
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA universal platform for selection and high-resolution phenotypic screening of bacterial mutants using the nanowell slide†
H.
Antypas
 a,
M.
Veses-Garcia
a,
E.
Weibull‡
b,
H.
Andersson-Svahn
b and
A.
Richter-Dahlfors
*a
a,
M.
Veses-Garcia
a,
E.
Weibull‡
b,
H.
Andersson-Svahn
b and
A.
Richter-Dahlfors
*a
aSwedish Medical Nanoscience Center, Department of Neuroscience, Karolinska Institutet, Stockholm, Sweden. E-mail: agneta.richter.dahlfors@ki.se
bDivision of Proteomics and Nanobiotechnology, Science for Life Laboratory, KTH-Royal Institute of Technology, Stockholm, Sweden
First published on 8th May 2018
Abstract
The Petri dish and microtiter plate are the golden standard for selection and screening of bacteria in microbiological research. To improve on the limited resolution and throughput of these methods, we developed a universal, user-friendly platform for selection and high-resolution phenotypic screening based on the nanowell slide. This miniaturized platform has an optimal ratio between throughput and assay complexity, holding 672 nanowells of 500 nl each. As monoclonality is essential in bacterial genetics, we used FACS to inoculate each nanowell with a single bacterium in 15 min. We further extended the protocol to select and sort only bacteria of interest from a mixed culture. We demonstrated this by isolating single transposon mutants generated by a custom-made transposon with dual selection for GFP fluorescence and kanamycin resistance. Optical compatibility of the nanowell slide enabled phenotypic screening of sorted mutants by spectrophotometric recording during incubation. By processing the absorbance data with our custom algorithm, a phenotypic screen for growth-associated mutations was performed. Alternatively, by processing fluorescence data, we detected metabolism-associated mutations, exemplified by a screen for β-galactosidase activity. Besides spectrophotometry, optical compatibility enabled us to perform microscopic analysis directly in the nanowells to screen for mutants with altered morphologies. Despite the miniaturized format, easy transition from nano- to macroscale cultures allowed retrieval of bacterial mutants for downstream genetic analysis, demonstrated here by a cloning-free single-primer PCR protocol. Taken together, our FACS-linked nanowell slide replaces manual selection of mutants on agar plates, and enables combined selection and phenotypic screening in a one-step process. The versatility of the nanowell slide, and the modular workflow built on mainstream technologies, makes our universal platform widely applicable in microbiological research.
Introduction
Development of the Petri dish, containing nutrient agar for bacterial cultivation, revolutionized the way bacteriologists performed their studies back in the 1880s.1,2 Over a century later, it still remains the golden standard to culture, select and screen bacteria to identify new genotypes and phenotypes.3 The large amount of genomic data generated by modern technologies requires, however, high resolution and throughput phenotypic analysis to understand the function of novel genes identified.4 This is not easily achieved by agar plate-based methods due to their inherent limitation in phenotypic resolution. On agar plates, bacterial growth is usually observed on the macroscale at the incubation endpoint. This restricts the analysis to the measurement of composite growth and disregards individual growth parameters that may be affected by gene mutations, such as growth rate, growth lag, and growth efficiency.5–7 Moreover, while agar plate screening is well-suited for qualitative pre-screening of catalytic activity using chromogenic or fluorogenic substrates, quantification is difficult to achieve because of incompatibility between agar plate-based and spectrophotometric methods.8 Introduction of time-lapse imaging of colonies growing on solid media, combined with computational analysis can increase the phenotypic resolution.9 Still, the sensitivity of such an approach is limited, because bacteria must grow to a considerable number before imaging can capture them on the macroscale. In addition, variations in solid media thickness, agar plate margins, but also nutrient and secreted molecule exchange between neighboring non-individually confined colonies, can lead to spatial bias during phenotypic analysis.9Platforms that use liquid media have a higher throughput and address some of the phenotypic screening limitations encountered on agar plates. The microtiter plate enables kinetic optical recordings and therefore accurate measurement of absorbance and fluorescence to monitor growth and biochemical reactions.10 The individual components of growth can be analysed and alterations can be identified. Despite these advantages, the well size of commonly used microtiter plates, such as the 96- and 384-well plate, is far from optimal for single-cell analysis.11 Moreover, manual handling of 384-plates can be time-consuming and automated liquid handling may be required for a smooth workflow. Miniaturization of these platforms has enabled high-throughput screening of bacteria on a single-cell level.12 One such example is the nanowell slide (nwSlide). This platform offers several advantages for microbiological applications compared with commonly used microtiter plates (Table S1, see Note S1†). The nwSlide features 672 wells holding 500 nl each within the dimensions of a microscopy slide.11 Each nanowell acts as an independent container that sustains long-term culturing and analysis of mammalian and bacterial cells.13–15
Here, we leverage the versatile features of the nwSlide to develop a platform for high-resolution phenotypic and genotypic analysis of bacterial mutants (Fig. 1). By integrating fluorescence-activated cell sorting (FACS), kinetic spectrophotometry, optical signal processing and microscopy on the same platform, we were able to screen bacterial mutants based on their growth and metabolic profile, as well as morphotype. Importantly, we established a recovery protocol to transfer selected mutants from nano- to macroscale cultures, and to perform genotyping screening to rapidly map the mutations.
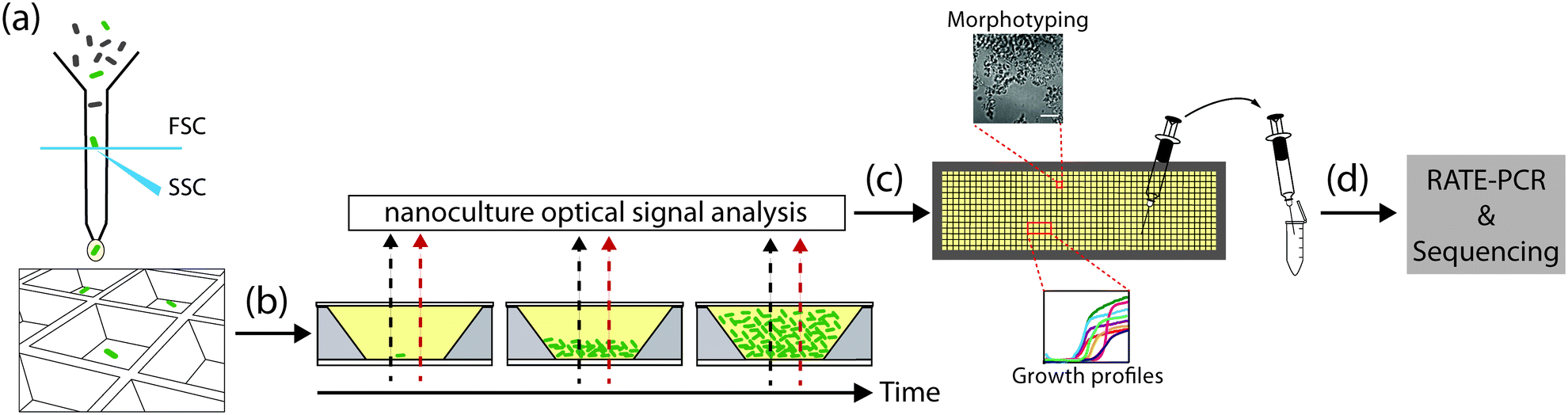 | ||
| Fig. 1 General workflow for selection and high-resolution phenotypic screening of bacterial mutants in the nwSlide. (a) FACS-based selection and sorting of single bacterial mutants in individual nanowells of the nwSlide, pre-filled with 500 nl of growth medium each. (b) While bacteria proliferate during incubation on the slide, absorbance (black dashed arrow) and fluorescence (red dashed arrow) are recorded at frequent intervals. Absorbance data are processed with the nanoculture optical signal analysis tool (nOSAT) to obtain the growth profile of each nanoculture. Fluorescence recordings in combination with a fluorescent substrate can identify mutants with decreased or increased enzymatic activity. (c) Guided by the nanowell identification number, bacteria showing the desired phenotype, based on growth profile, enzymatic activity or morphotype, are retrieved from the nwSlide (d) and subcultured in a larger-scale format to enable genotypic analysis and subsequent storage. | ||
Experimental
Materials
Enzymes were purchased from New England Biolabs (USA), and antibiotics and chemical reagents from Sigma Aldrich (USA), unless otherwise stated. Primers were synthesized by Eurofins Genomics (Germany) and used in all PCR reactions at a concentration of 10 pmol μl−1. Their sequences and PCR cycling parameters are presented in Table S2 and S3† respectively.Nanowell slide design and microfabrication
The nwSlide comprises a nanowell-etched silicon grid (75 × 0.5 × 25 mm) with tapered sides anodically bonded to a borofloat glass wafer (75 × 0.175 × 25 mm). This bonding results in a 14-column by a 48-row matrix of 672 wells, with center-to-center distance between 2 nanowells at 1500 μm and a volume capacity of 500 nl per nanowell. Due to the outward-tilted walls of each well, the surface area starts from 650 × 650 μm2 at the bottom of the well and increases up to 1360 × 1360 μm2 at the top.Standard microfabrication procedures were used to manufacture the nwSlide, as described in detail in ref. 11. Briefly, a low stress nitride (2000 Å) was used as masking material. Contact lithography was performed using a quartz mask. Nanowells were etched through 500 μm thick silicon wafers (Okmetic) by 30% potassium hydroxide at 70 °C for ≈800 min. The nitride was stripped by 50% hydrofluoric acid. The resulting silicon grid wafer was then anodically bonded to a 500 μm borofloat glass wafer (Planoptik), followed by dicing into the nwSlide format.
