
Unravelling the water adsorption in a robust iron carboxylate metal–organic framework†
Dirk
Lenzen
a,
Jakob G.
Eggebrecht
b,
Paulo G. M.
Mileo
c,
Dominik
Fröhlich
d,
Stefan
Henninger
d,
Cesare
Atzori‡
 e,
Francesca
Bonino
e,
Alexandra
Lieb
*b,
Guillaume
Maurin
*c and
Norbert
Stock
*a
e,
Francesca
Bonino
e,
Alexandra
Lieb
*b,
Guillaume
Maurin
*c and
Norbert
Stock
*a
aInstitute of Inorganic Chemistry, Christian-Albrechts-Universität Kiel, Max-Eyth Straße 2, 24118 Kiel, Germany. E-mail: stock@ac.uni-kiel.de
bOtto-von-Guericke-Universität Magdeburg, Institut für Chemie, Universitätsplatz 2, 39106 Magdeburg, Germany. E-mail: alexandra.lieb@ovgu.de
cICGM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: guillaume.maurin1@umontpellier.fr
dDepartment Heating and Cooling Technologies, Group Sorption Materials, Fraunhofer-Institut für Solare Energiesysteme ISE, Heidenhofstrasse 2, 79110 Freiburg, Germany
eDepartment of Chemistry, NIS and INSTM Reference Centre, Università di Torino, Via G. Quarello 15, I-10135 and Via P. Giuria 7, I-10125 Torino, Italy
First published on 13th July 2020
Abstract
A Fe-MOF was obtained from aqueous solution in high yield under reflux. The water sorption properties were studied by powder X-ray diffraction, volumetric and gravimetric sorption experiments and molecular simulations. The subsequent filling of hydrophobic and hydrophilic pores as well as the stability of the material are demonstrated.
Metal–organic frameworks (MOFs) are well known for their outstanding sorption properties.1 Recently, the adsorption of water vapour by MOFs has been a topic of very high interest2 and studies in the fields of water harvesting,3 air conditioning,4–6 indoor moisture control7 and water removal from gas mixtures8 have been reported. There are several prerequisites for the usability of a MOF as water adsorbent including high working capacity, high coefficient of performance, and regeneration at low temperature as well as thermal, mechanical and chemical stability and high water cycling stability for a long term use.6,9
A fundamental understanding of the water adsorption at the molecular level is mandatory for future developments of MOFs with desired sorption properties. Although this is an intensively explored field, only very few in situ studies have been reported, which correlate the investigation of water positions within the crystal structure using in situ powder X-ray diffraction (PXRD) under controlled humidity conditions and molecular simulations.10
MOFs containing tri- and tetravalent metal ions, especially a number of Al-MOFs,4,11 have been demonstrated as the most promising water adsorbents. Iron-based MOFs, would be even more preferable due to environmental issues, but only few Fe-MOFs have been studied for their water sorption properties, for example Fe-MIL-53-(COOH)2,12 and more intensively Fe-MIL-100.13 Based on our expertise on the use of non-linear linker molecules, we recently focused our attention on the investigation of isophthalic acid (C8H8O4, m-H2BDC) for the synthesis of Fe-MOFs. Here we report the water based synthesis of [Fe3(μ3-O)(C8O4H4)3(OH,Cl)(H2O)2], up to the 250 g scale under mild reaction conditions, its water cycling stability and water sorption properties. Additionally and most importantly, we discuss the sequence and mechanism of the water adsorption process by a combined PXRD and molecular simulations study.
