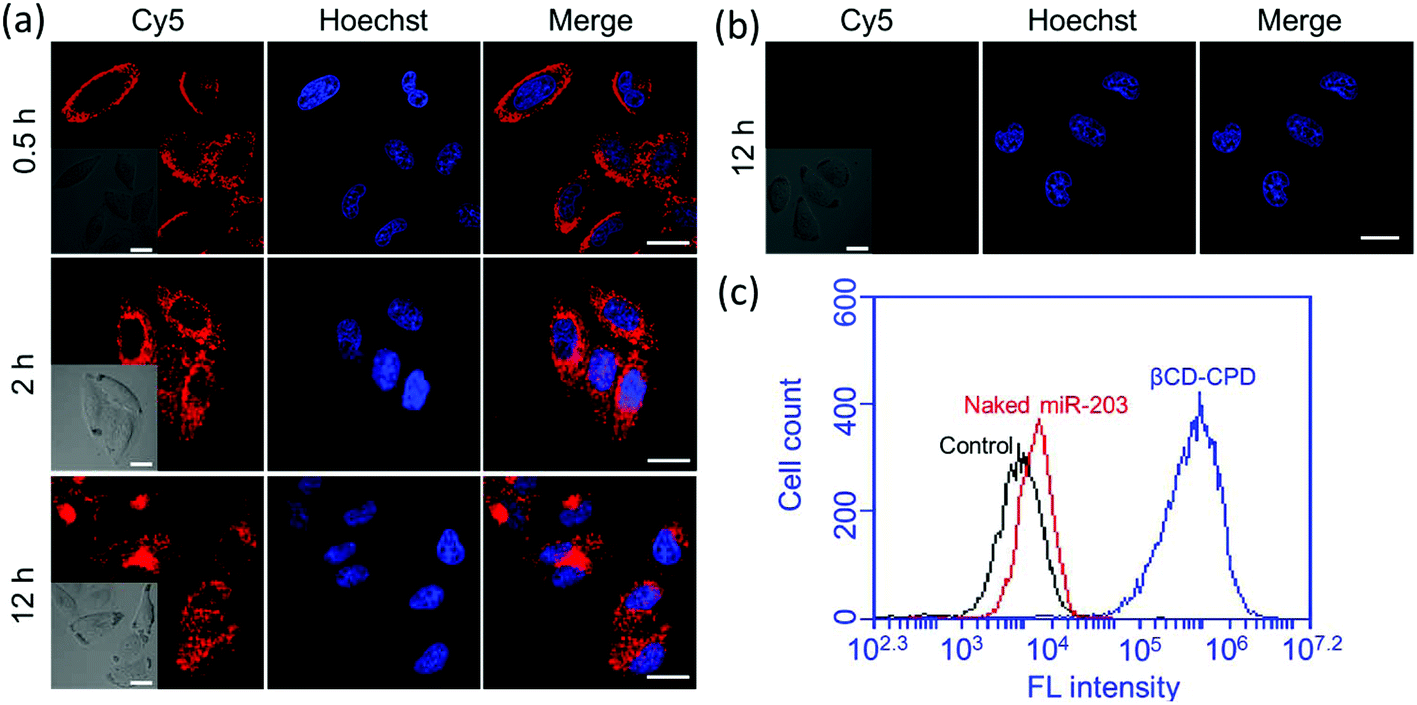 | ||
| Fig. 2 (a) CLSM images of HeLa cells incubated with βCD-CPD-miR-203Cy5 (10 μM; in red) for different periods of time. Cells were stained with Hoechst (in blue); (b) CLSM images of HeLa cells incubated with miR-203Cy5 (in red) for 12 h. Cells were stained with Hoechst (in blue). Scale bar = 20 μm. (c) Flow cytometry analysis of HeLa cells incubated with either βCD-CPD-miR-203Cy5 or naked miR-203Cy5 (10 μM each) for 2 h. | ||
Recent studies have shown that large CPD-assisted cargos could be rapidly delivered to mammalian cells via endocytosis-independent pathways.13–17 We decided to investigate whether or not the same holds true for our newly developed βCD-CPD-based drug delivery system. Initially, 3D CLSM was performed to track the intracellular distribution of the βCD-CPD-miR-203Cy5 complex. As shown in Fig. 3a and ESI Video,† after 2 h incubation with HeLa cells, the βCD-CPD-miR-203Cy5 complex has successfully been transported inside cells and was eventually distributed throughout the cytosolic space, with no evidence of endolysosomal trapping. Furthermore, the global Pearson's R value was calculated to be 0.31 between our complex (shown in Cy5 channel) and LysoTracker™, which is far below the threshold of >0.5 required for correlation.36 Next, flow cytometry analysis was further carried out to quantitatively assess the uptake studies in HeLa cells at different temperatures, as well as with known endocytosis inhibitors (including chlorpromazine, wortmannin and methyl-β-cyclodextrin). As shown in Fig. 3c, the cell uptake efficiency of our polymer complex was not significantly affected by endocytosis inhibitors. Previous cell uptake studies with CPD-loaded cargos showed that thiol-mediated CPD uptake mechanisms were temperature-dependent,13–17 and in the current βCD-CPD-miR-203Cy5 complex, we noted that a low temperature (4 °C) reduced the cell uptake ability on our complex but did not block the process completely. As expected, blocking free thiols on the cell surface by treating the cells with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) significantly suppressed the cell uptake efficiency, clearly supporting a thiol-mediated cargo translocation mechanism as previously reported.10,16,17
 | ||
| Fig. 3 (a) 3D CLSM projections showing Z-stack images at different viewpoints (step size, 0.163 μm) of HeLa cells incubated with βCD-CPD-miR-203Cy5 (10 μM) for 2 h, followed by staining with LysoTracker™ (green) and Hoechst (blue). Global Pearson's R value between Cy5 channel and LysoTracker™ was calculated to be 0.31 with ZEN software. See ESI Video and Fig. S8† for details. White arrows indicate no trapping between the Cy5 channel and LysoTracker™; (b) the corresponding orthogonal images of (a); (c) flow cytometry quantification of the βCD-CPD-miR-203Cy5 complex uptake (2 h incubation) by HeLa cells treated with different inhibitors including chlorpromazine (Chlor), wortmannin (Wort), methyl-β-cyclodextrin (M-βCD), and 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), and at low temperature (4 °C). | ||