Bacterial strains and electrocompetent cell preparation
All E. coli strains used in the study are listed in Table 1. Bacteria were cultured in either Müller-Hinton II (MHII) (BD, USA) or Luria-Bertani (LB) broth (Sigma Aldrich). All strains were streaked on LB agar plates from glycerol stocks stored at −80 °C, and grown at 37 °C for 18 h before use. Electrocompetent cells were either purchased from Invitrogen, USA (MegaX DH10B™ T1R) or prepared by diluting an overnight E. coli W3110 culture in LB 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 in 25 ml LB, which was grown to OD600 ≈ 0.4. Bacteria were concentrated 100-fold, washed 3 times with ice-cold MiliQ water and stored in 25 μl-aliquots at −80 °C until use.
:100 in 25 ml LB, which was grown to OD600 ≈ 0.4. Bacteria were concentrated 100-fold, washed 3 times with ice-cold MiliQ water and stored in 25 μl-aliquots at −80 °C until use.
| Strain | Genotype | Ref. |
|---|---|---|
| W3110 | E. coli K-12 F- λ- rph-1 INV(rrnD, rrnE) | 17 |
| JW1909 | BW25113 ΔfliD::kan | NBRP (NIG, Japan): E. coli |
| JW1910 | BW25113 ΔfliS::kan | NBRP (NIG, Japan): E. coli |
| JW1911 | BW25113 ΔfliT::kan | NBRP (NIG, Japan): E. coli |
| ARD219 | W3110 cyaA::Tn5 | This work |
| ARD220 | W3110 lacZ::Tn5 | This work |
| ARD230 | W3110 envC::Tn5 | This work |
| ARD232 | W3110 rodZ::Tn5 | This work |
| MegaX DH10B™ T1R | E.coli DH10B™ | Invitrogen, USA |
| D18 | E. coli DH10B™ tnpX::TnMHA | This work |
| D40 | E.coli DH10B™ yebB::TnMHA | This work |
| L43 | E.coli DH10B™ yhgE::TnMHA | This work |
| B5 | E.coli DH10B™ recF::TnMHA | This work |
| G13 | E.coli DH10B™ mocA::TnMHA | This work |
| A4 | E.coli DH10B™ rfaJ::TnMHA | This work |
| D14 | E.coli DH10B™ casE::TnMHA | This work |
Construction of pMHA transposon vector
The FRT-flanked kanamycin cassette was PCR-amplified from pKD4 with the SacI-Kan-FP and KpnI-Kan-RP primers to introduce SacI and KpnI restriction sites, using Phusion High-Fidelity (PHF) DNA polymerase. The PCR-amplified kanamycin cassette was purified with illustra™ GFX™ PCR and Gel Purification kit (GE Healthcare, UK) according to the manufacturer's instructions. The pMOD-2 Vector (Epicentre, USA) was isolated from an overnight E. coli W3110 culture in LB broth supplemented with 100 μg ml−1 ampicillin. The PCR-amplified kanamycin cassette and pMOD-2 were digested with SacI-HF and KpnI-HF, mixed at 1:2 ratio (vector:insert), and ligated with T4 DNA ligase at 16 °C overnight to construct pMOD-2-KAN. Electrocompetent E. coli W3110 cells were electroporated with 5 μl of the ligation reaction. Electroporated bacteria were recovered in 1 ml SOC broth for 2 h at 37 °C, with shaking at 200 r.p.m., then plated on LB agar plates supplemented with 50 μg ml−1 kanamycin and incubated overnight at 37 °C. Colonies transformed with pMOD-2-KAN were transferred in LB broth with 50 μg ml−1 kanamycin and incubated overnight at 37 °C. Overnight cultures were pelleted and pMOD2-KAN was isolated using the MiniPrep kit (Qiagen, Germany) according to manufacturer's instructions. To clone a GFP-expressing gene in pMOD2-KAN, gfpmut2 and its IPTG-inducible promoter were PCR-amplified from pKEN1-GFPmut2 with SalI-GFP-FP and HindIII-GFP-RV primers using PHF DNA polymerase, and purified as described above. The PCR-amplified gfpmut2 and pMOD-2-KAN were digested with SalI-HF and HindIII-HF, and ligated as described above to construct vector pMOD-2-KAN-GFPmut2, hereafter named as pMHA. Electroporation of pMHA into E. coli W3110, transformant selection and plasmid isolation was performed as described above.
Transposon mutagenesis
Transposon libraries using the custom-made transposon TnMHA were prepared by linearizing pMHA with ScaI-HF, and by PCR-amplifying the TnMHA sequence with PHF DNA polymerase using primers ME Plus 9-3′ and ME Plus 9-5′, followed by PCR purification in TE buffer as described above. The TnMHA transposon was prepared by mixing 2 μl of TnMHA amplicon (100 ng μl−1), 4 μl EZ-Tn5 transposase (Epicentre), and 2 μl glycerol, followed by incubation at RT for 30 min. 1 μl of the TnMHA transposon was electroporated in 40 μl MegaX DH10B™ T1R electrocompetent cells according to the manufacturer's instructions. After electroporation, 1 ml SOC medium was added and bacteria were incubated for 2 h at 37 °C. Bacteria were then diluted to 5 ml SOC supplemented with 50 μg ml−1 kanamycin and 1 mM IPTG and incubated overnight at 37 °C, under shaking conditions. The overnight culture containing TnMHA mutants was then single-sorted on a nwSlide with FACS as described below.Strains ARD219, ARD220, ARD230, and ARD232 were constructed using the EZ-Tn5 <KAN-2> Tnp transposome kit (Epicentre) according to the manufacturer's instructions using electrocompetent E. coli W3110.
