
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilizing nickel-rich layered oxide cathodes by magnesium doping for rechargeable lithium-ion batteries†
Hang
Li
a,
Pengfei
Zhou
ab,
Fangming
Liu
a,
Haixia
Li
a,
Fangyi
Cheng
 *a and
Jun
Chen
a
*a and
Jun
Chen
a
aKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: fycheng@nankai.edu.cn
bSchool of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo 255049, China
First published on 12th November 2018
Abstract
Nickel-rich layered transition metal oxides are attractive cathode materials for rechargeable lithium-ion batteries but suffer from inherent structural and thermal instabilities that limit the deliverable capacity and cycling performance on charging to a cutoff voltage above 4.3 V. Here we report LiNi0.90Co0.07Mg0.03O2 as a stable cathode material. The obtained LiNi0.90Co0.07Mg0.03O2 microspheres exhibit high capacity (228.3 mA h g−1 at 0.1C) and remarkable cyclability (84.3% capacity retention after 300 cycles). Combined X-ray diffraction and Cs-corrected microscopy reveal that Mg doping stabilizes the layered structure by suppressing Li/Ni cation mixing and Ni migration to interlayer Li slabs. Because of the pillar effect of Mg in Li sites, LiNi0.90Co0.07Mg0.03O2 shows decent thermal stability and small lattice variation until it is charged to 4.7 V, undergoing a H1–H2 phase transition without discernible formation of an unstable H3 phase. The results indicate that moderate Mg doping is a facile yet effective strategy to develop high-performance Ni-rich cathode materials.
Introduction
There is an increasing demand for rechargeable lithium-ion batteries (LIBs) to power consumer electronics and electric vehicles. Current prevailing commercial LIBs are based on lithiated transition metal oxide cathodes, LiCoO2 in particular, which however suffer from low capacity and high cost.1 As an alternative, the structural analogue Ni-rich LiNixCo1−xO2 (x ≥ 0.8) has received intensive attention because of its higher capacity (≥180 mA h g−1), lower cost, less environmental impact and more abundant resource of Ni.2 A higher Ni content in LiNixCo1−xO2 increases the deliverable capacity but imposes structural and thermal instabilities on charging.3 Meanwhile, Ni-rich layered oxides are often plagued by Li/Ni cation mixing that retards Li intercalation and induces lattice variation due to Ni redox.4 It remains a challenge to develop stable LiNixCo1−xO2 cathode materials with high Ni content (i.e., x ≥ 0.9).Cation substitution such as Mn, Al, Zr and Ti has been demonstrated as an effective strategy to improve the structural stability of LiNixCo1−xO2.5 These substituted cations are beneficial mainly because of stronger bonding with oxygen as compared to Ni,6 however, they exert less effect on the troublesome Li+/Ni2+ mixing and drastic lattice variation.7 Doping electrochemically inactive cations in the interlayer Li slabs instead of transition metal–oxygen layers is helpful.8 In this regard, Mg is attractive as its ionic radius is closest to that of Li+ (0.072, 0.076, 0.053, and 0.058 nm for Mg2+, Li+, Al3+ and Mn3+, respectively). Delmas's group proposed that the distribution of Mg in Li sites stabilizes the layered LiNiO2 through the pillar effect.8a,b Cho et al. also demonstrated a decreased cation mixing and a smaller exothermic peak in charged LiNi0.74Co0.26−xMgxO2.8c Recent studies confirmed the positive effect of Mg doping and Mn/Al/Mg co-doping in enhancing the cycling stability, but the electrode performance remains unsatisfactory relative to Ni–Co–Mn ternary oxides.9 Few studies have been directed to the atomic-scale microstructure of Mg-doped Ni-rich materials or structural evolution on cycling within different voltages. Furthermore, there is no report on Mg-doped LiNixCo1−xO2 with a high Ni content of x ≥ 0.9, leaving much room to develop advanced Ni-rich materials.
Here, we report the synthesis and application of LiNi0.90Co0.07Mg0.03O2 microspheres as a stable, high-capacity cathode for LIBs. The microstructure and electrochemical properties of this new material were investigated and compared with those of the benchmark LiNi0.8Co0.1Mn0.1O2 (NCM811). The prepared LiNi0.90Co0.07Mg0.03O2 microspheres exhibit high specific capacity (228.3 mA h g−1 at 0.1C), extended cyclability and superior rate capability (177 mA h g−1 at 2C, with 84.3% capacity retention over 300 cycles). A combined X-ray diffraction (XRD) and atomic-resolution microscopy analysis reveals largely suppressed Li/Ni cation mixing. Structural evolution upon cycling is elucidated by in situ diffraction and synchrotron X-ray absorption spectroscopy. The results suggest a two-phase transition mechanism (H1–H2) with small lattice variation (<1%) and without the formation of the unstable H3 phase in a wide voltage range of 2.8–4.7 V. The remarkable performance along with favourable thermal stability renders LiNi0.90Co0.07Mg0.03O2 as a promising cathode material for LIBs viable in large-scale applications.
