
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intermolecular states in organic dye dispersions: excimers vs. aggregates†
A. J.
Musser
 *a,
S. K.
Rajendran
b,
K.
Georgiou
a,
L.
Gai
c,
R. T.
Grant
a,
Z.
Shen
c,
M.
Cavazzini
d,
A.
Ruseckas
b,
G. A.
Turnbull
b,
I. D. W.
Samuel
b,
J.
Clark
a and
D. G.
Lidzey
a
*a,
S. K.
Rajendran
b,
K.
Georgiou
a,
L.
Gai
c,
R. T.
Grant
a,
Z.
Shen
c,
M.
Cavazzini
d,
A.
Ruseckas
b,
G. A.
Turnbull
b,
I. D. W.
Samuel
b,
J.
Clark
a and
D. G.
Lidzey
a
aDepartment of Physics and Astronomy, The University of Sheffield, Sheffield, UK. E-mail: a.musser@sheffield.ac.uk
bOrganic Semiconductor Centre, SUPA, School of Physics and Astronomy, University of St. Andrews, St. Andrews, UK
cState Key Laboratory of Coordination Chemistry, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, China
dInstituto di Scienze e Tecnologie Molecolari (ISTM), Consiglio Nazionale delle Ricerche (CNR), via C. Golgi 19, 20133 Milan, Italy
First published on 5th July 2017
Abstract
Rapid excited-state quenching in the solid state is a widespread limitation for organic chromophores. Even when molecules are dispersed in neutral host matrices, photoluminescence quantum yields decrease sharply with increased concentration, pointing to efficient intermolecular non-radiative decay pathways that remain poorly understood. Here we study the nature of the intermolecular states formed in dispersions of the prototypical BODIPY dyes. Using temperature-dependent and time-resolved photoluminescence measurements, we describe the processes of energy transfer into excimer states and, in materials with suitable chemical structure, excitonically coupled dimers. These dimer states exhibit remarkable near-unity quantum yield.
Introduction
Organic chromophores such as rhodamine and BODIPY are famed for their photostability and high quantum efficiency,1–5 which make them excellent candidates for labelling and sensing applications.6,7 It is typical of such high-efficiency dyes to exhibit negligible relaxation in the excited state and thus very small Stokes shifts. The large consequent overlap of absorption and emission spectra results in the condensed phase in substantial re-absorption of emitted light through the inner-filter effect. The rigid, planar geometry of these chromophores also leads to a propensity to aggregate through π–π stacking, often resulting in new quenching pathways. These factors together severely reduce the applicability of such dyes in solid-state plastic electronics such as solar cells and light-emitting diodes.5 Significant effort has been put into the modification of dyes such as BODIPY, both to reduce intermolecular interactions within neat films in order to suppress aggregation and to increase the Stokes shift to reduce self-absorption.8–11 These attempts have yielded some improvements, though the PL quantum efficiency from modified dye films often remains low. Indeed, even when the molecules are dispersed in neutral polymer matrices to prevent direct inter-chromophore interactions, rapid excited-state quenching results in a substantial reduction in the PL quantum yield.10,12In contrast to synthetic efforts to improve condensed-phase dye efficiency, relatively little attention has been paid to the detailed mechanism of singlet exciton quenching in solid films. In the case of BODIPY dyes, it has been well documented in a wide range of derivatives that dispersion within a polymer matrix or deposition of pure films results in significant PL quenching accompanied by spectral shifts.9,10,12–21 Analogous effects have also been described in a wide range of covalent dimers,11,15,21–26 though the geometries are typically a poor match for the structures obtained in the solid state. Quenching in these systems is often attributed to the formation of non-emissive H-aggregates, identified by a broadening of the absorption spectrum at higher energies.10,15–17,26,27 Red-shifted PL spectral features have been variously attributed to excimers and J-aggregates, typically on the basis of their spectral position alone.6,9,12–14,19–24 The properties of these emissive states have not been analyzed in detail, nor the mechanism of their formation. We have recently demonstrated that the excimer state formed in a high-concentration BODIPY dispersion enables exciton-polariton formation through a novel radiative pumping mechanism,28 highlighting the potential of these low-energy states for applications if their properties can be better understood.
To develop a clearer understanding of the intermolecular states that can form in such organic chromophore dispersions, and with the aim of eventually controlling them to mitigate quenching effects, we have studied a family of prototypical BODIPY dyes. The dyes, presented in Fig. 1, were dispersed in polystyrene matrices at a range of loading concentrations from 0.1% to 20%. These films were studied with a range of temperature-dependent and time-resolved PL spectroscopies, enabling description of the interplay of emissive states with unprecedented detail. We find that the excited-state dynamics are dominated by long-range singlet energy transfer through the film to a small population of low-energy emissive sites, and the nature of these sites divides the dyes into two distinct classes (Fig. 1). In class E (excimer), the singlet is primarily quenched by energy transfer to excimer-forming sites. These excimer states have high non-radiative decay rates and thus low quantum efficiency, but this is significantly improved at low temperatures. In class A (aggregate), the decay of singlet excitons is again dominated by transfer to excimer-forming sites, but there is parallel transfer to distinct, strongly red-shifted aggregates. Similar species are often attributed to BODIPY J-aggregates,6,12–17 but the observed time-resolved behaviour and temperature dependence reveal this to be an over-simplification. The states must be described specifically as J-coupled dimers, exhibiting exceptional near-unity quantum efficiency.
 | ||
| Fig. 1 Two classes of BODIPY dye. Chemical structures and normalised UV-Vis absorption (filled) and PL spectra (lines) of BODIPY derivatives used in this study. Molecules were dispersed in polystyrene matrix, each at three different loading ratios (1–20% by weight in solution). In the ‘E’ class, increasing the concentration results in a broadening at long wavelengths, consistent with the formation of excimers. High concentrations in the ‘A’ class yield strongly red-shifted, highly structured spectral features consistent with ground-state aggregates. PL spectra were acquired from excitation at 473 nm. | ||
Experimental
BODIPY dyes E1, E2, E3, A1, A2 and A3 were synthesised as per previous reports (see ESI†).9,29–35 Solutions for spin-coating were prepared from a stock solution of 35 mg mL−1 polystyrene in toluene. BODIPY dye powders were dissolved in this to final concentrations of 0.1–20 wt%, and the solutions were spin-coated on cleaned quartz substrates (2000 rpm, 55 s) to afford films of ∼200 nm thickness.Steady-state photoluminescence measurements were acquired on two systems, each equipped with a N2 bath cryostat. Temperature-dependent PL excitation maps were acquired using a Fluoromax fluorimeter (Horiba), with excitation and emission wavelength bandpass of 1 nm. Higher-sensitivity measurements were performed with a FLS980 fluorimeter (Edinburgh Instruments) utilizing double monochromators for excitation and emission, using similar wavelength bandpass. Absorption spectra were detected on the Fluoromax system through the transmission channel. Higher-resolution PL spectra were acquired on a custom setup using 480 nm excitation from a SC400 Fianium supercontinuum laser filtered by a SPEX 270M monochromator and detection with a Newton CCD coupled to a Shamrock spectrograph (Andor). During temperature-dependent measurements, the sample was allowed to equilibrate at each temperature for >15 minutes prior to measurement. Concentration-dependent PL spectra were acquired using a 473 nm laser diode. PL quantum yield was measured following excitation at 400 nm using a frequency-doubled mode-locked Coherent Mira 900 Ti:Sapphire crystal pumped by a 532 nm Verdi V10, focused onto the films inside an integrating sphere. PLQY was calculated by comparing the relative intensities of the incident laser with and without the sample present.