FACS of bacteria on the nwSlide
Sorting was performed in a BD Influx™ cell sorter fitted with a 100 μm nozzle. Sheath fluid and sample pressure, amplitude and frequency were calibrated for small particle sorting using beads between 0.22–1.34 μm (SPHERO™ Flow Cytometry Nano Fluorescent Size Standard Kit, Spherotech, USA). Sheath fluid was autoclaved and filtered with a 0.2 μm filter to ensure it is sterile and particle-free. Forward scatter (FSC) and side scatter (SSC) parameters were adjusted so that the calibration beads are on scale in FSC-W vs. FSC-H and SSC-W vs. SSC-H plots. This enabled us to detect bacteria based on their size, without additional labeling. To prepare bacteria for FACS, overnight cultures were subcultured in fresh media and incubated at 37 °C under shaking at 200 r.p.m. until OD600 = 0.4. 1 ml of culture was pelleted for 10 min at 8000 r.p.m. and resuspended in 1 ml of 0.2 μm-filtered PBS. Bacteria were diluted 1:10 in PBS and loaded in the sorter. Bacteria identified in the FSC-W vs. FSC-H and SSC-W vs. SSC-H plots were gated and further interrogated in a FSC-H vs. SSC-H plot to discriminate single bacteria from aggregates. The main cluster of events in FSC-H vs. SSC-H plot was gated as singlets. To identify TnMHA transposon mutants, we first adjusted the voltage of the GFPmut2 parameter (λem = 530 ± 40 nm) for wt bacteria so that their autofluorescence peaked within the first decade of a GFP-mut2 histogram. Subsequently, wt singlets were interrogated in a FSC-H vs. GFPmut2 plot to set the GFPmut2-negative gate. When samples containing both wt and TnMHA mutants were loaded, events with high GFPmut2 fluorescence, located above the GFPmu2-negative gate, were designated as TnMHA mutants.
Bacteria seeding precision in the nanowells was tested by calibrating the instrument to sort E. coli samples on solid media LB agar plates in a 672-point format (14 rows × 48 columns), with 1500 μm point–point distance, within a 76 × 26 mm area in total. After overnight incubation at 37 °C, the alignment of bacterial colony forming units on the LB agar plates was examined visually as exemplified in Fig. S2.†
Right before sorting bacteria, nwSlides were filled with LB or MHII broth by manually spreading a total volume of ≈500 μl broth in a single pipetting step, under sterile conditions. Events in selected gates were then sorted in the pre-filled nwSlides. When mixed samples of wt E. coli W3110, and strains ARD219, ARD220, ARD230, ARD232 were sorted, MHII broth was supplemented with 10 mM of 4-methylumbelliferyl β-D-galactopyranoside (MUG). When sorting the TnMHA transposon library, MHII broth was supplemented with 50 μg ml−1 kanamycin and 1 mM IPTG. Directly after sorting, the nwSlide was sealed to prevent evaporation with a sterile polyester acrylate membrane (Thermo Fisher Scientific, USA). The porosity of this membrane enables gas exchange to sustain cell culture and its transparency is compatible with microscopy and spectrophotometry.
Phenotypic screening of single-sorted bacteria for growth parameters
Growth parameters were assessed based on spectrophotometric measurements and subsequent data analysis. nwSlides inoculated with single bacteria via FACS were placed in a custom-designed microtiter plate adaptor as previously described,13 and incubated for 16 h at 37 °C in an Infinite® M1000 PRO plate reader (TECAN, Switzerland) with OD600 measured every 30 min. The baseline OD600 was subtracted from all OD600 kinetic recordings, which is defined as the minimum absorbance recorded in the first 8 kinetic recordings. Then, the OD600 recordings were processed with the nanoculture Optical Signal Analysis Tool (nOSAT) to select for nanowells positive for growth and to calculate growth parameters (Tlag, growth efficiency and ODmax). A detailed explanation of this algorithm can be found in Note S2† and in Fig. S3.†Phenotypic screening for β-galactosidase activity
To define MUG hydrolysis in E. coli W3110 wt and lacZ::Tn5, we prepared cultures of each strain by touching 4–6 colonies from an agar plate and re-suspending them in 2 ml MHII broth to an OD600 = 0.05. The bacterial suspensions were then diluted 1:100 in broth supplemented with MUG. 500 μl of each culture was used to inoculate a nwSlide by spreading the sample in the nanowells with the membrane. The nwSlides were incubated in an Infinite® M1000 PRO plate reader at 37 °C, and OD600 and fluorescence (λex = 360 nm; λem = 450 nm) were recorded every 30 min for 16 h. 2 biological replicates were performed for each strain. Nanowells positive for bacterial growth were identified with the processing of OD600 recordings with nOSAT. The fluorescence recordings of nanowells positive for growth were then processed as follows. The baseline fluorescence was subtracted from all recordings, where baseline was defined the minimum fluorescence recording observed in the first 8 recordings. Then, we plotted the maximum RFU (RFUmax) of each nanoculture and calculated the 1st and 99th percentile of each distribution using GraphPad Prism software (version 6.0). The 99th percentile of the ARD220 samples was chosen as the phenotypic discrimination threshold to differentiate between strains with active and inactive β-galactosidase.
To prepare mixed cultures, equal ratios of ARD219, ARD230, ARD232 and wt E. coli were resuspended in PBS. Then, the lacZ::Tn5 mutant was added to these mixed cultures at a 20% (Mix20%), 2% (Mix2%) and 0.2% (Mix0.2%) representation. Mixed cultures were screened in the presence of MUG and spectrophotometric data were processed as described above. To screen for mutants with inactive β-galactosidase, the RFUmax values of all nanocultures were plotted and mutants with RFUmax below the phenotypic discrimination threshold (1878 RFU), defined by ARD220, were marked as potential mutants.
Genotypic screening of single-sorted bacteria on the nwSlide
Bacteria grown on the nwSlide were retrieved for genotypic screening by piercing the membrane of the nanowell containing bacteria with a 26G 1/2′′ needle (BD) or 30G 1/2′′, 0.45 × 13 mm needle (Terumo, USA) attached to a 5 or 1 ml syringe (BD). Then the needle was dipped in 100 μl of broth in a 1.5 ml microcentrifuge tube, which was incubated overnight at 37 °C shaking at 200 r.p.m. For sample storage, we plated the overnight culture in LB agar plates to obtain single colonies and prepare glycerol stocks. For genotypic screening, cultures were pelleted and resuspended in 100 μl sterile MiliQ water. Bacteria were lysed by heating at 95 °C for 10 min. To verify whether cultures derived from one, two or three different flagellar mutants, PCR reactions were prepared using illustraTaq Beads (GE Healthcare) by adding 1 μl of the bacterial lysates, 1 μl of Kan-FP and fliT-RP primers and MiliQ water to a total volume of 25 μl. To verify whether bacteria were wt or mutants for lacZ, PCR reactions were prepared using illustraTaq Beads by adding 1 μl of the bacterial lysates, 1 μl of T6D4_FW and T6D4_RV primers and MiliQ water to a total volume of 25 μl. A 632 bp PCR product was generated for strains with wt lacZ and 1853 bp for lacZ::Tn5. TnMHA insertion sites in mutants of interest were identified by performing rapid amplification of transposon ends (RATE) using a single primer (RATE-FP, RATE-FP2 or RATE-FP3). This PCR protocol consists of 3 amplification rounds with 30 cycles each (90 in total). We adapted the protocol previously described16 for the TnMHA sequence. Briefly, 1 μl of bacterial lysate prepared as described or 1 μl gDNA isolated using the DNeasy Blood&Tissue kit (Qiagen), 2 μl of 20 pmol μl−1 of a single primer and 22 μl of MiliQ water were added to illustraTaq Beads. To identify the insertion site in transposon mutants generated with the EZ-Tn5 <KAN-2> Tnp transposome kit, we performed RATE using primer Inv-1.As RATE generates several PCR products, the most intense PCR bands observed after electrophoresis on a 1% agarose gel were excised and purified using illustra™ GFX™ PCR and Gel Purification kit. PCR bands were sequenced (Eurofins Genomics) using the SqFP primer for RATE performed with RATE-FP, RATE-FP2, or RATE-FP3, and the KAN-2 RP1 primer for RATE performed with Inv-1.