Results and discussion
Materials synthesis and characterization
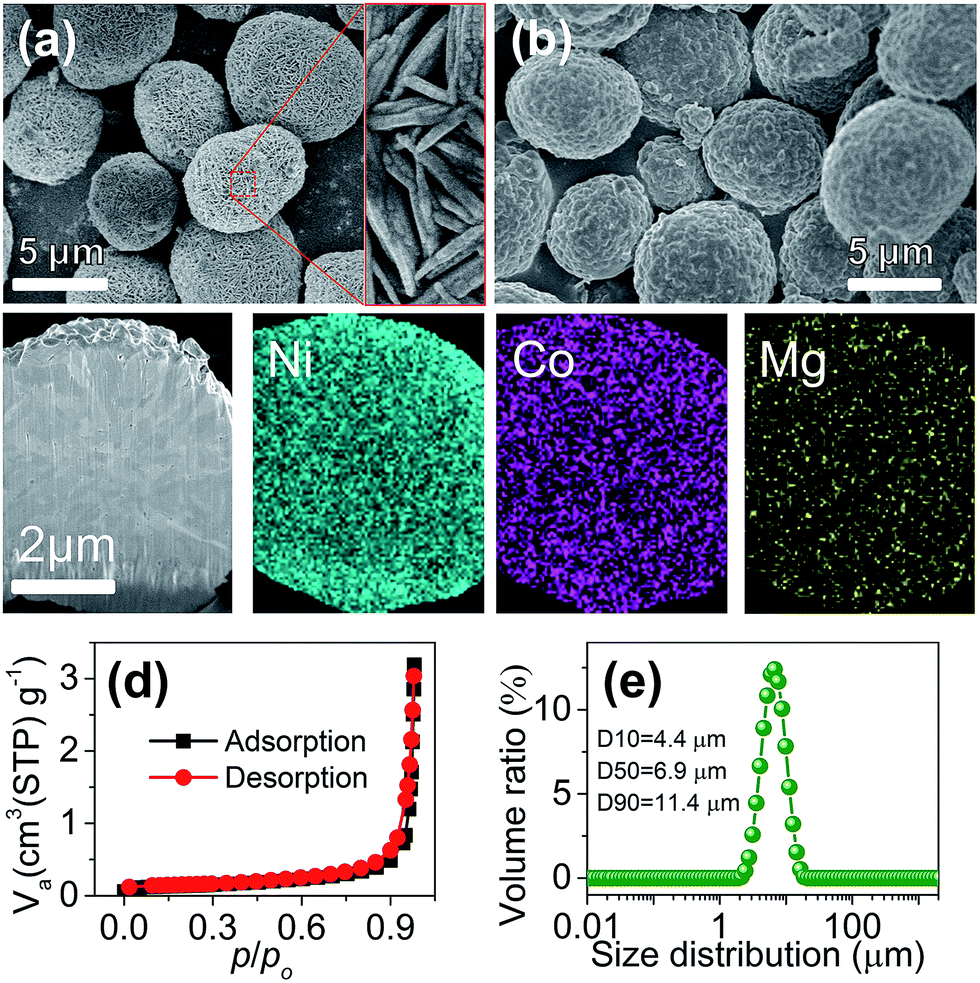
The synthesis of Mg-doped Ni-rich cathode materials involved two steps of coprecipitation and calcination, as shown in the schematic in ESI Fig. S1.† Details of the preparation are described in the ESI.† For the coprecipitation of the hydroxide precursor, control experiments were carried out to optimize reaction parameters. The main influential factors of the composition and morphology of the obtained precursor include the pH of the reactant solution and the concentration of ammonia in the continuous stirred tank reactor. A pH near 10.7 and ammonia concentration around 1.0 M favor the formation of a uniform ternary hydroxide with the target Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co:Mg ratio (ESI Table S1 and Fig. S2†). Fig. 1a shows the typical scanning electron microscopy (SEM) images of the prepared Ni0.9Co0.07Mg0.03(OH)2 precursor, which presents a spherical shape. The microspheres are assembled with needle-like primary grains and feature a porous texture with a high BET specific surface area of 176.1 m2 g−1 (ESI Fig. S3†), which would benefit the LiOH impregnation during calcination.10 The XRD pattern (ESI Fig. S4†) coincides with the profile of reference Ni(OH)2 (JCPDS card no. 14-0117), suggesting the formation of a homogeneous ternary hydroxide solid solution. Cross-sectional elemental mapping further evidences the uniform Ni/Co/Mg distribution inside the sphere (ESI Fig. S5†).
:Co:Mg ratio (ESI Table S1 and Fig. S2†). Fig. 1a shows the typical scanning electron microscopy (SEM) images of the prepared Ni0.9Co0.07Mg0.03(OH)2 precursor, which presents a spherical shape. The microspheres are assembled with needle-like primary grains and feature a porous texture with a high BET specific surface area of 176.1 m2 g−1 (ESI Fig. S3†), which would benefit the LiOH impregnation during calcination.10 The XRD pattern (ESI Fig. S4†) coincides with the profile of reference Ni(OH)2 (JCPDS card no. 14-0117), suggesting the formation of a homogeneous ternary hydroxide solid solution. Cross-sectional elemental mapping further evidences the uniform Ni/Co/Mg distribution inside the sphere (ESI Fig. S5†).
