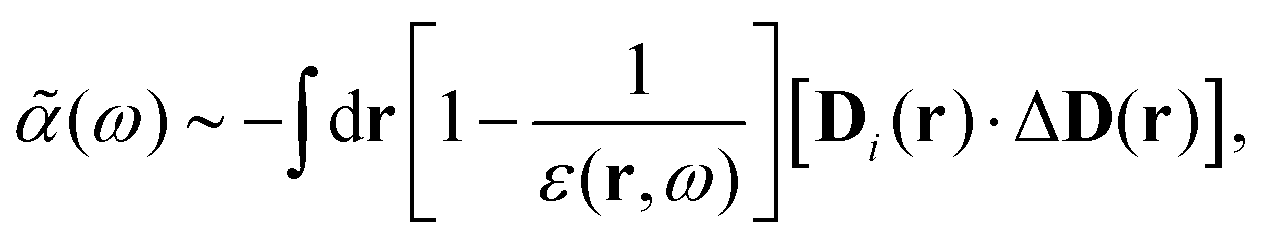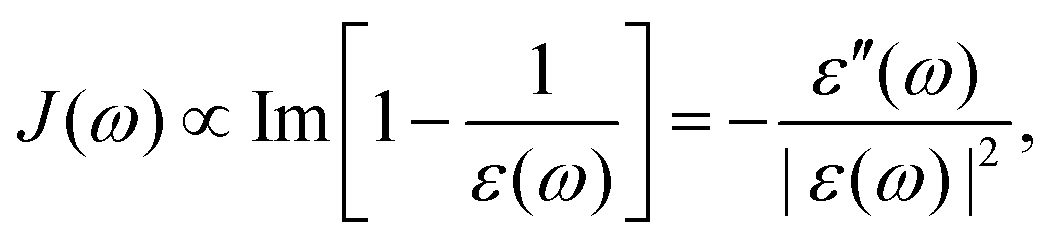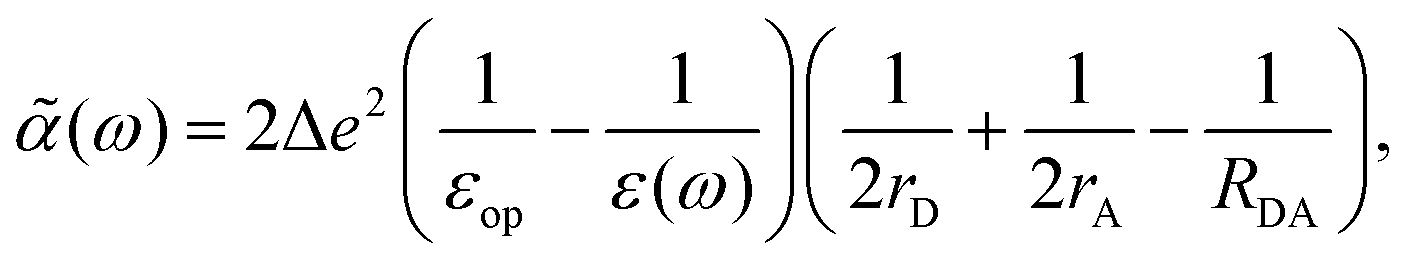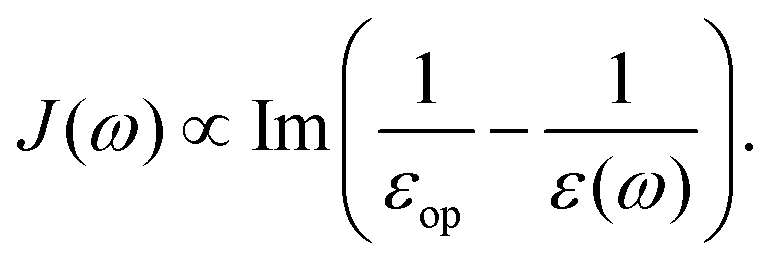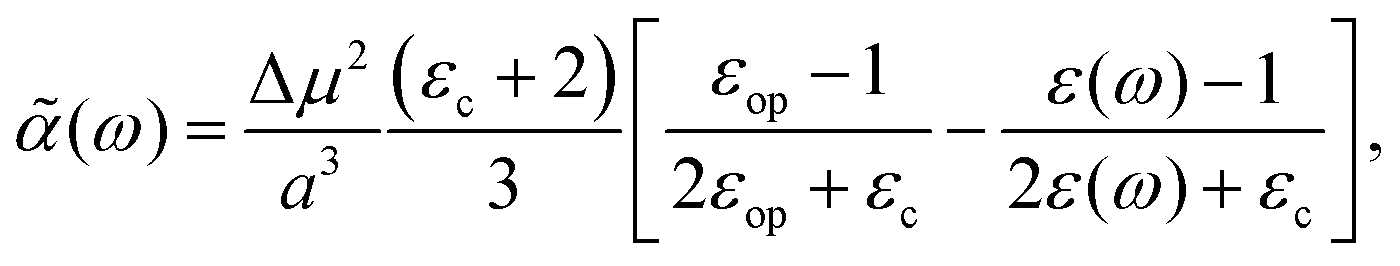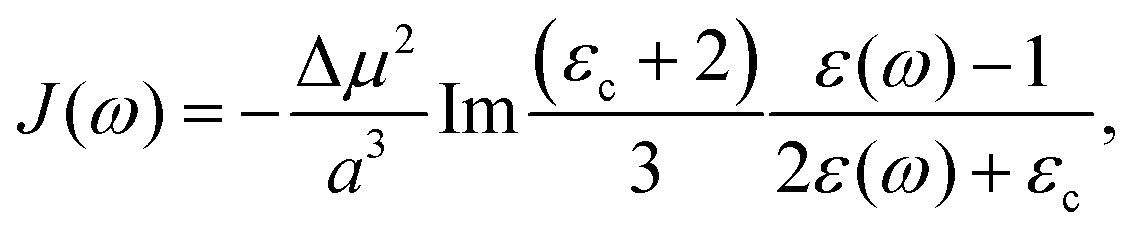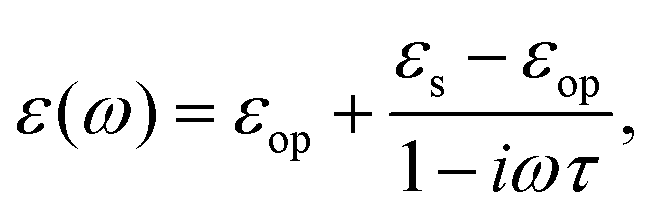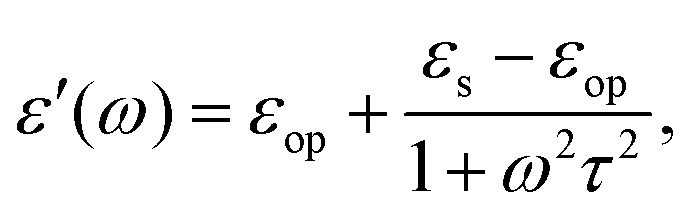DOI:
10.1039/D0CP02994G
(Perspective)
Phys. Chem. Chem. Phys., 2020,
22, 21630-21641
Reorganization energies and spectral densities for electron transfer problems in charge transport materials†
Received
3rd June 2020
, Accepted 13th August 2020
First published on 17th August 2020
Abstract
In describing the dynamics of electron transfer or charge transport, the reorganization energy and the spectral density function describe the influence of nuclei motion to the transporting electron. The spectral density can be obtained using various theoretical approaches: from a model of dielectric response, or calculated with various computational means. With the vast advancement of modern computational techniques, many details in vibronic coupling can be obtained, including those described in the early literature. In this work, we provide a comprehensive understanding of the nature of vibronic coupling in light of some early literature. The theoretical connection among different quantities for vibronic coupling is discussed, followed by a brief review of the spectral density function. Various approaches and some of the results for the spectral density function are also reviewed. The importance of low-frequency bands in nonpolar systems that can be overlooked is also discussed, for both Holstein and Peierls types of electron–phonon couplings.

Chao-Ping Hsu
| Chao-Ping (Cherri) Hsu is a research fellow at the Institute of Chemistry, Academia Sinica located in Taiwan. She developed computational schemes for electronic coupling in electron transfer, energy transfer and singlet fission with first-principles quantum chemistry computation. She is also interested in developing mathematical models for understanding noises and dynamics systems biology. Major awards and recognitions she has received include the Pople Medal from the Asia-Pacific Association of Theoretical & Computational Chemists in 2010, the Outstanding Research Award from the National Science Council in 2011, and the Investigator Award from Academia Sinica 2017. |
1 Introduction
The theory of electron transfer (ET) was developed more than half a century ago.1,2 The rich insights provided, and the great potential in many applications, have triggered enormous progress in many different fields. One area that brought great impact to our society has been the development of charge transport materials, especially in organic molecular devices. Organic semiconductors are important for many optoelectronic applications,3–5 including in field-effect transistors,6 light-emitting diodes,7 photovoltaic cells,8etc. As compared with their silicon-based counterparts, organic semiconductors are advantageous for their low power consumption, low cost and compatibility for large-area fabrication.9 Organic semiconductors are also important for solar energy conversion, in that hole- and electron-transporting layers are now commonly used in perovskite solar cells.10 In these applications, charge mobility has been one of the key properties of semiconducting devices.11 Theoretically predicting the charge mobility is highly desirable for physical insights and the chance to further improve the design of new materials.
To develop systems with good charge transport, non-polar (or less dipolar) molecules with low band gaps have been commonly considered, for the benefit of having low reorganization energy (often denoted as λ). One of the key insights of Marcus theory is that the activation energy of the reaction arises essentially from the reorganization of polarization from the environment. The less polar molecular device offers an environment with low dielectric response, which is quite different from systems discussed in the traditional electron transfer (ET) literature, where electrolytes are mainly in a polar or even aqueous solution. Most of the insights derived previously with polar environments in mind might still be valid in the nonpolar systems. Therefore, one of the goals of the present work is to lay out aspects of the reorganization energy with a stress on our current understanding of a nonpolar system.
With the vast advancement of computational hardware and software, it is now routinely possible to calculate and dissect the reorganization energy in various forms. Reorganization energy, electron–phonon coupling, and spectral density function are frequently discussed in the ET literature or related topics. Modern works simulating the charge transport dynamics have been developed using various schemes.12–18 However, because of the complex nature of the problem, there are inevitable discrepancies among different theoretical models and simulation approaches. For example, to date, most charge transport simulation works rarely considered the feedback of the polaron to the classical nuclear motions, whereas recent improvements have been developed19–21 to include such feedbacks. Some early literature, mainly in the field of non-equilibrium statistical mechanics, has offered rigorous and insightful bases, such that modern computational work can be relied on, helping us derive further understanding for the system.22–26 Many of these works are well-written, with great insights; some do not have apparent relevance, whereas others might appear to be difficult to understand especially in the theoretical derivations. For facilitating the development of modern simulation, a summary and overview on the theoretical grounds is helpful, which is what we aim to provide in the present work. Therefore, another goal of the present work was to introduce such works and connect their results to the modern computational chemistry. Therefore, this work is far from any comprehensive review for modern computational chemistry work on charge-transport materials. Instead, we aim to provide an integrated and yet modest set of relevant information from the traditional statistical mechanics field, for deriving more insights from computational results.
In this work, we first review various similar quantities for the reorganization energy and their background theoretical models, followed by a section on the dielectric continuum models and their theoretical predictions. We then outline some of the computational chemistry results and conclude with speculation on future perspectives.