Microscopy
Nanowells were microscopically examined for bacterial growth under phase contrast microscopy (Nikon TS 100, Japan) at 100× magnification. Images were captured using a camera (Hamamatsu, Japan) adjusted to the microscope. Fluorescence confocal microscopy (Olympus FV1000, USA) was performed at 60× magnification at λex = 488 nm and λem = 545 ± 20 nm.Results and discussion
Genotypic screening of single-cell derived bacterial cultures on the nwSlide
To elucidate the bacterial genotype–phenotype relationship, it is essential to select a monoclonal population harboring the genotype of interest. Similar to the agar plate where each colony originates from one bacterium, it is important to ensure the monoclonality of liquid nanocultures. To inoculate each nanowell with a single bacterium, we utilized fluorescence-activated cell sorting (FACS), since the 1.5 mm well-to-well distance in the nwSlide is compatible with the x- and y-movement of the motorized stage of FACS instruments.14 Following calibration of the FACS instrument, E. coli cultures were loaded and gates were designated to discriminate single bacteria from bacterial aggregates (Fig. S1†). To validate that this gating strategy selects for single bacteria, we used three isogenic E. coli K-12 flagella mutants, ΔfliD, ΔfliS, and ΔfliT (strains JW1909, JW1910, JW1911, Table 1). These mutants harbor well-defined gene deletions that can be easily differentiated by PCR when using primer pairs that anneal to the kanamycin cassette and the 3′ end of the flagella operon (Fig. 2a & Table S2†). After mixing the three mutants at a 1:1:1 ratio, we sorted the sample into nanowells prefilled with LB medium in less than 15 min. The nwSlide was immediately sealed with a porous membrane and incubated at 37 °C. After overnight incubation, we identified nanowells with and without bacterial growth using phase contrast microscopy. Based on three biological replicates, we found an average of 562 ± 35 nanowells/nwSlide with growing E. coli. This corresponds to 83.6% ± 5.2% of the total number of nanowells. To investigate the monoclonality of the growing nanocultures, we transferred bacteria from 40–60 nanowells per replicate into individual microcentrifuge tubes containing fresh LB media using syringe needles. Following overnight incubation at 37 °C, bacterial lysates were prepared and used for PCR analysis in order to identify the number of mutants that originated the population. Nanocultures deriving from one mutant generated a single PCR product of defined size, whereas doublets or triplets generated two and three PCR products respectively (Fig. 2b). We found that 82.8% ± 7.0% of the nanowell cultures originated from a single mutant, whereas 13.57% ± 2.4% and 3.63% ± 5.4% originated from two and three mutants, respectively. Despite the inability of PCR to discriminate between identical mutants, there is only a low risk that some single-mutant nanocultures may actually derive from doublets of the same mutant. Therefore, the relative frequency of single mutants reflects quite accurately the expected single-bacterium frequency.
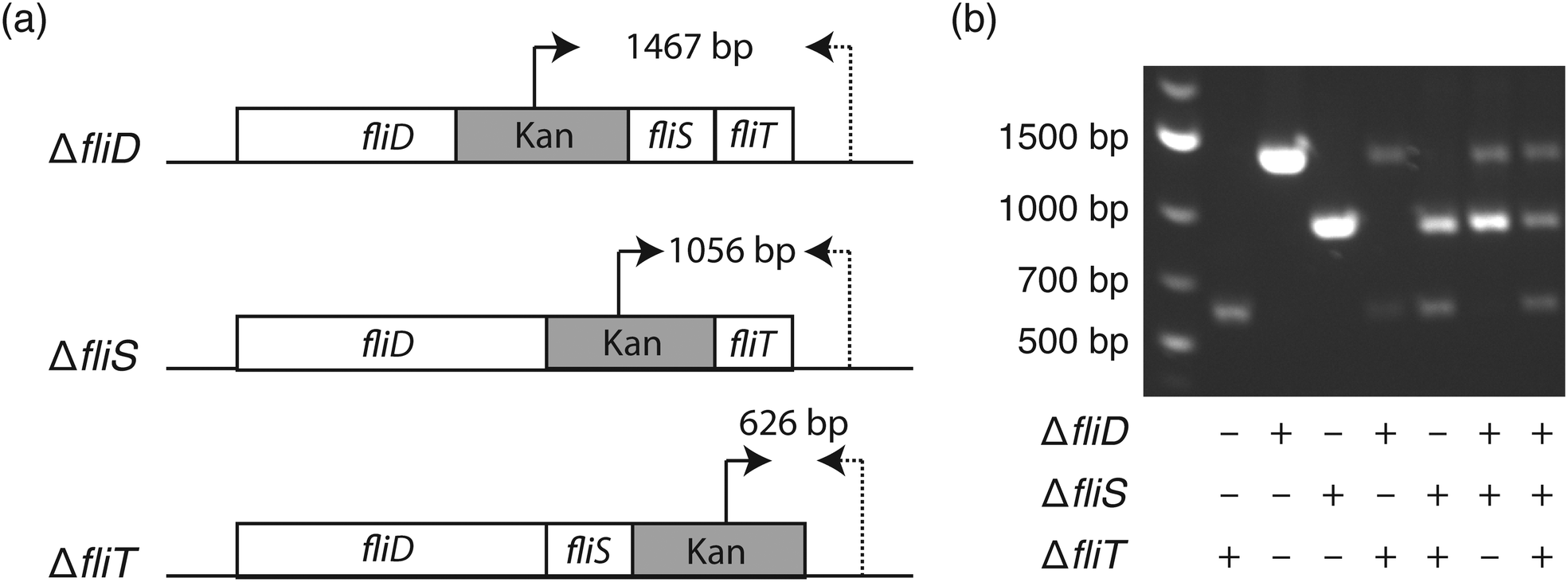 | ||
| Fig. 2 Genetic construction of flagellar mutants. (a) Schematic representation of the flagellar mutants ΔfliD, ΔfliS, and ΔfliT in strain W3110, with annealing sites of primers Kan-FP (arrow) and fliT-RP (dotted arrow) shown. The size of their respective PCR product, indicated in base pairs (bp), is used to differentiate the three mutants from each other. (b) The expected size of PCR bands generated by primers Kan-FP and fliT-RP in monoclonal and mixed cultures as indicated shows the feasibility of this approach for validating the FACS gating strategy. | ||
Overall, these results demonstrate that the FACS instrument is excellently suited for controlled inoculation of individual nanowells in the nwSlide. By applying a label-free gating strategy that effectively sorted droplets containing single bacteria, we generated a large number of monoclonal cultures, all initiated from the smallest inoculum possible – a single bacterium. Depending on the bacterial sample and the estimated representation of the mutant of interest, higher dilution of the culture loaded in the sorter could further improve the single-bacterium frequency. The ability to recover bacteria from specific nanowells enables translation of bacterial cultures from the nanoliter scale to any larger volume-sized cultures. This is essential for downstream analysis by standard molecular techniques.