The title compound [Fe3(μ3-O)(C8O4H4)3(OH,Cl)(H2O)2], previously synthesized under harsh solvothermal reaction condition,14 can easily be obtained under reflux within 6 h from a water-based synthesis procedure starting from aqueous solutions of iron(III) chloride and sodium isophthalate (ESI,† Section 1). Washing of the reaction product with hot water leads to the removal of Cl− ions from the sample. Thus, a highly crystalline product of orange colour is obtained in high yield (ca. 95%). The synthesis protocol and characterization of the material are detailed in the ESI.†
The title compound is isostructural to the vanadium form of MIL-59 that was first described in 2002 by Barthelet et al.15 Hence, we will denote the title compound as Fe-MIL-59 and focus our discussion of the structure mainly on the features of the pores which are formed and the location of water molecules. The crystal structure of a fully hydrated sample [Fe3(μ3-O)(C8O4H4)3(OH)(H2O)2]·10H2O, Fe-MIL-59·10H2O, was refined against PXRD data (ESI,† Section 6, structural details are given in ESI,† Section S2.2). The framework, contains the well-known trinuclear complex of μ3-oxygen connected [FeO6] octahedra, as the inorganic building unit (IBU, Fig. 1).16 For charge balance one of the three terminal positions (O5) within the trimer is statistically occupied by an OH− or Cl− ion, depending on the washing procedure (see ESI†), and the other two positions are taken by terminal water molecules. Each IBU is connected via six linker molecules to six other IBUs. Above and below the plane, defined by the Fe atoms of the IBUs, the linkers are arranged closer or further apart from each other (Fig. 1a), which leads to two different types of pores (Fig. 1c). Although they exhibit similar dimensions of approx. 7 Å, i.e. 7.2 Å (denoted as large pore, lp) and 6.8 Å (denoted as small pore, sp), theses pores exhibit different hydrophilicities. The pores are interconnected by windows with a diameter of ∼2.8 Å, expected to allow the diffusion of water molecules through the framework (kinetic diameter of water = 2.65 Å), while they are too small to be accessible to N2 molecules (kinetic diameter = 3.64 Å).17
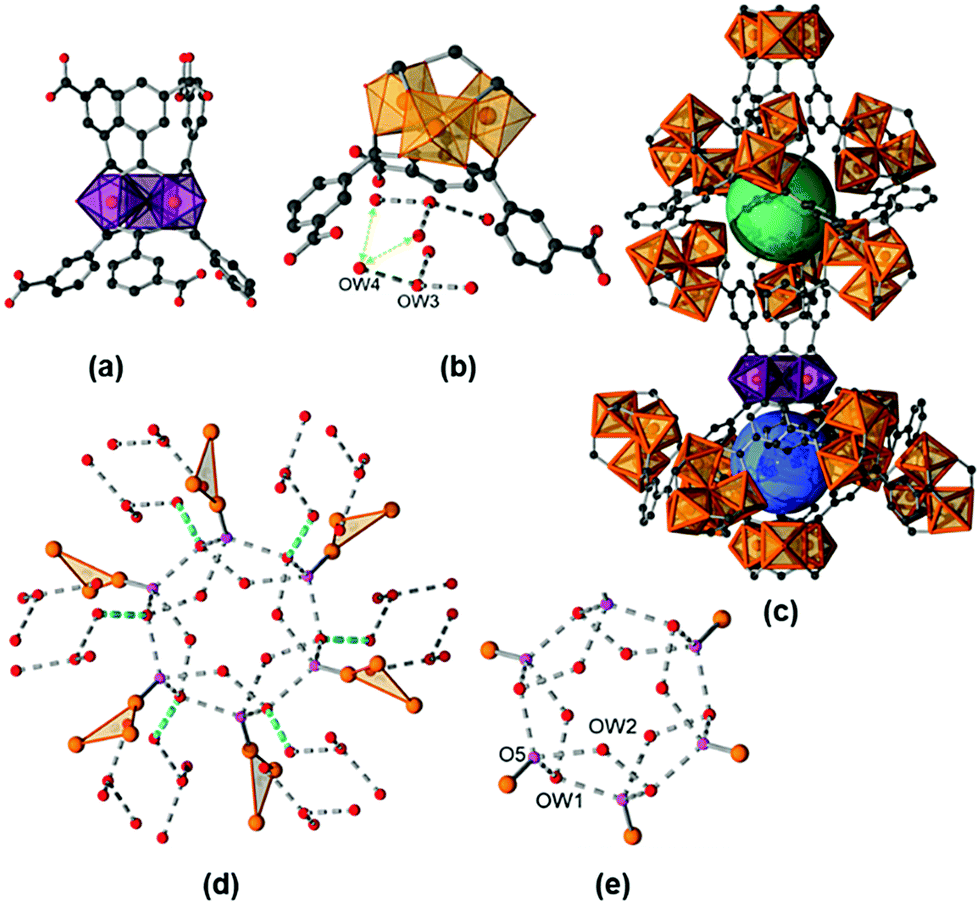 | ||
| Fig. 1 Crystal structure of Fe-MIL-59 and arrangement of the water molecules refined using in situ PXRD data. Trimeric unit (a), large pore (green) and small pore (blue) (c), possible H-bonds (grey) between adsorbed water molecules (red) and terminal oxygen atoms of the IBU (pink) in the large pore (e), the small pore (b) and in light blue through the window between the large pore and small pore (d). The trinuclear IBUs are schematically shown in (d) as triangles of iron atoms. | ||