2 Reorganization energy and its similar quantities in various models
For describing the process of charge transfer, the Marcus ET theory proposed a rate expression,1,2 which is determined by 3 parameters: the Gibbs standard free energy for the reaction ΔG0, the electronic coupling HRP, and the reorganization energy λ:| | 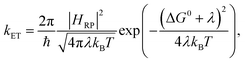 | (1) |
where kB is the Boltzmann constant. One of the most important insights for the Marcus ET rate theory is in the treatment of the solvent and its influence. Because ET involves a change in the location of a charge, it drives changes in many degrees of freedom of a presumably polar environment, and these changes in the environment feed back to the system. Such changes in the environment collectively form the reaction coordinate. The free energy of the reactant and product state in this reaction coordinate was assumed to be a parabola, an assumption that was later confirmed to hold until achieving very high energy.27 We outline several different charge transfer/transport models below, with a stress on their definitions and treatments of the quantities that are similar to the reorganization energy.
2.1 Reorganization energy in the Marcus theory
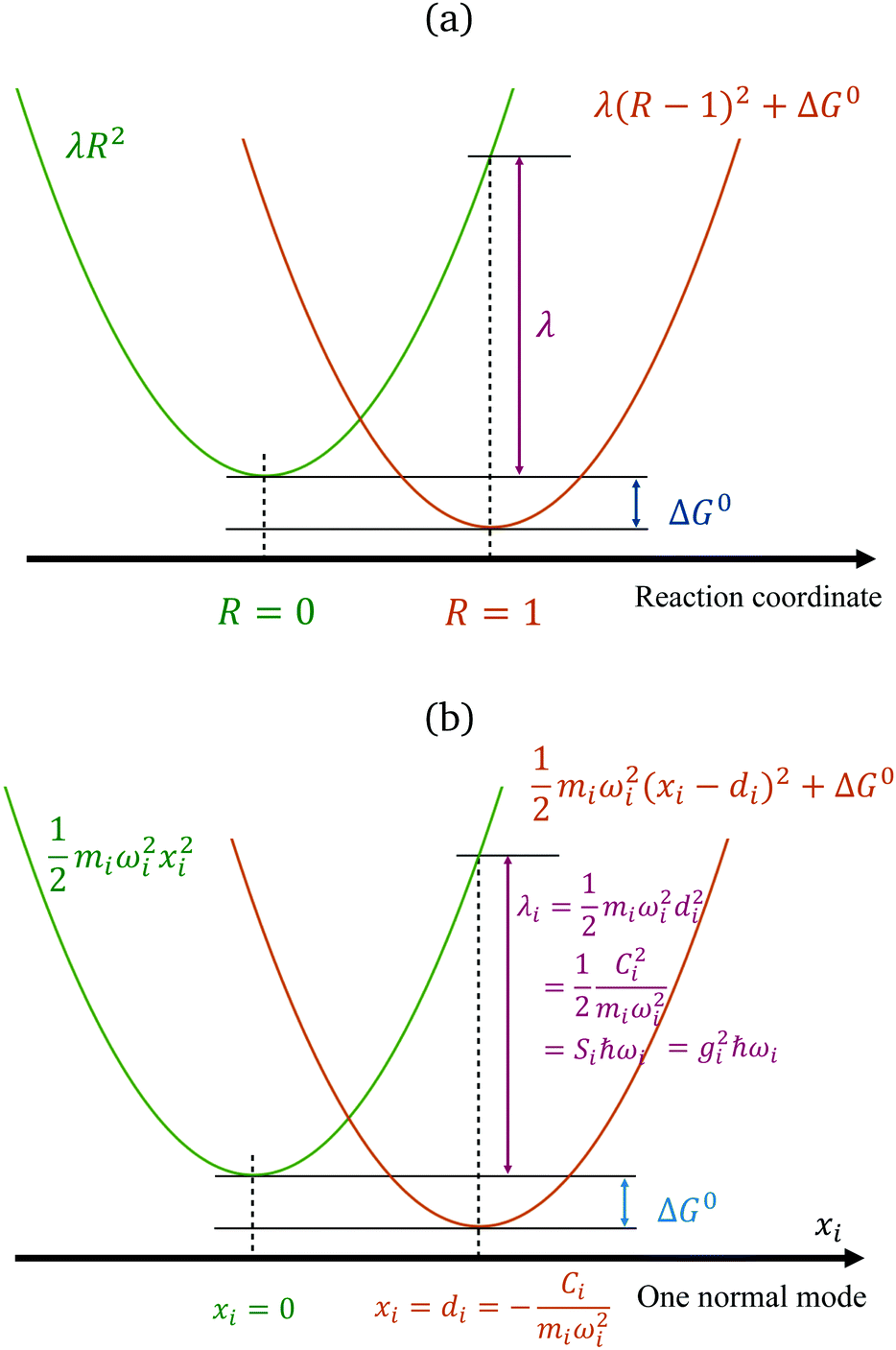
The reorganization energy is a critical quantity in ET theory. It is the energy required to reach the relaxed product nuclear configurations from a relaxed reactant configuration, while keeping the electronic state in the reactant (or vice versa). If the difference in equilibrium positions of the two parabolas is defined as 1 (arbitrary unit in the reaction coordinates), then one potential energy curve can be λR2, and the other is λ(R − 1)2 + ΔG0, with R being the position in the generalized reaction coordinate. Therefore, λ can also be considered as the curvature for the Marcus parabola (Fig. 1(a)).
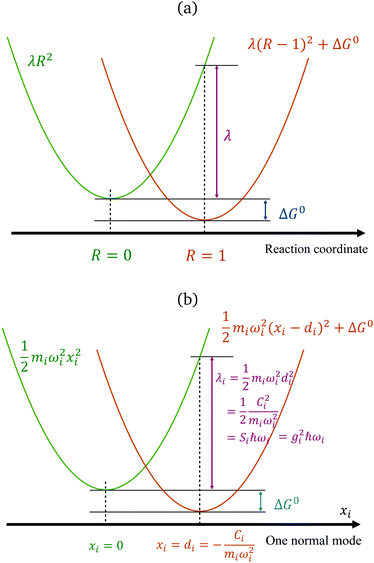 |
| | Fig. 1 Marcus' parabolic free energy curves (a), broken down into harmonic oscillators, showing one of them as xi as shown in (b), with various quantities labelled. | |
We note that, for a classical harmonic oscillator, a standard theoretical model does not need any parameter, because the units for displacement and the energy can be chosen to fit any harmonic oscillator in application. For ET problems, the minimal number of parameters describing the set of two parabola with the same curvature is 2, with one being the free-energy difference ΔG0, measuring the vertical shift of the two curves, and another can be defined by various means: the curvature, the displacement (horizontal shift), or the coefficient for the difference of the two states (which is a linear function), depending on the setting of the theoretical models.
The reorganization energy is further divided into two different contributions: one arising from the surrounding solvent, the outer sphere component, (λout) and the other from internal degrees of freedom (or the first solvation shell, λin).
For λout, because the process of ET involves a change in charge distribution of the donor–acceptor pair, the most important effect from the environment is its polarization, or the dielectric response. An expression considering such changes, in a simplified spherical cavity model, was given by Marcus:28,29
| | 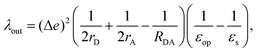 | (2) |
where Δ
e is the amount of charge transferred in the reaction;
rD(A) is the radius of the donor (acceptor), when a spherical cavity is used to model the molecule or fragment;
RDA is the center-to-center distance between the donor and the acceptor;
εop is the optical dielectric constant; and
εs is the static dielectric constant for the solvent. The expression in
eqn (2) was derived from a model where the dielectric solvation energy change was estimated by two independent spheres (infinitely separated) containing the donor and the acceptor, and the changes in the interaction energy between the two (the term involving 1/
RDA). The optical dielectric response follows the electron being transferred instantaneously, and this part does not contribute to the reorganization energy. In practice,
λout can be quite sensitive to the parameters
rD(A) and
RDA chosen.
rD(A) can be estimated from the sphere that has the same van der Waals volume of the donor or acceptor fragments, and
RDA is the distance between the centers of mass of the fragments.
Another component for reorganization is the internal contribution, which is derived from the internal degrees of freedom for a molecule, or the inner solvation shell for an ion in a polar solution. In this case it is common to express the reorganization energy in terms of the structural difference (Δ![[q with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0071_20d1.gif) ) in the reactant and product states and project such a structural difference to the normal modes (in mass-weighted coordinates {Qi}) of the reactant (or the product).
) in the reactant and product states and project such a structural difference to the normal modes (in mass-weighted coordinates {Qi}) of the reactant (or the product).
| | 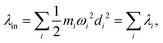 | (3) |
with
di being the projection of the structural difference, the displacement, to the
i-th normal mode,
| | 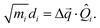 | (4) |
It is common to use the dimensionless Huang–Ruy factor {
Si} to describe the contribution of each mode,
with
Si =
miωidi2/2
ħ (as summarized in
Fig. 1(b)). Because the vibrational quanta can be closer to or higher than the thermal energy
kBT, such a breakdown is convenient for including quantum statistics.
23
2.2 Spin-boson Hamiltonian
The Spin-Boson Hamiltonian (SBH) has been used to study a broad range of dissipative problems.30 In SBH, a set of harmonic oscillators are included to model the environment, affecting the dynamics of a simple two-state system.| | 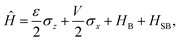 | (6) |
where the Pauli spin matrices σx and σz are used to denote the 2 × 2 Hamiltonian, with |+〉 and |−〉 as their basis. In this case, σx = |+〉〈−| + |−〉〈+| and σz = |+〉〈+| − |−〉〈−|. HB is the Hamiltonian for the environment (bath), and HSB is the interaction of the system and bath,| | 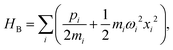 | (7) |
| | 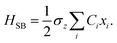 | (8) |
The ladder operators in the second quantization of harmonic oscillator, b†i and bi can also be used in the expression:| | 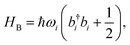 | (9) |
| | 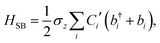 | (10) |
where| | 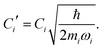 | (11) |
Namely, Ci is the coefficient of the linear vibonic coupling in the classical mechanical expression, and Ci′ is essentially the same quantity but with dimensions adjusted for the second quantization expression. With Cixi being the coupling from mode i, the displacement of the two states is| | 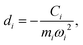 | (12) |
leading to a contribution to the reorganization energy| | 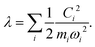 | (13) |
Fig. 1(b) summarizes the quantities in the parabola for each mode i. The two-state SBH is similar to the many-state Holstein models developed for describing polaron transport, as outlined below. Breaking down to Harmonic oscillators is intuitively straightforward for λin, because it arises from displaced vibrational modes of the donor and the acceptor. However, for SBH (and also the Holstein and/or Peierls models), such a break-down can be generalized to all the external influences to the states, both inner and outer-sphere contributions.
2.3 Holstein and Peierls models
For an array of organic semiconducting molecules, it is common to consider a Hamiltonian31–35 as| | 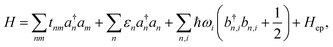 | (14) |
which contains electronic interaction between sites tmn, electronic site energy εn, the Hamiltonian for vibrations or phonons, and the electron–phonon interaction Hep, respectively. In eqn (14), a†n and an are the electronic creation and annihilation operators at site n, and εn is the site energy, b†n,i and bn,i are the creation and annihilation operators for the vibration associated with site n and the ith mode, and ωi denotes vibration frequencies. It is common to include the local coupling, or the Holstein's Hamiltonian, in the following form,| | 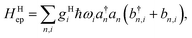 | (15) |
giωi is the electron–phonon coupling, and where the nth electronic basis is essentially the site (position) of the charge, and its energy is coupled to vibrational mode i at site n.33 Typically, in modeling organic semiconductors, each organic molecule constitutes a site, with vibrational modes i being assigned to each site. Here it is assumed that the vibrational modes are localized to one or a finite cluster of molecules, such that frequency ωi can be used for site n. In a periodic crystal system, it is common to describe the system in the Fourier transformed bases, turning vibration into phonons of wave vector q,| | 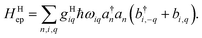 | (16) |
with biq being the annihilation operator for the phonon mode i with wavevector q.