3.4. Synergistic delivery of miRNAs and small molecule drugs with CPT@βCD-CPD-miR-203 complex and subsequent therapeutic effects
Having successfully demonstrated the in vitro GSH-responsive miR-203 release and excellent cell uptake efficiency of our newly developed βCD-CPD-based complex, we next investigated its ability to cause RNA interference (RNAi). Up-regulation of the miR-203 was previously shown to suppress cancer cell migration and proliferation.37 In a wound-healing and Matrigel invasion assay with HeLa cells treated with the βCD-CPD-miR-203 complex, successful delivery of miR-203 followed by concurrent inhibition of cell migration/invasion was clearly observed in Fig. 4. However, βCD-CPD only and the negative control βCD-CPD-21A (loaded with Poly21A DNA) did not affect the cell migration/invasion, which successfully demonstrated that our βCD-CPD could serve as a potential platform for gene delivery. | ||
| Fig. 4 (a) Cell migration analysis of HeLa cells upon treatment with different complex systems (15 μM) for 24 h and 48 h, respectively; (b) analysis of migration of HeLa cells after treatment with different complex systems (15 μM) for different periods of time. Error bar was calculated from three independent data. | ||
We next investigated the CPT@βCD-CPD-miR-203 system for simultaneous co-delivery of two mechanistically distinct drugs to achieve combination treatment in cancer cells. As shown in Fig. 5a, the HeLa cells treated with the CPT@βCD-CPD-miR-203Cy5 complex showed successful intracellular CPT and miR-203 release after 2 h incubation. Furthermore, MTT cell viability and FACS experiments were performed to assess subsequent cell killing effects of the released miR-203 and CPT in the HeLa cells treated with the CPT@βCD-CPD-miR-203 complex. As shown in Fig. 5b & Fig. S9,† a synergistic enhanced cell killing effect by our CPT@βCD-CPD-miR-203 complex treatment was achieved in comparison with single-cargo delivery systems (CPT@βCD-CPD & βCD-CPD-miR-203). Notably, our βCD-CPD exhibited minimal toxicity up to a concentration of 20 μM as shown in Fig. 5b. Thus, these results clearly indicate that our βCD-CPD system could serve as a robust platform for synergistic intracellular cargo delivery/release, as well as a combination therapeutic effect for potential disease treatments with two mechanistically distinct inhibitors.