 | ||
| Fig. 1 (a) SEM images of the Ni0.9Co0.07Mg0.03(OH)2 precursor. (b) SEM image, (c) cross-sectional elemental mapping, (d) N2 adsorption/desorption isotherms and (e) particle size distribution of the synthesized LiNi0.9Co0.07Mg0.03O2. | ||
The LiNi0.9Co0.07Mg0.03O2 microspheres were prepared via calcination of Ni0.9Co0.07Mg0.03(OH)2 and LiOH. Different samples were obtained by adjusting the calcination temperature and 700 °C was found to be optimum for obtaining a well-crystallized layered structure and the desirable intact spherical morphology (ESI Fig. S6, S7 and Table S2†). As shown in Fig. 1b, the average size and spherical shape of the obtained LiNi0.9Co0.07Mg0.03O2 are well inherited from the precursor, but more compact agglomeration of primary nanoparticles is observed (ESI Fig. S8†) after calcination. Homogeneous distribution of Ni, Co and Mg is also evidenced from the cross-sectional mapping of the microsphere (Fig. 1c). The Li:Ni:Co:Mg molar ratio measured by inductively coupled plasma atomic emission spectrometry (ICP-AES) is 1.000:0.892:0.070:0.029, in good agreement with the Ni:Co:Mg ratio determined by energy dispersive spectroscopy (EDS) analysis (ESI Fig. S9†). The BET specific surface area determined from N2 adsorption isotherms (Fig. 1d) is 0.453 m2 g−1, significantly lower than that of the precursor. Fig. 1e shows a narrow particle size distribution of the microspheres with a median diameter (D50) of 6.9 µm. The relative span (defined as (D90 − D10)/D50) was 1.014, suggesting the homogeneous size of LiNi0.9Co0.07Mg0.03O2 microspheres.11 These characteristics and the respectable tap density (2.48 g cm−3) would favour practical application of the synthesized LiNi0.9Co0.07Mg0.03O2 microspheres. Moreover, the presence of surface residual lithium species such as LiOH and Li2CO3 is problematic for the Ni-rich cathode. Titration results (detailed procedures in the ESI†) indicate a Li2CO3 and LiOH amount of 13750 and 15620 ppm, respectively, which is a moderate level for Ni-rich materials as compared with previous studies (15257 ppm Li2CO3 and 11285 ppm LiOH for LiNi0.85Co0.075Mn0.075O2).7b Surface modification with an acidic agent could lower the residual lithium content.
Fig. 2a shows the Rietveld refined XRD pattern of LiNi0.9Co0.07Mg0.03O2 microspheres. The peaks are assignable to the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (no. 166) and agree with the hexagonal LiNiO2 (JCPDS no. 09-0063). A well-crystallized layered structure with a low degree of Ni2+/Li+ cation mixing is indicated by the clear splitting of (006)/(012) and (018)/(110) peaks, the large I003/I104 (1.343), and the small (I006 + I102)/I101 (0.474, denoted as the R-factor).12 The cationic distribution and lattice parameters from XRD refinement are listed in ESI Table S3.† Around 2.1% Mg and 1.5% Ni reside in Li sites, giving a formula of (Li0.964Mg0.021Ni0.015)(Ni0.881Mg0.009Li0.040Co0.070)O2. Transmission electron microscopy (TEM) imaging of an individual nanoparticle reveals single crystalline character with clear (003) lattice fringes and electron diffraction spots (Fig. 2b). The high-angle annular-dark-field (HAADF) image shown in Fig. 2c clearly evidences that trace metal cations are located in the Li slabs.13 A high degree of crystallization and a well-developed layered structure can be further viewed from the annular-bright-field (ABF) image with the proposed atom arrangement (Fig. 2d). The contrast of the HAADF and ABF images respectively shows a rough Z1.7 and Z1/3 dependence on the atomic number, which makes it possible to discriminate Li, TM and O atom columns simultaneously.14 Based on the visualized contrast, there is a low degree of mixed locations of Mg or Ni in Li slabs. Thus, the atomic-scale TEM analysis and the Rietveld refinement results evidence the substitution of Li+ by Mg in Li–O slabs and the mitigation of Li/Ni cation mixing.
m (no. 166) and agree with the hexagonal LiNiO2 (JCPDS no. 09-0063). A well-crystallized layered structure with a low degree of Ni2+/Li+ cation mixing is indicated by the clear splitting of (006)/(012) and (018)/(110) peaks, the large I003/I104 (1.343), and the small (I006 + I102)/I101 (0.474, denoted as the R-factor).12 The cationic distribution and lattice parameters from XRD refinement are listed in ESI Table S3.† Around 2.1% Mg and 1.5% Ni reside in Li sites, giving a formula of (Li0.964Mg0.021Ni0.015)(Ni0.881Mg0.009Li0.040Co0.070)O2. Transmission electron microscopy (TEM) imaging of an individual nanoparticle reveals single crystalline character with clear (003) lattice fringes and electron diffraction spots (Fig. 2b). The high-angle annular-dark-field (HAADF) image shown in Fig. 2c clearly evidences that trace metal cations are located in the Li slabs.13 A high degree of crystallization and a well-developed layered structure can be further viewed from the annular-bright-field (ABF) image with the proposed atom arrangement (Fig. 2d). The contrast of the HAADF and ABF images respectively shows a rough Z1.7 and Z1/3 dependence on the atomic number, which makes it possible to discriminate Li, TM and O atom columns simultaneously.14 Based on the visualized contrast, there is a low degree of mixed locations of Mg or Ni in Li slabs. Thus, the atomic-scale TEM analysis and the Rietveld refinement results evidence the substitution of Li+ by Mg in Li–O slabs and the mitigation of Li/Ni cation mixing.