Photoluminescence dynamics <2 ns were measured using a streak camera (Hamamatsu C10910, resolution ∼2 ps) coupled to a Pharos femtosecond laser system (Light Conversion). Excitation pulses at 400 nm (180 fs) were generated using an Orpheus OPA. Excitation power was kept in the range 50–300 μW (0.5–3 nJ per pulse) using neutral density filters, and control measurements were performed at higher and lower excitation density to ensure dynamics are representative. PL dynamics at longer time delays were collected using a time-correlated single-photon counting system, with detection wavelengths scanned across the full emission bandwidth using a monochromator. Transient absorption measurements were performed on a Harpia system pumped with the output of a Pharos femtosecond laser system, reduced to 50 kHz with a pulse picker and frequency-doubled with an Orpheus OPA to 400 nm (Light Conversion).
Results
Fig. 1 presents the absorption spectrum of each BODIPY dye at low concentration (filled) and normalised PL spectra of polystyrene films with progressively higher loading content. At low loading the emission resembles that of isolated BODIPY molecules in solution, enabling its assignment to monomer emission. At high concentration, we observe clear signatures of the inner-filter effect. The energetic position of the highest-energy emission peak or shoulder progressively red-shifts as the concentration increases, as any photons emitted near the absorption edge are increasingly likely to be re-absorbed before escaping the film. This self-absorption behaviour is a well-known consequence of the small Stokes shift in BODIPYs, but it is unable to fully explain the spectral shapes in Fig. 1. In addition to the shifting PL edge, we observe spectral changes in all dyes at high concentration indicative of new, lower-energy emissive species. The effects are particularly striking in the A class, where a new state with a sharp vibronic progression is formed. These features are similar to some previous reports of BODIPY dimer or J-aggregate emission, though in our films they are unusually strongly shifted and well-defined.13,15–17,25 In none of the films did we observe changes in the main BODIPY absorption band at 2.3–2.8 eV, aside from slight broadening and a possible enhancement of the 0–1 absorption band (Fig. S1, ESI†). Nor did we detect any new features in the absorption that could correspond to the emissive species seen at high loading: if there are aggregates present, they must be present in very low concentration. We note that in agreement with previous reports, the formation of both types of lower-energy emitters is accompanied by partial quenching of the PL due to the introduction of new pathways for non-radiative decay (Fig. S2, ESI†).9,10,12–21 However, in both film types this quenching saturates for high loading at remarkably high efficiencies (>0.10) for a concentrated BODIPY film.We have observed that the emissive species in films of E1 and A1 exhibit different temperature dependence, enabling easy identification of the distinct spectral components (Fig. 2). In high-concentration films of both E1 and A1, we measure a strong enhancement of the total PL upon cooling (2–4×), marked by the evident growth and progressive blue-shift of one of the lower-energy emissive peaks. We have extracted the ‘intermolecular’ component of this emission by performing the same measurement on low-concentration (i.e. monomer-like) films of each, correcting for the effect of self-absorption near the band edge and subtracting the temperature-dependent monomer contribution (Fig. 2c, d and Fig. S3, S4, ESI†). In both cases, virtually the entire temperature dependence is contained within the ‘intermolecular’ component, though interestingly the highly-structured J-aggregate-type peaks in A1 are completely unaffected by cooling. We will return to this unusual observation below. It is clear that the dominant intermolecular emission in both films exhibits a slight blue-shift during cooling, in contrast to the red-shift found in monomeric films (Fig. S3, ESI†). Moreover, we highlight that the spectral shape of the temperature-dependent component does not change significantly. Indeed, as shown in Fig. 2e, the normalised intermolecular PL spectra of E1 can be shifted to overlap closely. Similar behaviour is observed in A1 films (Fig. S3–S5, ESI†). Based on this consistent spectral shape, we attribute these spectra to the same emissive species at all temperatures. This species contains virtually the entire temperature dependence exhibited in Fig. 2a and b, namely an increase in intensity and energetic position (Fig. 2e, inset) upon cooling.
 | ||
| Fig. 2 Isolation of intermolecular species. Temperature-dependent PL spectra of high-concentration films of (a) E1 and (b) A1 show a strong increase in intensity upon cooling. This effect is almost entirely intermolecular, with no significant shifts or enhancement in dilute films (see ESI†). Subtraction of the monomer contribution, deduced from low-concentration films and corrected for high-concentration self-absorption effects, affords the (c) E1 and (d) A1 intermolecular component, which contains the full temperature dependence. (e) The E1 intermolecular components, normalised and shifted to match peak position, exhibit the same spectral shape at every temperature. This emission arises from a single electronic species, with temperature-dependent energy and PL efficiency. Inset shows the degree each spectrum was shifted to achieve overlap. (f) PL excitation scan (filled) of the low-energy emissive peaks reveals sub-gap absorption bands with distinct vibronic structure mirrored in the PL. Sub-gap excitation at 2.14 eV (squares) and 2.0 eV (circles) yields the same low-energy emission as following excitation of the main BODIPY absorption (red line). Black and grey lines are Gaussian fits to excitation and emission spectra, giving a vibronic spacing of 170 meV. | ||
We have used PL excitation mapping over a range of temperatures on low- and high-concentration films of E1 and A1 (Fig. S6 and S7, ESI†) to investigate whether these intermolecular emitters correspond to ground-state aggregates. No sub-gap absorption features could be identified for the temperature-dependent component, even at low temperatures when its emission is relatively strong. Instead, the PL excitation spectrum in this region resembles that of a dilute film at all wavelengths: the broad, low-energy, temperature-dependent state emitting ∼2.0–2.3 eV is only generated through excitation of monomer-like sites. This behaviour is characteristic of excimers in solution and thin film, and we make the same assignment here. In both film types, these excimer states show high rates of non-radiative decay which are suppressed upon cooling.