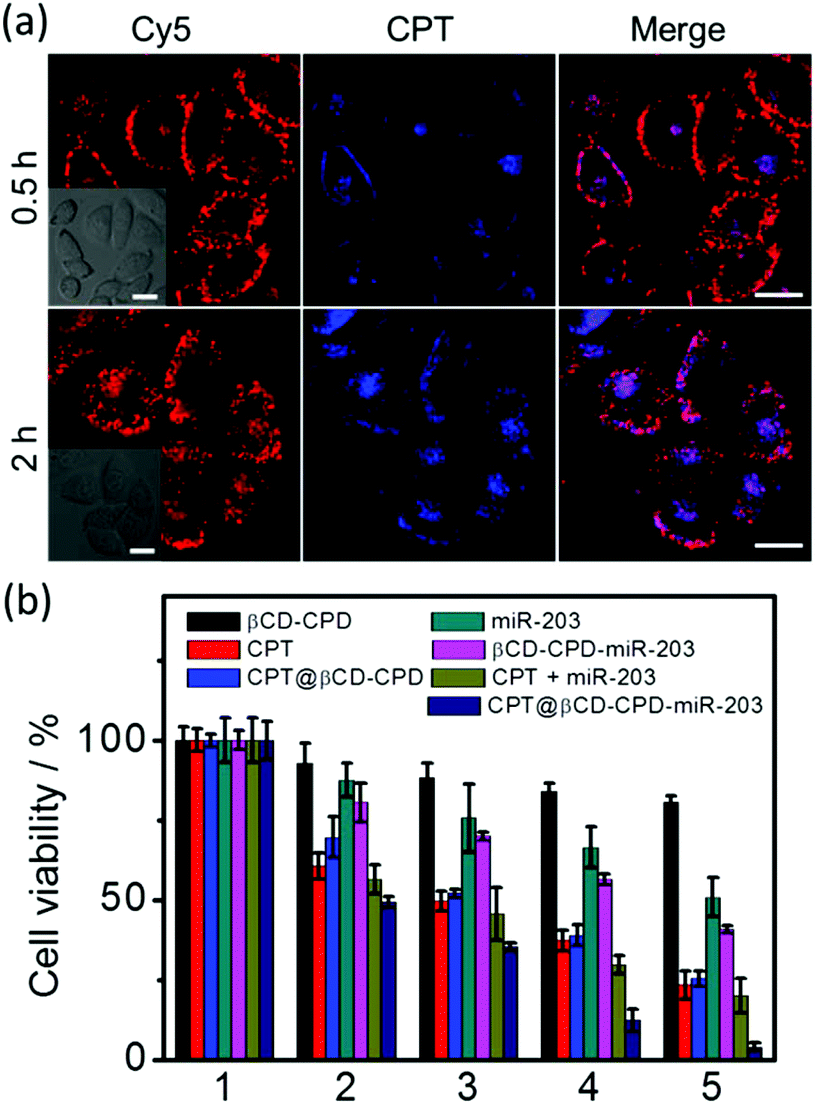 | ||
| Fig. 5 (a) CLSM images of HeLa cells incubated with CPT@βCD-CPD-miR-203Cy5 (10 μM; in red) for different periods of time. Scale bar = 20 μm. (b) Cell viability of HeLa cells treated with different complexes at various concentrations for 24 h. The concentrations are 1: control, 2: 1 μM, 3: 5 μM, 4: 10 μM, and 5: 20 μM of βCD-CPD complex. The same loading amounts of CPT and miR-203 were added as positive controls. All experiments were conducted in triplicate. | ||
Previous studies showed that successful delivery of either miR-203 or CPT to cancer cells led to down regulation of survivin protein expression.38 Herein, we prepared different complexes and incubated them with HeLa cells for 24 h. Western blotting (WB) assay was next performed to assess the endogenous survivin expression levels. As shown in Fig. 6, apparent enhanced inhibition of endogenous survivin protein expression upon treatment of the CPT@βCD-CPD-miR-203 complex was successfully observed. Interestingly, we found that our delivery system displayed more effective inhibition on survivin than Lipofectamine 3000 (a commercial transfection reagent), which was probably due to more intracellular release of miR-203 as a result of endocytosis-independent pathways and GSH-triggered CPD degradation endowed in our delivery system. All these lines of evidence lend further support that our newly developed polymers could be a promising platform for highly effective intracellular cargo delivery.