 | ||
| Fig. 2 (a) Rietveld refined XRD pattern of the prepared LiNi0.9Co0.07Mg0.03O2. (b) HRTEM image. Insets in (b) show the SAED pattern (1) of a single particle (2). (c) HAADF image along the [−110] zone axis. (d) ABF image along the [−110] zone axis. The inset in (d) illustrates the proposed atomic arrangement, with Li, transition metal and O atoms indicated in yellow, blue and red, respectively. | ||
A batch of Ni-rich oxide samples with different compositions (LiNi0.9Co0.1O2, LiNi0.89Co0.09Mg0.02O2, and LiNi0.9Co0.05Mg0.05O2) were also prepared via similar procedures to investigate the effect of Mg content on the electrode performance (ESI Fig. S10†). Structural refinement of the obtained samples suggests that the optimum Mg proportion is around 2–3%, which gives rise to lower Ni2+/Li+ mixing and better hexagonal ordering (ESI Table S4†). Furthermore, commercially available LiNi0.8Co0.1Mn0.1O2 (NCM811) powders were also characterized for comparison. ESI Fig. S11† shows high crystallinity and spherical morphology of NCM811 while EDS elemental analysis (ESI Fig. S12†) indicates the presence of Cu that arises from material modification by the supplier. Compared with LiNi0.9Co0.07Mg0.03O2, NCM811 has a slightly lower degree of lithium deficiency but a higher degree of Li/Ni mixing (ESI Table S5†).
Electrode performance
For Ni-based cathode materials, elevating the charging voltage can improve deliverable capacity but results in poor cycling stability.15 The effect of Mg doping on the electrode properties of Ni-rich cathodes at high charge voltages deserves investigation. We comparatively tested the performance of LiNi0.9Co0.07Mg0.03O2 and NCM811 electrodes from 2.8 V to 4.3, 4.5, and 4.7 V (ESI Fig. S13†). As expected, the initial charge capacity increases with voltage. However, charging to 4.7 V results in decreased discharge capacity and coulombic efficiency of LiNi0.9Co0.07Mg0.03O2 (ESI Tables S6 and S7†), which is attributed to severe electrode/electrolyte side reactions resulting in a thicker interface layer (ESI Fig. S14†). Morphological changes, electrode polarization and charge transfer resistance of cycled NCMg electrodes were also studied (ESI Fig. S15–S17†). The results indicate that cycling the LiNi0.9Co0.07Mg0.03O2 electrode within 2.8–4.5 V helps to attain a balanced capacity and stability.Fig. 3a displays the charge/discharge curves of LiNi0.9Co0.07Mg0.03O2 and NCM811 at 2.8–4.5 V. The specific charge and discharge capacities of LiNi0.9Co0.07Mg0.03O2 are 251.3 and 228.2 mA h g−1, respectively, which are slightly higher than those of NCM811 (244.3 and 218.4 mA h g−1). The coulombic efficiency is 90.8% and 89.4% for LiNi0.9Co0.07Mg0.03O2 and NCMC811, respectively, indicating the superiority of Mg over Mn in doping the Ni-rich LiNixCo1−xO2 cathode. As shown in ESI Fig. S18,† LiNi0.9Co0.07Mg0.03O2 delivers a considerably high capacity of 142.8 mA h g−1 at 10C, which slightly exceeds that of NCM811 (137.8 mA h g−1). After galvanostatic charge/discharge at 2C for 300 cycles, a capacity of 148.7 mA h g−1 is attained for LiNi0.9Co0.07Mg0.03O2, corresponding to a capacity retention of 83.9% (Fig. 3b). Extended cycling at a high rate of 5C affords a capacity retention of over 60% after 600 cycles (ESI Fig. S19†). Further enhancement of long-term cyclability could be expected by electrolyte optimization. Comparatively, the benchmark NCM811 shows a faster capacity fade of 30.1% after 300 cycles at 2C, which could be ascribed to its large structural change on cycling that induces the formation of cracks.15b,16
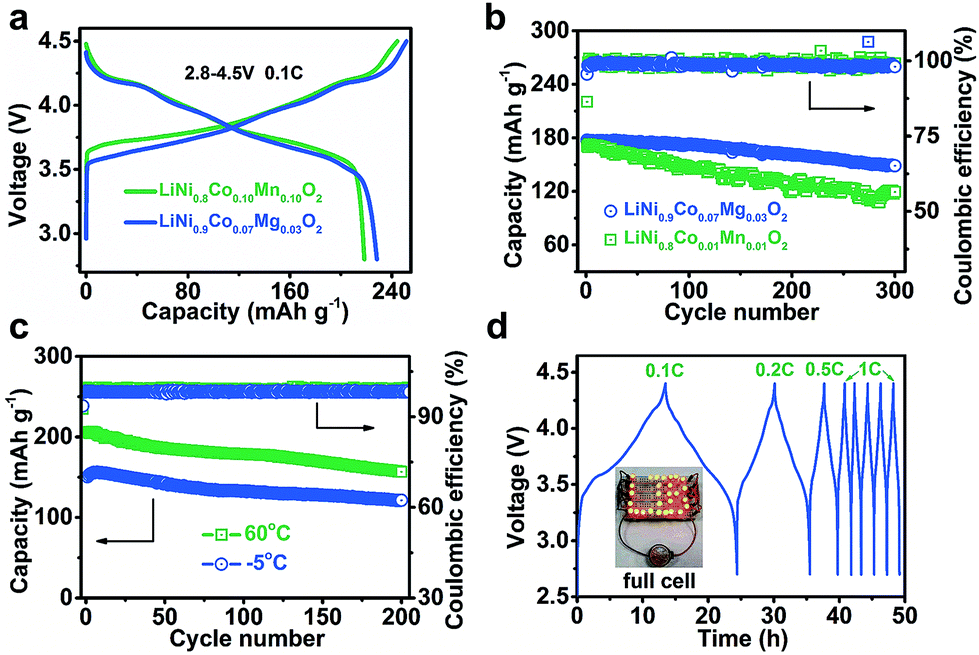 | ||
| Fig. 3 (a) Initial charge and discharge curves at 0.1C rate and (b) cycling performance of LiNi0.9Co0.07Mg0.03O2 and NCM811 at 2C rate for 300 cycles. (c) LiNi0.9Co0.07Mg0.03O2 performance at a temperature of −5 and 60 °C at 1C. (d) Charge/discharge profiles of the full LIB cell assembled with the LiNi0.9Co0.07Mg0.03O2 cathode and graphite anode. The inset shows a cell powering a string of LIB-shaped LED lights. | ||
We also tested the electrode performance at the temperatures of −5 and 60 °C. As shown in Fig. 3c, the corresponding initial capacity reaches 150.7 and 206.2 mA h g−1, with a retention of 81.2% and 76.0% after 200 cycles, respectively. Thus, the Mg-doped Ni-rich electrode is applicable in a wide temperature range. The superior performance of LiNi0.9Co0.07Mg0.03O2 motivates us to further evaluate its application in a full LIB cell with a coupled commercial graphite anode. Fig. 3d shows the charge/discharge curves of the assembled cell. At 0.1C, the average working voltage is 3.73 V. The corresponding energy density is 508.7 W h kg−1 based on the total active mass of the cathode and anode. Furthermore, the full cell operates well at higher rates (starting from 0.1C to 1C and maintaining at 1C in subsequent cycles) and shows good rechargeability.
The synthesized Ni-rich oxide microsphere samples with different compositions of LiNi0.9Co0.1O2, LiNi0.89Co0.09Mg0.02O2 and LiNi0.9Co0.05Mg0.05O2 were tested for comparison (ESI Fig. S20 and Table S8†). The results indicate that the optimal Mg content is within 2–3% and a higher degree of Mg doping exerts a negative effect, which is due to the fact that too many Mg2+ ions in Li sites hinder Li+ diffusion and decrease the deliverable capacity.8a,9c,17 Furthermore, LiNi0.9Co0.07Mg0.03O2 microspheres outperform other representative Ni-rich layered cathode materials in terms of reversible capacity and rate capability (ESI Table S9†), indicating the superiority of Mg-doping in improving the electrode performance of the LiNixCo1−xO2 cathode. Galvanostatic intermittent titration technique (GITT) method was employed to determine the diffusion coefficients of Li+ (DLi+) during the charge/discharge process (ESI Fig. S21 and S22†). For both NCM811 and LiNi0.9Co0.07Mg0.03O2, favourable kinetics can be observed upon charging and discharging, except for the stage close to the end of discharging. Furthermore, the values of DLi+ determined from the peak currents of CVs at various potential scanning rates are of the same order of magnitude (ESI Fig. S23†). These results indicate that a moderate Mg doping of the layered compound retains the Li+ diffusivity at a preferable level.
Structural evolution and thermal stability
To analyse the structural evolution of Ni-rich cathode materials, in situ XRD data of LiNi0.9Co0.07Mg0.03O2 and NCM811 were recorded at the first cycle (ESI Fig. S24 and S25†). Fig. 4a shows the representative profiles of LiNi0.9Co0.07Mg0.03O2 on charging in the 2θ region including Bragg reflections from (003) to (018), with the corresponding galvanostatic curve plotted to the left of the XRD patterns. The observed excursion of reflection peaks is indicative of changes in lattice parameters. The variation of c-axis in the crystal cell could be monitored by the skewing of (003) positions, while the (110) reflection parallel to the c-axis could mirror the change of the a- or b-axis.18Fig. 4c shows the plots of lattice parameters by refining the XRD data. The gradual shift of the (003) peak to a lower angle during deintercalation indicates the enlargement of parameter c, which is ascribed to the increase of electrostatic repulsion between adjacent oxygen layers.12a,15a,18b,19 In contrast, the (101), (012) and (018) peaks concomitantly skew to higher angles. The parameter a decreases as the higher oxidation state of Ni shrinks the Ni–O bonds in the MO6 octahedron,18a,20 which was evidenced by ex situ synchrotron X-ray absorption spectroscopy (XAS, ESI Fig. S26†). It is noteworthy that the (003) peak shifts back to higher angles in Fig. 4b, indicating a sudden decrease of the c-axis for NCM811 cathodes that originates from the collapse of LiO2 layers due to deep delithiation. In short, the amplitude of the (003) peak shift on LiNi0.9Co0.07Mg0.03O2 is significantly lower than that of NCM811 (Fig. 4b) and other reported in situ XRD results of Ni-rich cathodes.7a,18a,21c Such behaviour results in a maximum unit cell volume variation of <1%, much lower than that of NCM811 (>5%, Fig. 4b). The phase transition process of LiNi0.9Co0.07Mg0.03O2 largely differs from that of NCM811. Notably, the phase transition from the hexagonal phase H1 to H2 (with longer c) was extended to a charge voltage of 4.2 V. The formation of H2 is indicated by the appearance of the new reflections on the left of the (003) peak and on the right of the (101) peak. The intensity of H2 signals evolves slightly and no discernible H3 phase peaks emerge until 4.7 V. Comparatively, NCM811 experiences a H1–H2–H3 phase transition at lower voltages. This phase transformation has been proposed to induce an abrupt anisotropic lattice volume change, nonuniform internal stresses and subsequent numerous cracks along the grain boundaries.7a,15b,16a The mechanical damage is partly responsible for the thermal instability and poor cyclability of Ni-rich cathodes.22 Notably, the phase transition in LiNi0.9Co0.07Mg0.03O2 is extended to a higher charge voltage as compared to NCM811 and other Ni-rich cathode materials,7a,18a,21c indicating a significant effect of Mg doping in enhancing the structural stability. The drastic exothermic reaction occurring at an elevated charging voltage due to the high oxidation state of Ni is one of the stumbling drawbacks of Ni-rich cathodes.3a,7b To evaluate the thermal stability, the charged LiNi0.9Co0.07Mg0.03O2 electrodes were investigated by differential scanning calorimetry (DSC) analysis (ESI Fig. S27†). With increasing charge voltage, the peak exothermic temperature shifts negatively from 243.7 to 237.2 and 211.6 °C. The total generated heat is 543.2, 596.8, and 667.5 J g−1 for the electrode charged to 4.3, 4.5, and 4.7 V, respectively. Note that the LiNi0.9Co0.07Mg0.03O2 microspheres exhibit admirable thermal stability as compared with NCM811 and other Ni-rich cathode materials (ESI Table S10†).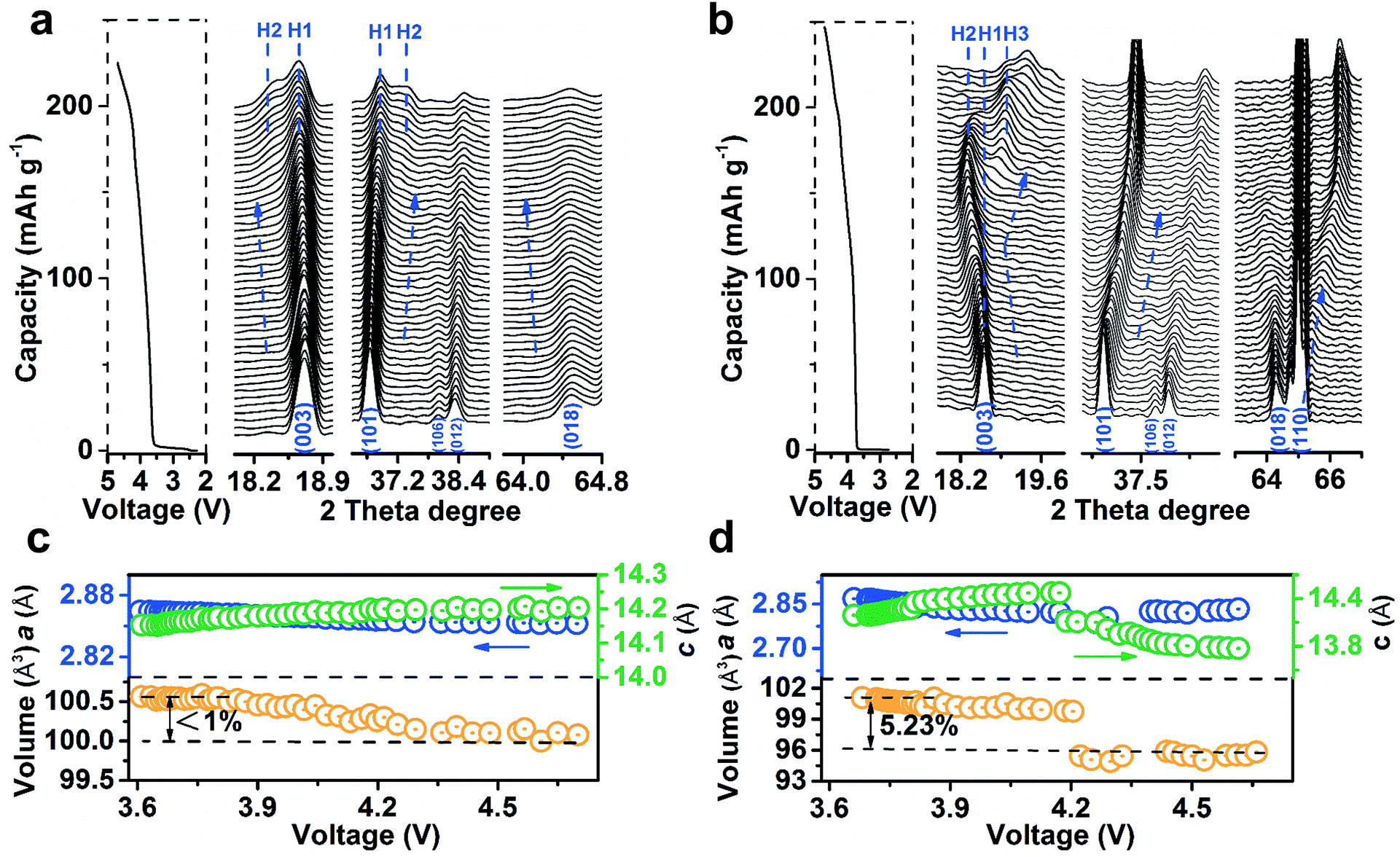 | ||
| Fig. 4 Structural evolution of LiNi0.9Co0.07Mg0.03O2 (a and c) and NCM811 (b and d). (a and b) In situ XRD patterns at selected 2θ regions and the corresponding galvanostatic curves on first charging. (c and d) The variation of lattice parameters and cell volumes as a function of voltage. | ||