By contrast, the sharply structured low-energy emission in concentrated A1 films can be associated with a pair of distinct vibronic PL excitation peaks at 2.13 eV and 1.96 eV (Fig. 2f), well below the A1 band gap. These exhibit close mirror-image symmetry with the structured PL peaks at 1.94 eV and 1.77 eV observed following excitation of the main A1 absorption band (red line). Precisely the same low-energy PL is observed following excitation directly into these sub-gap states at 2.14 eV and 2 eV (red circles). The presence of these PL excitation bands is a hallmark of ground-state aggregation. Another, distinct sub-gap species can be identified in the weak PL band 2.0–2.2 eV. This feature is selectively enhanced following excitation at 2.20–2.25 eV (Fig. S8, ESI†), demonstrating that it too corresponds to a unique ground-state species. It cannot be distinguished in PL excitation due to spectral congestion. For simplicity we refer to the species emitting in this region collectively as the sub-gap emitters (SGE). Such sites should be present in very low concentration in the film, as they are undetectable in linear absorption and >100× weaker than the primary monomer band in PL excitation. Nonetheless, these features dominate the steady-state PL spectrum in high-concentration films, indicating efficient population through energy transfer.
PL spectral dynamics
To better understand the nature and formation of these states, we have performed detailed time-resolved PL measurements of all dyes using a streak camera, scanning a range of concentrations and temperatures. Representative results are presented in Fig. 3 and support the identification of multiple emissive species above. The spectrum detected on the earliest timescales in both classes of film can be identified as monomer emission from comparison to steady-state measurements of dilute films, though the spectrum of this initial species is curtailed by self-absorption. Even at high loading of 10–20%, the predominant absorbing species in these films is isolated, un-coupled BODIPY monomers. In E-class films, the initial monomer signature is rapidly quenched, leaving a broadened and red-shifted species that persists on much longer timescales. In the case of E1 this species closely matches the temperature-dependent excimer described above, and indeed temperature-dependent streak-camera measurements show the same blue-shift and increase in intensity upon cooling (Fig. S14, ESI†). We propose that the similar behaviour observed in E2 and E3 likewise arises from long-lived excimer emission. | ||
| Fig. 3 Fast PL dynamics. (a) Streak-camera measurements of high-concentration E-class films, taken at 200 K (E1) or 295 K (E2 and E3). Normalised spectral cuts (lines; light blue: 50 ps, blue: 200 ps, dark blue: 1500 ps) highlight the evolution of emissive species. The initial spectrum in all cases corresponds to monomer emission detected in dilute films. The final spectrum in E1 matches the excimer component identified above, and equivalent red-shifts and broadening are observed in E2 and E3. (b) Streak-camera measurements of high-concentration A-class films at 295 K. Normalised spectral cuts (lines; pink: 50 ps, red: 200 ps, dark red: 1500 ps) highlight the evolution of emissive species. Initial spectra in all films can be assigned to monomer emission similar to dilute films. The final spectrum in A1 corresponds to the temperature-independent portion of the intermolecular spectrum identified above. The spectral shape at intermediate times indicates the presence of a third, distinct species emitting in the range of the excimer identified from steady-state measurements. Comparable multi-species evolution is evident in A2 and A3. Spectral maps are presented with logarithmic intensity scale to highlight the contribution of weak, long-lived features. | ||
Equivalent measurements in E1 films over a range of dye loading concentrations reveal strong variation of the rate and yield of excimer formation consistent with the steady-state results (Fig. 4). At all concentrations we observe the same initial monomer emission (solid lines), with slight concentration-dependent shifts due to self-absorption. A small contribution from excimer emission can be detected on long timescales (dashed) at 5% and 10% loading, but only at 20% is the yield of excimers high enough that this species dominates the spectrum at 1500 ps. Correspondingly, the quenching of monomer emission (Fig. 4b) becomes increasingly rapid as the concentration increases and average inter-chromophore distance decreases, pointing to an important role for exciton migration through energy transfer to sites capable of excimer formation. The case of the 10% film is instructive, as the very rapid quenching of monomer emission seen in Fig. 4b nonetheless yields only weak overall excimer emission. This result suggests that the excimer state has a low quantum yield, with high rates of non-radiative decay.
 | ||
| Fig. 4 Loading-dependent excimer formation. (a) Streak-camera spectra taken at 50 ps (solid) and 1500 ps (dashed) for E1 films from 0.1% to 20% dye content. The same initial state is detected at all concentrations, with a minor red-shift for 5% and above due to the inner filter effect. Slight signs of the red-shifted excimer emission can be detected at 5% loading, but only at the highest concentration does it dominate the long-time emission. (b) Corresponding normalised PL decay kinetics taken at the high-energy edge of the monomer emission. The initial singlet state is quenched progressively faster as the concentration is increased, indicating a process governed by intermolecular energy transfer. Full data and equivalent measurements of A1 presented in Fig. S11 and S12 (ESI†). | ||
Comparable spectral evolution is detected in A-class films (Fig. 3b), albeit with much more pronounced differences between initial and final states. The initial monomer emission in all films is again rapidly quenched, leaving on long timescales strongly red-shifted and vibronically structured SGE bands consistent with the features highlighted in steady-state measurements. In the case of A1, the strong spectral separation between monomer and SGE peaks enables direct observation at ∼2.0 eV of the SGE signal growth from zero background, demonstrating that this population is not directly photoexcited. As in E-class films, we find instead that the dynamics are most consistent with concentration-dependent energy transfer through the film (Fig. S12, ESI†).