 | ||
| Fig. 6 (a) WB analysis of inhibition of endogenous survivin protein expression in HeLa cells treated with different complexes (10 μM) or transfection of naked miR-203 with Lipo 3000; (b) intensity of endogenous survivin protein expression in the WB analyzed with ImageJ software. Error bar was calculated from three independent experiments. | ||
4. Conclusions
In summary, we have successfully developed a novel βCD-CPD-based star-like system for synergistic delivery of chemotherapeutic drugs and miRNAs. This system showed several advantages, including rapid cell uptake without apparent endolysosomal trapping, intracellular controlled release of miRNAs triggered by endogenous stimuli, and combination treatment via chemo- and gene therapy. In particular, the intracellular CPD arms of the βCD-CPD polymer underwent rapid degradation under a highly reductive cytosolic environment and caused subsequent in situ release of miRNAs, thus providing superior performance in gene transfection efficiency. In this study, by having successfully confirmed this “smart” multi-drug carrier with promising advantages, we believe that our newly developed βCD-CPD platform will show potential for future development of personalized polymers for different biomedical applications.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Project no. 51403065), the Science and Technology Planning Project of Guangzhou (Project no. 201607020015), the Science and Technology Planning Project of Guangdong Province (Project no. 2014A010105009), the Open Project of State Key Laboratory of Supramolecular Structure and Materials (sklssm201730), the State Key Laboratory of Pulp and Paper Engineering (201706), the Natural Science Foundation of Guangdong Province (2016A030312002) and the Fundamental Research Funds for the Central Universities (2015ZY013, D2174290), National Medical Research Council (CBRG/0038/2013) and an NUS-NNI collaborative grant (R-143-000-658-133), Singapore. We also thank Dr Jianxun Ding (Changchun Institute of Applied Chemistry) for the GPC measurement and discussion.Notes and references
- R. S. Pillai, S. N. Bhattacharyya and W. Filipowicz, Trends Cell Biol., 2007, 17, 118–126 CrossRef CAS PubMed.
- W. Filipowicz, Cell, 2005, 122, 17–20 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef CAS PubMed.
- G. A. Calin and C. M. Croce, Nat. Rev. Cancer, 2006, 6, 857–866 CrossRef CAS PubMed.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- (a) Y. Huang, D. Wang, X. Zhu, D. Yan and R. Chen, Polym. Chem., 2015, 6, 2794–2812 RSC; (b) C. Yu, X. Li, F. Zeng, F. Zheng and S. Wu, Chem. Commun., 2013, 49, 403–405 RSC; (c) J. Xu, F. Zeng, H. Wu, C. Yu and S. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 9287–9296 CrossRef CAS PubMed.
- (a) D. Wang, T. Zhao, X. Zhu, D. Yan and W. Wang, Chem. Soc. Rev., 2015, 44, 4023–4071 RSC; (b) J. Sun, F. Zeng, H. Jian and S. Wu, Polym. Chem., 2013, 4, 5810–5818 RSC; (c) H. Xie, F. Zeng, C. Yu and S. Wu, Polym. Chem., 2013, 4, 5416–5424 RSC.
- (a) C. Yu, Y. Wu, F. Zeng, X. Li, J. Shi and S. Wu, Biomacromolecules, 2013, 14, 4507–4514 CrossRef CAS PubMed; (b) Y. Wang and X. Gong, J. Mater. Chem. A, 2017, 5, 3759–3773 RSC; (c) Y. Wu, J. Wang, F. Zeng, S. Huang, H. Xie, C. Yu and S. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 1511–1519 CrossRef CAS PubMed.
- (a) E. K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed; (b) Y. Wu, S. Huang, F. Zeng, J. Wang, C. Yu, J. Huang, H. Xie and S. Wu, Chem. Commun., 2015, 51, 12791–12794 RSC.
- (a) N. Chuard, G. Gasparini, D. Moreau, S. Lörcher, C. Palivan, W. Meier, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 2947–2950 CrossRef CAS PubMed; (b) P. Liu, J. Xu, D. Yan, P. Zhang, F. Zeng, B. Li and S. Wu, Chem. Commun., 2015, 51, 9567–9570 RSC.
- (a) G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed; (b) B. Li, P. Liu, D. Yan, F. Zeng and S. Wu, J. Mater. Chem. B, 2017, 5, 2635–2643 RSC.
- D. Abegg, G. Gasparini, D. G. Hoch, A. Shuster, E. Bartolami, S. Matile and A. Adibekian, J. Am. Chem. Soc., 2017, 139, 231–238 CrossRef CAS PubMed.
- G. Gasparini, G. Sargsyan, E.-K. Bang, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 7238–7331 CrossRef PubMed.
- G. Gasparini, E.-K. Bang, J. Montenegro and S. Matile, Chem. Commun., 2015, 51, 10389–10402 RSC.
