
WO3 nanosheets rich in oxygen vacancies for enhanced electrocatalytic N2 reduction to NH3†
Wenhan
Kong
ab,
Rong
Zhang
b,
Xiaoxue
Zhang
c,
Lei
Ji
b,
Guangsen
Yu
b,
Ting
Wang
 c,
Yonglan
Luo
c,
Xifeng
Shi
d,
Yuanhong
Xu
*a and
Xuping
Sun
*b
c,
Yonglan
Luo
c,
Xifeng
Shi
d,
Yuanhong
Xu
*a and
Xuping
Sun
*b
aCollege of Materials Science and Engineering, College of Life Sciences, Qingdao University, Qingdao 266071, Shandong, China. E-mail: yhxu@qdu.edu.cn
bInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China. E-mail: xpsun@uestc.edu.cn
cChemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, College of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637002, Sichuan, China
dCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, Shandong, China
First published on 10th June 2019
Abstract
The Haber–Bosch process for industrial-scale NH3 production suffers from harsh conditions and serious CO2 release. Electrochemical N2 reduction is an alternative approach to synthesize NH3 under ambient conditions, but it requires highly-efficient electrocatalysts for the N2 reduction reaction (NRR). In this Communication, we demonstrate that WO3 nanosheets rich in oxygen vacancies (R-WO3 NSs) exhibit greatly enhanced NRR performances. In 0.1 M HCl, such R-WO3 NSs achieve a large NH3 yield of 17.28 μg h−1 mgcat.−1 and a high faradaic efficiency of 7.0% at −0.3 V vs. a reversible hydrogen electrode, much superior to the WO3 nanosheets deficient in oxygen vacancies (6.47 μg h−1 mgcat.−1 and 1.02%). Remarkably, R-WO3 NSs also show high electrochemical stability.
As an indispensable chemical in today's chemical industry, NH3 is an activated nitrogen feedstock for fertilizers, plastics, pharmaceuticals, dyes etc.1–3 It is also regarded as a green energy carrier.4 The synthesis of NH3 from the hydrogenation of atmospheric N2, however, is extremely difficult due to the inherent nature of the strong N
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond.5 To date, industrial NH3 production is still dominated by the Haber–Bosch process under harsh conditions.6,7 This process however consumes large energy and releases enormous CO2. Therefore, it is needed to develop sustainable and eco-friendly routes for efficient N2 fixation under milder conditions.
N bond.5 To date, industrial NH3 production is still dominated by the Haber–Bosch process under harsh conditions.6,7 This process however consumes large energy and releases enormous CO2. Therefore, it is needed to develop sustainable and eco-friendly routes for efficient N2 fixation under milder conditions.
Electrochemical reduction, which can be operated under ambient conditions using renewable electric energy, is regarded as a promising choice for artificial N2 fixation.8,9 However, the N2 reduction reaction (NRR) must be driven by efficient electrocatalysts.8–11 Precious metal-based catalysts (Au, Ag, Pd, Ru, etc.) show favourable NRR activity,12–16 but their high price and low abundance impede their large-scale applications. Considerable attention has thus been focused on Earth-abundant alternatives.17–41 Recent density functional theory (DFT) calculations suggest that a single tungsten atom anchored on N-doped graphyne can activate the inert NN triple bond.42 As an important transition metal oxide, tungsten oxide (WO3) has been widely applied in electrochemical fields due to its Earth abundance, highly tunable composition, and good electrochemical stability,43 but its use as a NRR electrocatalyst still remains unreported.
Introducing oxygen vacancies (OVs) into electrocatalysts stands out as an effective strategy to manipulate the electronic structure, lower the activation energy barrier and further improve the electrocatalytic activities.44,45 Recent studies emphasize the significant role of VOs in electrocatalytic N2 adsorption and activation.46–49 Furthermore, two-dimensional (2D) nanosheets with a large number of active sites are a promising structure for realizing high electrocatalytic NRR performance. Herein, we report the first experimental demonstration of using 2D WO3 nanosheets rich in OVs (R-WO3 NSs) as an active and selective electrocatalyst for artificial NH3 synthesis under ambient conditions. In 0.1 M HCl, such R-WO3 NSs achieve a large NH3 yield (VNH3) of 17.28 μg h−1 mgcat.−1 and a high faradaic efficiency (FE) of 7.0% at −0.30 V vs. the reversible hydrogen electrode (RHE), much superior to the WO3 nanosheets deficient in OVs (D-WO3 NSs: 6.47 μg h−1 mgcat.−1 and 1.02%). Remarkably, R-WO3 NSs also show excellent electrochemical and structure stability.
