
A rhenium catalyst with bifunctional pyrene groups boosts natural light-driven CO2 reduction†
Li-Qi
Qiu‡
,
Kai-Hong
Chen‡
 ,
Zhi-Wen
Yang
and
Liang-Nian
He
*
,
Zhi-Wen
Yang
and
Liang-Nian
He
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: heln@nankai.edu.cn; Fax: (+86) 022-23503878; Tel: (+86) 022-23503878
First published on 18th November 2020
Abstract
Developing effective sunlight-driven systems for CO2 reduction is one of the most promising subjects from the perspective of sustainably producing solar fuels. Herein, we develop a strategy to boost CO2 reduction performance by enhancing intermolecular electron transfer efficiency and visible light-absorption ability by introducing bifunctional pyrene groups on the ligand. This catalyst exhibits high-efficiency performance for natural light-powered CO2 reduction (TONCO up to 350 ± 36, ΦCO up to 46.6 ± 3%). This is the first report on using a single-molecule photocatalyst for CO2 reduction under natural conditions. Through the combination of experimental results and DFT calculations, the appending pyrene groups have been proven to promote the catalyst's ability to harness visible light as well as facilitate electron transfer (ET) through intermolecular π–π interactions. Due to the accelerated intermolecular ET, TONCO can be further boosted up to 1367 ± 32 in the presence of the ruthenium photosensitizer. Moreover, an enhancement in CO2 electroreduction performance can also be observed for the pyrenyl-functionalized rhenium catalyst further highlighting the versatile applications of this methodology.
Introduction
Controlling the total amount of carbon dioxide (CO2) in the atmosphere and responding to global warming and energy crisis positively is a critical research topic for scientists.1,2 Therefore, the conversion of CO2 into value-added carbon products or fuels has been a significant research field.3–9 The thermodynamic stability and kinetic inertness of CO2 make it necessary to overcome a high activation barrier and energy-intensive limitation for performing CO2 conversion.10,11 Converting CO2 into carbon monoxide (CO) using solar energy is an ideal and sustainable way to reduce the concentration of CO2 in the atmosphere while enabling the solar energy-to-fuel conversion schemes, because CO is an important raw material in the industrial syntheses based on Fischer–Tropsch chemistry.12 However, the intermittency of solar energy from variable atmospheric conditions is one of the major challenges that make the large scale application of this technology difficult.13–15 In this context, almost all of the photochemical CO2 reduction reactions (CO2RR) were conducted under simulated sunlight sources instead of the sun.16 No report is available for CO2RR directly promoted by the sun in homogeneous systems, presumably due to the great challenge for simultaneously overcoming both limitations associated with the visible-light absorption ability and, especially, intermolecular electron transfer (ET) efficiency.As a widespread event in nature, ET between molecules plays a key role not only in biology but in semiconductors, catalysts, and photoactive materials.17–21 A persisting fundamental challenge in the design of material with high intermolecular ET efficiency is that the intermolecular ET is sensitive to the material concentration because the non-covalent interactions can form only in a relatively high concentration.22–24 In the field of CO2RR, CO2 is reduced using metal complexes25–30 through ET from the photosensitizer (PS) to the catalyst (Cat) to drive the reaction (Scheme 1A). Clearly, enhancing ET will be a powerful strategy to design an efficient and selective catalyst for CO2RR. Accordingly, the Ishitani group developed a series of efficient supramolecular catalysts with short bridging linkers to connect different PS and rhenium (Re) catalysts.31–34 Because of the acceleration of intramolecular ET between PS and Cat, such supramolecular catalysts exhibit impressive performance in CO2RR. More recently, an elegant bimolecular system has been reported by Kubiak and coworkers, who utilized hydrogen-bonding interactions between ruthenium (Ru) PS and Re Cat to facilitate intermolecular ET (Scheme 1C), thereby enhancing CO2RR activity.35 However, most of these systems suffer from multiple synthetic steps and additional cost of PS. Although a single-molecular catalyst (Scheme 1B) can avoid these problems, presently, developing a single-molecule photocatalyst is still a challenge.36 As shown in previous studies,37–40 one of the most significant points is how to maintain or even enhance the visible light-absorption ability of the targeted catalysts.
 | ||
| Scheme 1 Strategies for the design of catalysts for photochemical CO2RR. | ||
Known as an effective noncovalent intermolecular force, strong van der Waals π–π interaction is widely found in life-related biomacromolecules such as proteins, DNA and RNA,41 and commonly promotes molecular recognition or self-assembly processes.42,43 Moreover, the π–π stacking also facilitates the intermolecular carrier mobility in organic π-conjugated material systems.44,45 Indeed, it has been found that noncovalent immobilization of pyrene-modified electrocatalysts on graphite surfaces or carbon nanotube electrodes for the conversion of CO2 to fuel renders notable efficiency and selectivity.46–48 At this point, we envision that the bifunctional pyrene group, as a photosensitizing moiety and extended π-conjugated unit, can not only enhance the visible light-harvesting capability, but also facilitate the intermolecular ET through π–π interactions, exceptionally promoting the visible light-induced CO2 photoreduction efficiency. In this work, the widely used rhenium bipyridine complexes,37–40,49e.g. Re(bpy)(CO)3Cl, are used as the model catalyst to investigate the effect of the appended pyrene groups in the ligand on the CO2RR. The novel and easy-to-prepare catalyst, i.e. Re[4,4′-di(pyren-1-yl)-2,2′-bipyridine](CO)3Cl (4,4′-dipyrenyl-Re), is found to demonstrate outstanding performance for the natural light-driven CO2RR under different weather conditions (Scheme 1D). Notably, a dramatically enhanced efficiency of this pyrene functionalized catalyst in CO2RR can be found even at a low concentration (0.0125 mM). As expected, further improvement in the turnover number for CO (TONCO) is observed after adding Ru PS, which underlines the versatile application of this methodology in photocatalytic CO2RR.