- P. Yuan, X. Mao, K. C. Chong, J. Fu, S. Pan, S. Wu, C. Yu and S. Q. Yao, Small, 2017 DOI:10.1002/smll.20170056.
- J. Fu, C. Yu, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153–12160 CrossRef CAS PubMed.
- C. Yu, L. Qian, J. Ge, J. Fu, P. Yuan, S. C. Yao and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 9272–9276 CrossRef CAS PubMed.
- J. Li, Y. Wang, Y. Zhu and D. Oupicky, J. Controlled Release, 2013, 172, 589–600 CrossRef CAS PubMed.
- (a) M. Brewster and M. E. Davis, Nat. Rev. Drug Discovery, 2004, 3, 1023–1035 CrossRef PubMed; (b) C. Yu, M. Luo, F. Zeng, F. Zheng and S. Wu, Chem. Commun., 2011, 47, 9086–9088 RSC.
- K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045–2076 CrossRef CAS PubMed.
- M. Qi, S. Duan, B. Yu, Y. Hao, T. Wei and F. Xu, Polym. Chem., 2016, 7, 4334–4341 RSC.
- J. Sheng, Y. Wang, L. Xiong, Q. Luo, X. Li, Z. Shen and W. Zhu, Polym. Chem., 2017, 8, 1680–1688 RSC.
- D. B. Pacardo, B. Neupane, S. M. Rikard, Y. Lu, R. Mo, S. R. Mishra, J. B. Tracy, G. Wang, F. S. Ligler and Z. Gu, Nanoscale, 2015, 7, 12096–12103 RSC.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- F. Zhao, H. Yin and J. Li, Biomaterials, 2014, 35, 1050–1062 CrossRef CAS PubMed.
- J. Deng, N. Li, K. Mai, C. Yang, L. Yan and L.-M. Zhang, J. Mater. Chem., 2011, 21, 5273–5281 RSC.
- Z. Luo, K. Cai, Y. Hu, J. Li, X. Ding, B. Zhang, D. Xu, W. Yang and P. Liu, Adv. Mater., 2012, 24, 431–435 CrossRef CAS PubMed.
- K. Liu, X. Jiang and P. Hunziker, Nanoscale, 2016, 8, 16091–16156 RSC.
- A. Steffen, C. Thiele, S. Tietze, C. Strassnig, A. Kamper, T. Lengauer, G. Wenz and J. Apostolakis, Chem. – Eur. J., 2007, 13, 6801–6809 CrossRef CAS PubMed.
- V. J. Venditto and E. E. Simanek, Mol. Pharmaceutics, 2010, 7, 307–349 CrossRef CAS PubMed.
- E. A. Kaifer, T. M. Rojas, R. Koniger and J. F. Stoddart, J. Am. Chem. Soc., 1995, 117, 336–343 CrossRef.
- Y. Zhang, S. Zhou, H. Yan, D. Xu, H. Chen, X. Wang, X. Wang, Y. Liu, L. Zhang, S. Wang, P. Zhou, W. Fu, B. Ruan, D. Ma, Y. Wang, Q. Liu, Z. Ren, Z. Liu, R. Zhang and Y. Wang, Sci. Rep., 2016, 6, 19995 CrossRef CAS PubMed.
- M. J. Bueno, I. Pérez de Castro, M. Gómez de Cedrón, J. Santos, G. A. Calin, J. C. Cigudosa, C. M. Croce, J. Fernández-Piqueras and M. Malumbres, Cancer Cell, 2008, 13, 496–506 CrossRef CAS PubMed.
- Y. Yuan, Z. Zeng, X. Liu, D. Gong, J. Tao, H. Cheng and S. Huang, BMC Cancer, 2011, 11, 57–63 CrossRef CAS PubMed.
- X. Lu, F. Jia, X. Tan, D. Wang, X. Cao, J. Zheng and K. Zhang, J. Am. Chem. Soc., 2016, 138, 9097–9100 CrossRef CAS PubMed.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol.: Cell Physiol., 2011, 300, C723–C742 CrossRef CAS PubMed.
- W. Wang, W. Liu, H. Sun, D. Chen, X. Yao and J. Zhao, Cell Biochem. Funct., 2013, 31, 82–85 CrossRef PubMed.
- K. Mizutani, K. Matsumoto, N. Hasegawa, T. Deguchi and Y. Nozawa, Exp. Oncol., 2006, 28, 209–215 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic schemes, 1H NMR spectra, determination of graft ratio, mass spectra, photographs and FACS data. See DOI: 10.1039/c7py00666g |
| This journal is © The Royal Society of Chemistry 2017 |