The final spectrum observed in A1 films closely resembles the temperature-independent intermolecular component identified in Fig. 2d, while on intermediate timescales we detect additional PL intensity in the range 2.1–2.4 eV. The spectra in this intermediate regime cannot be described as a linear combination of the early-time and long-lived emitters and demonstrate the presence of a third emissive species, in the range of the temperature-dependent excimer. We gain further insight into these states through temperature-dependent streak-camera measurements, over the range 100–295 K. In Fig. 5a we have normalised the data at each temperature to the initially photogenerated monomer signal, at 50 ps. Precisely the same spectral evolution of the SGE band at ∼2.0 eV is observed over the full temperature range, with no variation in the formation rate (Fig. 5b), yield or apparent SGE spectral shape. Likewise, the monomer dynamics monitored at the high-energy spectral edge (∼2.4 eV) show no change. Similarly temperature-independent monomer decay was observed in high-concentration E1 films (Fig. S14, ESI†). In both classes of BODIPY film the mechanism of monomer quenching must be independent of thermal activation, namely resonance energy transfer. Indeed, the entire temperature dependence in A1 is observed within the intermediate excimer emission band, as anticipated from the steady-state results. The dynamics in Fig. 5c show that in addition to a slight blue-shift of the average excimer PL intensity, cooling results in an enhancement of the excimer PL lifetime. The primary effect of cooling is thus to reduce the non-radiative decay rate in the excimer state.
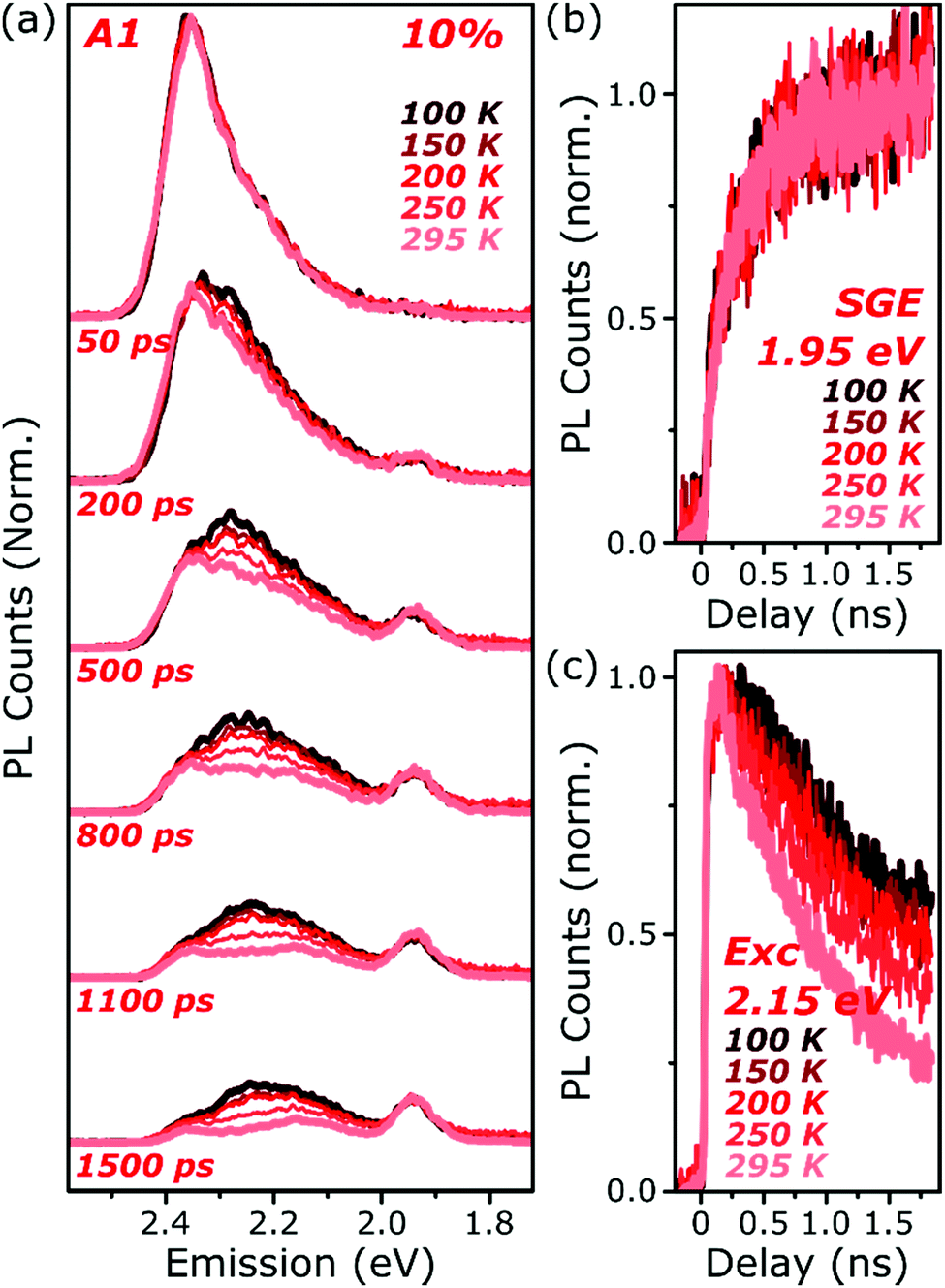 | ||
| Fig. 5 Temperature-dependent excimer and aggregate dynamics. (a) Streak-camera spectra taken at the indicated delay times following photoexcitation, for an A1 film at 10% dye content held at 100–295 K. The datasets were normalised to the intensity of the initial spectral slice; no further scaling was applied for longer time delays. The yield and shape of the SGE band at 1.95 eV and shape of the monomer edge at 2.4 eV are invariant across the temperature series. Only in the intermediate excimer region is a strong dependence observed. (b) Integrated PL kinetics centered on the peak of the SGE band reveal, within measurement limits, identical dynamics at all temperatures. (c) Integrated PL kinetics from the excimer region at 2.15 eV reveal progressive enhancement of the lifetime upon cooling, consistent with a reduction in non-radiative decay rates. Full data and equivalent measurements of E1 presented in Fig. S14 and S15 (ESI†). | ||
Spectral decomposition
Because of the distinct spectral features exhibited by the intermolecular species in all films, we are able to describe the full time-resolved PL evolution using simple spectral decomposition techniques (Fig. 6 and Fig. S9, S10, ESI†). Singular value decomposition of the full streak-camera matrices confirms the presence in E-class films of two distinct spectral species (Fig. S9a, ESI†), and these can be well described as monomer and excimer emission. In all three materials the latter is broadened and slightly red-shifted and exhibits a clear delayed rise that matches the primary decay of the monomer PL. The longest component of the excimer decay is beyond the limits of the measurement; TCSPC measurements of concentrated E1 exhibit complicated multiexponential decays with final lifetime >5 ns (Fig. S16, ESI†). There is, however, a fast component to the excimer decay which is also well resolved in the streak-camera measurements, and this regime reveals good correlation between excimer decay and the residual tail of monomer decay. These results suggest a distribution of excimer lifetimes, with some continual feeding of the shorter-lived excimer population by a reservoir of long-lived monomer excitations. In the context of the energy-transfer-mediated model of monomer exciton quenching, this picture would entail a relatively low density of excimer-capable sites. | ||
| Fig. 6 PL spectral decomposition. (a) Decomposition of time-resolved PL maps of high-concentration E-class BODIPY films affords a good match with two distinct species: the initially photo-generated monomer and weakly red-shifted, structureless excimer emission. E1 data was taken from a 20% film at 200 K, E2 and E3 from 10% films at 295 K. (b) The corresponding population kinetics reveal fast monomer decay (blues) and delayed excimer formation (greens) over 10–100's of ps, consistent with energy migration through the film. Time constants result from global multiexponential fitting. (c) Decomposition of the PL maps of high-concentration (10%) A-class BODIPY films at 295 K requires three distinct species for an adequate fit: the photo-generated monomer (reds), weakly red-shifted excimer (oranges) emission similar to that of the E class and strongly red-shifted SGE (yellows) with vibronic structure. (d) Relative population kinetics of monomer (reds), excimer (oranges) and SGE (yellows) show parallel population of long-lived SGE and shorter-lived excimer emission sites from the rapidly quenched monomer, over 10–100's of ps. Traces reflect relative spectral intensity. Time constants result from global multiexponential fitting. The time constant of excimer growth is similar to SGE, but the fast subsequent decay results in a markedly earlier population peak. | ||
Singular value decomposition of the dynamics in A-class films reveals the presence of three distinct spectral species (Fig. S9b, ESI†). From the analysis above these can be ascribed to the initially photoexcited monomer; a broad and slightly red-shifted excimer, similar to those in E-class films; and a long-lived, sharply structured SGE band. Fitting to the kinetics reveals that the excimer and SGE are not formed sequentially but rather in parallel. The SGE signal observed in time-resolved experiments is very small, though it can make up a significant proportion of the steady-state spectrum due to its long lifetime (>5 ns). The excimer signal is also small, but can be greatly enhanced at low temperature through the suppression of non-radiative decay channels. This indicates that a substantial proportion of excimer states formed are ‘dark’, especially at room temperature, and suggests that excimer formation may dominate the monomer decay dynamics.