Algorithm for rapid growth profiling of bacterial nanocultures
The ability to quickly identify nanowells with and without bacterial growth, or with growth artifacts, is important for any downstream analysis. This requires automated methods since growth monitoring of 672 nanowells generates tens of thousands of data points. To facilitate this, we developed the nanoculture Optical Signal Analysis Tool (nOSAT). The nOSAT algorithm comprises three checkpoints by which growth parameters of bacterial nanocultures are assessed (Fig. 3). For details of the algorithm development and specifics of checkpoint determination, see Note S2† and Fig. S3.† At the first checkpoint, each nanoculture is analysed to determine whether a transition from lag to logarithmic phase has occurred. This is performed by analysing absorbance recordings and calculating the Tlag, which represents the exact time point when a bacterial culture transitions from lag to logarithmic phase.13 The second checkpoint is designed to filter out growth artifacts. This is achieved by analysing maximum absorbance at 600 nm of each nanoculture, which should fall within 0.006–0.075. A culture that fails to reach the lower limit contains usually no bacteria, whereas those exceeding the higher limit are usually artifacts. The third checkpoint calculates growth efficiency in each nanoculture. The nOSAT algorithm controls that the net absorbance increase of the nanoculture is ≥0.006. This excludes any nanowell containing air bubbles, since their net absorbance increase is usually negative.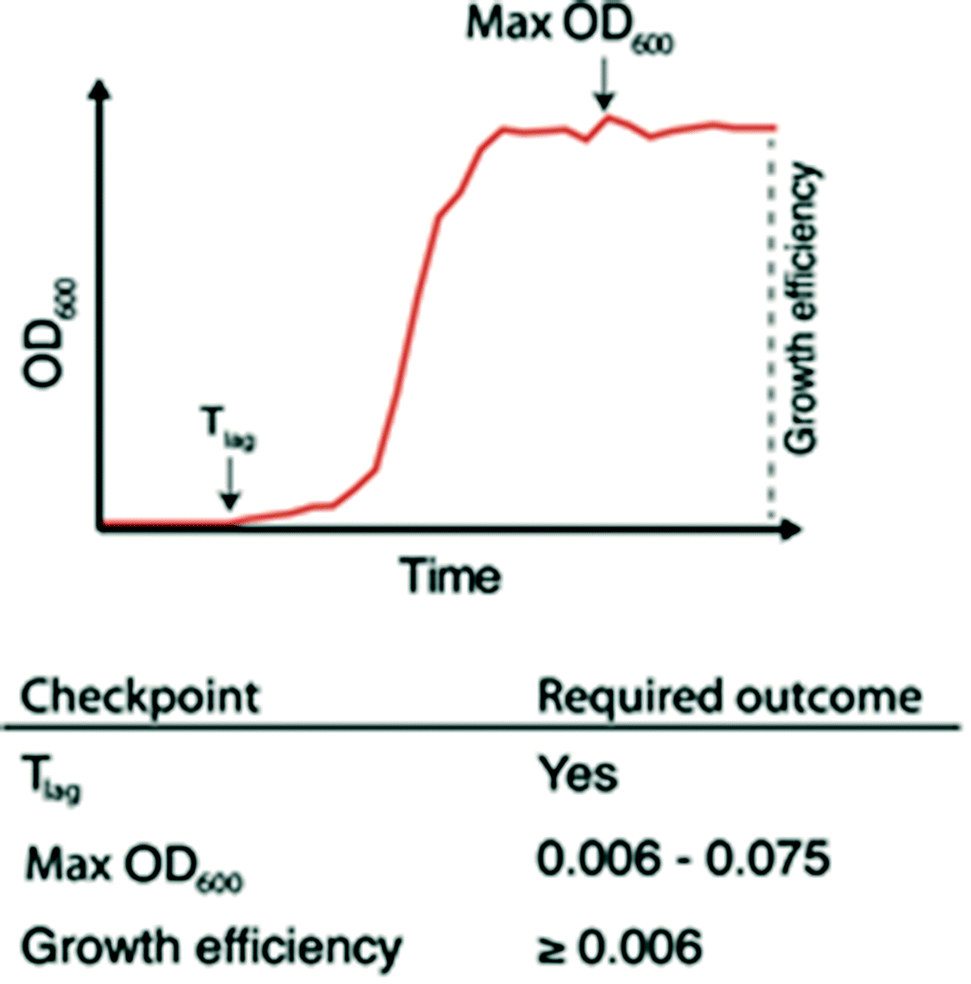 | ||
| Fig. 3 Schematic representation of checkpoints forming the basis of the nOSAT algorithm. During incubation, optical recordings from each nanoculture are processed and controlled for three checkpoints with the nOSAT algorithm. Generation of Tlag represents the first checkpoint, which controls whether a nanoculture has transitioned from lag to log phase. The second checkpoint controls whether maximum OD (ODmax) is ranging within 0.006–0.075. The third checkpoint controls whether growth efficiency is ≥0.006. If a nanoculture passes all 3 checkpoints, then it is considered as positive for bacterial growth. | ||
To test the specificity and sensitivity of nOSAT, we used absorbance data from the wild type (wt) E. coli strain W3110 (Table 1). The bacterial cultures were single-sorted onto nwSlides, then incubated for 16 h at 37 °C in a plate reader. Bacterial growth of each nanowell was monitored by OD600 readings every 30 min. Absorbance data from 997 randomly selected nanowells from three independent experiments were processed with nOSAT, which identified 644 nanowells as positive and 353 as negative for growth. To confirm these results, we examined the same 997 nanowells by phase contrast microscopy. A comparison of the nOSAT results with the microscopy observations showed 640 nanowells true positives for growth, 321 true negatives, 4 false positives and 32 false negatives. Based on these results, we determined the sensitivity of nOSAT to be 95.24% with a 95% confidence interval (C.I.) of 93.34–96.72%. The specificity was 98.77% with a 95% C.I. of 96.88–99.66%.
Overall, nOSAT enables rapid identification of the growth profile of each nanoculture and in parallel, extracts information about growth efficiency and the duration of the lag phase (Tlag). The latter is of particular interest when considering that a single bacterium is used as inoculum in each nanoculture. Starting with an identical inoculum allows precise comparisons of growth between bacterial cultures.
Phenotypic screening of bacterial nanocultures
The ability to synchronously initiate hundreds of bacterial cultures with one bacterium each and monitor their growth profiles by optical recordings prompted us to exploit the nwSlide as a novel tool for high throughput analysis in microbiological research. Phenotypic screening is a method used to identify a strain with a specific phenotypic trait and isolate it from the thousands of mutants present in a mutagenized bacterial culture. Traditionally, phenotypic screening is a step-wise process where individual mutants are first isolated as bacterial colonies on selective agar plates. A quantitative phenotypic analysis is then performed; subculturing of monoclonal bacteria in microtiter plates, sometimes in the presence of colorimetric or fluorometric substrates, enables monitoring of bacterial growth and phenotype in parallel by spectrophotometry.With an ultimate goal of developing a one-step process for simultaneous selection and phenotypic screening in the monoclonal liquid nanocultures, we first designed a phenotypic assay aimed to discriminate between positive and negative β-galactosidase (β-gal) phenotypes using the wt E. coli strain and a lacZ::Tn5 mutant (strains W3110 and ARD220, Table 1). Expression of β-gal enables wt E. coli to hydrolyse the substrate 4-methylumbelliferyl β-D-galactopyranoside (MUG), which in hydrolysed form emits fluorescence at 450 nm. In contrast, the lacZ::Tn5 mutant is unable to hydrolyse MUG. To define the basal levels of MUG hydrolysis in each strain, we cultured bacteria in the presence of MUG on two nwSlides for each strain. During the 16 h incubation at 37 °C, bacterial growth was monitored in each nanowell at OD600 and MUG hydrolysis at OD450 every 30 min. Processing of the OD600 recordings with nOSAT identified 1161 nanowells with growth for the wt E. coli and 1257 nanowells with growth for the lacZ::Tn5 mutant. To define the range of fluorescence intensity expected from MUG hydrolysis, we plotted the RFUmax values from each nanowell with bacterial growth. Fig. 4a shows a wide distribution of RFUmax for the wt E. coli, with the 1st percentile at 4638 RFU and the 99th percentile at 21833 RFU. Nanocultures with the lacZ::Tn5 mutant show, as expected, much lower RFUmax and a narrow distribution with the 1st percentile at 766.6 RFU and the 99th percentile at 1878 RFU. Close inspection reveals a very minor overlap between the RFUmax distribution of the two strains, with only two wt E. coli nanocultures appearing below the 99th percentile of the lacZ::Tn5 mutant distribution (Fig. 4b). Based on these results, we defined the 99th percentile of the lacZ::Tn5 mutant (1878 RFU) as the fluorescence threshold to discriminate between positive and negative β-gal phenotypes.