It is also possible to further generalize the electron–phonon coupling to the transfer integral, leading to the Peierl electron–phonon coupling, which is also studied in the literature36–38
| | 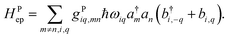 | (17) |
In both electron–phonon coupling models, gi is a dimensionless parameter for electron–phonon coupling. It can be shown that
| | 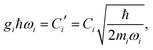 | (18) |
where coefficients
C and
C′ share the same definition for the SBH (
eqn (11)). The contribution to the reorganization energy is, with
eqn (13)| | 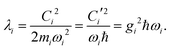 | (19) |
We note that all the parameters for electron–phonon coupling,
g and
C (
C′) have their counterparts in the off-diagonal Peierls coupling.
2.4 On the locality of electron–phonon coupling
In the literature, the term “local” electron–phonon coupling often refers to the Holstein model, whereas “nonlocal” indicates the Peierl electron–phonon coupling.39,40 For this purpose, the terms “diagonal” and “off-diagonal” coupling might be more intuitive.41,42 With an array of molecules in mind, the idea of “local” coupling can also refer to the physical location of the electron and vibration, assuming that they are described in a local basis. Because it is natural to develop physical pictures with real space and molecules in mind, here we discuss the idea of “locality” in this aspect. For example, a “non-local” Holstein model can be defined as:| | 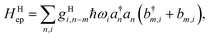 | (20) |
where a movement or vibration at site m could affect the energy at site n, and thus g carries an index for mode i, as well as the relative position in the site indexes m and n. Because a nuclear movement (vibration) at a certain location can affect a limited range of electrons nearby, both in the diagonal or off-diagonal Hamiltonian, the coupling factor g has some local nature in the real space. A very local, single-site coupling (eqn (15)) leads to a “universal” coupling in the reciprocal space, where gHiq is a constant of q.43 Such a simplified setting certainly allows for much easier theoretical manipulation. In practice, because electrostatic interaction that dominates the site energy fluctuation is long range in nature, the coupling should be range-dependent.
However, we note that, for an array of molecules, periodic or not, assigning a single-site location for a vibrational mode in general can be difficult. As eigenvectors of the Hessian matrix, the motion of nuclei can be distributed all over the system. Expressions such as eqn (15) and (20) were written when the coupling among local vibrations is not large, or, in an amorphous condensed phase where vibrational modes are somewhat localized to certain sites. Therefore, the frequently used expression in eqn (16) is more general and could better describe the long-range interaction between a general vibrational mode q and site n.
A great proportion of interesting problems in chemistry are not in the gas phase, nor do they take place in a periodic environment. The classical redox systems mainly involve electrolytes in solutions, and the modern charge transporting devices are mostly in amorphous solid-state phase, not to mention that many interesting ET problems are in biomolecules such as proteins or DNAs, mainly in aqueous solution. A convenient treatment for the model is to keep electrons in a desirable basis,‡ and to use vibrational modes for the system as a whole. Depending on the computational setting, usually it is easy to separate the inner-shell contribution, the vibrational modes of the donor and acceptor molecules (or fragments) from those arising from the surrounding molecules. Here we note that the expression in eqn (17) can be generalized further, with an index for all vibrational modes of the whole system, collective or not.38,45 The Holstein–Peierls (HP) coupling term can be expressed as,
| | 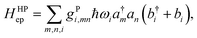 | (21) |
which allows both Holstein (
m =
n) and Peierls (
m ≠
n) types of coupling, regardless of the location of electrons and phonons/vibrations.
2.5 Spectral density function
For both the SBH and HP models, one commonly defines a spectral density function J(ω) to represent the coupling strength of modes of frequency ω. It can be defined with Ci, the strength of coupling for mode i, as,| | 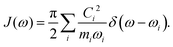 | (22) |
The propagation of the two-level dynamics requires a treatment for the vibronic coupling 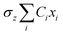 , which couples the nuclear and electronic degrees of freedom. This is often achieved by the a time-dependent perturbation theory under the interaction picture of quantum mechanics. Such techniques have been successful in treating many problems in quantum dynamics,46,47 including nonlinear spectroscopy48 and the two-state reaction rates with or beyond Fermi's golden rule.49 In these works, a central quantity X is defined as the energy difference between the two states in SBH, or the change in energy when the site carries a charge for the Holstein model,
, which couples the nuclear and electronic degrees of freedom. This is often achieved by the a time-dependent perturbation theory under the interaction picture of quantum mechanics. Such techniques have been successful in treating many problems in quantum dynamics,46,47 including nonlinear spectroscopy48 and the two-state reaction rates with or beyond Fermi's golden rule.49 In these works, a central quantity X is defined as the energy difference between the two states in SBH, or the change in energy when the site carries a charge for the Holstein model,| | 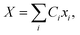 | (23) |
and the time evolution of this operator in the ensemble of one of the states is central in describing the dissipative two-state dynamics.| | | X(t) = eitH/ħXe−itH/ħ, | (24) |
where H is the Hamiltonian of the system. Typically the off-diagonal part of H is separated and treated with perturbation expansion.46,47 The time-correlation function for X(t) can be calculated and related to the spectral density function. With the correlation function of harmonic oscillators, we have,| | 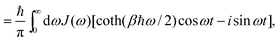 | (26) |
with β = 1/kBT. With the fluctuation–dissipation theorem, C(t) is also related to the response of the system when an external change takes place.
With Cixi, the displacement of each mode is Ci/miωi2, and the contribution to the reorganization energy is Ci2/2miωi2. Therefore, the reorganization can be related to J(ω) as well:
| | 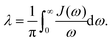 | (27) |
Just as λ is divided into the inner and outer-shell contribution, the spectral density can also be divided in the same way. The inner-shell component is much easier to define. As discussed in Section 2.1, the individual contribution λi can be defined and calculated routinely (eqn (3)). Therefore the “inner” spectral density function can be
| | 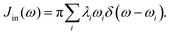 | (28) |
Thus, in principle, we should have
J(
ω) =
Jin(
ω) +
Jout(
ω). However, because
Jout(
ω) is much less trivial to obtain and will be the main focus in the present work, for the sake of simplicity and to follow the convention of the major literature, we will use
J(
ω) for
Jout(
ω) when it is not confusing.
In theoretical studies, one commonly uses analytical functions such as
with
s = 1 being the ohmic dissipation. Models for
s > 1 (super-ohmic) and
s < 1 (sub-ohmic) are also discussed.
30 Such expressions are convenient. With only 3 parameters, there is a good chance to take
J(
ω) fully into account in analytical theories. However, these Ohmic and similar models are more-or-less phenomenological, since there are a lot of important details in
J(
ω) that are not included, as outlined below. Realistic models can be developed for calculating
J(
ω) with known physical properties of the system (Section 3), or simulating it (Section 4). Such approaches eliminate the need to fit the parameters and offer chances to predict and design new systems with desirable properties.
We note that in Marcus theory, all the influence of the nuclei motion is integrated into λ. The internal vibrational modes of the donor and acceptor contribute to λin, which comes with a specific set of frequencies. The polarization of the environment constitutes λout, which usually forms a “band” of continuous modes. However, in the SBH and HP models, all the nuclei motions are treated with the harmonic oscillator models, with an infinite number of harmonic oscillators as the model onto which all influences can be projected.25 Such theoretical models are flexible for many different problems, but model setting for this infinite number of coupling coefficients becomes a problem. The dielectric continuum models introduced in the following section are practical solutions.
The hypothetical 1-dimensional reaction coordinate is also a central concept in Marcus theory. The harmonic oscillator model for the bath naturally leads to a many (or infinite) dimensional picture, with each dimension similar to that depicted in Fig. 1(b). However, the dynamics of the system can be described using a 1-dimensional model, and the simplest definition for this coordinate is the energy difference between the reactant and product states, ΔE(≡X). As will be shown in the following section and the references cited therein, ΔE offers a means to model, calculate and understand the dynamics of the system. The fluctuation–dissipation theorem and its corresponding (linear) response function can be built with ΔE. Moreover, a parabolic free energy curve along the reaction coordinate implies a Gaussian distribution in the ensemble.27 However, the linear assumption could be challenged, and it can be corrected with theoretical and numerical efforts.50
3 Dielectric continuum models
The mathematical form of infinite harmonic oscillators can also be derived from the linear dielectric response, without having to hypothesize with an infinite number of harmonic vibrations.25 With proper dielectric models and boundary conditions, the environmental contribution to J(ω) can be related to the dielectric response ε(ω). Here we lay out the theoretical ground for such a treatment, with data derived from experiments included, with which we further discuss the physics and requirements for the spectral densities.
3.1 Theoretical grounds
Suppose Δρ(r) is the change in charge distribution in the ET, which can be arbitrarily scaled by a time-dependent factor, Δρ(r,t) = f(t)Δρ(r). The system goes from an initial to final state with f(t) going from 0 to 1. The frequency-dependent dielectric constant of the media gives rise to an immediate response (modeled by the optical dielectric constant, εop) and a delayed dielectric solvation after a time-dependent change. The electrostatic interaction between a system and its surrounding dielectric solvent is also time-dependent, ΔEint(t). This energy difference is the vibronic coupling 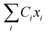 which is often named X as a major dynamic variable in many related works.25,46,51,52
which is often named X as a major dynamic variable in many related works.25,46,51,52
ΔEint(t) can be generally written as
| | 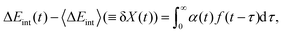 | (30) |
where
α(
t) is a generalized susceptibility function, and
f(
t) is an arbitrary time-denpendent perturbation function. With the fluctuation–dissipation theorem,
α(
τ) can be related to the time-correlation for Δ
Eint(
t),
53–55| | α(τ) = 〈[δX(τ),δX(0)]〉 = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ImC(τ)/ħ, ImC(τ)/ħ, | (31) |
where
C(
t) is the auto-correlation function for Δ
Eint(
t) (or
X(
t), as defined in
eqn (26)), spectral density
J(
ω). Therefore, we have,
| | J(ω) = −Im![[small alpha, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0dc.gif) (ω). (ω). | (32) |
Δ
Eint(
t) can be formulated using a range of methods, such as the product of the charge (from the solute in the ET system) and the electric potential generated by the polarized solvent, or the displacement field generated by the solute. Δ
Eint(
t) has its Fourier transform as follows,
| | 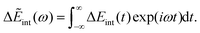 | (33) |
It can be formulated with dielectric solvation theories:
22,25| | ΔẼint(ω) = −![[f with combining tilde]](https://www.rsc.org/images/entities/i_char_0066_0303.gif) (ω)(ω), (ω)(ω), | (34) |
where
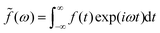
is a dimensionless function, which can be an arbitrarily chosen perturbation, and
α(
ω) is the response function with frequency
ω. The specific form of
α(
ω) depends on the setting in the electrostatic model.
22,25,56
1. An early version with the general formulation mainly for a uniform electric (and displacement) field developed in ref. 22, leading to,§
| | 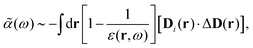 | (35) |
where
D(
r) is the displacement field at location
r, which implies that
| | 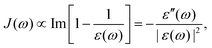 | (36) |
where the dielectric response is written in its real and imaginary parts, as,
| | | ε(ω) = ε′(ω) + iε′′(ω). | (37) |
We note that this model did not consider the boundary condition for the solute–solvent interface. Most similar models use a cavity or a boundary for different dielectric constants.