Discussion
The similarity of monomer decay kinetics in Fig. 6b and d is striking. It suggests a common mechanism for quenching of the initially photoexcited state robust against the differences in molecular energy levels and the presence of lower-energy molecular aggregates in the ground state. The timescales of the primary decay (typically 100–200 ps) are independent of temperature but vary strongly with concentration of BODIPY dye within the polymer matrix; the latter parameter is similar for all of the data in Fig. 6. As described above, these behaviours point to intermolecular energy transfer as the likely mechanism. The measured timescales are very slow for such a process, however. As a gauge of exciton diffusion in E1 and A1 films, we have measured PL anisotropy dynamics. This parameter decays to near zero on <50 ps timescales (Fig. S13, ESI†). Indeed, even at the low concentration of 1%, rapid loss of PL anisotropy demonstrates fast and efficient energy transfer between sites.Rapid energy migration through the film is to be expected for these dyes. The strong overlap of absorption and monomer PL spectra and typically high BODIPY PLQYs result in a large radius of homo-FRET of ∼5 nm.5,12,14 The 10% dispersions correspond to an average intermolecular separation of only ∼2 nm, ensuring that FRET strongly out-competes monomer fluorescence. Over the 100–200 ps timescale for monomer decay in concentrated films, the excitation should consequently be able to sample a large number of sites. Even a small population of excimer-forming or aggregated sites should then be sufficient to explain the observed behaviour. For instance, we estimate from Fig. 2f that no more than 1/200 of the A1 molecules at 10% loading are aggregated. Thus the dominant process in these BODIPY films is efficient energy transfer between isolated molecules, with eventual quenching through the formation of lower-energy excimer or SGE states.
From the spectral shapes extracted in Fig. 6, the corresponding population kinetics and steady-state quantum yield measurements, we are able to evaluate the population branching ratios and PLQY of each state. Full details are presented in ESI,† and here we summarise the key results. In concentrated E1, the rapid quenching of monomer emission means that 96% of excitations result in excimer formation. This state is very weakly emissive, with a quantum yield of only 1% at 295 K, rising to 10% at 100 K. Monomer quenching is very similar in concentrated A1, and again 96% of all excitations form either excimers or SGE states. The branching between these intermolecular species cannot be fully determined, but the parameter range most consistent with the spectral characteristics of excimers and J-aggregates indicates that weakly emissive excimers are favoured by over 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This in turn implies an SGE PLQY of >75%, and indeed the temperature-independence of this emission suggests it should be near unity.
:1. This in turn implies an SGE PLQY of >75%, and indeed the temperature-independence of this emission suggests it should be near unity.
Nature of the excimer
The excimer states we observe demonstrate that in all of these dispersed dye films some fraction of the molecules are sufficiently close together for direct electronic contact. The excimer is an excited-state dimer, in which mixing with delocalised configurations such as charge-transfer states induces strong geometric relaxation. This stabilised relaxed geometry is dissociative in the ground electronic state and thus no distinct vibronic features appear in its emission. Because the polymer matrix prevents actual molecular diffusion, the state cannot be fully dissociative and the molecules at excimer-forming sites should remain proximate in the ground state. Their interactions do not result in J-aggregation – hence the absence of corresponding sub-gap PL excitation features. The latent excimer pairs are either weakly-coupled ‘null aggregates’ with no spectral shift,36 or a very small population of H-coupled dimers. The latter species could be correlated with the slight enhancement of the 0–1 absorption peak at high concentrations (Fig. S1, ESI†), sometimes attributed to H-aggregates which are associated with cofacial overlap of the BODIPY π-systems.6,15,17,24 The precise ground-state signature of this state is of lesser significance, as it is predominantly populated via energy transfer rather than direct photoexcitation.Excimer states typically represent a major energy loss channel, though their PL efficiency can be substantially enhanced at low temperature (from ∼1% to 10% in E1, and from 0.3% to 23% in A1). There is thus scope for potentially harvesting this excimer emission by suppressing non-radiative decay channels, for instance by increasing the host matrix rigidity or density or tuning its vibrational properties. In the context of organic optoelectronic devices, such an approach would have an important advantage in the intrinsic red-shift of the excimer state. This has the effect of reducing the overlap between absorption and emission and thus the inner filter effect. Indeed, we have recently demonstrated the important role of excimer states to radiatively pump exciton-polariton states in a BODIPY microcavity.28 It is currently unclear why the excimer energy increases at low temperatures (Fig. 2 and Fig. S5, ESI†). We propose that increased conformational restriction as the matrix is constricted under cooling prevents complete relaxation to the same stabilised equilibrium geometry. Similar observations have been made for intermolecular charge-transfer states,37 suggesting that there may be a charge-transfer or charge-resonance contribution to the excimer wavefunction.