Results and discussion
Catalyst synthesis and characterization
4,4′-Dipyrenyl-Re was synthesized via the reaction of 4,4′-di(pyren-1-yl)-2,2′-bipyridine and Re(CO)5Cl. The structure of 4,4′-dipyrenyl-Re was verified by NMR spectroscopy and FT-IR spectroscopy, X-ray crystallography, and mass spectrometry measurement. Its crystal was recrystallized from dichloromethane and ether, and the molecular structure was refined, as shown in Fig. 1. Obviously, the intermolecular off-set π–π stacking is observed with a distance of about 3.48 Å in the packing structure of 4,4′-dipyrenyl-Re. | ||
| Fig. 1 X-ray crystal structure of 4,4′-dipyrenyl-Re with top (left) view and packing (right) view. Hydrogen atoms are omitted for clarity. Main bond lengths (Å) and angles (°): Re1–Cl1 2.480(3), Re1–C2 1.918(7), Re1–N1 2.176(5), N1–Re1–Cl1 84.03(15), C1–Re1–Cl1 175.7(3), and C1–Re1–N1 92.6(3). CCDC reference number: 1954434.† | ||
Visible light-driven CO2 reduction
To validate our proposal on designing new molecular photocatalysts originating from the effect of π–π interaction, we initially performed the photocatalytic experiments in a mixture of CO2-saturated N,N-dimethylformamide (DMF) solution with triethanolamine (TEOA) and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) driven by a white-light LED. Both 4,4′-dipyrenyl-Re and Re(bpy)(CO)3Cl exhibit excellent selectivity (>99%) towards CO, and only traces of H2 (<1%) were detected, probably because triethanolamine (TEOA) can be used as a proton source, inevitably accompanied by the generation of H2 as a byproduct (Table S6†).No formation of HCOOH in the liquid phase can be detected by 1H NMR experiments. The control experiments under an Ar atmosphere exhibit no CO production, demonstrating that CO originates from CO2 (Table S4†). As shown in Fig. 2, a relatively high TONCO (138 ± 5) is obtained when the 4,4′-dipyrenyl-Re complex is irradiated for 3 h, which then levels off to a terminal plateau. Compared to Re(bpy)(CO)3Cl (TONCO = 20 ± 1), the TONCO for 4,4′-dipyrenyl-Re (149 ± 2) shows a more than 7-fold increase after 12 h irradiation, thus proving our hypothesis of π–π interaction effect on the large enhancement of photocatalytic efficiency. Moreover, simply mixing pyrene with the Re(bpy)(CO)3Cl system has no effect on the catalytic efficiency, further demonstrating the significance of the π-expansive ligand (Table S5†). When more BIH (20 mM) is added, the amount of CO increases slightly (about 7.8 mM CO, TONCO = 157 ± 5) (Table S5†). This result shows that the photocatalytic performance of CO2 reduction is mainly limited by the decomposition and/or transformation of the Re–pyrene complex into inactive species upon two electron reduction.
 | ||
| Fig. 2 TON for CO production versus time for 0.05 mM 4,4′-dipyrenyl-Re and 0.05 mM Re(bpy)(CO)3Cl. Conditions: DMF (3 mL), TEOA (0.3 mL), 10 mM BIH; irradiated with a white LED lamp (0.60 mW cm−2). | ||
Light harvesting and redox properties
This superiority of pyrene-functionalized catalyst drove us to explore plausible reasons. At first, the visible-light absorption ability was taken into consideration. The UV-vis absorption spectra of 4,4′-dipyrenyl-Re and Re(bpy)(CO)3Cl in DMF solution are compared in Fig. 3A. It's worth noting that the molar absorptivity of 4,4′-dipyrenyl-Re (εmax = 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 323 M−1 cm−1) at about 400 nm is about six times larger than that of Re(bpy)(CO)3Cl (εmax = 3754 M−1 cm−1), which is mainly ascribed to the electron-rich expansion of the π-conjugated system through pyrene chromophores. The 4,4′-dipyrenyl-Re complex exhibits an absorption maximum at 395 nm and shows obvious bathochromic shifts in comparison with Re(bpy)(CO)3Cl, indicating that the extended conjugation increases the transition probability of the metal-to-ligand charge-transfer (MLCT).39
323 M−1 cm−1) at about 400 nm is about six times larger than that of Re(bpy)(CO)3Cl (εmax = 3754 M−1 cm−1), which is mainly ascribed to the electron-rich expansion of the π-conjugated system through pyrene chromophores. The 4,4′-dipyrenyl-Re complex exhibits an absorption maximum at 395 nm and shows obvious bathochromic shifts in comparison with Re(bpy)(CO)3Cl, indicating that the extended conjugation increases the transition probability of the metal-to-ligand charge-transfer (MLCT).39
 | ||
| Fig. 3 Optical and electrochemical properties of the Re(bpy)(CO)3Cl and 4,4′-dipyrenyl-Re. (A) UV-vis absorption of the Re(bpy)(CO)3Cl and 4,4′-dipyrenyl-Re in DMF at rt; (B) shape of HOMOs and LUMOs of Re(bpy)(CO)3Cl and 4,4′-dipyrenyl-Re; and (C) HOMO and LUMO energy levels of Re(bpy)(CO)3Cl and 4,4′-dipyrenyl-Re, together with redox potentials (eV) of CO2/CO and electron donors BIH and TEOA. | ||