Fig. 1a presents the X-ray diffraction (XRD) patterns for R-WO3 NSs and D-WO3 NSs. The XRD peaks for both R-WO3 NSs and D-WO3 NSs can be well distributed to monoclinic WO3 (JCPDS No. 83-0951). The transmission electron microscopy (TEM) images show that R-WO3 NSs (Fig. 1b) and D-WO3 NSs (Fig. S1a†) exhibit a sheet-like morphology. A slight lattice disorder and dislocations are locally observed in the R-WO3 NSs (Fig. 1c), highlighting the existence of numerous OVs. D-WO3 NSs have a relatively flat and smooth surface without OVs (Fig. S1b†). As shown in the high-resolution TEM (HRTEM) image (inset in Fig. 1c), the nanosheet displays clear lattice fringes with the lattice spacings of 0.38 and 0.36 nm, which correspond to the (002) and (200) planes of WO3, respectively. The polymorphic nature of R-WO3 NSs is supported by the selected area electron diffraction pattern (Fig. S2†). The energy-dispersive X-ray (EDX) spectrum (Fig. S3†) reveals the existence of W and O. TEM and the corresponding EDX elemental mapping images (Fig. 1d) further indicate the uniform distribution of W and O.
 | ||
| Fig. 1 (a) XRD patterns of R-WO3 NSs and D-WO3 NSs. (b, c) TEM and HRTEM images of R-WO3 NSs. (d) TEM and the corresponding EDX mapping images of W and O elements for R-WO3 NSs. | ||
The chemical states of R-WO3 NSs are characterized by X-ray photoelectron spectroscopy (XPS). The XPS survey spectrum further confirms the co-existence of W and O elements (Fig. 2a). As shown in the W 4f XPS spectra (Fig. 2b), two peaks at 35.5 and 37.6 eV correspond to the characteristic W 4f7/2 and W 4f5/2 peaks of W6+, respectively. The other two peaks at 36.4 and 34.3 eV are assigned to the typical binding energies of W5+.50 W5+ usually has a great relationship with OVs.50,51 In the O 1s region (Fig. 2c), two peaks at 529.9 and 531.7 eV are attributed to the lattice oxygen and the oxygen species chemisorbed at OVs, respectively.52 The OVs were further resolved by electron spin resonance (ESR) spectroscopy. Room-temperature ESR spectra (Fig. 2d) indicate that R-WO3 NSs rather than D-WO3 NSs exhibit a significant ESR signal at g = 2.002, suggesting the electron trapping at OVs.53 All of these results support the successful preparation of WO3 rich in OVs.
 | ||
| Fig. 2 (a) XPS survey spectrum of R-WO3 NSs. XPS spectra of R-WO3 NSs in the (b) W 4f and (c) O 1s regions. (d) Room-temperature ESR spectra of R-WO3 NSs and D-WO3 NSs. | ||
A carbon paper (CP) electrode was loaded with R-WO3 NSs (R-WO3/CP, R-WO3 loading: 0.1 mg cm−2) for NRR tests in N2-saturated 0.1 M HCl electrolytes. All potentials are reported on a RHE scale. We applied the indophenol blue method and the Watt and Chrisp's method to measure the produced NH3 and the possible by-product (N2H4), respectively.54 Fig. S4 and S5† present the corresponding calibration curves. NRR electrolysis experiments were conducted by applying controlled potentials from −0.25 to −0.45 V, as shown in Fig. S6.† The UV-Vis absorption spectra for electrolytes stained with the indophenol indicator at different potentials are shown in Fig. 3a. Fig. 3b presents the calculated results of VNH3 and FEs. Obviously, VNH3 and FE are enhanced as the negative potential increases until −0.30 V, where the values reach 17.28 μg h−1 mgcat.−1 and 7.0%, respectively. This outstanding performance is superior to most reported NRR electrocatalysts. Table S1† lists more detailed comparison. When applying a more negative potential, both the VNH3 and FEs decrease sharply because of the competition of the hydrogen evolution reaction.55 N2H4 was not detected at a series of potentials (Fig. S7†), indicating that this catalyst possesses good selectivity for NH3 formation. The electrocatalytic NRR performance of D-WO3 NSs on the CP (D-WO3/CP) was investigated for comparison. VNH3 and FE of D-WO3/CP can only achieve 6.47 μg h−1 mgcat.−1 and 1.02% at −0.30 V, respectively, which are much lower than those of R-WO3/CP (Fig. 3c and d).