Nature of the SGE
Analysis of the SGE state in A-class films is less straightforward, and is summarised for A1 in Fig. 7a. The steady-state PL spectrum (top) can be neatly separated into monomer (red), excimer (orange) and sub-gap emitter (green, SS SGE) components. The latter very closely matches the SGE spectrum (yellow) extracted from streak-camera measurements, and we take them to represent the same species; the discrepancy beyond 1.8 eV is due to reduced detector sensitivity in that range. These features are typically considered signatures of BODIPY J-aggregates, but this simple assignment cannot be correct: such aggregates should exhibit strongly enhanced 0–0 vibronic peaks, whereas we detect a markedly stronger apparent 0–1 peak.38,39 Moreover, it is well established that the spectral shape of J-aggregate PL should exhibit marked temperature dependence, due to the suppression of local geometric fluctuations and enhancement in the J-aggregate delocalisation.38,39 The temperature-dependent measurements in Fig. 2 and 5 reveal invariant spectral shape for this species. Most significantly, the PL excitation measurements of the SGE peaks at <2.0 eV (Fig. 7a, dashed) reveal a clear absorption peak at 1.96 eV. Thus, even though the PL peaks at 2.15 eV and 1.95 eV are correlated in concentration- and temperature-dependent and time-resolved measurements (see also Fig. S16 and 17 (ESI†) for longer-time dynamics), they must reflect distinct electronic species. The correlated rise dynamics are a simple consequence of the parallel population of these species from the same pool of migrating monomer excitons.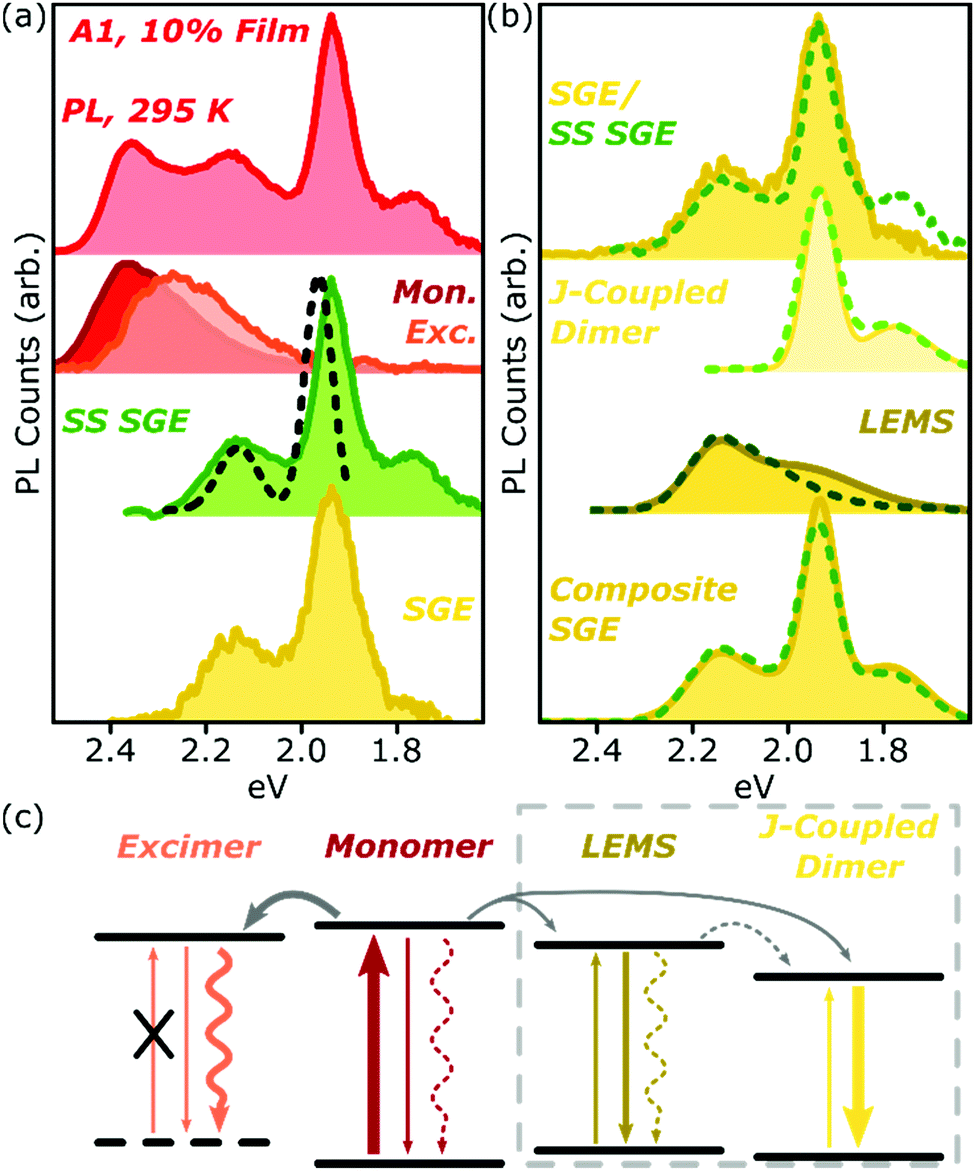 | ||
| Fig. 7 Analysis of A1 spectra. (a) Room-temperature PL spectrum of 10% film (top) can be decomposed into monomer (red), excimer (orange) and steady-state (SS) sub-gap emitter (green) sub-components. The latter closely matches the SGE species identified in time-resolved measurements (yellow). PL excitation spectrum of the main SGE band (dashed) reveals that peaks at 2.15 eV and 1.95 eV must have different electronic origin. (b) Both time-resolved (filled) and steady-state (dashed) SGE spectra can be decomposed into two components. J-coupled dimer consists of two vibronic peaks with 170 meV spacing as in Fig. 2f. Residual (LEMS) closely resembles a monomer PL spectrum red-shifted by 225 meV (dashed) and broadened (filled). Reconstruction from these spectra (composite SGE) closely matches measured emission. (c) Schematic of BODIPY film photophysics. Arrows represent photon absorption or emission (straight), non-radiative decay (wavy) or energy transfer (grey). Thickness of arrows indicates relative weight of processes; dashed arrows make a weak or negligible contribution. Excimer has no defined ground state, while LEMS and J-coupled dimers can be directly photoexcited. Processes in dashed box only occur in A-type films. | ||
As shown in Fig. 7b, we have found it is possible to decompose the steady-state (SS, dashed) or time-resolved SGE (filled) spectra into two contributions: a J-coupled dimer and a red-shifted monomer-like lineshape. The dimer can be described with a pair of Gaussian peaks with vibronic separation of 170 meV, a marked increase from the 140 meV spacing observed in the monomer. On the basis of the enhancement of the 0–0/0–1 peak ratio in PL and increase in the radiative rate (see ESI† for details), we can confirm that this species is J-aggregated. The 0–0/0–1 peak ratio is only changed by a factor of ∼2 from the monomer, suggesting that the aggregate is delocalised over only two molecules.38 We reach the same conclusion from the lack of temperature dependence (see below).