From the wavelength of the onsets of UV-vis absorption spectra, the optical energy gaps of 4,4′-dipyrenyl-Re and Re(bpy)(CO)3Cl are determined as 2.71 and 3.35 eV, respectively (Table S14†). The lowest unoccupied molecular orbital (LUMO) values calculated from the onsets of the reduction potential are −3.23 and −3.12 eV for 4,4′-dipyrenyl-Re and Re(bpy)(CO)3Cl, as determined by cyclic voltammetry (CV) analysis (Fig. S9†). Therefore, the highest occupied molecular orbital (HOMO) levels of both catalysts are estimated based on their LUMO levels and optical gaps, being in line with the DFT calculations (Table S14† and Fig. 3B).50
The next question is, what leads to the excellent optical properties of 4,4′-dipyrenyl-Re. Thus, DFT calculations were performed to compare energy levels and electron distribution from Frontier molecular orbitals of 4,4′-dipyrenyl-Re and Re(bpy)(CO)3Cl (Fig. 3B). For both Re complexes, the LUMOs are mainly localized on the bipyridine unit. However, the HOMOs are distributed on the Re center, such as Re(bpy)(CO)3Cl, as well as on the appending pyrene groups. A narrower energy gap between the LUMO and HOMO (Fig. 3C) is found for 4,4′-dipyrenyl-Re. Consequently, pyrene units with a large extended conjugation and electron-donating character can lower the LUMO level and effectively elevate the HOMO level for this catalyst, leading to the narrower energy gap, enhancing the light-harvesting capability. Moreover, the driving force for the sacrificial reductants was also discussed to study the thermodynamic process of CO2RR. For the 4,4′-dipyrenyl-Re complex, we found that the yield of CO was distinctly reduced in the absence of BIH through the control experiments (Table S7†), although TEOA itself was considered as a base in addition to being an electron donor.33 The main reason is that the redox potential of TEOA is almost close to the HOMO value of 4,4′-dipyrenyl-Re (Fig. 3C), which indicates that the ET from TEOA to pyrene moieties of this catalyst is limited.51 Thus, the use of BIH as the sacrificial reductant and TEOA, a “magic” reagent,52 is a feasible strategy to effectively avoid the thermodynamic limitation of photocatalytic CO2RR. According to previous reports, TEOA, acting as a base to capture a proton from BIH˙+, a proton source, or a CO2 absorbent to form the TEOA-CO2 adduct, was considered as a main component of the Re complexes during the photocatalytic formation of CO.53,54
Investigation of the intermolecular π–π interactions
As is well known, intermolecular π–π stacking has the ability to accelerate intermolecular ET.55,56 Despite π–π stacking being identified in the crystal of 4,4′-dipyrenyl-Re, the intermolecular force pattern of this catalyst in solution should be further investigated. Thus, the photoluminescence (PL) spectra of 4,4′-dipyrenyl-Re were measured at different concentrations in DMF solution. The fluorescence intensity of 4,4′-dipyrenyl-Re dramatically decreases when the concentration is increased from 0.0125 mM to 0.2 mM (Fig. 4A). At the same time, the appearance of an additional emission peak at 650 nm from the Re–pyrene excimer was confirmed by time-resolved emission, and the emission lifetime was found to be 0.141 ns (Fig. S4†). In sharp contrast, only a small decrease can be observed in the same concentration range for Re(bpy)(CO)3Cl (Fig. S3†). Clearly, the severe aggregation-caused quenching (ACQ) mainly originates from the π–π stacking of 4,4′-dipyrenyl-Re in DMF solution. | ||
| Fig. 4 Photoluminescence (PL) and variable-temperature 1H NMR characteristics of 4,4′-dipyrenyl-Re. (A) Photoluminescence experiments at different concentrations of 4,4′-dipyrenyl-Re and (B) partial variable temperature 1H NMR of 4,4′-dipyrenyl-Re in DMSO-d6. | ||
Furthermore, the intermolecular π–π interaction of 4,4′-dipyrenyl-Re in solution was verified by variable-temperature NMR spectroscopy. The 1H NMR spectra of 4,4′-dipyrenyl-Re in DMSO-d6 manifest obvious upfield shifts with increasing temperature for the pyrene protons. For instance, the typical peak for pyrene groups shifts from δ = 9.35 to 9.29 ppm when the temperature increases from 25 °C to 65 °C (Fig. 4B), indicating that the π–π stacking is impeded between pyrene moieties by heating. However, only inconspicuous shift can be found for the protons of bipyridine. The above observations suggest that the pyrene group leads to the π–π stacking of 4,4′-dipyrenyl-Re other than bipyridine.
It is well known that decreasing the catalyst concentration is an effective and feasible way to evaluate the activity, because intermolecular ET and non-covalent interactions (such as intermolecular π–π interactions) will be limited by the low catalyst concentration. We then investigated the concentration effects on the photocatalytic performance (Table S8†). Compared with Re(bpy)(CO)3Cl, 4,4′-dipyrenyl-Re showed a significant increase in CO yield at all concentrations examined in this work (Fig. 5). The main reason would be that the fraction of absorbed photons of Re–pyrene catalyst was significantly higher than that of Re(bpy)(CO)3Cl at different concentrations as shown in the Fig. S2A.† For example, when the concentration is 0.0125 mM, the CO yield of 4,4′-dipyrenyl-Re is 26 times higher than that of Re(bpy)(CO)3Cl.