The high structural and thermal stability of LiNi0.9Co0.07Mg0.03O2 is proposed to correlate with the pillar effect of Mg and the suppression of cation migration. In view of the ionic radii of Li+, Mg2+, Ni2+ and Ni3+ (0.076, 0.072, 0.069 and 0.056 nm), if extra cations are required to compensate for lithium deficiency, Mg occupies the Li sites preferentially to the Ni cations. Thus, the Ni/Li cation exchange could be suppressed in the formation of the layered structure, as evidenced from XRD and Cs-corrected TEM.8a,c Additionally, the cations residing in interlayers could increase the energy of TM ion migration to Li slabs and effectively restrain such migration on cycling due to electrostatic repulsion.13a,23 A recent first-principles calculation has also shown a larger Li/Ni exchange energy after Mg doping.24
The positive effect of Mg doping in suppressing Ni migration can be evidenced from the structural analysis of the electrode after 100 cycles (ESI Fig. S28 and S29†). From XRD Rietveld refinement, the cation mixing increases slightly from 1.95% to 2.0% for the Mg-doped cathode, while the NCM811 electrode experiences a larger variation from 2.0% to 3.4%. The cation mixing could trigger the increase of positive charges, decrease of repulsion force in the adjacent oxygen layers, phase transition and consequently capacity fading.4b,5c,15b,25 Furthermore, Ni in interslabs is oxidized to smaller Ni3+ (0.056 nm) or Ni4+ (0.048 nm) on deep charging, leading to a local collapse of the interlayer space.8a This local collapse and concomitant phase transition incur structural and thermal instabilities. In contrast, Mg cations are electrochemically inactive and could support the layered structure upon Li+ deintercalation at high charge voltages, preventing continuous migration and realizing the pillar effect (Fig. 5). The presence of Mg in Li slabs also helps to offset electrostatic repulsion between adjacent oxygen layers by Mg2+–O2− electrostatic attraction, thus slowing the change of lattice parameter c and suppressing the formation of the H3 phase on deep charging. The large lattice shrink/expansion in H2–H3 phase conversion would cause electrode cracks and pulverization.7a,15b,16a Consequently, Mg doping stabilizes LiNixCo1−xO2 and benefits cycling performance.
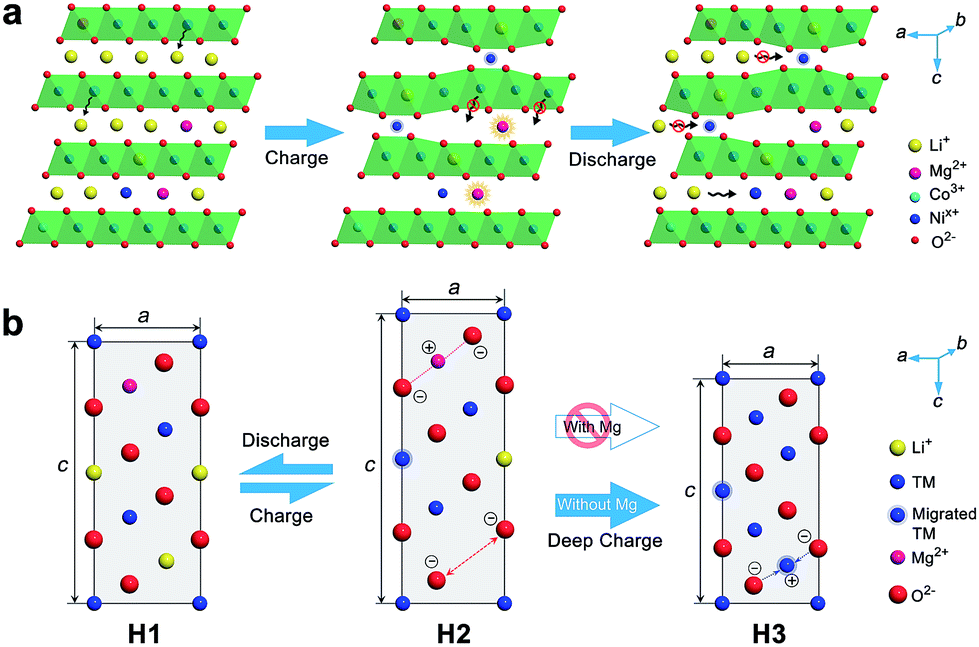 | ||
| Fig. 5 Schematic diagram of the pillar effect of Mg doping. (a) The interlayer Mg prevents TM migration and stabilizes the layered structure, while Li/Ni mixing incurs lattice collapse on charging and retards Li intercalation on discharge. (b) Structural evolution of Ni-rich cathodes with and without Mg doping. | ||
Conclusions
In summary, we have synthesized LiNi0.9Co0.07Mg0.03O2 microspheres as a new stable cathode material for LIBs. The obtained Mg-doped microspheres show a low degree of Li/Ni cation mixing and exhibit remarkable electrode performance in terms of high capacity (228.3 mA h g−1 at 0.1C) and cycling stability (84.3% capacity retention over 300 cycles at 2C). The remarkable performance is attributed to the electrochemically inactive Mg cations in interlayer Li sites that suppress Ni migration to Li slabs and stabilize the layered structure. Unlike the NCM811 cathode, LiNi0.9Co0.07Mg0.03O2 could withstand charging to a high voltage of 4.7 V and undergo <1% lattice variation without the formation of an unstable H3 phase. The superior cyclability and thermal stability make the Mg-doped Ni-rich materials promising for advanced LIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MOST (2016YFA0202503), MOE (B12015), Tianjin Project (18JCZDJC31100) and the Fundamental Research Funds for the Central Universities. Prof. Lin Gu at the Institute of Physics, Chinese Academy of Sciences is acknowledged for the assistance in TEM measurements. H. L. and P. F. Z. contributed equally to this work.Notes and references
-
(a) M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed
; (b) F. Y. Cheng, J. Liang, Z. L. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed
-
(a) W. Liu, P. Oh, X. Liu, M.-J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef CAS PubMed
-
(a) T. M. Bandhauer, S. Garimella and T. Fuller, J. Electrochem. Soc., 2011, 158, R1–R25 CrossRef CAS
-
(a) Y. Koyama, H. Arai, I. Tanaka, Y. Uchimoro and Z. Ogumi, Chem. Mater., 2012, 24, 3886–3894 CrossRef CAS
-
(a) W. D. Li, X. M. Liu, H. Celio, P. Smith, A. Dolocan, M. F. Chi and A. Manthiram, Adv. Energy Mater., 2018, 8, 1703154 CrossRef
-
(a) K. Kang and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 094105 CrossRef
-
(a) H.-H. Ryu, K.-J. Park, C. S. Yoon and Y.-K. Sun, Chem. Mater., 2018, 30, 1155–1163 CrossRef CAS
-
(a) C. Pouillerie, L. Croguennec, P. Biensan, P. Willmann and C. Delmas, J. Electrochem. Soc., 2000, 147, 2061–2069 CrossRef CAS
-
(a) T. Sasaki, V. Godbole, Y. Takeuchi, Y. Ukyo and P. Novak, J. Electrochem. Soc., 2011, 158, A1214–A1219 CrossRef CAS
-
(a) F. Y. Cheng, H. B. Wang, Z. Q. Zhu, Y. Wang, T. R. Zhang, Z. L. Tao and J. Chen, Energy Environ. Sci., 2011, 4, 3668–3675 RSC
- H.-H. Sun, W. C. Choi, J. K. Lee, I.-H. Oh and H.-G. Jung, J. Power Sources, 2015, 275, 877–883 CrossRef CAS
-
(a) J. R. Dahn, U. V. Sacken and C. A. Michal, Solid State Ionics, 1990, 44, 87–97 CrossRef CAS
-
(a) W. Liu, P. Oh, X. Liu, S. Myeong, W. Cho and J. Cho, Adv. Energy Mater., 2015, 5, 1500274 CrossRef
- S. D. Findlay, N. Shibata, H. Sawada, E. Okunishi, Y. Kondo and Y. Ikuhara, Ultramicroscopy, 2010, 110, 903–923 CrossRef CAS PubMed
-
(a) Q. Liu, X. Su, D. Lei, Y. Qin, J. G. Wen, F. M. Guo, Y. A. Wu, Y. C. Rong, R. H. Kou, X. H. Xiao, F. Aguesse, J. Bareno, Y. Ren, W. Q. Lu and Y. X. Li, Nat. Energy, 2018, 3, 936–943 CrossRef CAS
-
(a) D. J. Miller, C. Proff, J. G. Wen, D. P. Abraham and J. Bareno, Adv. Energy Mater., 2013, 3, 1098–1103 CrossRef CAS
- C. Pouillerie, L. Croguennec and C. Delmas, Solid State Ionics, 2000, 132, 15–29 CrossRef CAS
-
(a) R. Robert, C. Bunzli, E. J. Berg and P. Novak, Chem. Mater., 2015, 27, 526–536 CrossRef CAS
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS
-
(a) I. Saadoune and C. Delmas, J. Solid State Chem., 1998, 136, 8–15 CrossRef CAS
-
(a) W.-S. Yoon, K. Y. Chung, J. Mcbreen and X. Q. Yang, Electrochem. Commun., 2006, 8, 1257–1262 CrossRef CAS
-
(a) T. Ohzuku, A. Ueda and M. Kouguchi, J. Electrochem. Soc., 1995, 142, 4033–4039 CrossRef CAS
-
(a) Y. Cho, P. Oh and J. Cho, Nano Lett., 2013, 13, 1145–1152 CrossRef CAS PubMed
- A. Choi, J. Lim, H.-J. Kim, S.-C. Jung, H.-W. Lim, H. Kim, M.-S. Kwon, Y.-K. Han, S.-M. Oh and K. T. Lee, Adv. Energy Mater., 2018, 8, 1702514 CrossRef
- C. Delmas, J. P. Peres, A. Rougier, A. Demourgues, F. Weill, A. Chadwick, M. Broussely, F. Perton, P. Biensan and P. Willmann, J. Power Sources, 1997, 68, 120–125 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, additional figures, tables as mentioned in the text. See DOI: 10.1039/c8sc03385d |
| This journal is © The Royal Society of Chemistry 2019 |