Following subtraction of the J-coupled peaks, the residual closely resembles the PL spectral shape of the monomer, and we refer to this as the low-energy monomer-like state (LEMS). Indeed, we can well describe this species with a monomer spectrum red-shifted by 225 meV (dashed) with at most a slight broadening and enhancement of the 0–1 peak intensity (filled). It is important, however, to note that this state must also be some form of aggregate: it only appears at high concentration and exhibits a substantial red-shift, well beyond what might be expected from conformational disorder in the monomer. Indeed, the appearance of these well-defined bands suggests a surprisingly high degree of local order, in spite of the undirected nature of the sample preparation.
The very strong red-shifts observed in the LEMS (225 meV) and J-coupled dimer (400 meV) relative to the monomer transition are not easily reconciled with their spectral shapes. These shifts are of the order of 10× the reorganisation energy, which would typically correspond to extremely strong J-type coupling and yield extremely small 0–1 emission and absorption peaks. The spectra we observe are much more consistent with the weak or intermediate coupling regime, but this requires that the coupling strength be commensurate with the reorganisation energy.38 We must thus conclude that the large shifts observed are not primarily due to excitonic dipole–dipole coupling. Other contributions may include the gas-to-crystal shift, mixing with charge-transfer states36 or changes in molecular geometry due to dimerisation, for instance partial planarisation of the pendant phenyl rings to extend conjugation.
These cannot be uniquely determined from the available data, nor can standard structural characterisation techniques be applied to these very sparse sites. Nonetheless, the delineation into two photophysical classes suggests a link to molecular structure and thus intermolecular geometry. BODIPY dimerisation is typically guided by the electric dipole of the core chromophore. The highly polar NO2 group in E3 will introduce additional interactions and geometries, making close approach of the BODIPY cores less likely and thus resulting in relatively weak coupling, i.e. small shifts between monomeric and excimer states. The space of possible dimer geometries is strongly restricted in A1–3 and E1 by the need to accommodate the bulky out-of-plane phenyl groups, which may inhibit the cofacial packing associated with H-type coupling and thus excimer formation. The comparison of E1 and A3 is particularly informative, revealing that it is specifically the bulky ethyl chains that prevent the close intermolecular contact needed to observe sizeable excitonic coupling. This may be an indication that the aggregates exhibit side-by-side structural motifs.30 Indeed, some degree of side-by-side orientation or slip-stacking is necessary to yield J-type coupling of the BODIPY transition dipole moments, which are oriented along the long BODIPY axis. While detailed chemical structural design rules are beyond the scope of this work, it is clear from the molecular series presented here that formation of SGE sites requires careful balance of intermolecular forces, in order to suppress cofacial π–π stacking in favour of slip-stacked geometries.
Localisation on dimers
It remains to reconcile the observed J-type coupling with the complete invariance of the SGE spectrum upon cooling. As noted above, J-aggregates typically exhibit strong enhancement of the 0–0 vibronic peak upon cooling, due to suppression of disorder and extension of the exciton coherence. This is accompanied by a marked increase in the radiative rate, leading to super-radiance. In our films, the SGE features are remarkably stable over the full temporal and temperature range, indicating a long lifetime of >5 ns. As anticipated from the steady-state results, this lifetime appears to be unaffected by cooling, suggesting a constant radiative rate and temperature-invariant degree of delocalisation. The simplest explanation of this behaviour is that the red-shifted state thus corresponds to a ground-state J-coupled dimer, similar to what was proposed in protein-coupled dyes.6 The dimer state is by definition delocalised over both molecules. This is the maximum possible extent of the dimer wavefunction, so no increase in coherence is observed upon cooling and the spectra and radiative rates do not change. Intriguingly, our results require that the PL quantum efficiency of these dimer states must approach unity. This observation also explains the large contribution of the SGE state to the integrated PL, in spite of its very small relative signal in transient absorption suggestive of a small population. Efforts are currently underway to generate films with significantly enhanced dimer content, which should offer superior emission even in concentrated solid state.Conclusions
We have investigated in detail the photophysical pathways of singlet exciton quenching in condensed films of a prototypical organic dye, BODIPY (Fig. 7c). The significant overlap between monomer absorption and emission spectra enables rapid and efficient resonance energy transfer through the film, even at relatively low dye loading. This migration ends when the excitation reaches a site capable of excimer formation, suggesting close proximity of two molecules. These low-energy states emit with low quantum yield, but their non-radiative decay pathways can be effectively suppressed at low temperature. In the A-type class of films, the molecular structure enables closer packing, resulting in a low density of J-coupled dimer sites. Following photoexcitation, the formation of excimers proceeds in parallel with migration to these dimers, which are strongly red-shifted in spite of relatively weak J-type coupling. Surprisingly, these states exhibit long excited-state lifetimes with near-unity quantum yield. These results highlight the wide variety of intermolecular states in concentrated dispersions of organic chromophores. The system described here can be considered as a simple model for molecular crystals, where self-assembly into extended aggregates and other structures produces a range of comparable states, with similar spectral shifts and quenching behaviour. That the nature of states formed can vary so widely with subtle changes in side-group structure is testament to the richness, complexity and tuneability of intermolecular excited states.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council, U.K. (Grant Number EP/M025330/1, “Hybrid Polaritonics” and Grant Number EP/L017008). I. D. W. S. acknowledges support from a Royal Society Wolfson Research Merit Award.Notes and references
- D. Bloor, G. Cross, P. K. Sharma, J. A. Elliott and G. Rumbles, J. Chem. Soc., Faraday Trans., 1993, 89, 4013 RSC
.