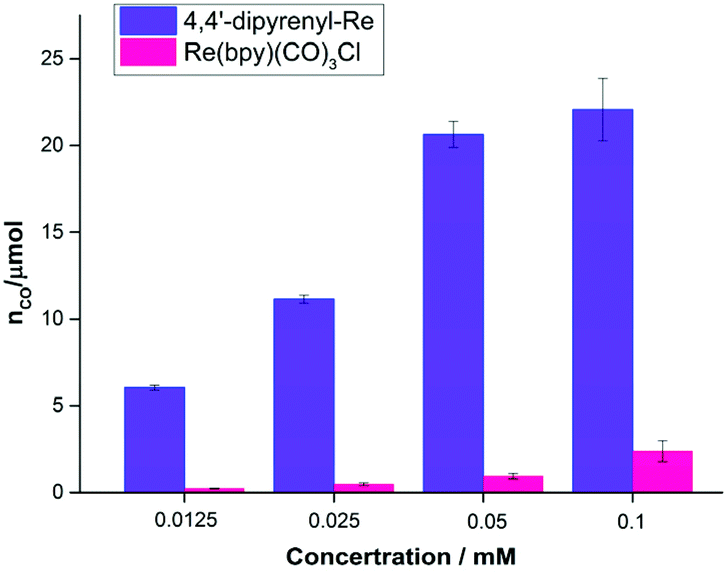 | ||
| Fig. 5 TON for CO production versus catalyst concentration for 4,4′-dipyrenyl-Re and Re(bpy)(CO)3Cl. Conditions: DMF (3 mL), TEOA (0.3 mL), 10 mM BIH; irradiated with a white LED (0.60 mW cm−2). | ||
More importantly, unlike Re(bpy)(CO)3Cl, 4,4′-dipyrenyl-Re exhibits less sensitivity to the concentration. When the concentration is reduced from 0.1 to 0.0125 mM, the yield of CO decreases only by 72.6% for 4,4′-dipyrenyl-Re that is even lower than the reduced concentration ratio (87.5%) (Table S9†).
The effect of fraction of absorbed photons on the molar amount of CO was further explored (Fig. S2B†), we found that Re–pyrene could produce approximately three times more CO than Re(bpy)(CO)3Cl at the same fraction of absorbed photons (such as 0.5), which indicated that the high activity of Re–pyrene may be due to the improvement of intermolecular ET. Thus, increasing the light harvesting ability and intermolecular π–π interaction of the Re catalyst leads to high performance at low concentrations, which is remarkably superior to previously reported molecular systems (Table S13†). In contrast, in the case of Re(bpy)(CO)3Cl, the CO yield is reduced by more than 90% when the concentration is decreased from 0.1 to 0.0125 mM (Table S9†), presumably owing to the impediment of ET and weak absorbance under low catalyst concentrations. A Re complex and another Re molecule interact less frequently and the potential degradation of either species is more competitive at low concentrations.38 In addition, it should be mentioned that the quantum yields of CO formation (ΦCO) are 46.6 ± 3% for 4,4′-dipyrenyl-Re (nCO = 29.08 ± 2 μmol) and 7.3 ± 2% for Re(bpy)(CO)3Cl (nCO = 4.56 ± 1.4 μmol) after irradiation for 0.5 h by a xenon lamp (λ = 420 nm, 0.82 mW cm−2) (Table S10†). These results suggest that 4,4′-dipyrenyl-Re has a significant enhancement in the photocatalytic activity compared to Re(bpy)(CO)3Cl.
Transient absorption spectra were investigated to further explore the mechanism. As shown in Fig. S8A,† 4,4′-dipyrenyl-Re in degassing DMF exhibited a strong bleaching peak at 300–400 nm upon pulsed laser excitation at 410 nm, corresponding to its ground state absorption. 4,4′-Dipyrenyl-Re showed two positive absorption bands at around 500 and 558 nm, and its lifetime was determined as 117.7 μs (Fig. S8D†), which may be attributed to the monomer excited state and the aggregated excited Re–pyrene species. By the addition of BIH, a species of reduced 4,4′-dipyrenyl-Re was formed with a decay of 35.1 μs under an Ar atmosphere (Fig. S8B and S8E†), supporting an electron transfer pathway from BIH to the excited Re–pyrene species. In addition, it is worth noting that reduced 4,4′-dipyrenyl-Re returned quickly to the baseline with a decay of 21.0 μs under a CO2 atmosphere (Fig. S8C and S8F†). Based on above experimental results and previous reports,57,58 the proposed pathway is depicted in Fig. 6. We speculate that the CO2RR most probably initiates via reductive quenching of the excited state of the Re(I) unit with the electron donor (BIH). The π–π stacking interaction, one of the common noncovalent forces, efficiently facilitates intermolecular ET of pyrene moieties of the 4,4′-dipyrenyl-Re complex. Thus, the one-electron-reduced Re-pyrene˙− could be consumed more quickly via intermolecular ET, inhibiting back-electron transfer from the reduced state of Re species to the oxidized state of the sacrificial electron donor, and ultimately improving the photocatalytic efficiency of CO2RR. Intermolecular ET may proceed from the photochemically formed Re-pyrene˙− to another proximal Re–pyrene catalytic center by intermolecular π–π interaction of pyrene units, and CO2RR takes place at the Re center to catalyze CO2-to-CO conversion. In short, π–π interaction and extension of the π-conjugated system dramatically improve ET and visible light-harvesting, thereby leading to efficient photocatalytic performance in solar energy-driven CO2 reduction.