2. For the Marcus' sufficiently separated donor and acceptor model, the dielectric image effect can be neglected, such that
| | 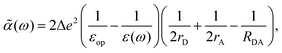 | (38) |
leading to
| | 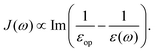 | (39) |
which is very similar to the results of the model above.
3. Using the Onsager model, the system can also be modeled as a closely spaced donor and acceptor in a spherical cavity of radius a and change in the dipole moment Δμ. In this case we can obtain,¶
| | 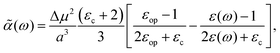 | (40) |
where
εc is the dielectric constant inside the spherical cavity, which has been modeled as 1 because it is in a vacuum, for a cavity that is occupied by the solute (donor and acceptor for ET). Therefore,
| | 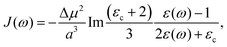 | (41) |
since
εop represents an infinitely fast dielectric response of the system and can be regarded as a real number.
3.2 Physical origin of dielectric solvation
In a dielectric material, various factors can contribute to the dielectric effect, roughly from slow to fast in their time scales:
• movement of the molecules (mainly charged particles),
• changes in the orientation of the polar molecule,
• changes in the bond angle or other internal degrees of freedom of the solvent molecules,
• stretching or suppression of polar bonds, and the polarization of electronic distribution,
• polarization of electrons.
For ET, the donor–acceptor system has a change in its charge distribution. The first term, the movement of charge particles does not really occur in the regular solvents composed of neutral molecules. The second item, the orientational effects in polarization, gives rise to Debye dielectric solvation. For ET, the electronic polarization is faster than the charge transfer process; thus an optical dielectric constant εop is used to represent the “infinite frequency” limit in the theories.
3.2.1 Low frequency: Debye dielectric relaxation.
For polar solvents, the Debye dielectric relaxation model can be used to describe the dielectric dispersion ε(ω), where the response of polarization was assumed to be an exponential decay function, with lifetime τ. τ can be considered as the orientational lifetime for a fluctuating solvent. This exponential response yields to a dielectric function as,| | 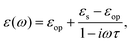 | (42) |
where τ is the characteristic relaxation time. We note that in most of the literature the dielectric response at the high-frequency limit is often denoted as ε∞, but here we use εop for better coherence with expressions shown above. With this model, it gives| | 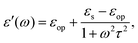 | (43) |
and| | 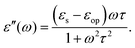 | (44) |
Therefore, there should be a diffuse peak at 1/τ for the spectral function J(ω) for a polar solvent.
3.2.2 Higher frequency: resonances.
When a dielectric material is placed under the oscillatory electric field, the oscillations at various levels, as listed above, affect the polarization of the material. If the frequency is low, the motion of the oscillation follows, leading to a cancellation of the electric field applied, and thus dielectric shielding is obtained (in the real part of ε(ω)). When the frequency of the applied field increases, to a point at which the frequency matches the intrinsic frequency of the oscillation, a resonance is observed and there is an absorption of electric energy. An absorption peak (dielectric loss) appears in the imaginary part of ε(ω). For frequencies above the resonance, the oscillation can no longer catch up with the external field, and thus this mode no longer contributes to the cancelation of the electric field, which leads to a lower value in the real part of ε(ω). Therefore, the real part, ε′(ω), steps down to a lower value with each resonance, whereas the imaginary part, ε′′(ω), has successive absorption peaks.
For systems composed of polar molecules, the lowest frequency motion that leads to a dielectric effect is perhaps the reorientation. Therefore, the system starts with a (large) static dielectric constant in the real part, with Debye-like relaxation accounting for the rotational relaxation, followed perhaps by low and high-frequency vibrations that can contribute to the polarity, finally reaching the optical frequency such that it is no longer relevant to the ET problem.
The mathematical description for the above process, (i.e., a resonance in the imaginary part together with a step down in the real part of the response function) can be described by the Kramer–Kronig relationship, which arises from the general causality of the response function,57,58 but will not be recapitulated here.
3.3 Implication from dielectric dispersion
The dielectric response for many commonly seen solvents has been studied extensively. The absorption spectra and dielectric constant at all the frequency ranges have been reported, offering an “experimental” version for the spectral density J(ω).51J(ω) can also be calculated from molecular dynamics (MD) simulation.59 Both offer useful information for charge-transfer behavior.
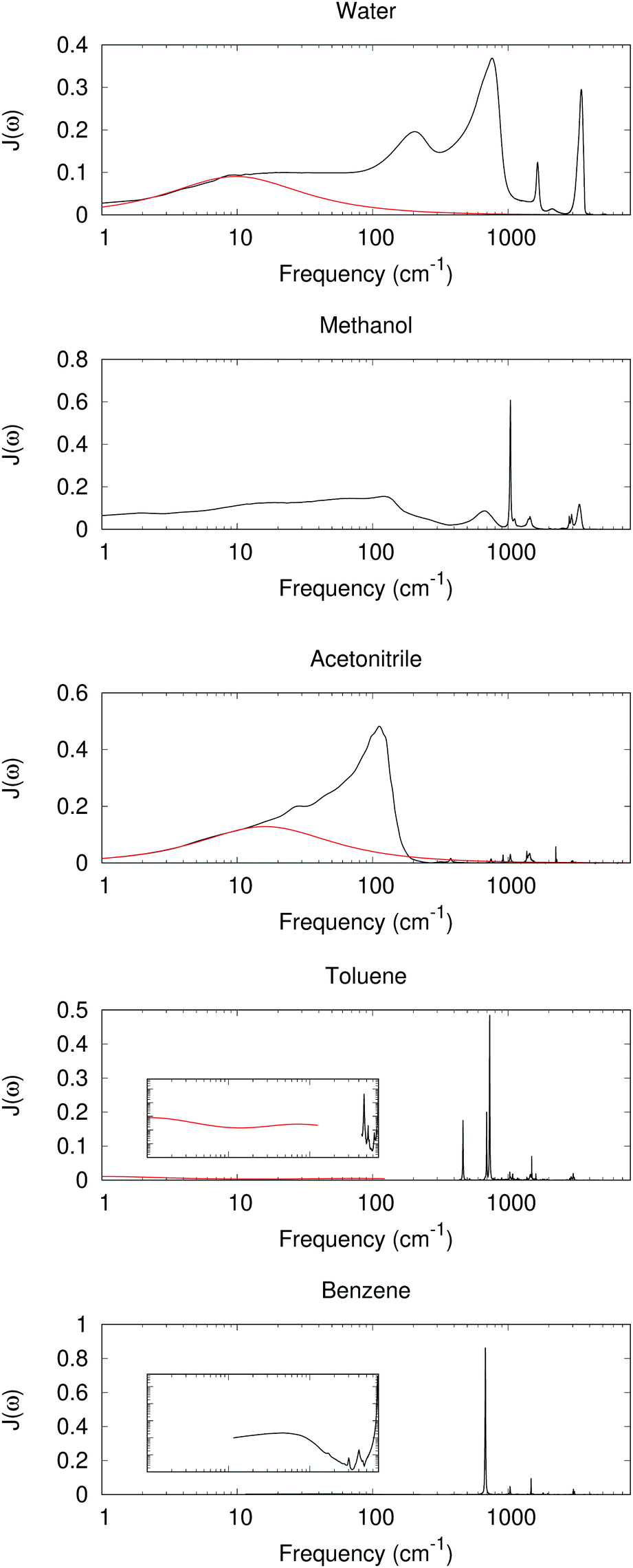
With experimental results, we include the spectral density function J(ω) for 5 different solvents as shown in Fig. 2. The experimental dielectric data were obtained from ref. 60–70. From Fig. 2 dielectric response spectra for the polar solvents have a large component for the low frequency region (≤1000 cm−1 for water, ≤2–300 cm−1 for methanol and acetonitrile). The low-frequency response consists of intermolecular modes such as constrained translational and rotational modes. For polar molecules, an external field may induce an orientational motion, leading to a dielectric response. These are known to contribute significantly to the dynamic Stokes shift in time-dependent fluorescence.51,52
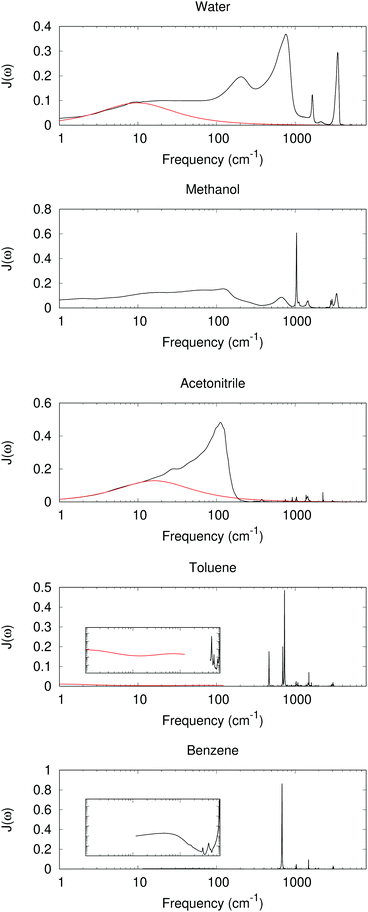 |
| | Fig. 2 Im[1/ε(ω)] as a model for J(ω) with experimentally measured ε(ω) (black lines, data derived from ref. 52, 60, 63, 64, 66–68 and 70). At low frequency, a red curve for the Debye or multiple Debye model is included, with parameters from ref. 62 and 65 for water and acetonitrile, and interpolation and adjustment to 25 °C according to ref. 69. Inset: log–log scaled plots between 1 and 1000 cm−1 and Im[1/ε(ω)] between 0.0001 and 1. | |
The intramolecular bending and OH stretching bands are also seen for both water and methanol, which are broader than the vibrational bands in the nonpolar system. All of these bands contribute to λout.
For nonpolar or weakly polar systems, sharp, narrow vibrational bands occur in the high-frequency area, and the low-frequency responses are very weak. For benezene and toluene, the largest contribution is from a band near 700 cm−1, which has been assigned as the out-of-plane CH bending. Indeed the C–H bond is weakly polar, and its bending can create a polarization in response to a change in the solute. With these results, one might conclude that the high-frequency intramolecular vibration is important for the nonpolar solvation, whereas the influence is not so great for the low-frequency, orientational, or acoustic phonon-like region. However, the dielectric response model has its own assumption and limitations. For periodic or MD simulations, some response is still observed in the low-frequency region, as outlined below.
4 Computational approaches and their findings
The electron–phonon coupling can be evaluated nowadays using various computational approaches, for both Holstein and Peierls types of coupling. Here we summarize the available observations and limitations of these approaches in Table 1, with the detailed discussion as follows.
Table 1 Contribution of and limitations to various computational approaches for electron–phonon coupling
|
|
Holstein type |
Peierls type |
| Isolated molecules |
Obtained |
• Breaking down λin to all intramolecular vibrational modes. |
• Scanning few intermolecular degrees of freedom for coupling; directly evaluating the nuclear derivative of electronic coupling is also seen. |
| • Mainly high-frequency, intramolecular contribution. |
• Most works reported intermolecular contributions with scanning. |
| Limitation |
• Difficult to obtain the low-frequency, intermolecular contribution. |
• With scanning: need to specify the mode and scan. Difficult to obtain all the modes. |
| • Difficult to obtain the corresponding frequencies and their dispersion in a condensed state. |
|
|
| Periodic models |
Obtained |
• General approaches can account for contributions from all phonons, including both optical (intramolecular) and acoustic (intermolecular) phonons |
• Can fully account for all modes for the off-diagonal matrix elements. |
| Limitation |
• Can be difficult to derive “chemical insights” in real space. |
• Involves 3 sets of indexes for the reciprocal vectors and corresponding electron and phonon bands. Even more complicated to gain chemical insights. |
|
|
| With MD simulation |
Obtained |
• Depending on the setting, can obtain purely outer-sphere component, or both inter and intra-molecular contribution |
• The off-diagonal coupling can be obtained from MD snap shots. |
| • Mainly arises from low-frequency, intermolecular motions |
| Limitation |
• Quality depends on the force field used. |
• Large cost of quantum chemistry calculation, and therefore, poor ensemble averaging quality. |
4.1 Gas-phase calculations
With the convenience of modern quantum chemistry computational programs, λin can be routinely computed with geometry optimization in both reactant and product states, followed by a projection of the structural difference to the vibrational modes, as mentioned above in Section 2.1. Such results have been seen in many works.37,71,72
The commonly seen charge transporting materials often involve the aromatic π moiety that has alternate C–C single and double bonds. Ionization or excitation in these molecules generally involves a change in this alternacy: shorter single bonds and longer double bonds. Therefore, it is common to see a large contribution from this alternating C–C stretch mode in the projection λin, leading to peaks in the region of 1300–1500 cm−1, whereas many other modes could have some contribution as well.73,74
For a mode with large vibration frequency, the population will mainly reside at the zero-point energy level, thus resulting in weak temperature dependence. Such a behavior was believed to contribute to the weak or even inverse temperature dependence reported in charge mobility in molecular crystals.72 However, such a model ignores the low-frequency modes which mainly arise from intermolecular libration or reorientation in response to a change in the charge position, an effect that exists predominantly in any condensed phase as further discussed below.
The electronic coupling calculation75 is becoming routinely available in many quantum chemistry packages, but the off-diagonal Peierls type of vibronic coupling has been also reported.73,74,76 In most of these works, the electronic coupling factor was scanned via a few degrees of freedom between two organic molecules. The scan is typically performed with planar π-conjugated organic molecules with one or two degrees of freedom in their relative position of π–π stacking, and oscillatory coupling is observed owing to the nature of wavefunctions in the HOMO or LUMO, thus showing the importance of the intermolecular modes in a condensed array of molecules. Another well-known property is the exponential intermolecular distance dependence of the electronic coupling.77 These observed behaviors contribute to important Peierls coupling for the intermolecular acoustic-like modes. A direct computation for the nuclear derivative for the electronic coupling is available nowadays,78,79 which allows for direct computation for the Peierls coupling with any given vibrational mode, which would facilitate such characterization without having to scan for the nuclear degrees of freedom.
4.2 Periodic boundary calculations
In addition to amorphous solid states, charge mobilities have also been studied with single molecular crystals,3,80–83 which allows for better insights and understanding because the intermolecular structures are known. Thus, mobilities in crystals are excellent testbeds for theoretical predictions. Much theoretical and computational works have been developed for predicting properties for polarons for molecular crystals.34,84–88
Computational schemes for electron–phonon coupling for periodic systems have also been developed.89–91 In general, the electron–phonon coupling is a function of the band and the reciprocal vector for the electron and the band and the reciprocal vector for the phonon. A lot of such works have been reported for covalent systems such as inorganic semiconductors or graphene,92,93 whereby the rigid covalent bonds are likely to offer a steep electron phonon coupling. Such approaches can be used for organic molecular crystals, but a careful treatment of the density functionals for a good intermolecular dispersion interaction would be necessary.
A direct evaluation for the electron–phonon coupling in a periodic system can also be achieved by using derivative coupling94 or directly accounting for the matrix elements.95 Both diagonal and off diagonal electron–phonon couplings were reported in these works. The diagonal Holstein type of electron–phonon coupling is affected by both the low- and high-frequency modes, whereas the coupling for off-diagonal elements, the Peierls type of coupling, is mostly in the low-frequency acoustic phonons.94,95
4.3 MD studies
MD simulation has been a useful tool to offer structures and many quantities from the ensemble. In principle, function J(ω) can be obtained by the Fourier transformation of the non-equilibrium trajectory ΔEint, equivalent to X defined in eqn (23), right after an electron is transferred to the acceptor. With the fluctuation–dissipation theorem, it is equivalently calculated by the autocorrelation function depicted in eqn (26). This approach has been commonly adopted and a good number of results have been reported. Such spectral density functions may be sensitive to the forcefield used,96 and thus the results should be taken cautiously. Such an approach has been important to elucidate the dynamics of charge-transfer systems.97,98
For example, the spectral densities have been reported for water and methanol.59,99 In these early works, the general features are remarkably similar to those depicted in Fig. 2: an intramolecular contribution as well as low-frequency intermolecular bands. Nevertheless, such results imply that both inter- and intra-molecular bands contribute to the dielectric response, or to the reorganizational energy for an ET process.
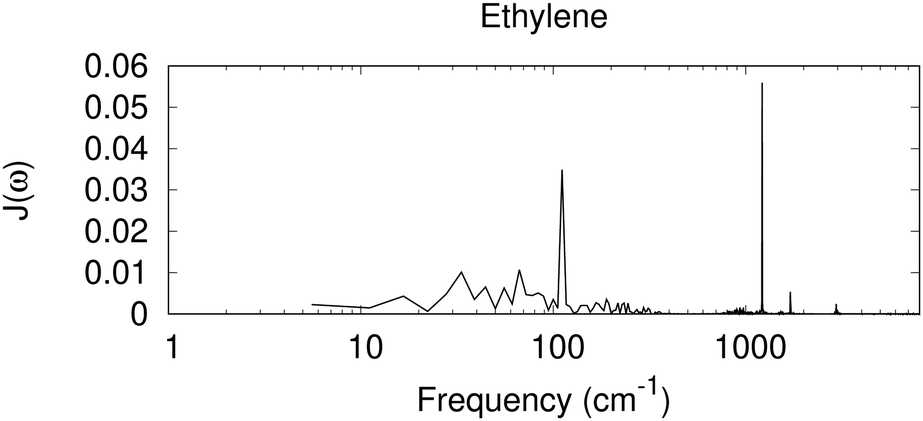
In Fig. 3, we have included the spectral density derived from a Fourier transformation of the autocorrelation function C(t), for ethylene, a nonpolar molecule in its liquid state,|| as a test case for the nonpolar system. In this case, sharp high-frequency peaks are seen, similar to those with the dielectric response for benzene and toluene. The 1200 cm−1 peak for the CH2 rocking mode, together with weaker peaks near 1700 cm−1 for possibly the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching, and 2900 cm−1 for C–H stretching modes, is observed.
C stretching, and 2900 cm−1 for C–H stretching modes, is observed.
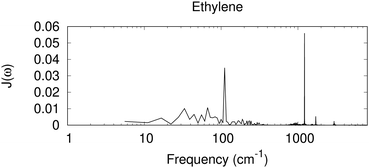 |
| | Fig. 3 Spectral density derived from a MD simulation for a periodic box of small nonpolar molecules of ethylene. Shown is the Fourier transformation of the autocorrelation function C(t), scaled by coth(βħω/2) (eqn (26)), in arbitrary units. | |
An interesting feature in Fig. 3 is the diffuse signal at the low-frequency region, at about 100 cm−1 and below. In contrast to the spectral density derived from the dielectric response for the nonpolar benzene that has nearly zero contribution in this region, with a similar nonpolar system, and similar to the direct calculation for electron–phonon coupling for nonpolar molecular crystals94,95 a low-frequency response is seen with MD. The current dielectric response model assumes that the surrounding medium is a homogeneous dielectric, and the solute charge is assumed to be in a spherical cavity. In MD, ΔE collected includes all the electrostatic interaction between a cationic ethylene and the surrounding neural molecules. Near the cation, a change in the orientation of a neutral ethylene leads to a different electrostatic interaction. It is also possible that higher order polarity effects could contribute (e.g. the quadruple moment could interact with a spatial change in the electric field). Therefore, low-frequency orientational or translational motion can contribute. Omitting such low-frequency contribution, even for nonpolar systems, might lead to an incorrect description for the system.
The off-diagonal electron–phonon coupling has also been studied with MD simulation and INDO/S semiempirical Hamiltonian calculations, or DFT.36,41,104 Again, the importance of low-frequency modes close or under 100 cm−1 is observed, similar to those observed with periodic first-principle calculations.94,95 With the off-diagonal element of the Hamiltionian, the electronic coupling is sensitive to the relative position and orientation of the two molecules where ET is being considered, and thus, the low-frequency modes are important in this case. A future possibility is to use machine learning for a better ensemble averaging and higher resolution in the spectral density function.105
The quality of MD simulation results highly depends on the force field used.96 A polarizable force field should offer a better solution. Meanwhile, the development for combined quantum mechanical/molecular mechanical (QM/MM) calculations106 is worth noting. QM/MM allows for treating one or several molecules using a high-level method while accounting for the effects of the rest, which will offer a much better description for the reorganization λ and its break-down, electron–phonon coupling factors g. By combining QM/MM with MD, a spectral density function can be obtained. This approach has been shown useful to simulate the spectra for pigment-binding proteins, and it can also be useful for describing ET systems.107
5 Conclusions
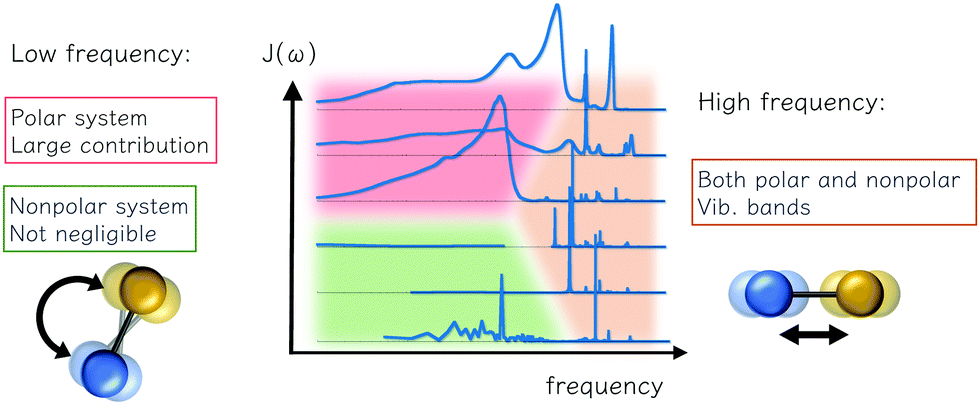
Here we summarize the consideration and description for reorganization energy, electron–phonon coupling, and spectral density for ET problems. The inner-shell contribution is from high-frequency intramolecular vibrational modes, and the outer-shell contribution is mainly low-frequency intermolecular modes. The traditional understanding of polar solvents includes an intense low-frequency Debye dielectric response and responses from high-frequency vibrational modes. However, for non-polar systems commonly seen in modern charge-transporting materials, a number of weak contributions from the vibration is predicted from the dielectric response model, and low-frequency contributions were reported via a direct account of electron–phonon coupling, or MD simulation. The low-frequency bands are also important for off-diagonal, Peierls couplings. As summarized in Fig. 4, these characteristics could facilitate future works in modeling and simulating with realistic electron–phonon settings for good predictions in charge-transporting dynamics.
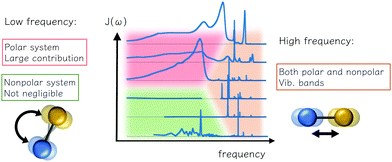 |
| | Fig. 4 Various contributions to the outer reorganization energy of an electron transfer system and their theoretical and computational aspects: the low-frequency reorientational and high-frequency vibrational contributions are observed. For polar environments both can be important, and for the nonpolar solvent, the low-frequency is important, depending on the model and computational settings, and such contribution could have bene overlooked. | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The support from the Academia Sinica Investigator award (AS-IA-106-M01) and the Ministry of Science and Technology of Taiwan (project 105-2113-M-001-009-MY4) is gratefully acknowledged. Data for Fig. 3 were kindly provided by Chun-I Wang and Yi-Siang Wang.
References
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 Search PubMed.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 Search PubMed.
- M. E. Gershenson, V. Podzorov and A. F. Morpurgo, Rev. Mod. Phys., 2006, 78, 973–989 Search PubMed.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 Search PubMed.
- J. D. Myers and J. Xue, Polym. Rev., 2012, 52, 1–37 Search PubMed.
- A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066 Search PubMed.
- S.-C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097 Search PubMed.
- H. Spanggaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2004, 83, 125 Search PubMed.
- S. R. Forrest, Nature, 2004, 428, 911 Search PubMed.
- C. H. Teh, R. Daik, E. L. Lim, C. C. Yap, M. A. Ibrahim, N. A. Ludin, K. Sopian and M. A. M. Teridi, J. Mater. Chem. A, 2016, 4, 15788–15822 Search PubMed.
-
S. Sze and K. N. Kwok, Physics of Semiconductor Devices, Wiley Online Library, 3rd edn, 2007 Search PubMed.
- L. Wang and D. Beljonne, J. Chem. Phys., 2013, 139, 064316 Search PubMed.
- G. Granucci and M. Persico, J. Chem. Phys., 2007, 126, 134114 Search PubMed.
- J. C. Tully, J. Chem. Phys., 2012, 137, 22A301 Search PubMed.
- L. Wang, A. Akimov and O. V. Prezhdo, J. Phys. Chem. Lett., 2016, 7, 2100–2112 Search PubMed.
- L. Wang, J. Qiu, X. Bai and J. Xu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1435 Search PubMed.
- H. Oberhofer, K. Reuter and J. Blumberger, Chem. Rev., 2017, 117, 10319–10357 Search PubMed.
- L. Wang, R. Long and O. V. Prezhdo, Annu. Rev. Phys. Chem., 2015, 66, 549–579 Search PubMed.
- J. Spencer, F. Gajdos and J. Blumberger, J. Chem. Phys., 2016, 145, 064102 Search PubMed.
- S. Giannini, A. Carof and J. Blumberger, J. Phys. Chem. Lett., 2018, 9, 3116–3123 Search PubMed.
- S. Giannini, A. Carof, M. Ellis, H. Yang, O. G. Ziogos, S. Ghosh and J. Blumberger, Nat. Commun., 2019, 10, 1–12 Search PubMed.
- A. A. Ovchinnikov and M. Y. Ovchinnikova, Sov. Phys. JEPT, 1969, 29, 688–693 Search PubMed.
- J. Ulstrup and J. Jortner, J. Chem. Phys., 1975, 63, 4358–4368 Search PubMed.
- J. N. Gehlen and D. Chandler, J. Chem. Phys., 1992, 97, 4958–4963 Search PubMed.
- Y. Georgievskii, C.-P. Hsu and R. A. Marcus, J. Chem. Phys., 1999, 110, 5307–5317 Search PubMed.
- M. Bixon and J. Jortner, Chem. Phys., 1993, 176, 467–481 Search PubMed.
- R. A. Kuharski, J. S. Bader, D. Chandler, M. Sprik, M. L. Klein and R. W. Impey, J. Chem. Phys., 1988, 89, 3248–3257 Search PubMed.
- R. A. Marcus, J. Chem. Phys., 1965, 43, 679–701 Search PubMed.
- R. A. Marcus, Faraday Discuss. Chem. Soc., 1982, 74, 7–15 Search PubMed.
- A.
J. Leggett, S. Chakravarty, A. T. Dorsey, M. P. A. Fisher, A. Garg and W. Zwerger, Rev. Mod. Phys., 1987, 59, 1–85 Search PubMed.
- T. Holstein, Ann. Phys., 1959, 8, 325–342 Search PubMed.
- T. Holstein, Ann. Phys., 1959, 8, 343–389 Search PubMed.
- Y. C. Cheng and R. J. Silbey, J. Chem. Phys., 2008, 128, 114713 Search PubMed.
- N. Prodanovicć and N. Vukmirovicć, Phys. Rev. B, 2019, 99, 104304 Search PubMed.
- E. Mozafari and S. Stafstroöm, Phys. Lett. A, 2012, 376, 1807–1811 Search PubMed.
- A. Troisi, Adv. Mater., 2007, 19, 2000–2004 Search PubMed.
- Z. Shuai, L. Wang and Q. Li, Adv. Mater., 2011, 23, 1145 Search PubMed.
- E. Mozafari and S. Stafstroöm, J. Chem. Phys., 2013, 138, 184104 Search PubMed.
- Y. Zhao, D. W. Brown and K. Lindenberg, J. Chem. Phys., 1994, 100, 2335–2345 Search PubMed.
- R. W. Munn and R. Silbey, J. Phys. Chem., 1985, 83, 1843–1853 Search PubMed.
- A. Troisi and G. Orlandi, Phys. Rev. Lett., 2006, 96, 086601 Search PubMed.
- Y. Yao, N. Zhou, J. Prior and Y. Zhao, Sci. Rep., 2015, 5, 1–10 Search PubMed.
- S. Fratini and S. Ciuchi, Phys. Rev. Lett., 2003, 91, 256403 Search PubMed.
- H. Ishii, N. Kobayashi and K. Hirose, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 205208 Search PubMed.
- K. Hannewald and P. A. Bobbert, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075212 Search PubMed.
-
S. Mukamel, Principles of Nonlinear Optical Spectroscopy, Oxford University Press, 1995 Search PubMed.
- S. Jang and Y.-C. Cheng, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 3, 84–104 Search PubMed.
- T. Joo, Y. Jia, J.-Y. Yu, M. J. Lang and G. R. Fleming, J. Chem. Phys., 1996, 104, 6089–6108 Search PubMed.
- A. A. Stuchebrukhov and X. Song, J. Chem. Phys., 1994, 101, 9354–9365 Search PubMed.
- T. Ichiye, J. Chem. Phys., 1996, 104, 7561–7571 Search PubMed.
- C.-P. Hsu, X. Song and R. A. Marcus, J. Phys. Chem. B, 1997, 101, 2546–2551 Search PubMed.
- C.-P. Hsu, Y. Georgievskii and R. A. Marcus, J. Phys. Chem. A, 1998, 102, 2658–2666 Search PubMed.
-
D. Chandler, Introduction to Modern Statistical Mechanics, Oxford University Press, 1987 Search PubMed.
- R. Kubo, Rep. Prog. Phys., 1966, 29, 255 Search PubMed.
-
R. Kubo, M. Toda and N. Hashitsume, Statistical Physics II – Nonequilibrium Statistical Mechanics, Springer-Verlag Berlin Heidelberg, 1991 Search PubMed.
- B. Bagchi, D. W. Oxtoby and G. R. Fleming, Chem. Phys., 1984, 86, 257–267 Search PubMed.
- J. S. Toll, Phys. Rev., 1956, 104, 1760–1770 Search PubMed.
- R. Kronig, J. Opt. Soc. Am., 1926, 12, 547–557 Search PubMed.
- K. Ando, J. Chem. Phys., 1997, 106, 116–126 Search PubMed.
- J. E. Bertie and Z. Lan, Appl. Spectrosc., 1996, 50, 1047–1057 Search PubMed.
- C. G. Malmberg and A. A. Maryott, J. Res. Natl. Bur. Stand., 1956, 56, 2641 Search PubMed.
- H. J. Liebe, G. A. Hufford and T. Manabe, Int. J. Infrared Millimeter Waves, 1991, 12, 659–675 Search PubMed.
- J. E. Bertie, S. L. Zhang, H. H. Eysel, S. Baluja and M. K. Ahmed, Appl. Spectrosc., 1993, 47, 1100–1114 Search PubMed.
- J. E. Bertie and Z. Lan, J. Phys. Chem. B, 1997, 101, 4111–4119 Search PubMed.
- J. Barthel, M. Kleebauer and R. Buchner, J. Solution Chem., 1995, 24, 1–17 Search PubMed.
- S. Ikawa, T. Ohba, S. Tanaka, Y. Morimoto, K. Fukushi and M. Kimura, Int. J. Infrared Millimeter Waves, 1985, 6, 287–306 Search PubMed.
- P. Firman, A. Marchetti, M. Xu, E. M. Eyring and S. Petrucci, J. Phys. Chem., 1991, 95, 7055–7061 Search PubMed.
- J. E. Bertie, Y. Apelblat and C. D. Keefe, J. Mol. Struct., 2005, 750, 78–93 Search PubMed.
- V. A. Santarelli, J. A. MacDonald and C. Pine, J. Chem. Phys., 1967, 46, 2367–2375 Search PubMed.
- J. E. Bertie and C. D. Keefe, J. Mol. Struct., 2004, 695–696, 39–57 Search PubMed.
- N. E. Gruhn, D. A. Da Silva Filho, T. G. Bill, M. Malagoli, V. Coropceanu, A. Kahn and J.-L. Breédas, J. Am. Chem. Soc., 2002, 124, 7918–7919 Search PubMed.
- G. Nan, X. Yang, L. Wang, Z. Shuai and Y. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115203 Search PubMed.
- J. Cornil, D. Beljonne, J.-P. Calbert and J.-L. Breédas, Adv. Mater., 2001, 13, 1053–1067 Search PubMed.
- J. L. Brédas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804 Search PubMed.
- C.-P. Hsu, Acc. Chem. Res., 2009, 42, 509–518 Search PubMed.
- B.-C. Lin, C.-P. Cheng, Z.-Q. You and C.-P. Hsu, Phys. Chem. Chem. Phys., 2011, 13, 20704–20713 Search PubMed.
- Z. Q. You, Y. H. Shao and C. P. Hsu, Chem. Phys. Lett., 2004, 390, 116–123 Search PubMed.
- A. F. Morrison and J. M. Herbert, J. Phys. Chem. Lett., 2017, 8, 1442–1448 Search PubMed.
- N. Bellonzi, E. Alguire, S. Fatehi, Y. Shao and J. E. Subotnik, J. Chem. Phys., 2020, 152, 044112 Search PubMed.
- V. C. Sundar, J. Zaumseil, V. Podzorov, E. Menard, R. L. Willett, T. Someya, M. E. Gershenson and J. A. Rogers, Science, 2004, 303, 1644 Search PubMed.
- V. Podzorov, E. Menard, A. Borissov, V. Kiryukhin, J. A. Rogers and M. E. Gershenson, Phys. Rev. Lett., 2004, 93, 086602 Search PubMed.
- J. Y. Lee, S. Roth and Y. W. Park, Appl. Phys. Lett., 2006, 88, 252106 Search PubMed.
- J. Takeya, M. Yamagishi, Y. Tominari, R. Hirahara, Y. Nakazawa, T. Nishikawa, T. Kawase, T. Shimoda and S. Ogawa, Appl. Phys. Lett., 2007, 90, 102120 Search PubMed.
- V. M. Kenkre, J. D. Andersen, D. H. Dunlap and C. B. Duke, Phys. Rev. Lett., 1989, 62, 1165–1168 Search PubMed.
- F. Ortmann, F. Bechstedt and K. Hannewald, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235206 Search PubMed.
- F. Ortmann, F. Bechstedt and K. Hannewald, New J. Phys., 2010, 12, 023011 Search PubMed.
- Y.-C. Wang and Y. Zhao, Phys. Rev. B, 2020, 101, 075205 Search PubMed.
- Y.-J. Zhong, C.-F. Lan, B.-C. Lin, C.-D. Hu, Y.-C. Cheng and C.-P. Hsu, Adv. Quantum Chem., 2020, 81, 219–241 Search PubMed.
- F. Giustino, M. L. Cohen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 165108 Search PubMed.
- F. Giustino, Rev. Mod. Phys., 2017, 89, 015003 Search PubMed.
- J. Noffsinger, F. Giustino, B. D. Malone, C.-H. Park, S. G. Louie and M. L. Cohen, Comput. Phys. Commun., 2010, 181, 2140–2148 Search PubMed.
- T. Gunst, T. Markussen, K. Stokbro and M. Brandbyge, Phys. Rev. B, 2016, 93, 035414 Search PubMed.
- C.-H. Park, N. Bonini, T. Sohier, G. Samsonidze, B. Kozinsky, M. Calandra, F. Mauri and N. Marzari, Nano Lett., 2014, 2014, 3 Search PubMed.
- A. F. Morrison and J. M. Herbert, J. Chem. Phys., 2017, 146, 224110 Search PubMed.
- P. Ordejoón, D. Boskovic, M. Panhans and F. Ortmann, Phys. Rev. B, 2017, 96, 035202 Search PubMed.
- P. Zarzycki and B. Gilbert, Phys. Chem. Chem. Phys., 2020, 22, 1011–1018 Search PubMed.
- J. N. Gehlen, M. Marchi and D. Chandler, Science, 1994, 263, 499–502 Search PubMed.
- J. S. Bader and D. Chandler, Chem. Phys. Lett., 1989, 157, 501–504 Search PubMed.
- K. Ando, J. Chem. Phys., 2001, 114, 9040–9047 Search PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 Search PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Paáll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 Search PubMed.
-
A. Savin, in Recent Developments and Applications of Modern Density Functional Theory, ed. J. Seminario, Elsevier Science, Amsterdam, 1996, pp. 327–358 Search PubMed.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540–3544 Search PubMed.
- A. Troisi and G. Orlandi, J. Phys. Chem. A, 2006, 110, 4065–4070 Search PubMed.
- C.-I. Wang, M. K. E. Braza, G. C. Claudio, R. B. Nellas and C.-P. Hsu, J. Phys. Chem. A, 2019, 123, 7792–7802 Search PubMed.
- H. Lin and D. G. Truhlar, Theor. Chem. Acc., 2006, 117, 185–199 Search PubMed.
- A. M. Rosnik and C. Curutchet, J. Chem. Theory Comput., 2015, 11, 5826–5837 Search PubMed.
Footnotes |
| † This article is dedicated to the memory of Prof. Ching-Liang Lin (1931–2019), for her warm love to students and her firm support to women physicists. |
| ‡ The electrons can be described with adiabatic13,14,16,21 or diabatic basis,44 but the discussion is out of the scope of the present work. |
| § α(ω) has a dimension of energy. Here, for the clarity of theories, an expression similar to eqn (12) of ref. 22 was loosely given without a rigorous ground. |
| ¶ The expression in eqn (40) was slightly different from that described in ref. 25, by a constant factor (εc + 2), and it is closer to those in ref. 51 and 52. |
| || The simulation was performed using the Berendsen NPT ensemble, with temperature set at 130 K (for a liquid state) and pressure set at 1 bar, under optimized potentials for the liquid simulations force field100 using GROMACS (v 2016.4).101 ΔE was simulated with the Coulomb interaction of a cationic ethylene (modeled by Mulliken population analysis via DFT at the LC-BLYP/DZ* level102,103) and the equilibrated surrounding molecules. |
|
| This journal is © the Owner Societies 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence

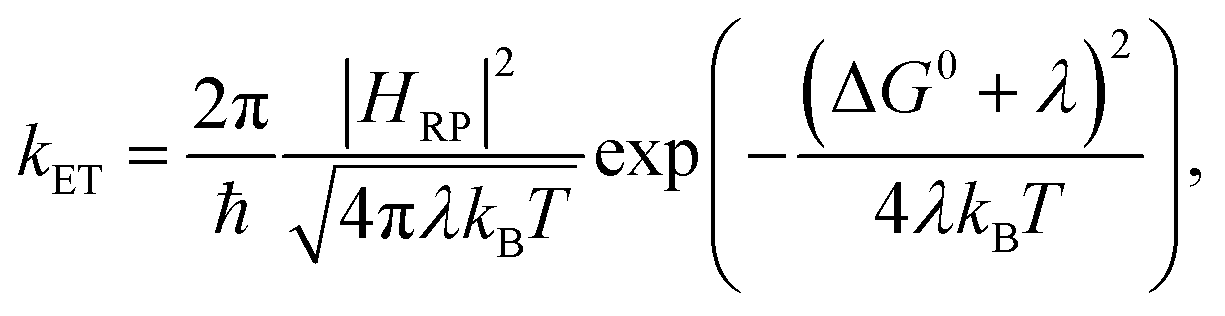
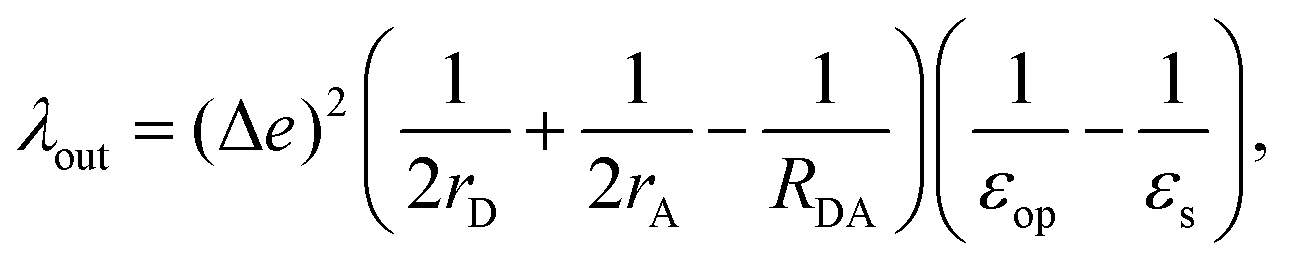
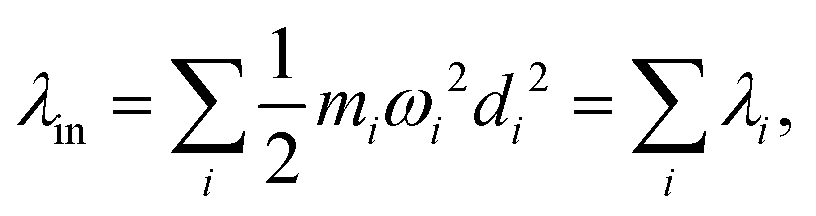
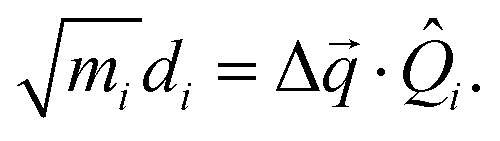
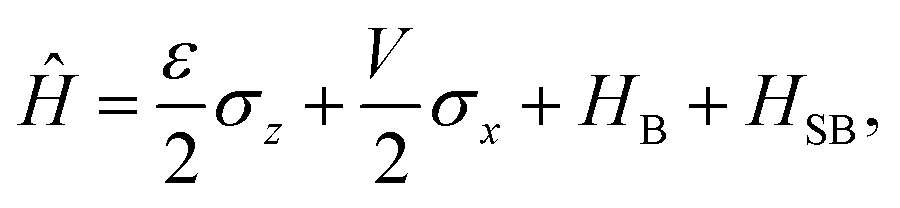
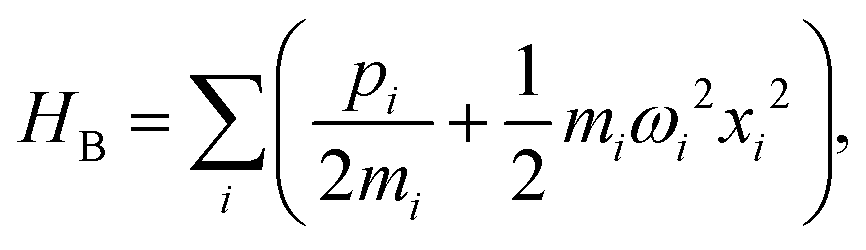
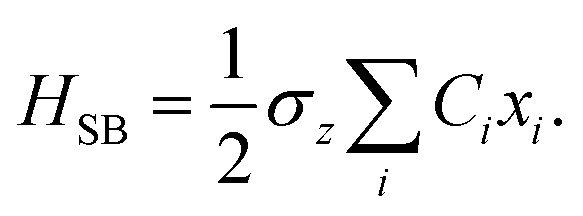
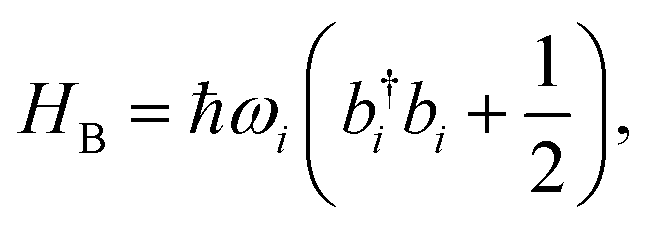
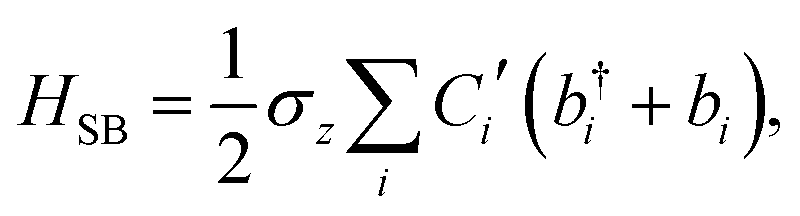
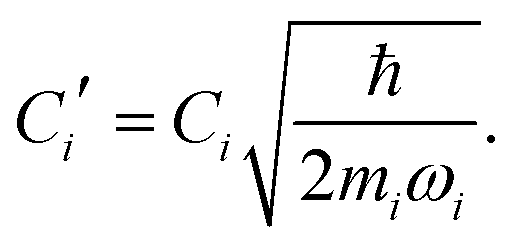
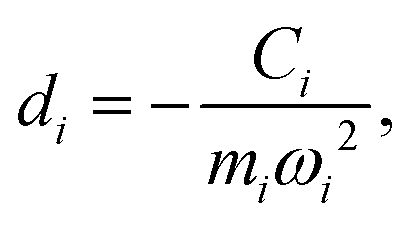
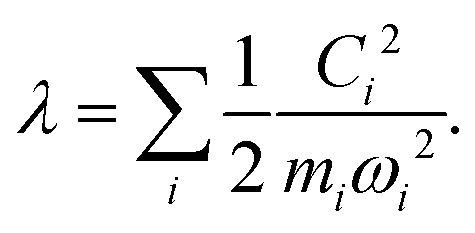
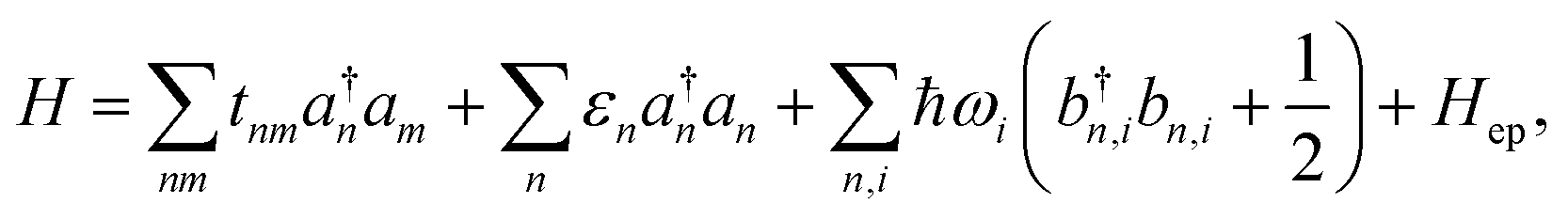
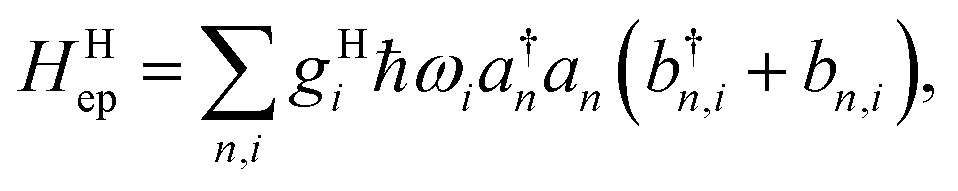
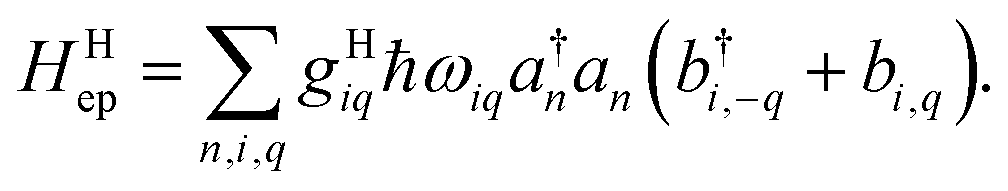
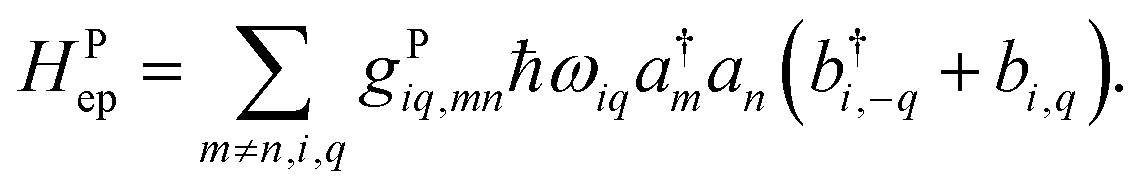
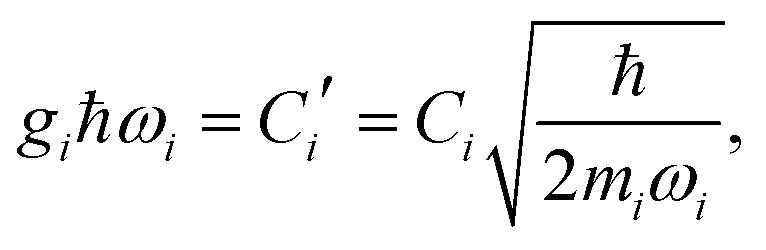
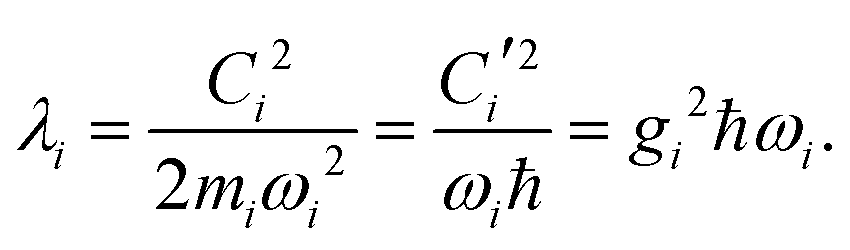
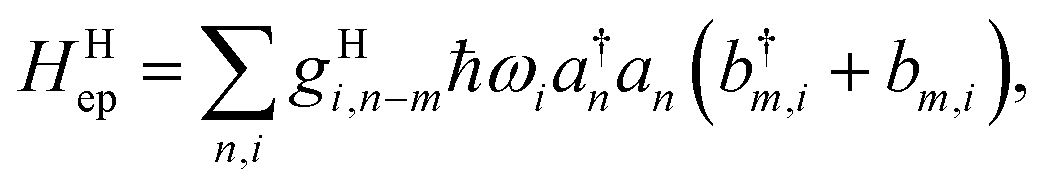
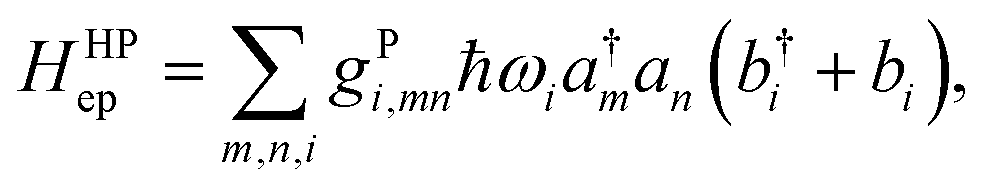
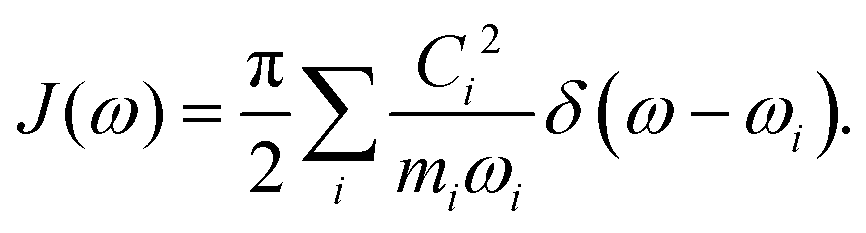
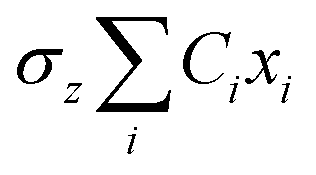 , which couples the nuclear and electronic degrees of freedom. This is often achieved by the a time-dependent perturbation theory under the interaction picture of quantum mechanics. Such techniques have been successful in treating many problems in quantum dynamics,46,47 including nonlinear spectroscopy48 and the two-state reaction rates with or beyond Fermi's golden rule.49 In these works, a central quantity X is defined as the energy difference between the two states in SBH, or the change in energy when the site carries a charge for the Holstein model,
, which couples the nuclear and electronic degrees of freedom. This is often achieved by the a time-dependent perturbation theory under the interaction picture of quantum mechanics. Such techniques have been successful in treating many problems in quantum dynamics,46,47 including nonlinear spectroscopy48 and the two-state reaction rates with or beyond Fermi's golden rule.49 In these works, a central quantity X is defined as the energy difference between the two states in SBH, or the change in energy when the site carries a charge for the Holstein model,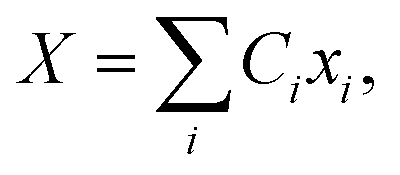
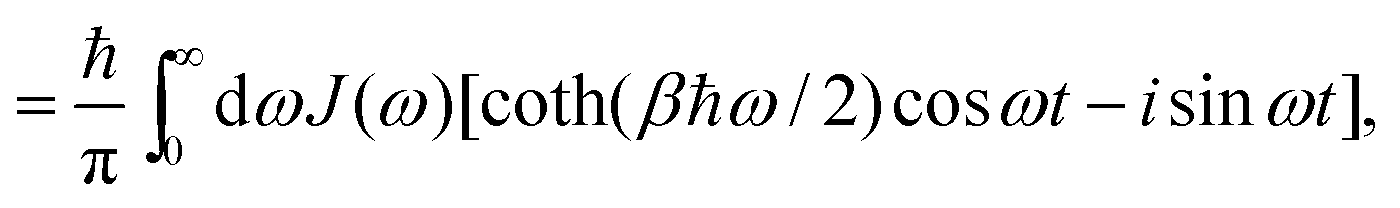
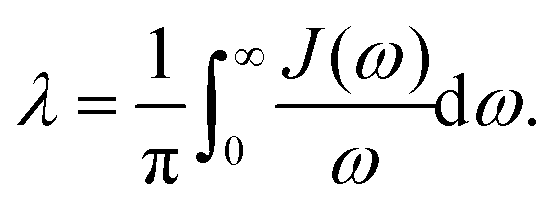
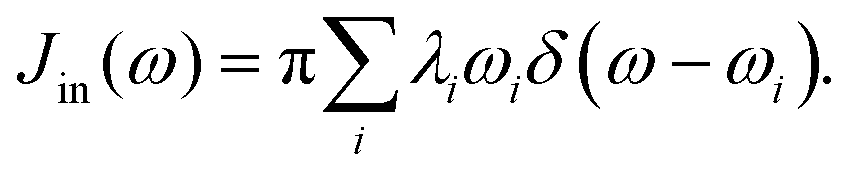
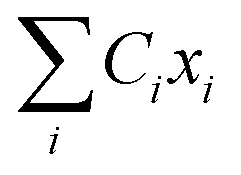 which is often named X as a major dynamic variable in many related works.25,46,51,52
which is often named X as a major dynamic variable in many related works.25,46,51,52
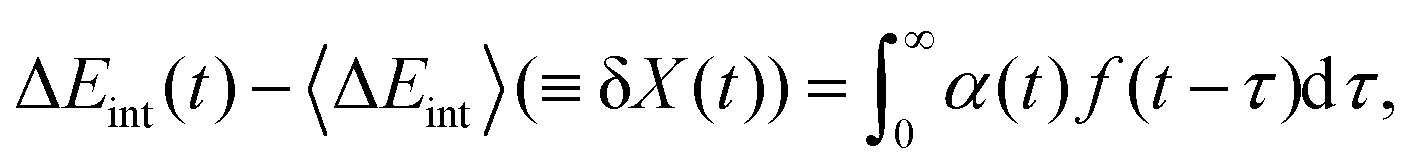
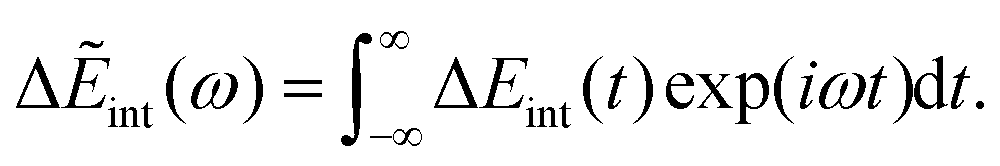
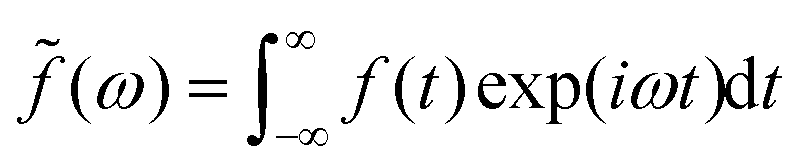 is a dimensionless function, which can be an arbitrarily chosen perturbation, and α(ω) is the response function with frequency ω. The specific form of α(ω) depends on the setting in the electrostatic model.22,25,56
is a dimensionless function, which can be an arbitrarily chosen perturbation, and α(ω) is the response function with frequency ω. The specific form of α(ω) depends on the setting in the electrostatic model.22,25,56