- J. L. Clark and G. Rumbles, Phys. Rev. Lett., 1996, 76, 2037–2040 CrossRef CAS PubMed
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed
- D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS PubMed
- F. Bergström, I. Mikhalyov, P. Hägglöf, R. Wortmann, T. Ny and L. B.-Å. Johansson, J. Am. Chem. Soc., 2002, 124, 196–204 CrossRef
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC
- T. Ozdemir, S. Atilgan, I. Kutuk, L. T. Yildirim, A. Tulek, M. Bayindir and E. U. Akkaya, Org. Lett., 2009, 11, 2105–2107 CrossRef CAS PubMed
- H. Lu, Q. Wang, L. Gai, Z. Li, Y. Deng, X. Xiao, G. Lai and Z. Shen, Chem. – Eur. J., 2012, 18, 7852–7861 CrossRef CAS PubMed
- T. T. Vu, M. Dvorko, E. Y. Schmidt, J.-F. Audibert, P. Retailleau, B. A. Trofimov, R. B. Pansu, G. Clavier and R. Méallet-Renault, J. Phys. Chem. C, 2013, 117, 5373–5385 CAS
- L. Gai, H. Lu, B. Zou, G. Lai, Z. Shen and Z. Li, RSC Adv., 2012, 2, 8840 RSC
- D. Okada, T. Nakamura, D. Braam, T. D. Dao, S. Ishii, T. Nagao, A. Lorke, T. Nabeshima and Y. Yamamoto, ACS Nano, 2016, 10, 7058–7063 CrossRef CAS PubMed
- L. Bonardi, H. Kanaan, F. Camerel, P. Jolinat, P. Retailleau and R. Ziessel, Adv. Funct. Mater., 2008, 18, 401–413 CrossRef CAS
- C. Spies, A.-M. Huynh, V. Huch and G. Jung, J. Phys. Chem. C, 2013, 117, 18163–18169 CAS
- I. Mikhalyov, N. Gretskaya, F. Bergström and L. B.-Å. Johansson, Phys. Chem. Chem. Phys., 2002, 4, 5663–5670 RSC
- H. Manzano, I. Esnal, T. Marqués-Matesanz, J. Bañuelos, I. López-Arbeloa, M. J. Ortiz, L. Cerdán, A. Costela, I. García-Moreno and J. L. Chiara, Adv. Funct. Mater., 2016, 26, 2756–2769 CrossRef CAS
- S. Choi, J. Bouffard and Y. Kim, Chem. Sci., 2014, 5, 751–755 RSC
- Y. Kubota, J. Uehara, K. Funabiki, M. Ebihara and M. Matsui, Tetrahedron Lett., 2010, 51, 6195–6198 CrossRef CAS
- Y. Liao, V. Génot, R. Méallet-Renault, T. T. Vu, J.-F. Audibert, J.-P. Lemaistre, G. Clavier, P. Retailleau and R. B. Pansu, Phys. Chem. Chem. Phys., 2013, 15, 3186 RSC
- S. Badré, V. Monnier, R. Méallet-Renault, C. Dumas-Verdes, E. Y. Schmidt, A. I. Mikhaleva, G. Laurent, G. Levi, A. Ibanez, B. A. Trofimov and R. B. Pansu, J. Photochem. Photobiol., A, 2006, 183, 238–246 CrossRef
- B. Ventura, G. Marconi, M. Bröring, R. Krüger and L. Flamigni, New J. Chem., 2009, 33, 428–438 RSC
- D. Marushchak, S. Kalinin, I. Mikhalyov, N. Gretskaya and L. B.-Å. Johansson, Spectrochim. Acta, Part A, 2006, 65, 113–122 CrossRef PubMed
- M. A. H. Alamiry, A. C. Benniston, G. Copley, A. Harriman and D. Howgego, J. Phys. Chem. A, 2011, 115, 12111–12119 CrossRef CAS PubMed
- N. Saki, T. Dinc and E. U. Akkaya, Tetrahedron, 2006, 62, 2721–2725 CrossRef CAS
- A. B. Nepomnyashchii, M. Bröring, J. Ahrens and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 19498–19504 CrossRef CAS PubMed
- J. Ahrens, A. Scheja, R. Wicht and M. Bröring, Eur. J. Org. Chem., 2016, 2864–2870 CrossRef CAS
- D. Tleugabulova, Z. Zhang and J. D. Brennan, J. Phys. Chem. B, 2002, 106, 13133–13138 CrossRef CAS
- R. T. Grant, P. Michetti, A. J. Musser, P. Gregoire, T. Virgili, E. Vella, M. Cavazzini, K. Georgiou, F. Galeotti, C. Clark, J. Clark, C. Silva and D. G. Lidzey, Adv. Opt. Mater., 2016, 4, 1615–1623 CrossRef CAS
- G. Nöll, J. Daub, M. Lutz and K. Rurack, J. Org. Chem., 2011, 76, 4859–4873 CrossRef PubMed
- A. B. Nepomnyashchii, M. Bröring, J. Ahrens and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 8633–8645 CrossRef CAS PubMed
- H. Imahori, H. Norieda, H. Yamada, Y. Nishimura, I. Yamazaki, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 100–110 CrossRef CAS PubMed
- D. Aydın Tekdaş, G. Viswanathan, S. Zehra Topal, C. Y. Looi, W. F. Wong, G. Min Yi Tan, Y. Zorlu, A. G. Gürek, H. B. Lee and F. Dumoulin, Org. Biomol. Chem., 2016, 14, 2665–2670 Search PubMed
- L. Gai, J. Mack, H. Lu, H. Yamada, D. Kuzuhara, G. Lai, Z. Li and Z. Shen, Chem. – Eur. J., 2014, 20, 1091–1102 CrossRef CAS PubMed
- Z. Yang, Y. He, J. Lee, N. Park, M. Suh, W.-S. Chae, J. Cao, X. Peng, H. Jung, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 9181–9185 CrossRef CAS PubMed
- N. Gupta, S. I. Reja, V. Bhalla, M. Gupta, G. Kaur and M. Kumar, Chem. Commun., 2015, 51, 10875–10878 RSC
- N. J. Hestand and F. C. Spano, J. Chem. Phys., 2015, 143, 244707 CrossRef PubMed
- A. P. Arndt, M. Gerhard, A. Quintilla, I. A. Howard, M. Koch and U. Lemmer, J. Phys. Chem. C, 2015, 119, 13516–13523 CAS
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Methods, full steady-state and time-resolved PL characterisation, spectral decomposition and radiative rates analysis. The data underlying this publication are available at DOI: 10.15131/shef.data.5179033. See DOI: 10.1039/c7tc02655b |
| This journal is © The Royal Society of Chemistry 2017 |