The water molecule arrangement as determined from Rietveld refinements (Fig. S6.2–S6.14, ESI†) is shown in Fig. 1 and a table of possible H-donor/acceptor distances is given in the ESI† (Table S2.3). The large pore (lp) (Fig. 1e) is occupied by two symmetry independent adsorbed water molecules on general positions (OW1, OW2) leading to twelve water molecules per pore, equivalent to six water molecules per formula unit. Based on the O⋯O distances H-bonds can be postulated including interactions with six terminal atoms of the trimers (O5) pointing towards the pore. These O5 atoms render the lp highly hydrophilic. The small pore (sp) contains two crystallographically different adsorbed water molecules OW3 and OW4 (Fig. 1b). Every sp is occupied by two OW3 and six OW4 molecules, equivalent to four water molecules per formula unit, which form hydrogen bonds between each other, but not with any framework atom. Hence, the sp is less hydrophilic compared to the lp. Nevertheless, H-bonds are found between the adsorbed water molecules located in the sp and the ones in the lp.
The atoms OW1 and OW4 represent the water molecules closest to the pore windows, with an OW1⋯OW4 distance of 3.12(2) Å (Fig. 1d). The thermal properties of Fe-MIL-59 were investigated by thermogravimetry, variable temperature (VT) PXRD and IR spectroscopy (ESI,† Sections 3–5) in order to determine the temperature that can be used to remove water molecules without loss of structural integrity. According to the TG measurements, up to 200 °C adsorbed water molecules are lost and at higher temperatures the terminal water molecules are desorbed, the oxidation of the linker takes place and hematite (Fe2O3, #ICSD = 15840) is formed as the only crystalline decomposition product. VT-PXRD in air confirms the structural integrity upon removal of the adsorbed water molecules, the formation of an unknown crystalline intermediate at 400 °C and the decomposition to hematite at 440 °C. The in situ IR spectroscopic study of a sample thermally treated at 100 °C and 10−4 mbar revealed the presence of small amounts of guest species, i.e. residual linker and adsorbed water molecules that are removed by this treatment. The addition of water vapour leads to a quite sharp band at about 3660 cm−1 followed by a broader signal at about 3475 cm−1. These signals may be assigned to Fe–OH species, as well as coordinating and adsorbed water molecules interacting through H-bonds.18
Sorption properties (ESI,† Section 7) of Fe-MIL-59 were determined using different gases (N2, CO2) and vapours (CH3OH and H2O). The compound is non-porous upon nitrogen adsorption at 77 K, but an uptake of CO2, CH3OH (Fig. S7.1, ESI†) and H2O is observed (Fig. 2). While the CO2 and CH3OH isotherms exhibit a type-1 shape, a two-step uptake is found for water. Activation up to 150 °C under reduced pressure does not lead to changes in the H2O sorption isotherm, while under harsher conditions the loss of long-range order is observed by PXRD (Fig. S7.2, ESI†).