 | ||
| Fig. 6 Proposed pathway for such a photochemical CO2 conversion. | ||
Application under natural conditions
Sunlight-driven CO2 reduction could be promising, but still remains an elusive goal for photocatalytic CO2 reduction. Having identified the superiority of 4,4′-dipyrenyl-Re in visible-light absorption and intermolecular ET, we subjected 4,4′-dipyrenyl-Re and Re(bpy)(CO)3Cl to CO2 reduction under sunlight under different weather conditions (Table S11†). These experiments were conducted near a window inside the laboratory from 11:00 am to 2:00 pm (Fig. 7). We found that sunny sunlight (about 0.75 mW cm−2) and the long-arc xenon lamp (λ > 400 nm, 8.34 mW cm−2) and xenon lamp (λ = 420 nm, 0.82 mW cm−2) give rise to almost the same results for both catalysts, while cloudy sunlight (about 0.32 mW cm−2) performs similar to a white LED. Notably, TONCO for 4,4′-dipyrenyl-Re on a sunny day reaches up to 350 ± 36, but Re(bpy)(CO)3Cl exhibits much lower activity (TONCO = 22 ± 2) under identical conditions. Although performance is reduced on a cloudy day, TONCO for 4,4′-dipyrenyl-Re is 40-fold higher than that for Re(bpy)(CO)3Cl. Then, the reactions for longer time (6 hours) at a Re–pyrene concentration of 0.0125 mM under a white LED light as well as a long-arc xenon lamp were also performed. We found that the amount of CO increased slightly when the reaction time was prolonged driven by white LED light (0.60 mW cm−2) or long-arc xenon lamp (8.34 mW cm−2) (Table S11†). These results suggest that the change of light intensity more/less leads to the decomposition of the Re–pyrene catalyst. Therefore, the main factors affecting photocatalytic efficiency of Re–pyrene catalyst would be light intensity and light absorption performance. | ||
| Fig. 7 Photocatalytic CO2 reduction driven by natural light. CO2 photo-reduction by 4,4′-dipyrenyl-Re (0.0125 mM) and Re(bpy)(CO)3Cl (0.0125 mM) in 3 h driven by different light sources. | ||
To further broaden the application of this method in photocatalytic CO2RR, multicomponent systems using Ru(bpy)3(Cl)2·6H2O (RuBPY) as PS were also investigated. First, without RuBPY, Re(bpy)(CO)3Cl has been deactivated when the concentration is as low as 3.125 μM, while 4,4′-dipyrenyl-Re can still give a TONCO of 76 ± 3 (Fig. 8 and Table S12†). The activities are greatly boosted by additional PS (RuBPY), and 4,4′-dipyrenyl-Re (TONCO = 1367 ± 32) shows higher efficiency than Re(bpy)(CO)3Cl (TONCO = 840 ± 28). Then, the rates of the kinetic process for both systems were determined through Stern–Volmer analysis (Fig. S5 and S6†). It is found that the fluorescence emission of RuBPY was oxidatively quenched by both catalysts with quenching constants (kq) of 3.45 × 1010 M−1 s−1 for 4,4′-dipyrenyl-Re, which is much larger than that for Re(bpy)(CO)3Cl (1.07 × 109 M−1 s−1). Moreover, Stern–Volmer analysis also revealed that BIH quenches RuBPY fluorescence with corresponding quenching rate constants of 5.1 × 109 M−1 s−1 (Fig. S7†). This pathway could be dominant because the concentration of BIH is much greater than that of the Re–pyrene catalyst. These results imply that the mechanism with reductive quenching in this multi-component system would be favourable.
 | ||
| Fig. 8 CO2 photo-reduction by 4,4′-dipyrenyl-Re or Re(bpy)(CO)3Cl (3.125 μM) as catalyst and RuBPY (0.5 mM) as PS in 3 h driven by a long-arc xenon lamp (8.34 mW cm−2). | ||
The ET process also plays a significant role in the electroreduction of CO2. We wonder if our method can be used to improve the efficiency of electrochemical CO2 reduction. First, the redox behaviors of 4,4′-dipyrenyl-Re and Re(bpy)(CO)3Cl were analyzed by CV in DMF solutions under an Ar atmosphere, displaying initial 1e− reduction potentials at −1.69, −1.79 V, respectively (Fig. S9 and Table S15†). By introducing pendant pyrene groups, the reduction potential of 4,4′-dipyrenyl-Re with a 0.1 V positive shift compared to that of Re(bpy)(CO)3Cl, and thus the 4,4′-dipyrenyl-Re complex is more easily reduced. Moreover, the catalytic current response of both Re complexes for electrocatalytic CO2 reduction was investigated under saturated CO2 conditions (Fig. 9). Most notably, the pyrene functionalized catalyst exhibits reductive current enhancement with a higher icat (catalytic peak current)/ip (initial 1e− reduction under Ar conditions) value of 13.6 as compared to Re(bpy)(CO)3Cl (3.3) (Table 1), strongly indicating the remarkable role of the pendant pyrene groups to facilitate ET during CO2 reduction.
 | ||
| Fig. 9 Electrocatalytic performances of rhenium complexes. Cyclic voltammetry of 1 mM 4,4′-dipyrenyl-Re (A) and Re(bpy)(CO)3Cl (B) in argon- and CO2-saturated DMF/0.1 M NBu4PF6 solutions recorded at 0.1 V s−1. | ||
| Catalyst | E cat/2 | η | i cat/ipc | k cat (s−1) |
|---|---|---|---|---|
a The value at half of the catalytic current.
b Overpotentials were determined by the expression  , ,  .59
c Ratio between the peak current under CO2 (icat) and the initial 1e− reduction under argon conditions (icat/ip) at a scan rate of 0.1 V s−1.
d Scan rate is given in parentheses (V s−1). .59
c Ratio between the peak current under CO2 (icat) and the initial 1e− reduction under argon conditions (icat/ip) at a scan rate of 0.1 V s−1.
d Scan rate is given in parentheses (V s−1).
|
||||
| 4,4′-Dipyrenyl-Re | −2.40 | 1.67 | 13.6 | 967 (25) |
| Re(bpy)(CO)3Cl | −2.23 | 1.50 | 3.3 | 150 (5) |
The quantity icat/ip can act as a useful tool for determining catalytic performance by CVs, but this is a rough measure. Ideally, the catalytic CV assumes an “S-shaped” wave independent of the scanning rate, where the platform current can be used to determine the intrinsic catalytic rate constant (kcat). However, a peak-shaped response occurs commonly due to interference of “secondary phenomena” such as the catalyst deactivation or substrate consumption.60 One method to counter these phenomena is to increase the scan rate and thereby reduce the passing charge, so as to get back to an S-shaped current potential response and derive the rate constant kcat (s−1) from the S-shaped current plateau, as shown in eqn (1),61,62 where n′ is the catalyst equivalents required per turnover, n is the number of electrons transferred in the catalytic process, ν is the scan rate, R is the ideal gas constant, and T is the temperature. The kcat (s−1) values are equal to the maximum turnover frequency (TOFmax) under these conditions. For 4,4′-dipyrenyl-Re and Re(bpy)(CO)3Cl complexes through increasing the scan rate to obtain the plateau (S-shaped) current, the kcat (s−1) values are calculated as 967 and 150 s−1, respectively (Fig. 10 and Table 1). Therefore, with faster ET efficiency, the pyrenyl functionalized catalyst results in a higher TOFmax relative to unmodified Re(bpy)(CO)3Cl.