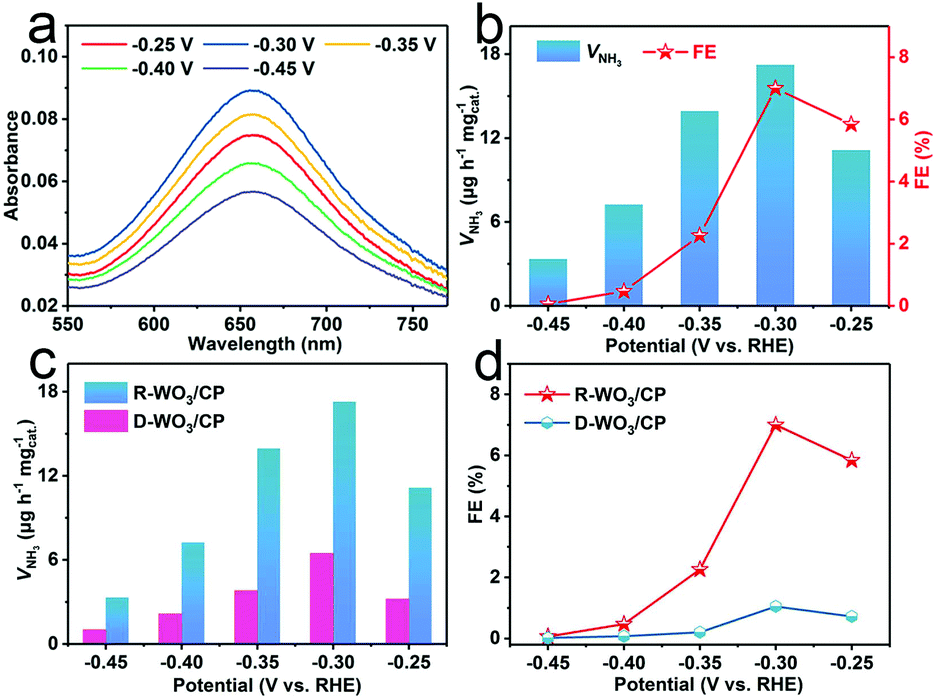 | ||
| Fig. 3 (a) UV-Vis absorption spectra of the 0.1 M HCl electrolytes stained with the indophenol indicator after NRR electrolysis. (b) VNH3 and FEs at each given potential. (c) VNH3 and (d) FEs at different potentials on R-WO3/CP and D-WO3/CP. | ||
The enhancement of VNH3 and FE as an NRR electrocatalyst could be attributed to the following reasons: (1) the existing OVs in R-WO3 NSs could act as the active sites for boosting the adsorption and activation of N2; (2) the introduction of OVs could advantageously affect the electronic structure of WO3 NSs, enabling enhanced electron transfer. Fig. S8† shows that R-WO3/CP possesses a much lower impedance56 and thus obviously faster NRR kinetics than D-WO3/CP; (3) the creation of OVs significantly increases the surface area (Fig. S9†) and provides more active sites for catalytic reactions (Fig. S10†).
Stability is another crucial indicator to evaluate NRR performance. As shown in Fig. 4a, this catalyst reveals a slight fluctuation in VNH3 and FEs during consecutive recycling experiments for 6 times. As observed in Fig. 4b, the current density shows almost no fluctuation during 24 h NRR electrolysis. We also tested the NRR performance of R-WO3/CP after 24 h electrolysis. Fig. S11† shows that there is only a slight decrease of VNH3 after 24 h electrolysis. XRD (Fig. S12a†) confirms that this catalyst is still WO3 in nature. The TEM image shows that the R-WO3 NSs still kept their vacancy structure post-NRR (Fig. S12b†). These results indicate that R-WO3 NSs show stable NRR performance under ambient conditions. Moreover, we executed a series of control experiments to verify the N source of the detected NH3. The amount of NH3 (mNH3) produced on blank CP was examined at −0.30 V for 2 h (Fig. S13a and b†). Note that blank CP shows no obvious activity to the NRR. We also carried out NRR tests in a N2-saturated electrolyte at the open circuit potential (OCP) for 2 h and an Ar-saturated electrolyte at −0.30 V for 2 h. As expected, no apparent NH3 was detected in either case (Fig. S13c and d†). We conducted a total of 12 h cycles with the intervals of 2 h in N2- and Ar-saturated electrolytes at −0.30 V and NH3 was formed only in the N2-saturated electrolyte (Fig. 4c). Fig. 4d shows that the mNH3 nearly linearly increases with increased electrolysis time. All the above results prove that the N source of the detected NH3 originates from N2. We also explored the influence of electrolytes with different concentrations on NRR performance. Fig. S14† shows the VNH3 and FEs of R-WO3/CP at different concentrations of the electrolyte, indicating that the optimum NRR performance is obtained when the HCl electrolyte concentration is 0.1 M.
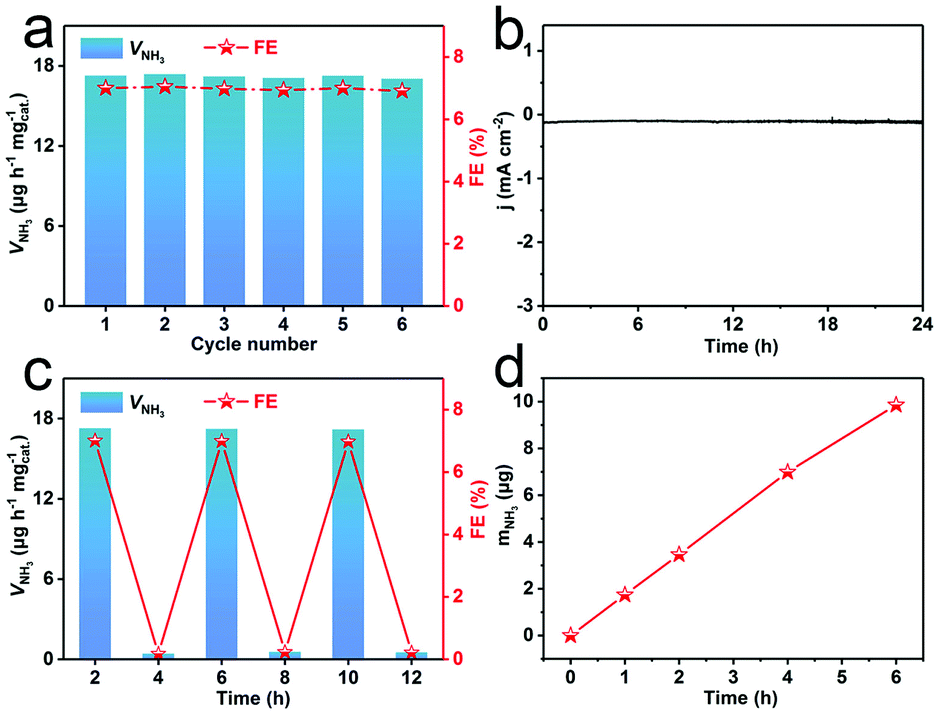 | ||
| Fig. 4 (a) Recycling experiments for R-WO3/CP during NRR at −0.30 V. (b) Time-dependent current density curve of R-WO3/CP at −0.30 V. (c) VNH3 and FEs of R-WO3/CP with alternating 2 h cycles between N2- and Ar-saturated electrolytes for a total of 12 h at −0.30 V. (d) The amount of produced NH3vs. time recorded at −0.30 V. | ||
In summary, R-WO3 NSs are successfully proven as a highly-active 2D electrocatalyst for NRR under ambient conditions. R-WO3 NSs exhibit a high activity and selectivity with a NH3 yield of 17.28 μg h−1 mgcat.−1 and a FE of 7.0% at −0.30 V vs. RHE, outperforming D-WO3 NSs (NH3 yield: 6.47 μg h−1 mgcat.−1; FE: 1.02%). Notably, R-WO3 NSs show excellent electrochemical stability as well. This study not only provides us with an attractive non-noble-metal catalyst for electrocatalytic NH3 synthesis, but also guides the rational design of W-based catalysts with high performance for applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21575137). The Science & Technology Fund Planning Project of Shandong Colleges and Universities (J16LA13 & J18KA112), the Qingdao Basic & Applied Research Project (15-9-1-100-jch), and the Qingdao Science & Technology Planning Project (17-6-3-15-gx).References
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- T. Vegge, R. Z. Sørensen, A. Klerke, J. S. Hummelshøj, T. Johannessen, J. K. Nørskov and C. H. Christensen, Indirect Hydrogen Storage in Metal Ammines, British Welding Research Association, 2008, pp. 533–564 Search PubMed.
- M. D. Fryzuk, J. B. Love, S. J. Rettig and V. G. Young, Science, 1997, 275, 1445–1447 CrossRef CAS.
- M. Appl, Ammonia – Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2002 Search PubMed.
- G. Ertl, Catalytic Ammonia Synthesis, ed. J. R. Jennings, Plenum, New York, 1991 Search PubMed.
- C. Guo, J. Ran, A. Vasileff and S. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- M. Shi, D. Bao, B. R. Wulan, Y. Li, Y. Zhang, J. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- D. Bao, Q. Zhang, F. Meng, H. Zhong, M. Shi, Y. Zhang, J. Yan, Q. Jiang and X. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- H. Huang, L. Xia, X. Shi, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 11427–11430 RSC.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- H. Liu, S. Han, Y. Zhao, Y. Zhu, X. Tian, J. Jiang, B. Xia and Y. Chen, J. Mater. Chem. A, 2018, 6, 3211–3217 RSC.
- L. Zhang, X. Ren, Y. Luo, X. Shi, A. M. Asiri, T. Li and X. Sun, Chem. Commun., 2018, 54, 12966–12969 RSC.
- X. Zhang, Q. Liu, X. Shi, A. M. Asiri, Y. Luo, X. Sun and T. Li, J. Mater. Chem. A, 2018, 6, 17303–17306 RSC.
- J. Zhao, L. Zhang, X. Xie, X. Li, Y. Yong, Q. Liu, W. Fang, X. Shi, G. Cui and X. Sun, J. Mater. Chem. A, 2018, 6, 24031–24035 RSC.
- J. Kong, A. Lim, C. Yoon, J. H. Jang, H. C. Ham, J. Han, S. Nam, D. Kim, Y. Sung, J. Choi and H. S. Park, ACS Sustainable Chem. Eng., 2017, 5, 10986–10995 CrossRef CAS.
- X. Ren, J. Zhao, Q. Wei, Y. Ma, H. Guo, Q. Liu, Y. Wang, G. Cui, A. M. Asiri, B. Li, B. Tang and X. Sun, ACS Cent. Sci., 2019, 5, 116–121 CrossRef CAS PubMed.
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Energy Mater., 2018, 8, 1801357 CrossRef.
- X. Li, X. Ren, X. Liu, J. Zhao, X. Sun, Y. Zhang, X. Kuang, T. Yan, Q. Wei and D. Wu, J. Mater. Chem. A, 2019, 7, 2524–2528 RSC.
- Y. Fang, Z. Liu, J. Han, Z. Jin, Y. Han, F. Wang, Y. Niu, Y. Wu and Y. Xu, Adv. Energy Mater., 2019, 9, 1803406 CrossRef.
- X. Zhu, Z. Liu, Q. Liu, Y. Luo, X. Shi, A. M. Asiri, Y. Wu and X. Sun, Chem. Commun., 2018, 54, 11332–11335 RSC.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- R. Zhang, X. Ren, X. Shi, F. Xie, B. Zheng, X. Guo and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 28251–28255 CrossRef CAS PubMed.
- J. Han, Z. Liu, Y. Ma, G. Cui, F. Xie, F. Wang, Y. Wu, S. Gao, Y. Xu and X. Sun, Nano Energy, 2018, 52, 264–270 CrossRef CAS.
- Y. Liu, M. Han, Q. Xiong, S. Zhang, C. Zhao, W. Gong, G. Wang, H. Zhang and H. Zhao, Adv. Energy Mater., 2019, 9, 1803935 CrossRef.
- S. Zhang, C. Zhao, Y. Liu, W. Li, J. Wang, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Chem. Commun., 2019, 55, 2952–2955 RSC.
- Y. Zhang, W. Qiu, Y. Ma, Y. Luo, Z. Tian, G. Cui, F. Xie, L. Chen, T. Li and X. Sun, ACS Catal., 2018, 8, 8540–8544 CrossRef CAS.
- X. Zhu, H. Wang, Z. Liu, R. Zhao, H. Chen, T. Wang, F. Wang, Y. Luo, Y. Wu and X. Sun, Chem. Commun., 2019, 55, 3987–3990 RSC.
- W. Kong, Z. Liu, J. Han, L. Xia, Y. Wang, Q. Liu, X. Shi, Y. Wu, Y. Xu and X. Sun, Inorg. Chem. Front., 2019, 6, 423–427 RSC.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- Z. Wang, F. Gong, L. Zhang, R. Wang, L. Ji, Q. Liu, Y. Luo, H. Guo, Y. Li, P. Gao, X. Shi, B. Li, B. Tang and X. Sun, Adv. Sci., 2018, 5, 1801182 Search PubMed.
- H. Chen, X. Zhu, H. Huang, H. Wang, T. Wang, R. Zhao, H. Zheng, A. M. Asiri, Y. Luo and X. Sun, Chem. Commun., 2019, 55, 3152–3155 RSC.
- J. Zhao, B. Wang, Q. Zhou, H. Wang, X. Li, H. Chen, Q. Wei, D. Wu, Y. Luo, J. You, F. Gong and X. Sun, Chem. Commun., 2019, 55, 4997–5000 RSC.
- Y. Luo, G. Chen, L. Ding, X. Chen, L. Ding and H. Wang, Joule, 2019, 3, 279–289 CrossRef CAS.
- J. Zhao, X. Ren, X. Li, D. Fan, X. Sun, H. Ma, Q. Wei and D. Wu, Nanoscale, 2019, 11, 4231–4235 RSC.
- X. Li, L. Li, X. Ren, D. Wu, Y. Zhang, H. Ma, X. Sun, B. Du, Q. Wei and B. Li, Ind. Eng. Chem. Res., 2018, 57, 16622–16627 CrossRef CAS.
- X. Ren, G. Cui, L. Chen, F. Xie, Q. Wei, Z. Tian and X. Sun, Chem. Commun., 2018, 54, 8474–8477 RSC.
- T. He, S. K. Matta and A. Du, Phys. Chem. Chem. Phys., 2019, 21, 1546–1551 RSC.
- Z. Huang, J. Song, L. Pan, X. Zhang, L. Wang and J. Zou, Adv. Mater., 2015, 27, 5309–5327 CrossRef CAS PubMed.
- Q. Wang, Y. Lei, D. Wang and Y. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC.
- Y. Wan, J. Xu and R. Lv, Mater. Today, 2019 DOI:10.1016/j.mattod.2019.03.002.
- C. Lv, C. Yan, G. Chen, Y. Ding, J. Sun, Y. Zhou and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 6073–6076 CrossRef CAS PubMed.
- L. Yang, T. Wu, R. Zhang, H. Zhou, L. Xia, X. Shi, H. Zheng, Y. Zhang and X. Sun, Nanoscale, 2019, 11, 1555–1562 RSC.
- G. Zhang, Q. Jia, K. Zhang, Y. Chen, Z. Li, H. Liu, J. Li and J. Qu, Nano Energy, 2019, 59, 10–16 CrossRef CAS.
- L. Zhang, X. Xie, H. Wang, L. Ji, Y. Zhang, H. Chen, T. Li, Y. Luo, G. Cui and X. Sun, Chem. Commun., 2019, 55, 4627–4630 RSC.
- G. Wang, Y. Ling, H. Wang, X. Yang, C. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2012, 5, 6180–6187 RSC.
- L. Cheng, Y. Hou, B. Zhang, S. Yang, J. Guo, L. Wu and H. Yang, Chem. Commun., 2013, 49, 5945–5947 RSC.
- K. Banger, Y. Yamashita, K. Mori, R. Peterson, T. Leedham, J. Rickard and H. Sirringhaus, Nat. Mater., 2011, 10, 45–50 CrossRef CAS PubMed.
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS PubMed.
- X. Zhang, T. Wu, H. Wang, R. Zhao, H. Chen, T. Wang, P. Wei, Y. Luo, Y. Zhang and X. Sun, ACS Catal., 2019, 9, 4609–4615 CrossRef CAS.
- W. Qiu, X. Xie, J. Qiu, W. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui, B. Tang and X. Sun, Nat. Commun., 2018, 9, 3485 CrossRef PubMed.
- P. Chen, N. Zhang, S. Wang, T. Zhou, Y. Tong, W. Chu, C. Wu and Y. Xie, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6635–6640 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: 10.1039/c9nr03678d |
| This journal is © The Royal Society of Chemistry 2019 |