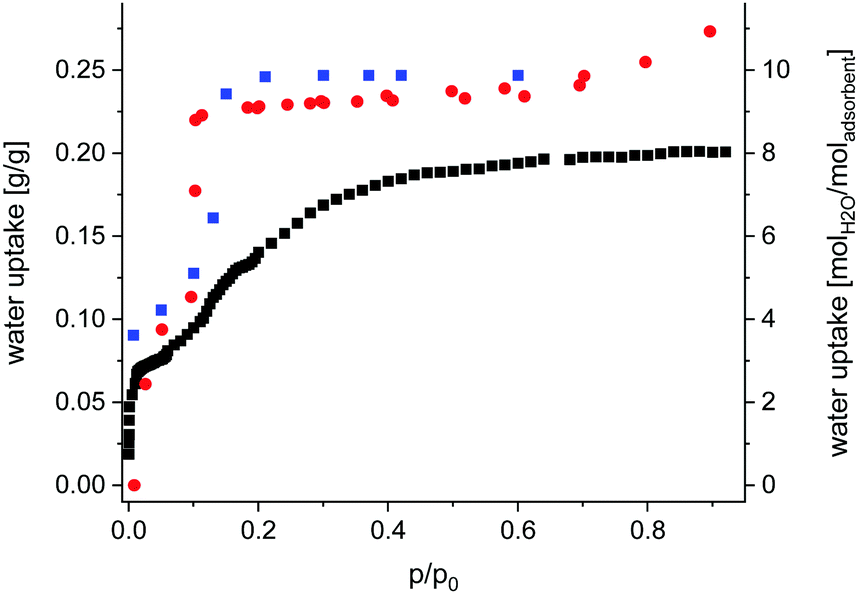 | ||
| Fig. 2 Gravimetric water adsorption isotherm collected at 298 K for Fe-MIL-59 (red dots), activated at 100 °C and 10−2 mPa; water content obtained from Rietveld analyses (Table S1.1, ESI†) of PXRD data collected at different relative humidity values at 298 K (blue squares) and water uptake obtained from GCMC simulations (black squares). | ||
A total uptake of ∼0.25 gH2O/gsorbent was obtained corresponding to ∼10 H2O molecules per formula unit (Fig. 2). Thus compared to other water MOF adsorbents Fe-MIL-59 represents a rather hydrophilic material with intermediate uptake capacity (Table S10.1, ESI†).
The long-term stability of Fe-MIL-59 was studied in multi cycle H2O sorption experiments involving 20 ad- and desorption cycles in a humidified atmosphere. The stability of this MOF was confirmed by thermal cycling (40–140 °C) TG experiments (Fig. 3 and ESI,† Section 8) using a sample subjected to a constant relative water pressure (p/p0 = 0.76).19 The loading difference of the cycles is constant at 18 wt%, which correlates to approximately seven water molecules per formula unit. The other three water molecules cannot be desorbed during the performed cycle experiment in the presence of water vapour, which is in accordance with the high water uptake at low p/p0 values observed in the sorption isotherms. Within the equilibrium steps at the beginning and the end of the measurements approximately three adsorbed water molecules per formula unit are also desorbed leading to the expected total loading difference of ∼25 wt% or 10 water molecules per formula unit. PXRD before and after the cycles confirm the structural integrity of the MOF material.
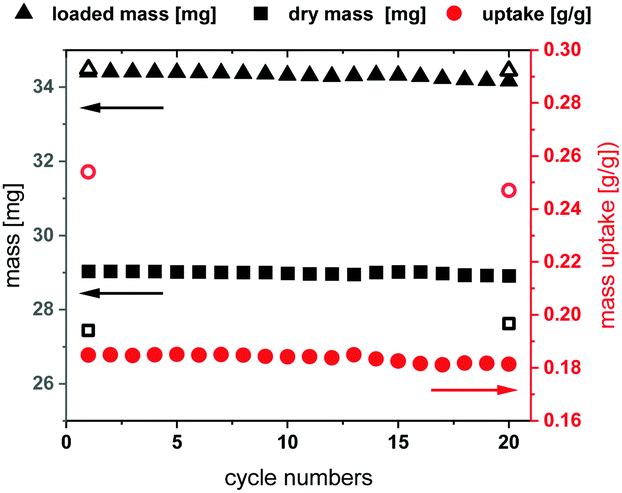 | ||
| Fig. 3 Results of the thermogravimetric cycling measurement showing the absolute mass of the empty (black squares) and filled structure (black triangles) and the loading difference (red circles) of every step (filled symbols) and of equilibrium steps at the beginning and the end of the measurement (empty symbols). | ||
To understand the processes that take place during the water adsorption, in situ PXRD measurements were carried out on a sample humidified at different relative water vapour pressures (p/p0) at 25 °C.