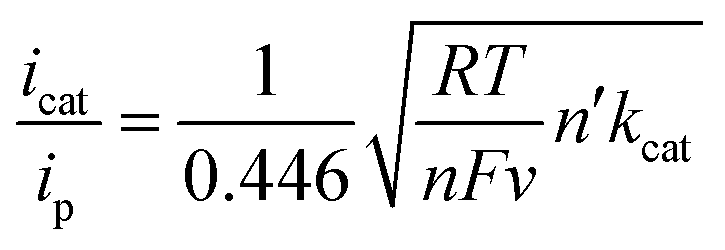 | (1) |
 | ||
| Fig. 10 Determination of the catalytic rate constant. Catalytic scan rate dependence studies for the 4,4′-dipyrenyl-Re (A) and Re(bpy)(CO)3Cl (B) complexes in CO2-saturated DMF/0.1 M NBu4PF6 solutions. | ||
Conclusions
In conclusion, we have designed an easy-to-prepare single-molecule catalyst system comprising a Re complex with covalently linked pyrene groups without additional photosensitizers for natural light-driven photocatalytic CO2 reduction under different weather conditions. A series of experimental, spectroscopic and theoretical studies demonstrate that π–π interactions and extension of the π-conjugated system originating from pyrene groups dramatically improve the electron transfer efficiency and the visible light-harvesting ability. To our delighted, 4,4′-dipyrenyl-Re exhibits high efficiency in the sunny sunlight-driven CO2-to-CO conversion with a high turnover number (TONCO = 350 ± 36) and ΦCO is up to 46.6 ± 3%. To the best of our knowledge, this is the first example to realize CO2RR directly under the sun by using a single-molecule catalyst. More importantly, this methodology can also be applied to multicomponent systems with a remarkable increase in the TONCO of up to 1367 ± 32. In addition, due to the accelerated ET efficiency, pyrenyl functionalized Re catalyst with higher electrocatalytic activity (TOFmax = 967 s−1) toward CO2 reduction compared to Re(bpy)(CO)3Cl. We believe that this strategy will pave the way for designing catalysts in photo- and electro-chemical CO2RR. Our laboratory will pay more attention to novel intermolecular ET related materials using CO2 from flue gas or atmospheric CO263,64 for photocatalytic CO2 reduction.
Experimental section
Materials
N,N-Dimethylformamide (DMF, J&K, 99.8%), 4,4′-dibromo-2,2′-bipyridine (Meryer, 97%), 1-pyrenylboronic acid (Bide, 97%), Pd(PPh3)4 (J&K, 98%), and rhenium pentacarbonyl chloride (Acros Organics, 98%) were commercially available and used without further purification. The solvents (toluene and tetrahydrofuran) used for reactions were distilled under argon after drying over an appropriate drying agent. Argon (purity ≥99.998%) and 12CO2 (purity ≥99.999%) were from Tianjin HAOLUN.Synthesis of Re(bpy)(CO)3Cl and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]-imidazole (BIH) were prepared according to the literature.65,66
Synthesis of 4,4′-di(pyren-1-yl)-2,2′-bipyridine
A mixture of 4,4′-dibromo-2,2′-bipyridine (157 mg, 0.5 mmol), 1-pyrenylboronic acid (270 mg, 1.1 mmol), Na2CO3 (159 mg, 1.5 mmol) and Pd(PPh3)4 (29 mg, 0.025 mmol) in THF/H2O (6 mL/3 mL) was stirred at 75 °C for 12 h under an Ar atmosphere. The reaction mixture was allowed to cool to room temperature, and water (5 mL) and dichloromethane (10 mL) were added. The organic phase was collected, and the solid was suspended in dichloromethane and filtered to give crude 4,4′-di(pyren-1-yl)-2,2′-bipyridine as a pale yellow powder, which was used without further purification. Due to the extremely low solubility of 4,4′-dipyrenyl-2,2′-bipyridine in common NMR solvents, such as CDCl3, CD3CN, DMSO-d6, or C6D6, a detailed spectroscopic NMR investigation was not possible. The precursor 4,4′-di(pyren-1-yl)-2,2′-bipyridine was confirmed by high-resolution mass spectrometry and FT-IR spectroscopy. 4,4′-Di(pyren-1-yl)-2,2′-bipyridine: pale yellow solid (232 mg, 83% yield). HRMS (ESI) Calcd for C24H42N2: 556.1939; Found: 556.2768. IR (KBr) ν(C–H): 3038 cm−1, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N): 1590 cm−1.
N): 1590 cm−1.
Synthesis of Re[4,4′-di(pyren-1-yl)-2,2′-bipyridine](CO)3Cl (4,4′-dipyrenyl-Re)
A mixture of Re(CO)5Cl (18 mg, 0.05 mmol) and 4,4′-di(pyren-1-yl)-2,2′-bipyridine (28 mg, 0.05 mmol) was dissolved in 40 mL toluene solution under an Ar atmosphere. The mixture was refluxed at 110 °C overnight. The resulting mixture was extracted with dichloromethane (3 × 15 mL), dried over anhydrous Mg2SO4, filtered and the solvent was removed in vacuo, and the residue was subjected to column chromatography to afford the pure product (hexane:dichloromethane = 1:2, TLC: Rf = 0.25). Re[4,4′-di(pyren-1-yl)-2,2′-bipyridine](CO)3Cl: orange yellow solid (32 mg, 74% yield). 1H NMR (400 MHz, DMSO) δ 9.35 (s, 2H), 9.26 (d, J = 5.7 Hz, 2H), 8.45 (d, J = 8.0 Hz, 2H), 8.37 (d, J = 14.7 Hz, 2H), 8.37 (s, 2H), 8.30–8.25 (m, 10H), 8.24–8.10 (m, 4H) ppm. HRMS (ESI) Calcd for C45H24N2O3Re (M − Cl)+ 827.1344; Found: 827.1344. IR (KBr) ν(CO): 2017 cm−1, 1910 cm−1, 1888 cm−1. Anal. Calcd for C45H24ClN2O3Re: C, 62.68; H, 2.81; N, 3.25. Found: C, 62.36; H, 3.15; N, 2.87.