 | ||
| Fig. 4 Phenotypic screening to detect mutants with impaired β-galactosidase activity. (a) Distribution of RFUmax from nanowells inoculated with E. coli W3110 wt and the lacZ::Tn5 mutant. Data is pooled from two biological replicates, representing 1161 (E. coli W3110 wt) and 1257 (lacZ::Tn5 mutant) nanocultures. Whiskers define 1st and 99th percentile. (b) Magnification of data in the range 0–5000 RFUmax from panel (a), showing in greater detail the narrow RFUmax distribution of lacZ::Tn5 mutant nanocultures, and their minor overlap with the E. coli W3110 wt. (c) Distribution of RFUmax from nanowells inoculated with single bacteria from mixed cultures containing equal ratio of strains E. coli W3110 wt, ARD219, ARD230 and ARD232 that are all positive for lacZ, as well as 20% (Mix20%), 2% (Mix2%) and 0.2% (Mix0.2%) of the lacZ::Tn5 mutant strain. Data originate from one biological replicate. (d) Magnification of data in the range 0–1878 RFUmax from panel (c), showing in greater detail the number of mutants with RFUmax below the phenotypic discrimination threshold in each Mix. | ||
Next, we analysed whether this discrimination threshold could be used to identify the lacZ::Tn5 mutant strain, based on its phenotype, within a mixed bacterial population. To make a complex yet controlled mix of strains, we generated isogenic mutants by Tn5 transposon mutagenesis of the wt E. coli strain W3110. We randomly selected three of the Tn5 transposon mutant strains (ARD219, ARD230, ARD232, Table 1) and mixed them with the wt E. coli strain at equal ratios. The lacZ::Tn5 mutant was then added at a representation of 20% (Mix20%). The mixed culture was single-sorted into nanowells containing MUG-supplemented medium, and the nwSlide was incubated while bacterial growth and MUG hydrolysis were recorded every 30 min. By applying the nOSAT, we identified all nanowells with growth and plotted the RFUmax of corresponding cultures (Fig. 4c). Mix20% showed 427 nanocultures with growth, whose RFUmax varied between 206 and 22854 RFU. By applying the phenotypic discrimination threshold (1878 RFU), we identified 108 nanocultures with lower RFU as likely candidates to contain the lacZ::Tn5 mutant (Fig. 4d). To test whether phenotypic screening correctly discriminated lacZ::Tn5 mutants from the additional four strains in Mix20%, we randomly retrieved and sub-cultured 24 nanocultures with RFUmax below and 11 nanocultures above the phenotypic discrimination threshold, and defined their lacZ genotype by PCR analysis. Agreement between genotype and phenotype was 87.12%. This showed that our screening platform could adequately discriminate the lacZ::Tn5 mutant in a complex sample based on its phenotype alone.
To define the sensitivity of the phenotypic screening assay, we prepared mixed cultures as above, this time adding only 2% (Mix2%) and 0.2% (Mix0.2%) of lacZ::Tn5 mutant cells. When repeating the above procedure on Mix2%, nOSAT identified growth of 502 monoclonal nanocultures, with RFUmax varying between 877–43879 RFU (Fig. 4c). Close inspection revealed 68 nanocultures with RFUmax below the phenotypic discrimination threshold (Fig. 4d). PCR-based confirmation of the lacZ genotype in randomly retrieved nanocultures below and above the RFU threshold showed 98% agreement with the LacZ phenotype. In Mix0.2%, nOSAT identified 416 nanocultures with growth, whose RFUmax varied between 717 and 43637 RFU (Fig. 4c). Five nanocultures appeared below the phenotypic discrimination threshold (Fig. 4d). Genotypic confirmation of strains above and below the discrimination threshold revealed 100% agreement with the phenotype. Collectively, this demonstrates that our method can successfully identify individual bacteria based on their phenotype even when their representation is as low as 0.2% of the population.
Simultaneous selection and phenotypic screening in the nwSlide enabled by the dual-selection transposon TnMHA
Having shown that the FACS-linked nwSlide works excellently to generate monoclonal nanocultures for phenotypic screening, we next extended this method to develop a one-step process for simultaneous selection and phenotypic screening of bacterial mutants. Given that FACS can select and sort single bacteria based on fluorescence, a fluorescent marker can be used to distinguish the fraction of mutated bacteria from the large population of wt cells in a mutagenized bacterial culture. Sorting of mutants would result in liquid monoclonal nanocultures corresponding to single-cell colonies formed on selective agar plates after an overnight incubation.To test the feasibility of this approach, we constructed a transposon harboring an antibiotic resistance gene along with a fluorescent protein-encoding gene (see Experimental for details). Briefly, the gene encoding the enhanced GFP protein, gfpmut2, and its IPTG-inducible promoter were cloned in the multiple cloning site of plasmid pMOD2, where it is flanked by the mosaic ends recognized by the EZ-Tn5 transposase (Fig. 5a). To enable antibiotic selection of transposon mutants, a kanamycin resistance gene with its cognate promoter was cloned upstream gfpmut2. The new transposon-containing vector was named pMHA and the transposon TnMHA. To our knowledge, this is the first transposon that contains two independently expressed genes encoding an antibiotic resistance cassette and a fluorescent protein. With both genes being under the control of their cognate promoters, there is no need for in-frame insertion in the bacterial chromosome, which greatly simplifies the use of the construct.
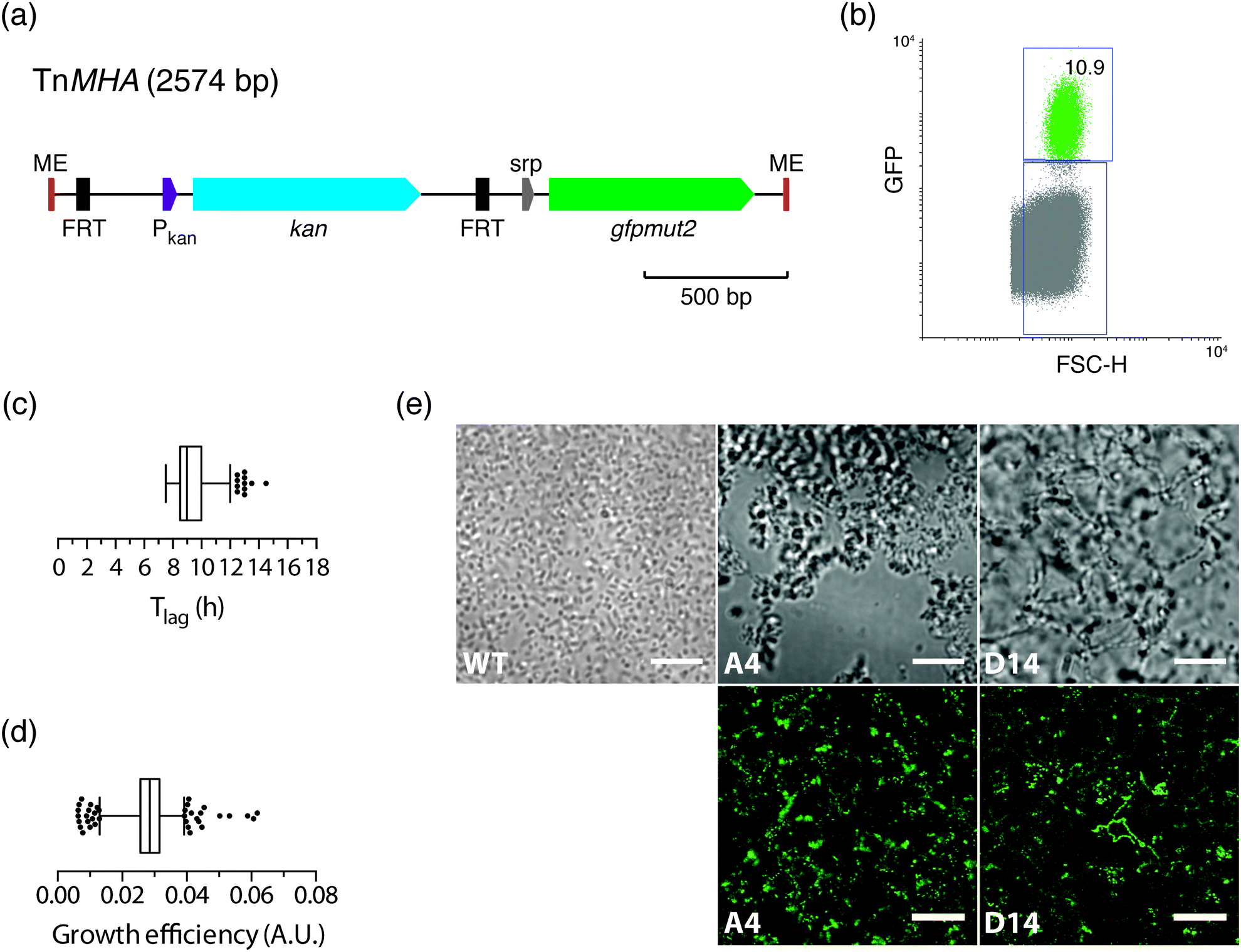 | ||
| Fig. 5 Design of TnMHA to enable FACS-based selection and phenotypic screening of transposon mutants in the nwSlide (a) schematic representation of the transposon TnMHA, which provides dual selection based on GFP fluorescence (gfpmut2) and kanamycin resistance (kan). ME = mosaic ends, Pkan = kanamycin promoter, srp = sterically repressed promoter. To enable optional removal of the kan gene, it is flanked by flippase recognition target (FRT) sequences. (b) FACS analysis of bacteria electroporated with TnMHA, showing 10.9% mutant cells in the total population. (c and d) Distribution of Tlag (c) and growth efficiency (d) of the TnMHA mutants sorted in (b). (e) Phase contrast (upper panel) and confocal fluorescence (lower panel) microscopy of TnMHA mutants in nanowells A4 and D14 show how their morphotypes differ from E. coli W3110 wt. Scale bars = 10 μm (phase contrast microscopy) and 25 μm (confocal fluorescence microscopy). | ||
The dual-selection transposon was next used to generate TnMHA mutants. PCR-amplified TnMHA mixed with EZ-Tn5 transposase was electroporated in E. coli cells (MegaX DH10B™ T1R, Table 1), and bacteria were allowed to recover during a 2 h incubation. This procedure typically generates 1 × 105 transposon mutants, which constitute ≤0.002% of the total bacterial population. As this low number classifies the mutant cells as rare events for FACS, we enriched for mutants in the bacterial culture by overnight incubation in SOC medium supplemented with kanamycin. Moreover, IPTG was added to maximize bacterial expression of GFP for subsequent fluorescence sorting. The bacterial culture was loaded onto the FACS instrument, where the GFP negative gate had been previously set using non-transformed E. coli as a negative control. The GFP vs. FSC-H plot showed that the GFP positive fraction was 10.9% of the total sample (Fig. 5b). This indicated a successful generation and enrichment for TnMHA mutants. To isolate and subculture the individual mutants, we single-sorted the GFP positive bacteria on a nwSlide pre-filled with Müller-Hinton medium with kanamycin. After overnight incubation with OD600 recordings, data processing by nOSAT identified 371 nanocultures with TnMHA mutants. This shows that TnMHA successfully enables fluorescence-based selection of transposon mutants in the FACS-linked nwSlide system. A recovery rate of 56% (371 mutants out of 672 nanowells) suggests that a number of wt cells also were FACS sorted onto the nwSlide, but as they were exposed to medium containing kanamycin, they were unable to grow. A more stringent gating strategy for GFP+ mutant selection in the FACS instrument would increase the recovery of transposon mutants.
Following the dual-selection based on GFP expression during sorting and kanamycin resistance during growth, we screened the 371 TnHMA mutants for a phenotype of interest. As a proof-of-concept, we performed growth phenotyping based on data generated by the nOSAT analysis of the monoclonal nanocultures. When analysing the length of the lag phase, Tlag, in the mutant population, we found a median of 9 h (Fig. 5c). By applying the 95th percentile (12 h) as an upper cut-off, we identified 11 mutants with unusually long lag phase. From these, we recovered mutants in nanowells D18, D40 and L43, and mapped the transposon insertion sites using a cloning-free strategy, rapid amplification of transposon ends (RATE), based on single-primer PCR. Sequencing of RATE-generated PCR products revealed transposon insertions in the genes tnpX (D18), in yebB (D40) and in yhgE (L43) (Table 1). Similarly, we applied the 5th percentile (7.5 h) as a lower cut-off to identify mutants with short lag phase (Fig. 5c). Out of 31 mutants, we recovered the mutant with shortest lag phase (nanowell B5), and located the transposon insertion site in recF (Table 1).
As a second approach, we performed phenotypic screening based on the nanocultures' growth efficiency defined by nOSAT. The mutant population showed a median growth efficiency of 0.029 A.U. (Fig. 5d). By applying the 95th percentile (0.039 A.U.) as cut off, we selected the mutant in nanowell G13 as it showed increased growth efficiencies (0.052 A.U.), and recovered bacteria for subsequent RATE analysis. Sequencing of RATE-generated PCR products located the transposon insertion site at mocA. Taken together, our experiments demonstrate that the FACS-linked nwSlide provide a novel method to identify potential new roles for genes based on multiple parameter analysis that could otherwise go unnoticed. Further work, however, would be required to unravel the link between gene function and the phenotypes discovered, but this falls outside the scope of the present investigation.
Morphotyping of transposon mutants
With the physical dimensions of a microscopy slide, a 500 μm thick glass bottom and a total slide thickness of only 1 mm, the nwSlide is well suited for microscopic analysis of the 672 nanowells. This provides an opportunity to image the monoclonal liquid nanocultures in a phenotypic screen for bacterial morphotypes. Phase contrast microscopy of wt E. coli reveals the typical short rod morphotype of bacteria freely dispersed in the nanowell (Fig. 5e). To analyse whether any mutation generated a deviating morphotype, we also screened the 371 TnMHA mutants sorted on the nwSlide with phase contrast microscopy and observed several mutants whose phenotype differed markedly from the wt E. coli. The mutant in nanowell A4 showed a rare morphotype, with clumps of tightly aggregated bacteria. Another rare morphotype was observed in nanowell D14, where bacteria formed highly irregular aggregates. The altered morphotypes of the two mutants were also visualized by fluorescence microscopy. Fluorescence imaging of the mutants was possible because they harbor the GFP-expressing transposon construct. Recovery of the two mutants followed by RATE PCR and sequencing, identified the transposon insertion sites in the genes rfaJ (nanowell A4) and in casE (nanowell D14) (Table 1). As with the rest of the mutants identified in this study, defining the link between genotype and phenotype requires further investigation.Conclusions
Similar to the omic technologies, miniaturized devices for bacterial culturing and measurement have great potential to revolutionize microbiological research. A significant engagement in this field has generated a plethora of miniaturized, often technologically advanced, devices. Their use may, however, be somewhat restricted, as many devices require dedicated equipment and technological expertise rarely seen in traditional microbiological laboratories. This prompted us to develop a universal platform for high resolution, high throughput phenotypic and genotypic analysis of bacterial cells. To establish a user-friendly interface, we combined the nwSlide with mainstream technologies present in most microbiological laboratories, such as FACS, spectrophotometry, and microscopy. As this generates great versatility in experimental designs, the nwSlide becomes a miniaturized substitute for both the agar and microtiter plate.Merging the nwSlide with FACS and spectrophotometry greatly shortens the time for selection and screening protocols. Plating of bacterial cultures on selective agar plates, isolation of monoclonal colonies the following day, and sub-cultivation in liquid medium for screening purposes can now be omitted. The FACS-linked nwSlide allows up to 672 single bacteria to be sorted in individual wells in 15 min. Placement of the inoculated nwSlide in a temperature-controlled plate reader enables immediate spectrophotometric monitoring of growth and phenotypic screening. The nOSAT algorithm, specifically developed for efficient handling of large amount of data, achieves rapid characterization of all cultures with high sensitivity and specificity. As nOSAT identifies mutants with differing growth parameters, our method is able to provide quantitative information already from the first experimental round. Compatibility of the nwSlide with microscopy provides yet another value for screening purposes, as morphologies can be assessed directly on bacteria growing in the nanowells. Bacterial morphotyping is otherwise rarely used in phenotypic screens since preparation of bacterial samples for microscopic analysis is a lengthy and laborious process using current workflows. Finally, the nwSlide enables easy translation of bacterial cultures from the nanoliter format to larger volumes. This allows downstream subculturing for mutant retrieval and genetic analysis by standard molecular biology techniques.
The universal design of our platform is further manifested by its ability to host a wide variety of bacterial screens, given that the output can be measured as an optical signal. We demonstrated label-free sorting of single bacteria from a mixed culture to phenotypically identify strains with mutations in enzymatic genes, using a nwSlide that contained medium supplemented with a fluorescent enzymatic substrate. Alternatively, FACS can be used to achieve direct selection of mutants. To demonstrate this, we designed the FACS-based transposon mutagenesis workflow. Contrary to transposons carrying a single antibiotic resistance cassette as a selection marker, TnMHA carries a dual selection system for emission of fluorescence and antibiotic resistance. The former enabled direct selection of transposon mutants, as fluorescent transposon-containing bacteria are selectively single-sorted into individual nanowells. While our transposon was stably integrated in the chromosome, this selection principle can also be applied to other constructs, such as fluorescently detectable plasmid-containing bacteria. Once deposited in the nanowells, bacteria are exposed to a second round of selection, as growth under antibiotic exposure selects for transposon-containing bacteria harboring an antibiotic resistance cassette. Non-fluorescent phenotypic screens, shown here by identification of mutants with altered growth parameters and morphotypes using nOSAT, can be performed along the selection. Alternatively, fluorescent or colorimetric screens can be performed, where relevant substrate in the nanowells enables phenotypic screens for mutations in enzymatic genes, or mutants containing transcriptional and translational gene fusions, among others. The ability to perform high throughput selection and phenotypic screening in a one-step process in nanoliter-sized liquid cultures saves time and reduces the need for plastic consumables, the amount of growth medium and supplements, such as antibiotics.
The versatility and modular workflow provide easy adaptation of the nwSlide to a wide range of laboratory settings. Its use in basic research laboratories can be readily extended to industrial and clinical laboratories, where novel applications, such as pathogen surveillance, can be addressed by high-throughput phenotypic screening of clinical or environmental samples. By reinventing traditional microbiology assays in miniaturized formats, the output of bacterial culturing and phenotypic screening in diverse environments can be greatly increased.
Author contributions
H. A., E. W., H. A. S., and A. R.-D. conceived the presented idea. H. A. and E. W. performed a preliminary investigation to test the feasibility of the project. H. A. and M. V. G. designed and carried out all experiments. H. A. designed and applied the analysis algorithm on generated data. A. R.-D. and H. A. S. supervised the project and contributed to result interpretation. H. A., M. V. G., and A. R.-D. wrote the manuscript and E. W. and H. A. S. provided critical feedback.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr. J. Mikes at the National Mass Cytometry Facility at ScilifeLab, Stockholm, for his help in optimizing FACS. This work was supported by grants from the Swedish Research Council, Stockholm County Council (ALF project), European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement No. 615458.Notes and references
-
R. J. Petri, Cent. für bacterriologie und Parasittenkd., 1887, vol. 1, pp. 279–280 Search PubMed
.
- W. Hesse, ASM News, 1992, 58, 425–428 Search PubMed
-
C. Ingham and P. M. Schneeberger, in The Role of New Technologies in Medical Microbiological Research and Diagnosis, ed. J. P. Hays and W. B. Leeuwen, Bentham e Books, Rotterdam, 2012, pp. 3–15 Search PubMed
- M. Y. Galperin and E. V. Koonin, Trends Biotechnol., 2010, 28, 398–406 CrossRef PubMed
- A. Blomberg, Curr. Opin. Biotechnol., 2011, 22, 94–102 CrossRef PubMed
- J. Warringer, E. Ericson, L. Fernandez, O. Nerman and A. Blomberg, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15724–15729 CrossRef PubMed
- J. Warringer, D. Anevski, B. Liu and A. Blomberg, BMC Chem. Biol., 2008, 8, 3 CrossRef PubMed
- R. Martinez and U. Schwaneberg, Biol. Res., 2013, 46, 395–405 CrossRef PubMed
- M. Zackrisson, J. Hallin, L.-G. Ottosson, P. Dahl, E. Fernandez-Parada, E. Ländström, L. Fernandez-Ricaud, P. Kaferle, A. Skyman, S. Stenberg, S. Omholt, U. Petrovič, J. Warringer and A. Blomberg, G3: Genes, Genomes, Genet., 2016, 6, 3003–3014 Search PubMed
- Y. Qiao, X. Zhao, J. Zhu, R. Tu, L. Dong, L. Wang, Z. Dong, Q. Wang and W. Du, Lab Chip, 2018, 8, 62 Search PubMed
- S. Lindström, R. Larsson and H. A. Svahn, Electrophoresis, 2008, 29, 1219–1227 CrossRef PubMed
- S. Lindström and H. Andersson-Svahn, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 308–316 CrossRef PubMed
- E. Weibull, H. Antypas, P. Kjäll, A. Brauner, H. Andersson-Svahn and A. Richter-Dahlfors, J. Clin. Microbiol., 2014, 52, 3310–3317 CrossRef PubMed
- S. Lindström, M. Eriksson, T. Vazin, J. Sandberg, J. Lundeberg, J. Frisén and H. Andersson-Svahn, PLoS One, 2009, 4, 1–9 Search PubMed
- S. Lindstrom, M. Hammond, H. Brismar, H. Andersson-Svahn and A. Ahmadian, Lab Chip, 2009, 9, 3465–3471 RSC
- A. Karlyshev and M. J. Pallen, BioTechniques, 2000, 28, 1078–1082 Search PubMed
- M. Karow and C. Georgopoulos, J. Bacteriol., 1992, 174, 702–710 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc00190a |
| ‡ Current address: Vironova AB, Gävlegatan 22, 113 30 Stockholm, Sweden. |
| This journal is © The Royal Society of Chemistry 2018 |