The PXRD measurement started at a low relative pressure of 0.007 p/p0 which was stepwise increased to 0.6 p/p0 followed by two desorption steps (0.2 and 0.1 p/p0) to check for reversibility. Rietveld refinements against PXRD data were carried out to determine the occupancy of the atomic sites assigned to adsorbed water molecules and the results are given in the ESI† (Table S2.1 and Fig. S6.1–S6.14). At low relative pressure only the hydrophilic lp is partially filled. The OW1 atom shows a shortened O5–OW1 distance to one of the two neighbouring O5 atoms, whereas at p/p0 > 0.13 both O5–OW1 distances are nearly the same. At low relative pressure, the OW2 atom is shifted to the middle of the pore allowing the formation of OW2–OW2 hydrogen bonds. At a complete occupation of OW1 and OW2 sites, at p/p0 >![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0.13 no OW2–OW2 interactions are observed. After the complete filling of the hydrophilic lp, the OW3 and OW4 sites are populated at higher p/p0 values. Only intermolecular OW3-OW4 but no H-bonds to the framework are observed. Through the connecting windows between the two pore types, OW1 and OW4 form a weak H-bond (D⋯A ∼ 3 Å). A complete list of distances at different p/p0 values is given in Table S2.2 (ESI†). Most importantly, the obtained occupancies of OW1–OW4 match the loading detected in an isothermal water sorption measurement at 25 °C (Fig. 2). The small differences at p/p0 > 0.15 are due to kinetic factors.
0.13 no OW2–OW2 interactions are observed. After the complete filling of the hydrophilic lp, the OW3 and OW4 sites are populated at higher p/p0 values. Only intermolecular OW3-OW4 but no H-bonds to the framework are observed. Through the connecting windows between the two pore types, OW1 and OW4 form a weak H-bond (D⋯A ∼ 3 Å). A complete list of distances at different p/p0 values is given in Table S2.2 (ESI†). Most importantly, the obtained occupancies of OW1–OW4 match the loading detected in an isothermal water sorption measurement at 25 °C (Fig. 2). The small differences at p/p0 > 0.15 are due to kinetic factors.
Grand canonical Monte Carlo simulations were carried out to model the water adsorption behaviour of Fe-MIL-59 (see computational details in ESI,† Section 9). First, these calculations were able to reproduce well the profile of the experimental adsorption isotherm up to p/p0 = 0.15 while the adsorption uptake is underestimated at higher relative pressure, as depicted in Fig. 2. This deviation suggests that the terminal water molecules are subject to significant reorientations upon adsorption (not taken into account in the calculations), which allow more molecules to accommodate the pores.
Our calculations show that in the first adsorption step up to p/p0 = 0.11, the water molecules preferentially populate the hydrophilic lp as illustrated in Fig. 4. These water molecules (labelled as OW1 and OW2 in the Rietveld refinement) strongly interact with the hydrophilic sites of the lp, i.e. the terminal water molecules as well as the hydroxide ions of the Fe-trimers. The radial distribution functions calculated for the corresponding Ow–O5 pairs (Fig. S9.2, ESI†) reveal strong hydrogen bond interactions associated with characteristic short donor–acceptor distances of 2.57 Å, even shorter than those between Ow–Ow (2.88 Å). This high affinity is consistent with a high adsorption enthalpy simulated in this range of relative pressure (−73 kJ mol−1, see Fig. S9.3, ESI†) thus confirming the high hydrophilicity of the lp. For p/p0 > 0.11, the increase in the water uptake corresponds to the filling of the sp (Fig. S9.4b, ESI†), consistent with the Rietveld refinement conclusions (OW3 and OW4). This concurs with a significant enhancement of the self-organization of the adsorbed water molecules with the formation of water clusters (Fig. S9.4a, ESI†) and a high number of hydrogen bonds per water molecules (2.31) at saturation (Fig. S9.4b, ESI†).
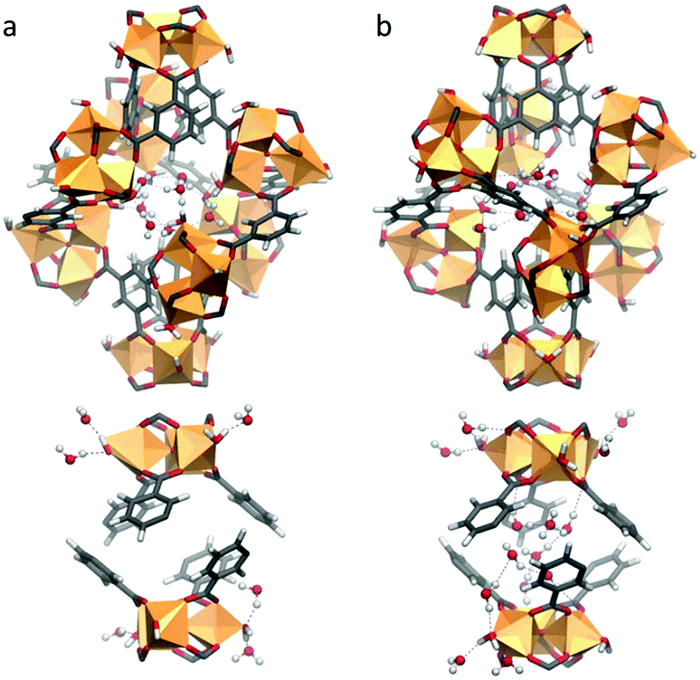 | ||
| Fig. 4 Representative GCMC snapshots at 298 K; (a) occupation of the hydrophilic large pores (lp) up to p/p0 = 0.11; (b) filling of the hydrophilic small pores (sp) at p/p0 > 0.11. | ||
The two distinct water adsorption regimes can be also discretised from an energetic standpoint. The simulated adsorption enthalpy shows a decreasing profile as one passes from the filling of the lp to the less hydrophilic sp (from −73 to −55 kJ mol−1) followed by a plateau related to the aforementioned formation of water clusters for which water/water interactions contribute strongly to the overall adsorption enthalpy.
To summarize, the synthesis of the iron carboxylate Fe-MIL-59 on the 250 g scale in water under reflux conditions as well as its water sorption properties were reported. Through the utilisation of multiple characterisation methods including water sorption measurements, PXRD and molecular simulations, the water adsorption mechanism was elucidated at the atomistic scale. The two types of micropores, the hydrophilic large pores (lp) and the hydrophobic small pores (sp), are sequentially filled, which is also reflected in the calculated adsorption enthalpy profile. This in-depth understanding of the water adsorption in such a complex hybrid material is expected to boost the development of prominent materials for application fields such as adsorptive heat transformation, indoor moisture control or water removal from gas mixtures.
N. S. acknowledges the support by the state of Schleswig-Holstein, Germany and the German Science Foundation (STO 643/11-1). A. L. thanks the German Ministry of Education and Science BMBF for financial support (FKZ 03SF0450). The authors thank Dr Helge Reinsch for the Rietveld refinements and Philipp Hügenell for the sorption experiments carried out at Fraunhofer ISE.
Conflicts of interest
There are no conflicts to declare.References
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC.
- (a) M. F. de Lange, K. J. F. M. Verouden, T. J. H. Vlugt, J. Gascon and F. Kapteijn, Chem. Rev., 2015, 115, 12205–12250 CrossRef CAS PubMed; (b) J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- (a) H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS PubMed; (b) M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30, 1704304 CrossRef PubMed; (c) F. Fathieh, M. J. Kalmutzki, E. A. Kapustin, P. J. Waller, J. Yang and O. M. Yaghi, Sci. Adv., 2018, 4, eaat3198 CrossRef PubMed; (d) N. Hanikel, M. S. Prévot, F. Fathieh, E. A. Kapustin, H. Lyu, H. Wang, N. J. Diercks, T. G. Glover and O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1699–1706 CrossRef CAS PubMed; (e) A. J. Rieth, A. M. Wright, G. Skorupskii, J. L. Mancuso, C. H. Hendon and M. Dincǎ, J. Am. Chem. Soc., 2019, 141, 13858–13866 CrossRef CAS PubMed; (f) R. G. AbdulHalim, P. M. Bhatt, Y. Belmabkhout, A. Shkurenko, K. Adil, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2017, 139(31), 10715–10722 CrossRef CAS PubMed.
- (a) D. Lenzen, J. Zhao, S.-J. Ernst, M. Wahiduzzaman, A. Ken Inge, D. Fröhlich, H. Xu, H.-J. Bart, C. Janiak, S. Henninger, G. Maurin, X. Zou and N. Stock, Nat. Commun., 2019, 10, 3025 CrossRef PubMed; (b) F. Jeremias, D. Fröhlich, C. Janiak and S. K. Henninger, New J. Chem., 2014, 38, 1846–1852 RSC; (c) S. K. Henninger, F. Jeremias, H. Kummer and C. Janiak, Eur. J. Inorg. Chem., 2012, 2625–2634 CrossRef CAS.
- J. S. Lee, J. W. Yoon, Paulo G. M. Mileo, K. H. Cho, J. Park, K. Kim, H. Kim, M. F. de Lange, F. Kapteijn, G. Maurin, S. M. Humphrey and J.-S. Chang, ACS Appl. Mater. Interfaces, 2019, 11, 25778–25789 CrossRef CAS PubMed.
- A. Cadiau, J. S. Lee, D. D. Borges, P. Fabry, T. Devic, M. T. Wharmby, C. Martineau, D. Foucher, F. Taulelle, C. H. Jun, Y. K. Hwang, N. Stock, M. F. De Lange, F. Kapteijn, J. Gascon, G. Maurin, J. S. Chang and C. Serre, Adv. Mater., 2015, 27, 4775–4780 CrossRef CAS PubMed.
- R. G. AbdulHalim, P. M. Bhatt, Y. Belmabkhout, A. Shkurenko, K. Adil, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2017, 139, 10715–10722 CrossRef CAS PubMed.
- A. Cadiau, Y. Belmabkhout, K. Adil, P. M. Bhatt, R. S. Pillai, A. Shkurenko, C. Martineau-Corcos, G. Maurin and M. Eddaoudi, Science, 2017, 356, 731–735 CrossRef CAS PubMed.
- S. K. Henninger, F. Jeremias, H. Kummer, P. Schossig and H.-M. Henning, Energy Procedia, 2012, 30, 279–288 CrossRef CAS.
- M. Wahiduzzaman, D. Lenzen, G. Maurin, N. Stock and M. T. Wharmby, Eur. J. Inorg. Chem., 2018, 3626–3632 CrossRef CAS.
- (a) D. Fröhlich, E. Pantatosaki, P. D. Kolokathis, K. Markey, H. Reinsch, M. Baumgartner, M. A. van der Veen, D. E. de Vos, N. Stock, G. K. Papadopoulos, S. K. Henninger and C. Janiak, J. Mater. Chem. A, 2016, 4, 11859–11869 RSC; (b) D. Lenzen, P. Bendix, H. Reinsch, D. Fröhlich, H. Kummer, M. Möllers, P. P. C. Hügenell, R. Gläser, S. Henninger and N. Stock, Adv. Mater., 2018, 30, 1705869 CrossRef PubMed; (c) F. Jeremias, D. Fröhlich, C. Janiak and S. K. Henninger, RSC Adv., 2014, 4, 24073–24082 RSC.
- A. Shigematsu, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2011, 133, 2034–2036 CrossRef CAS PubMed.
- (a) P. Küsgens, M. Rose, I. Senkovska, H. Fröde, A. Henschel, S. Siegle and S. Kaskel, Microporous Mesoporous Mater., 2009, 120, 325–330 CrossRef; (b) F. Jeremias, A. Khutia, S. K. Henninger and C. Janiak, J. Mater. Chem., 2012, 22, 10148–10151 RSC; (c) Y.-K. Seo, J. W. Yoon, J. S. Lee, Y. K. Hwang, C.-H. Jun, J.-S. Chang, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, S. Bourrelly, P. L. Llewellyn, P. Horcajada, C. Serre and G. Férey, Adv. Mater., 2012, 24, 806–810 CrossRef CAS PubMed.
- (a) D. Feng, K. Wang, Z. Wei, Y.-P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T.-F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H.-C. Zhou, Nat. Commun., 2014, 5, 5723 CrossRef CAS PubMed; (b) B. Li, S.-T. Zheng and G.-Y. Yang, Z. Anorg. Allg. Chem., 2009, 635, 1345–1347 CrossRef CAS.
- K. Barthelet, D. Riou and G. Férey, Chem. Commun., 2002, 1492–1493 RSC.
- (a) S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Férey and N. Stock, Inorg. Chem., 2008, 47, 7568–7576 CrossRef CAS PubMed; (b) P. Horcajada, S. Surblé, C. Serre, D.-Y. Hong, Y.-K. Seo, J.-S. Chang, J.-M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820–2822 RSC; (c) C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS PubMed.
- N. W. Ockwig and T. M. Nenoff, Chem. Rev., 2007, 107, 4078–4110 CrossRef CAS PubMed.
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS PubMed.
- S. K. Henninger, G. Munz, K.-F. Ratzsch and P. Schossig, Renewable Energy, 2011, 36, 3043–3049 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The synthetic procedure, results of the Rietveld refinements, detailed structural information, IR spectra, VT-PXRD patterns, results of the sorption and thermogravimetric cycling experiments as well as details on the computational studies. See DOI: 10.1039/d0cc03489d |
| ‡ Present address: European Synchrotron Radiation Facility, 71 Avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France. |
| This journal is © The Royal Society of Chemistry 2020 |