Photocatalytic experiments
Photocatalytic CO2 reduction was performed on a 10 mL Schlenk tube containing the Re catalyst, TEOA (0.3 mL), BIH (10 mM), and 3 mL DMF mixed solution under a CO2 atmosphere at room temperature. After the reaction system was purged with CO2 (1 atm) for three freeze–pump–thaw cycles, the reaction mixture was stirred with a magnetic bar and irradiated under a white LED light (9 W) or a long-arc xenon lamp or near a window inside a room under different weather conditions. After the reaction, partial gaseous products (1 mL) were taken from the tube using a syringe, and then detected by gas chromatography (GC, FuLi 9790II) with a TCD detector. The volume of carbon dioxide can be calculated by injecting water into the 10 mL Schlenk tube, and then we used the ideal gas law (PV = nRT) to figure out the moles of CO2 in the Schlenk tube. All the photocatalytic reactions were repeated three times to confirm the reliability of the data. The turnover numbers (TONs) are based on the Re catalysts. Only traces of H2 were detected. In the liquid phase, no formation of HCOOH can be observed by 1H NMR experiments. Products below the detectable limit are labelled as NA (see the ESI†).Determination of the quantum yield
The photon flux was determined by standard ferrioxalate actinometry.67 To determine the photon flux of the xenon lamp (0.82 mW cm−2), the ferrioxalate solution was placed in a cuvette and irradiated at λ = 420 nm. After irradiation, the phenanthroline solution was added to the cuvette and the mixture was allowed to stir in the dark for 1 h to promote the ferrous ions to completely coordinate with phenanthroline. The absorbance of the solution was measured at 510 nm. A nonirradiated sample was also prepared and the absorbance at 510 nm was measured. It is noteworthy that CO formation from CO2 requires two electrons, one-electron-reduced Re-pyrene˙− should be produced by the second excitation. The radical species BI˙ that is produced via deprotonation from the one-electron-oxidized BIH˙+ is also a second-electron donor candidate. Therefore, the reaction path should be a photon absorption, followed by an electron transfer. The quantum yield (Φ) was determined using eqn (2), where the photon flux is 6.93 × 10−8 einstein per s. | (2) |
Single-crystal X-ray analysis and photoelectrochemical measurements
X-ray crystallographic analyses were performed on a Rigaku Saturn 724 CCD diffractometer using graphite-monochromated Mo Kα radiation. UV-visible spectra were recorded at room temperature on a Shimadzu UV-2450 spectrophotometer. Fluorescence spectra were obtained with a Varian Cary Eclipse spectrometer. Emission intensities used for Stern–Volmer analysis were taken at 630 nm, i.e., the emission maximum of Ru(bpy)3(Cl)2·6H2O. Infrared (IR) spectra were recorded on a Bruker Tensor 27 FT-IR spectrophotometer with KBr pellets. Cyclic voltammograms were recorded on BAS Epsilon with a three electrode setup: 3 mm diameter glassy carbon working electrode, non-aqueous reference electrode (Ag/AgNO3 in CH3CN solution), and platinum foil counter electrode. CVs of 1.0 mM catalyst recorded at 0.1 V s−1 in DMF (0.1 M NBu4PF6). Transient absorption spectra were measured on the LP980 laser flash photolysis instrument (Edinburgh, UK). FL lifetime was determined using a time-resolved confocal FL instrument (MicroTime 200, PicoQuant, Berlin, Germany). All potentials were referenced internally at the end of the experiments to the ferrocenium/ferrocene (Fc+/0) couple.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21672119 and 21975135), the National Key R&D Program of China (2016YFA0602900), and the China Postdoctoral Science Foundation (2018M641624). We do greatly appreciate the support from Prof. Tong Bu Lu, Dr Song Guo, and Ping Wang (Tianjin University of Technology) in photoluminescence lifetime and transient absorption measurement.References
- M. Meinshausen, N. Meinshausen, W. Hare, S. C. B. Raper, K. Frieler, R. Knutti, D. J. Frame and M. R. Allen, Nature, 2009, 458, 1158–1162 CrossRef CAS.
- T. R. Karl and K. E. Trenberth, Science, 2003, 302, 1719–1723 CrossRef CAS.
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ACS Catal., 2020, 10, 302–310 CrossRef CAS.
- Q. Guo, S.-G. Xia, X.-B. Li, Y. Wang, F. Liang, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2020, 56, 7849–7852 RSC.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- L. Lu, X.-F. Sun, J. Ma, D.-X. Yang, H.-H. Wu, B.-X. Zhang, J.-L. Zhang and B.-X. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CrossRef CAS.
- X. He, Y. Cao, X.-D. Lang, N. Wang and L.-N. He, ChemSusChem, 2018, 11, 3382–3387 CrossRef CAS.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS.
- Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC.
- M.-Y. He, Y.-H. Sun and B.-X. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS.
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS.
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS.
- I. Dincer and C. Acar, Int. J. Energy Res., 2015, 39, 585–606 CrossRef.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- G. Lan, Z. Li, S. S. Veroneau, Y.-Y. Zhu, Z. Xu, C. Wang and W. Lin, J. Am. Chem. Soc., 2018, 140, 12369–12373 CrossRef CAS.
- M. Zhang, M. Lu, Z.-L. Lang, J. Liu, M. Liu, J.-N. Chang, L.-Y. Li, L.-J. Shang, M. Wang, S.-L. Li and Y.-Q. Lan, Angew. Chem., 2020, 59, 6500–6506 CrossRef CAS.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J.-L. Wang, D.-Q. Yuan and Y.-Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS.
- A. V. Akimov, A. J. Neukirch and O. V. Prezhdo, Chem. Rev., 2013, 113, 4496–4565 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS.
- S. Horiuchi, Y. Okimoto, R. Kumai and Y. Tokura, Science, 2003, 299, 229–232 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS.
- K. V. Rao and S. J. George, Chem. – Eur. J., 2012, 18, 14286–14291 CrossRef CAS.
- D. Hong, T. Kawanishi, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2019, 141, 20309–20317 CrossRef CAS.
- D.-C. Liu, H.-J. Wang, T. Ouyang, J.-W. Wang, L. Jiang, D.-C. Zhong and T.-B. Lu, ACS Appl. Energy Mater., 2018, 1, 2452–2459 CrossRef CAS.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2016, 7, 70–88 CrossRef.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Chem. Commun., 2014, 50, 1491–1493 RSC.
- J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS.
- Y. Yamazaki, K. Ohkubo, D. Saito, T. Yatsu, Y. Tamaki, S. Tanaka, K. Koike, K. Onda and O. Ishitani, Inorg. Chem., 2019, 58, 11480–11492 CrossRef CAS.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS.
- P. L. Cheung, S. C. Kapper, T. Zeng, M. E. Thompson and C. P. Kubiak, J. Am. Chem. Soc., 2019, 141, 14961–14965 CrossRef CAS.
- T. J. Whittemore, C. Xue, J. Huang, J. C. Gallucci and C. Turro, Nat. Chem., 2020, 12, 180–185 CrossRef CAS.
- K.-H. Chen, N. Wang, Z.-W. Yang, S.-M. Xia and L.-N. He, ChemSusChem, 2020 DOI:10.1002/cssc.202000698.
- N. P. Liyanage, W. Yang, S. Guertin, S. S. Roy, C. A. Carpenter, R. E. Adams, R. H. Schmehl, J. H. Delcamp and J. W. Jurss, Chem. Commun., 2019, 55, 993–996 RSC.
- D. R. Whang, D. H. Apaydin, S. Y. Park and N. S. Sariciftci, J. Catal., 2018, 363, 191–196 CrossRef CAS.
- A. Maurin, C.-O. Ng, L. Chen, T.-C. Lau, M. Robert and C.-C. Ko, Dalton Trans., 2016, 45, 14524–14529 RSC.
- C. A. Hunter, J. Mol. Biol., 1993, 230, 1025–1054 CrossRef CAS.
- M. P. Parker, C. A. Murray, L. R. Hart, B. W. Greenland, W. Hayes, C. J. Cardin and H. M. Colquhoun, Cryst. Growth Des., 2018, 18, 386–392 CrossRef CAS.
- W. Wang, J. J. Han, L.-Q. Wang, L.-S. Li, W. J. Shaw and A. D. Q. Li, Nano Lett., 2003, 3, 455–458 CrossRef CAS.
- Y. Liu, W. Hao, H. Yao, S. Li, Y. Wu, J. Zhu and L. Jiang, Adv. Mater., 2018, 30, 1705377 CrossRef.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 CrossRef CAS.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 53, 8709–8713 CrossRef CAS.
- S. Sinha, A. Sonea, W. Shen, S. S. Hanson and J. J. Warren, Inorg. Chem., 2019, 58, 10454–10461 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- M. J. Frisch, et al., Gaussian 09, revision D.01, Gaussian, 2009 Search PubMed.
- F. M. Wisser, M. Duguet, Q. Perrinet, A. C. Ghosh, M. Alves-Favaro, Y. Mohr, C. Lorentz, E. A. Quadrelli, R. Palkovits, D. Farrusseng, C. Mellot-Draznieks, V. de Waele and J. Canivet, Angew. Chem., 2020, 59, 5116–5122 CrossRef CAS.
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS.
- T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS.
- H. Koizumi, H. Chiba, A. Sugihara, M. Iwamura, K. Nozaki and O. Ishitani, Chem. Sci., 2019, 10, 3080–3088 RSC.
- C.-Q. Jiao, Y.-S. Meng, Y. Yu, W.-J. Jiang, W. Wen, H. Oshio, Y. Luo, C.-Y. Duan and T. Liu, Angew. Chem., Int. Ed., 2019, 58, 17009–17015 CrossRef CAS.
- Q.-F. Gu, J.-H. He, D.-Y. Chen, H.-L. Dong, Y.-Y. Li, H. Li, Q.-F. Xu and J.-M. Lu, Adv. Mater., 2015, 27, 5968–5973 CrossRef CAS.
- P. Lang, R. Giereth, S. Tschierlei and M. Schwalbe, Chem. Commun., 2019, 55, 600–603 RSC.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS.
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Inorg. Chem., 2015, 54, 11883–11888 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS.
- M. L. Clark, P. L. Cheung, M. Lessio, E. A. Carter and C. P. Kubiak, ACS Catal., 2018, 8, 2021–2029 CrossRef CAS.
- A. Barthel, Y. Saih, M. Gimenez, J. D. A. Pelletier, F. E. Kühn, V. D'Elia and J.-M. Basset, Green Chem., 2016, 18, 3116–3123 RSC.
- A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS.
- X.-Q. Zhu, M.-T. Zhang, A. Yu, C.-H. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS.
- H. J. Kuhn, S. E. Braslavsky and A. R. Schmidt, Pure Appl. Chem., 2004, 76, 2105–2146 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1954434. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc03111a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |