
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic pathways to tetrahydrocannabinol (THC): an overview
Victor R. L. J.
Bloemendal
 ab,
Jan C. M.
van Hest
b and
Floris P. J. T.
Rutjes
*a
ab,
Jan C. M.
van Hest
b and
Floris P. J. T.
Rutjes
*a
aInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: Floris.Rutjes@ru.nl
bBio-Organic Chemistry, Eindhoven University of Technology, P.O. Box 513 (STO 3.31), 5600 MB Eindhoven, The Netherlands. E-mail: J.C.M.v.Hest@tue.nl
First published on 31st March 2020
Abstract
The therapeutic effects of molecules produced by the plant species Cannabis sativa have since their discovery captured the interest of scientists and society, and have spurred the development of a multidisciplinary scientific field with contributions from biologists, medical specialists and chemists. Decades after the first isolation of some of the most bioactive tetrahydrocannabinols, current research is mostly dedicated to exploiting the chemical versatility of this relevant compound class with regard to its therapeutic potential. This review will primarily focus on synthetic pathways utilised for the synthesis of tetrahydrocannabinols and derivatives thereof, including chiral pool-based and asymmetric chemo- and biocatalytic approaches.
 Bloemendal | Victor R. L. J. Bloemendal received his MSc in Chemistry from Radboud University in 2016, during which he focused on the synthesis of bio-orthogonally functionalized carbohydrates. Currently, he is a PhD-candidate working on a collaboration between Eindhoven University of Technology and Radboud University. As a researcher on the EU-FETOPEN One-Flow project, his goal is to develop one-flow cascade reactions for the synthesis of cannabinoids. |
 van Hest | Jan C. M. van Hest obtained his PhD from Eindhoven University of Technology in 1996 in macro-organic chemistry with prof. E. W. Meijer. He worked as a postdoc with prof. D. A. Tirrell on protein engineering. In 2000 he was appointed full professor in Bio-organic chemistry at Radboud University. As of September 2016 he holds the chair of Bio-organic Chemistry at Eindhoven University of Technology. He is the recipient of an ERC Advanced grant and member of the Royal Netherlands Academy of Arts and Sciences. The group's focus is to develop well-defined compartments for nanomedicine and artificial cell research. |
 Rutjes | Floris P. J. T. Rutjes received his PhD from the University of Amsterdam in 1993 with prof. W. N. Speckamp and H. Hiemstra and conducted postdoctoral research in the group of prof. K. C. Nicolaou at The Scripps Research Institute, La Jolla, USA. In 1995 he was appointed assistant professor in Amsterdam, and in 1999 he became full professor in organic synthesis at Radboud University. Awards include the Gold Medal of the Royal Netherlands Chemical Society (KNCV, 2002), the AstraZeneca award for research in organic chemistry (2003), and most Entrepreneurial Scientist of the Netherlands (2008). He is currently director of the Institute for Molecules and Materials at Radboud University and President-Elect of the European Chemical Society (EuChemS). |
1. Introduction
The plant species Cannabis sativa has captured for centuries the attention of society because of its therapeutic and psychoactive properties. Dating back to 440 B.C., the first literary mention by the philosopher Herodotus was about the use of hemp by the Scythians.1 The plant was processed into clothing fibres and the hemp-seed was thrown on red-hot stones producing vapours for delight and joy. In the following centuries, cannabis remained a useful plant for society and has often been described in literature.2 In the 17th century, cannabis-based therapies were prescribed to treat disorders such as depression, joint pain and epilepsy. Centuries later, in 1964 Mechoulam et al. described the isolation of tetrahydrocannabinols and a partial synthesis of the molecular scaffold.3 This seminal paper sparked more intensive research into cannabinoids and led to the scientific field that we know today. The major phytocannabinoids isolated from Cannabis sativa include cannabidiol (CBD, 5), Δ8- and Δ9-tetrahydrocannabinol (Δ8-THC (9) and Δ9-THC (7), respectively). These compounds have been thoroughly investigated from biological, chemical and medicinal perspectives, and have been employed in several clinical trials. Phytocannabinoids are ligands for a specific class of G-protein-coupled receptors which belong to the endocannabinoid system: cannabinoid receptors 1 and 2 (CB1 and CB2).4,5 In 1975, the first structure–activity relationship studies involving small molecules were conducted on the THC-scaffold to increase affinity and selectivity for both cannabinoid receptors.6,7 Despite intensive research on these receptors, the biological function to date has not yet fully been unveiled, and there are even indications of a third cannabinoid receptor present in mammals.8,9The biosynthesis of phytocannabinoids and structurally related molecules in Cannabis sativa has been described extensively over the past decades.7,10–12 In most cannabinoids a structural similarity can be identified comprising a resorcinol (A-ring) and terpinoid moiety (C-ring, Fig. 1).13 Terpenoids are formed starting from the (non)-mevalonate pathways which produce dimethylallyl pyrophosphate (DMAPP) and isopentenyl pyrophosphate (IPP).14 Both components are coupled by geranyl pyrophosphate synthase to form geranyl pyrophosphate (GPP, 1) which is a precursor for a broad range of terpenoids (Scheme 1). Coupling of olivetolic acid (2) with GPP by geranyl transferase yields cannabigerolic acid (CBGA, 3), which is the biosynthetic starting point for most cannabinoids.3,12Via a subsequent stereoselective ring-closure induced either by CBDA or THCA synthase, cannabidiolic acid (CBDA, 4) and Δ9-tetrahydrocannabinolic acid (Δ9-THCA, 6) are formed, respectively. In addition, numerous other cannabinoid subvariants are formed from CBGA (3) including cannabidiol (CBD, 5), Δ9-tetrahydrocannabinol (Δ9-THC, 7) and cannabinol (CBN, 8). Until now various synthetic cannabinoids have been synthesised and high affinity CB1 and CB2 agonists and antagonists have been identified, which hold considerable promise for pharmaceutical applications.4,5
 | ||
| Fig. 1 Chiral pool and asymmetric catalytic approaches to THC showing the official numbering. | ||
 | ||
| Scheme 1 Biosynthesis of several major cannabinoids from Cannabis sativa. | ||
Although a large collection of synthetically prepared cannabinoids have been reported, the synthetic approaches employed have not yet been systematically reviewed for tetrahydrocannabinol.15 Here we will provide an overview of the most prominent synthetic pathways to the THC scaffold. This is of interest from both a medicinal chemistry viewpoint, as it will shed light on possible new cannabinoid derivatives to be synthesised, but also from a synthetic organic chemistry angle, as the synthesis of the terpenoid C-ring of THCs is especially challenging requiring creative synthetic solutions (Fig. 1).
2. Terpinoid chiral pool approaches
The biosynthesis of THC relies on stereoselective enzymatic conversions using linear, achiral polyketides and alkenyl phosphates. From a synthetic point of view, utilising chiral pool feedstocks is preferred over asymmetric approaches since it avoids typically more complex asymmetric transformations to control the stereochemistry.2.1 Cannabinoid synthesis using (−)-verbenol
The first isolation and structural NMR characterisation of THC and (−)-CBD was reported by Gaoni and Mechoulam in 1964.3 This pioneering research is considered as one of the most influential publications since it sparked the scientific field of cannabinoid chemistry and biology. Previously, Fahrenholtz and co-workers attempted to obtain enantiomerically pure THC, but were unsuccessful in resolving a THC racemate.16,17 Three years after the initial publication by Mechoulam and Gaoni, the first fully stereoselective synthesis of (−)-trans-Δ8-THC was described by Mechoulam, Braun and Gaoni.18 Friedel–Crafts alkylation of olivetol (11) with (−)-verbenol (10), catalysed by p-TSA or boron trifluoride to produce the intermediate allylic cation, provided olivetylverbenyl (12), which after repeated treatment with boron trifluoride provided (−)-trans-Δ8-THC (9) (Scheme 2). A direct approach using Lewis acid catalysis, combining both steps into one, led to similar overall yields of up to 35% of (−)-trans-Δ8-THC (9). Subsequent isomerisation through chlorination and base-induced elimination provided (−)-trans-Δ9-THC (7).19 | ||
| Scheme 2 Mechoulam's synthesis of Δ8- and Δ9-THC using (−)-verbenol (10). | ||
Interestingly, five years later the authors gave a mechanistic rationale describing the allylic carbocation intermediate.20 By using deuterated (−)-verbenol (10) the corresponding labelled (−)-trans-Δ8-THC (9) product was prepared, effectively demonstrating the stereochemistry of the prepared C–C bond. The authors stated that due to the challenging isolation of pure cannabinoids from plants, they considered stereoselective synthesis a preferred approach to obtain single, well-defined cannabinoids in enantiopure form.
Recently, Dethe and co-workers also demonstrated the use of (−)-verbenol (10) in the synthesis of phytocannabinoids (−)-murrayamine-O (16) and (−)-murrayamine-P (17, Scheme 3).21 Previous reports showed that BF3-mediated activation of verbenol leads to the thermodynamic Δ8-isomer.18 In this synthesis, however, the authors describe that with small amounts of BF3·OEt2 and short reaction times the kinetic Δ9-isomer was obtained. This was demonstrated by Friedel–Crafts alkylation of carbazole 13 with verbenol (10) to give the regioisomeric cannabinoids 14 and 15 selectively using different amounts of catalyst. The natural products 16 and 17 were obtained upon m-CPBA oxidation of olefin 15 to give the corresponding epoxide isomers, which were both subsequently ring opened using LiAlH4. In addition, the authors successfully extended this approach to (+)-verbenol leading to ent-murrayamine-O and -P (not shown).
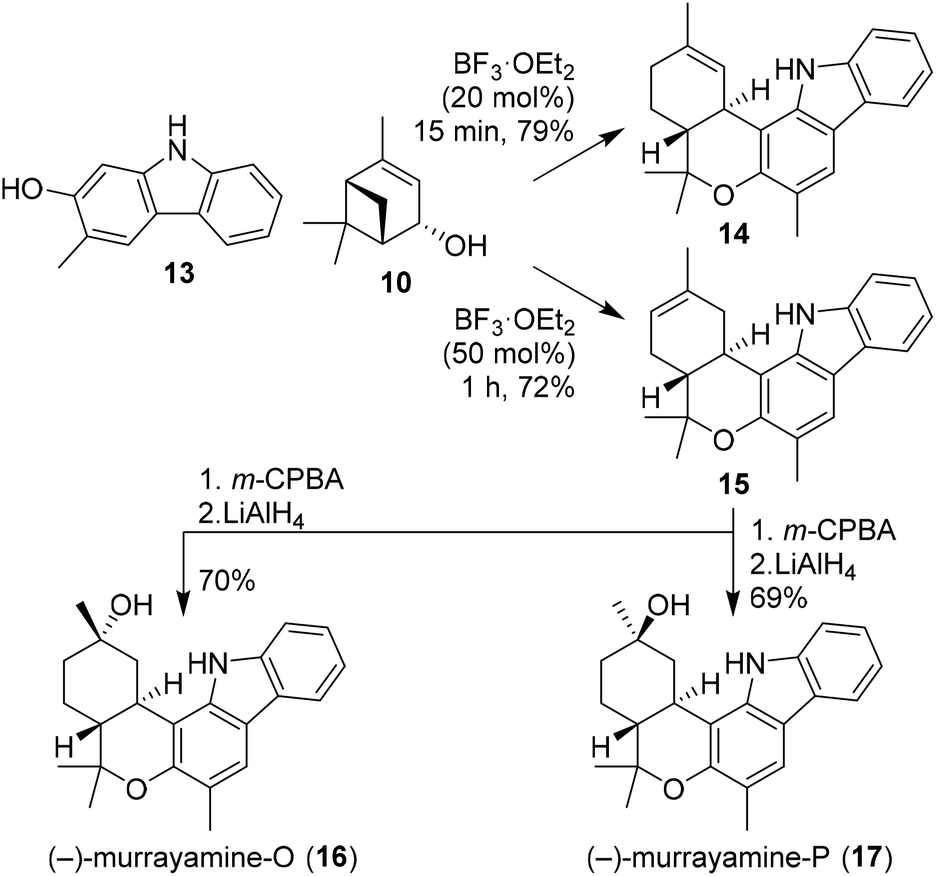 | ||
| Scheme 3 Synthesis of murrayamines-O and -P using (−)-verbenol. | ||
2.2 p-Mentha-2,8-dien-1-ol for both THC isomers
Shortly after the publication of Mechoulam, the use of enantiopure chiral pool terpenoids became common practice in cannabinoid synthesis. Elaborating on an earlier procedure,22 Petrzilka and co-workers reported a stereoselective synthesis of (−)-trans-Δ8-THC (9) starting from p-mentha-2,8-dien-1-ol (18).23 Using this terpenoid, olivetol (11) and p-TSA, electrophilic aromatic substitution provided (−)-trans-CBD (5), which upon renewed exposure to p-TSA cyclised to (−)-trans-Δ9-THC (7, not shown), followed by in situ isomerisation to the thermodynamically more stable (−)-trans-Δ8-THC (9) in 53% yield (Scheme 4A).19 | ||
| Scheme 4 Use of p-mentha-2,8-dien-1-ol by Petrzilka in the synthesis of Δ8-THC (A) and Razdan's approach in the synthesis of Δ9-THC (B). | ||
The successful isolation and direct synthesis of (−)-trans-Δ9-THC (7) was reported shortly thereafter by Razdan and co-workers.24 Instead of the isomerisation of (−)-trans-Δ8-THC (9) to its Δ9-isomer using gaseous hydrochloric acid as shown by Mechoulam, Razdan described a direct synthesis starting from the same starting materials but now using 1% of boron trifluoride as catalyst and magnesium sulfate as drying agent. Besides bis-adducts and iso-THC derivatives, there was no Δ8-THC encountered under these conditions (Scheme 4B). Despite the challenging purification and rather low yield, this pioneering work is still used in the production of (−)-trans-Δ9-THC derivatives because of the high efficiency and mild reaction conditions of the one-pot process.
The stereoselective preparation of Δ8/9-THC scaffolds using p-mentha-2,8-dien-1-ol can also be achieved by using a direct C-ring conjugation and acetate 19. Previous Lewis or Brønsted acid approaches often lacked regiochemical selectivity, were highly sensitive to reaction conditions and afforded many side products.25 In contrast, most C-ring arylation strategies rely on metal-catalysed activation of resorcinol structures and allow for a broad range of aryl substrates.26–29
Rickards and Rönneberg introduced the organometallic synthesis of Δ9-THC brominated analogues (Scheme 5).30 It commenced with the preparation of homocuprate olivetol 20 from the corresponding lithiated olivetol dimethyl ether. Cuprate 20 was coupled in an SN2′-fashion to p-mentha-2,8-dien-1-ol acetate (19) using boron trifluoride etherate and afforded (−)-trans-CBD dimethyl ether 21 in 78% yield. Deprotection of the methyl ethers appeared challenging and was unsuccessful using boron tribromide. Alternatively, the authors proposed to mask the alkene bonds via addition of hydrogen bromide, forming the highly unstable product 22. The brominated isopropenyl moiety unexpectedly underwent mono-demethylation and concomitant formation of the B-ring system at ambient temperature to afford a methyl ether protected THC scaffold. Fortunately, the final demethylation could be completed using boron tribromide, to give halogenated product 23. (−)-trans-Δ9-THC (7) was successfully obtained by following Mechoulam's intermolecular dehalogenation with potassium tert-butoxide (Scheme 2). The authors also investigated a one-pot procedure to convert 21 into 23 without intermediate purification. Compound 21 was subjected to a stoichiometric excess of HBr at −15 °C, then mono-demethylated, cyclised at ambient temperature, and finally deprotected using boron tribromide to afford crude 23. The crude product was subjected to potassium tert-butoxide to form (−)-trans-Δ9-THC in 75% overall yield from (−)-trans-CBD dimethyl ether 21.
 | ||
| Scheme 5 p-Mentha-2,8-dien-1-ol acetate (19) arylation using olivetolic homocuprate (20) as reported by Rickards and Rönneberg. | ||
2.3 p-Menth-2-ene-1,8-diol for 6-hydroxy-CBD and Δ9-THC
In continuation of previous research, Razdan and co-workers investigated the use of other chiral monoterpenoids in the preparation of (−)-trans-Δ9-THC (7). A structurally comparable starting material is p-menth-2-ene-1,8-diol (25), which is activated in a similar fashion using Brønsted or Lewis acid catalysis.31 Conversions of up to 51% were observed and isolated yields of 28% of (−)-trans-Δ9-THC (7) were obtained using anhydrous ZnCl2 instead of the earlier reported boron trifluoride etherate, (Scheme 6).18,24 Notably, with this zinc-based Lewis acid the reactions were successfully conducted on multi-gram scale. Despite the one-step procedures of both p-mentha-2,8-dien-1-ol (18) and p-menth-2-ene-1,8-diol (24), the reaction produced a large variety of side products which limits large scale application.32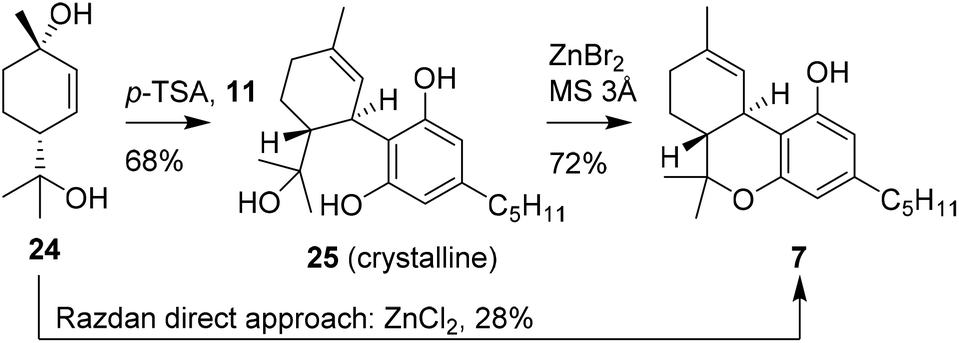 | ||
| Scheme 6 Preparation of Δ9-THC using p-menth-2-ene-1,8-diol (24). | ||
Stoss and Merrath attempted to optimise the synthesis of (−)-trans-Δ9-THC (7) by crystallising the (−)-trans-6-hydroxy-CBD (25) intermediate.32 This crucial step allowed a cleaner cyclisation reaction with ZnBr2 to form (−)-trans-Δ9-THC (7). Despite the two-step sequence, the product was obtained in higher isolated yield, because the final product purification was more efficient. Recently, Cabaj and co-workers published a new synthetic route for the large scale preparation of p-menth-2-ene-1,8-diol (24). This discovery may contribute to industrial application of 24 in the synthetic preparation of cannabinoids.33
2.4 Syntheses using (+)-trans-2- and (+)-trans-3-carene epoxides
Razdan and co-workers additionally investigated other methods to readily prepare enantiopure THC, and were inspired by the seminal work of Ohloff and Giersch.34 By utilising (+)-trans-2-carene epoxide (26), structurally similar to the strained bicyclic (−)-verbenol (10), another one-step synthesis to (−)-trans-Δ9-THC (7) was discovered (Table 1).35 Both boron trifluoride and p-TSA were effective in the activation of carene epoxide, but both catalysts resulted in mixtures of the (−)-trans-Δ9-THC (7) and (−)-cis-Δ9-THC (23) stereoisomers. The observed stereochemical outcome was explained via a carbocation intermediate or concerted pathway by Brønsted or Lewis acid catalysis, respectively. Later, Jamieson and co-workers investigated the mechanism of (+)-trans-2-carene epoxide (21) and compared it with p-mentha-2,8-dien-1-ol (18).36 Interestingly, reactions of 2-carene epoxides (26) with olivetol (11) were lower yielding and produced more byproducts than p-mentha-2,8-dien-1-ol. Reactions using (+)-trans-2-carene epoxides did not afford cannabidiols, and it was hypothesised that the initial Friedel–Crafts alkylation did not occur. The authors postulated a phenolic nucleophilic substitution reaction of olivetol (11) on the cyclopropane moiety of carene, directly followed by an acidic cyclisation reaction.37
Additionally, the isomeric (+)-trans-3-carene epoxide (27) was reacted with p-TSA and the authors observed a p-menthadienol-like pathway.35 The proposed mechanism proceeds through acid-catalysed epoxide ring-opening forming a tertiary cation, followed by elimination to an allylic carbocation.38 The (+)-trans-3-carene (27) only gave (−)-trans-Δ8-THC (9) in low yield, while forming a large variety of side products.39
2.5 cis-Chrysanthenol in the synthesis of (−)-trans-Δ9-THC
Razdan and co-workers also sought to start from a different enantiopure verbenol-based terpenoid, which would allow a direct synthesis of Δ9-THC products. Via a photochemical isomerisation of (−)-verbenone (29)40,41 and subsequent reduction, cis-chrysanthenol (30) was prepared.42 Brønsted or Lewis acid activation of cis-chrysanthenol should lead to the formation of a secondary carbocation for Friedel–Crafts alkylation of olivetol (11). A second activation of the alkene moiety resulted in ring-opening and cyclisation (Scheme 7). Although (−)-trans-Δ9-THC (7) was formed, the reactivity of chrysanthenol was considerably lower than of (−)-verbenol (10) under similar reaction conditions.38 This led the authors to conclude that this particular approach was not the most viable synthetic route towards Δ9-THC. | ||
| Scheme 7 Preparation and conversion of cis-chrysanthenol (30) by Razdan to afford (−)-trans-Δ9-THC (7). | ||
2.6 (+)-Apoverbenone for the preparation of oxidized Δ8/9-THC
Huffman and co-workers demonstrated the use of (+)-apoverbenone (31) to efficiently prepare (±)-trans-11-nor-Δ9-THCs.26 Previous syntheses of oxidised C-ring Δ8/9-THC derivatives were suffering from lack of regio- and stereoselectivity and low yields, and the authors aimed to design a fast, efficient and stereoselective route (Scheme 8A). The synthesis commenced with (+)-apoverbenone (31) which was coupled to lithiated protected olivetol 36, and thereafter directly oxidized to enone 32. Phenolic deprotection, p-TSA-mediated cyclisation and methyl ether deprotection finally afforded enone 33. This was transformed into the corresponding enol triflate to access the oxidised C-ring of Δ9-THCs. Due to the sp2-hybridised alkene in enone 33, the final stereochemistry was installed through a Birch reduction with poor selectivity. Ultimately, triflated (−)-trans and (−)-cis-Δ9-THC derivatives were obtained in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (not shown).29,43 Despite the low stereoselectivity in the apoverbenone route, the resulting building block thus provides straightforward access to oxidised C-ring cannabinoids. Alternatively, the synthesis of enantiomerically enriched oxidised (−)-trans-Δ8-THC (9) using (+)-apoverbenone (31) was published by Tius and Kannangara (Scheme 8B).27 After successfully preparing olivetolic lithiocuprate from ethoxyethyl-protected olivetol (37), (+)-apoverbenone (31) was reacted with the lithiocuprate to form a ketone intermediate. This was then transformed into an enol triflate and deprotected to obtain enol ether 34. Treatment with boron trifluoride etherate led to synthon 35, which is a precursor for C-ring-modified (−)-trans-Δ8-THC derivatives.
:1 ratio (not shown).29,43 Despite the low stereoselectivity in the apoverbenone route, the resulting building block thus provides straightforward access to oxidised C-ring cannabinoids. Alternatively, the synthesis of enantiomerically enriched oxidised (−)-trans-Δ8-THC (9) using (+)-apoverbenone (31) was published by Tius and Kannangara (Scheme 8B).27 After successfully preparing olivetolic lithiocuprate from ethoxyethyl-protected olivetol (37), (+)-apoverbenone (31) was reacted with the lithiocuprate to form a ketone intermediate. This was then transformed into an enol triflate and deprotected to obtain enol ether 34. Treatment with boron trifluoride etherate led to synthon 35, which is a precursor for C-ring-modified (−)-trans-Δ8-THC derivatives.
 | ||
| Scheme 8 (+)-Apoverbenone (31) based preparation of THC-precursors 32 and 34 as demonstrated by Huffman (A) and Tius and Kannangara (B). | ||
2.7 CBD synthesis using (R)-2,9-dibromocamphor
Vaillancourt and Albizati developed a one-step pathway to cannabidiols with a higher efficiency than earlier methods.28 The monoterpenoid (R)-2,9-dibromocamphor (39) was postulated as a suitable starting point, as it would facilitate stereoselective arylation and allow for high regioselectivity. Methoxy-protected olivetol 36 was transformed into homocuprate 38 and reacted with dibromide 39 to form adduct 40 (Scheme 9). Keto–enol isomerisation, ring-opening and subsequent debromination were conducted in one-pot using sodium naphthalenide and diethyl chlorophosphate in a high yield of 89%. The resulting enolphosphate 41 was demethylated and reduced with an excess of lithium in methylamine to form (−)-trans-CBD (5) and its monomethylated CBD counterpart in 35 and 43% yield, respectively.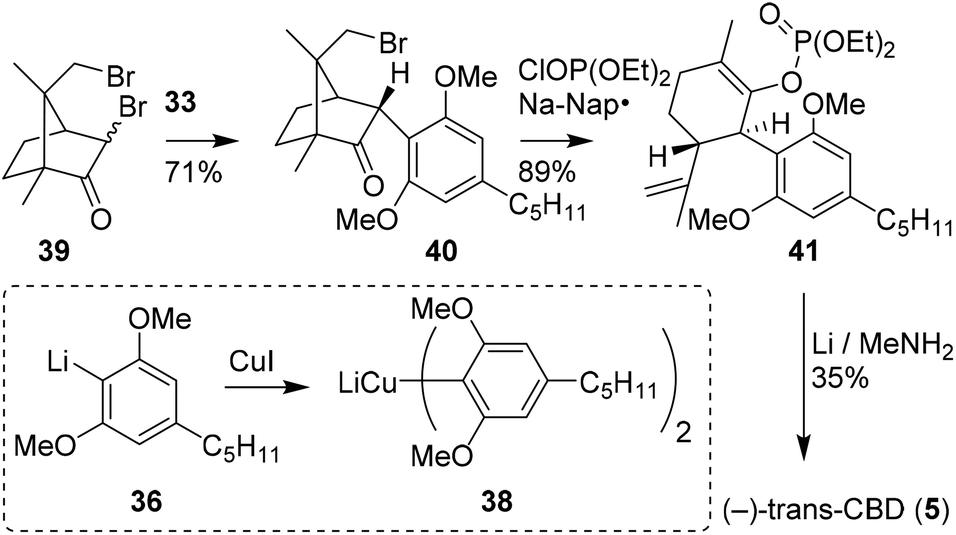 | ||
| Scheme 9 Synthesis of (−)-trans-CBD (5) using 2,9-dibromo-(R)-camphor (39) demonstrated by Vaillancourt and Albizati. | ||
2.8 Carveol for the synthesis of hexahydrocannabinols
Hexahydrocannabinols, such as (+)-machaeriol and (+)-machaeridiol were prepared using aryl cuprates and chiral pool substrates, demonstrated by She and co-workers. In several publications enantiopure silyl enol ether 43 appeared an effective substrate in the preparation of C-ring-substituted cannabinoids (Scheme 10).44–46 The synthesis commenced with epoxidized (+)-cis-carveol ((+)-42) which was oxidised and isomerised to silyl enol ether (+)-43. Initial attempts with sterically hindered resorcinol cuprates 44 were effective in the preparation of cannabinoid-like structures 45 and 46. In a variety of different methods using reductions, deoxygenations and Lewis acid cyclisation reactions, (+)-machaeridiol B (47), (+)-machaeriol B (48), (+)-machaeriol D (49), and other aryl-substituted hexahydrocannabinols were obtained.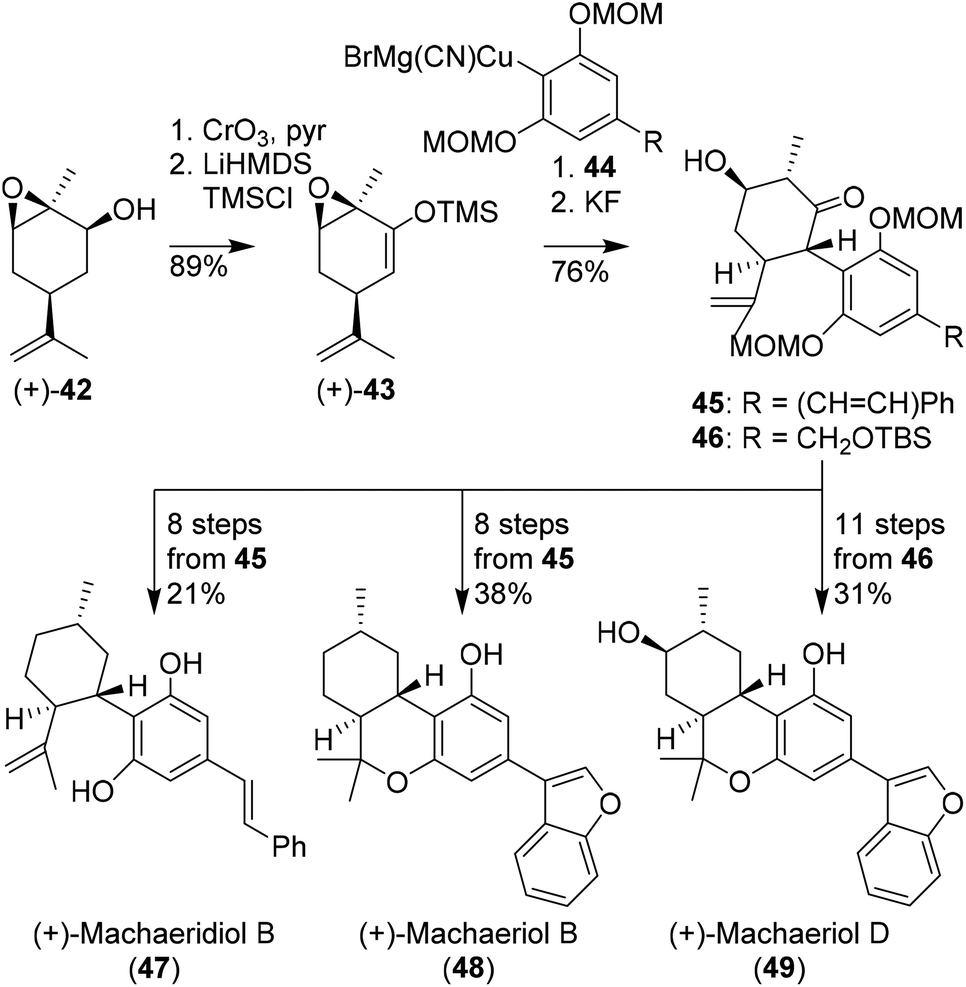 | ||
| Scheme 10 Synthesis of enantiopure machaeridiol B, machaeriol B and machaeriol D (47, 48, 49, respectively) reported by She and co-workers. | ||
Additionally, She and co-workers aimed to prepare (−)-trans-Δ8-THC to demonstrate the versatility of silyl enol ether 43 (Scheme 11).46 By using MOM-protected olivetolic cuprate 44 and (−)-43, the authors were successful in obtaining C-ring oxidised cannabinoid 50 after acidic hydrolysis. Subsequent keto reduction, radical Barton–McCombie deoxygenation and acidic cyclisation reaction afforded (−)-trans-Δ8-THC (9).
 | ||
| Scheme 11 Synthesis of (−)-trans-Δ8-THC (9), starting from (−)-carveol, demonstrated by She and co-workers. | ||
2.9 α-Iodocyclohexenones
Kobayashi and co-workers revisited the preparation of (−)-trans-Δ9-THC and (−)-trans-CBD using aryl cuprates. The authors aimed to use α-iodocyclohexenones and a variety of resorcinols and unveil the arylation of these cyclohexenones (Scheme 12).47 They observed that 1,4-addition of bulky diaryl cuprates to enones was only successful in the presence of BF3OEt2. The stereochemistry of α-iodocyclohexenone 52 facilitated the exclusive formation of trans-configured arylated products. After preparing α-iodoketone 52 from 51, the product was transformed into a dehalogenated enol Grignard, which opened synthetic strategies towards various 9′ derivatisations (not shown). Thereafter, the reactive enol Grignard intermediate was transformed into the corresponding enol phosphate 53. A sequence of methylation with MeMgBr in the presence of Ni(acac)2, methyl ether deprotection using sodium ethanethiolate and cyclisation with ZnBr2 afforded (−)-trans-Δ9-THC (7) in 28% yield. Notably, the 1,4-arylation to enones was reported and optimised a year later.48 This invaluable publication broadened the aryl substrate scope, led to new C-ring oxidised substrates, and the synthesis of α-iodocyclohexenones using (+)-β-pinene. | ||
| Scheme 12 Kobayashi's boron trifluoride-promoted arylation using α-iodocyclohexenones and diaryl cuprates. | ||
The successful stereoselective route to tetrahydrocannabinol using α-cyclohexenone was extended to (−)-trans-CBD (5), C-ring oxidised derivatives, and even alkenyl derivatives on the B-ring (Scheme 12).49 The route started from cyclohexenediol monoacetate 54, which appeared more versatile and allowed for more facile preparation of different α-cyclohexenones. The alkylation of monoacetate 54 was investigated using various reagents, additives, and catalysts. Isopropenylmagnesium bromide, zinc chloride, TMEDA and NiCl2(tpp)2 were shown to be most reactive, resulting in yields of up to 95% with minimal regioisomeric sideproducts. Subsequent Jones oxidation and iodination afforded 55. The consecutive coupling of diaryl cuprate 38, transformation to enol Grignard and final coupling to diethyl chlorophosphate afforded 56, the precursor of (−)-trans-CBD (5).
The same group also published direct 1,4-arylation of diaryl cuprates onto non-iodinated cyclohexenones, promoted solely by boron trifluoride.50 This direct approach is considered advantageous for organic synthesis, but synthetically challenging because of the strongly stabilised boron enolate. To circumvent the low reactivity, MeLi was used to create the lithio enolate intermediates which are more reactive towards electrophiles. To demonstrate the principle, cyclohexenone 51 was activated with boron trifluoride, arylated with 38, transformed into the enolate using MeLi, and finally phosphorylated using diethyl chlorophosphate to afford 53.
3. Concerted approaches
The earliest approaches to the tetrahydrocannabinol scaffold were based on chiral pool syntheses and hence afforded enantiopure products, albeit often with formation of side products and in moderate to low yields. Alternatively, more elaborate synthetic pathways using asymmetric catalysts were employed, leading to high levels of enantioselectivity and additional access to unnatural THC enantiomers. Synthesis of unsaturated six-membered rings can be achieved using Diels–Alder [4 + 2] cycloadditions, which have also been used to prepare cannabinoids.51 The earliest report of a Diels–Alder cyclisation in the preparation of CBD was by Korte, Dlugosch and Claussen in 1966.52 They conducted an intermolecular reaction with methyl vinyl ketone and dimethyl ether-protected 3-methyl-1,3-butadiene-olivetol. Without the use of any chiral catalyst present, an unseparable complex mixture of all possible CBD analogues was obtained.53,54The first asymmetric synthesis of the unnatural isomer (+)-trans-Δ9-THC (59) using a Diels–Alder reaction was reported by Evans, Shaughnessy and Barnes (Scheme 13).55 Key-step was the synthesis of the acetylated cycloadduct 57 on 50 mmol scale. Employing the enantiopure Cu(II)-bisoxazoline catalyst C1 led to an endo-transition state, resulting in a trans-configuration of crystalline cycloadduct 57.56–58 Transesterification into the benzyl ester, nucleophilic methylation and ester hydrolysis provided p-menth-1-ene-3,8-diol (58). Activation of the diol using p-TSA in the presence of olivetol (11) resulted in the cannabidiol analogue which was cyclised using anhydrous ZnBr2 to obtain (+)-trans-Δ9-THC (59) in modest yield. Two years later, the key-step Diels Alder cyclisation catalysed by C1 was further investigated in a mechanistic study with a significant larger substrate scope.59
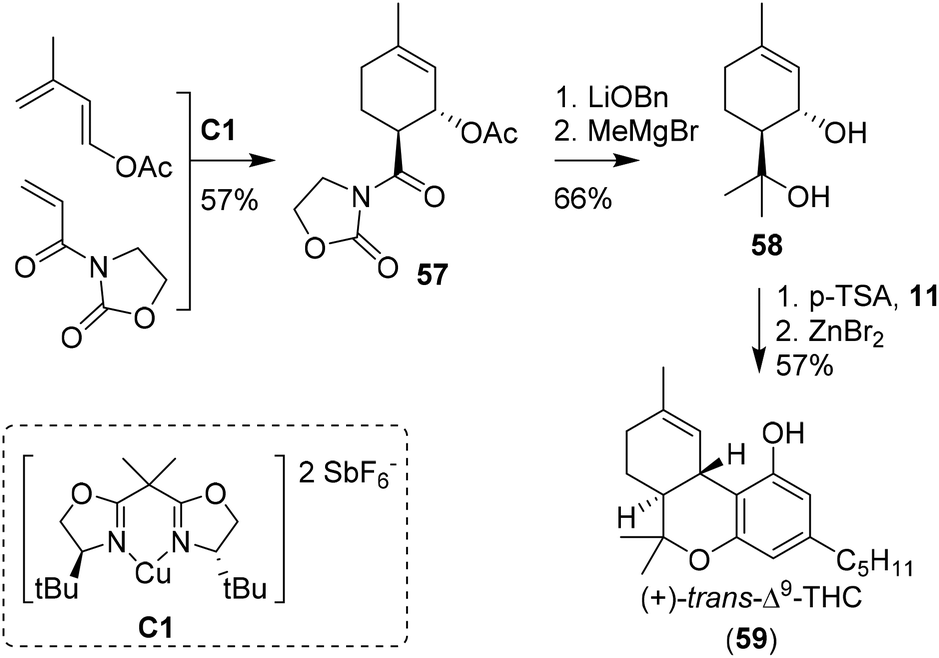 | ||
| Scheme 13 Seminal work of Evans, Shaughnessy and Barnes utilising a Diels–Alder cyclisation to form cycloadduct 57. | ||
Subsequent seminal work of Avery and co-workers represented the first entry into a direct synthesis of the B- and C-rings of hexahydrocannabinol derivatives through a Diels–Alder cyclisation reaction (Scheme 14).60 Though this approach utilises (S)-citronellal, a chiral pool terpinoid, a concerted double cyclisation approach was required to provide the tricyclic cannabinoid scaffold. Olefin 61 was obtained as a 1:1 mixture of diastereomers via the reaction of lithiated methoxymethyl ether (MOM) protected triol 60 with (S)-citronellal. The following hetero-Diels–Alder cyclisation reaction surprisingly proceeded using 4% aqueous HCl in methanol at ambient temperature in moderate yield, while normally these reactions are conducted at elevated temperatures. The stereoselectivity of the cyclisation reaction was explained by invoking intermediate 62, and afforded tricyclic scaffold 63. The subsequent phenolic triflation provided hexahydrocannabinol 64, suitable for further functionalisation. To ensure successful palladium-catalysed cross-coupling, Avery and co-workers first protected the phenolic position with a MOM group. Finally, hexahydrocannabinols (+)-machaeriol A and B (65 and 66, respectively) were obtained via Suzuki–Miyaura cross-coupling reactions and subsequent hydrolysis. Notably, Avery and co-workers did not comment on the stereospecificity, and specific rotations were not described.61 Nevertheless this pioneering work demonstrated the possibility to perform late-stage derivatisation of a cannabinoid scaffold.
 | ||
| Scheme 14 First stereoselective synthesis of (+)-machaeriol A (65) and B (48) based on a Diels–Alder cyclisation as demonstrated by Avery and co-workers. | ||
The group of Sherburn demonstrated the synthesis of Δ9-THC with a late stage trans-selective Diels–Alder cyclisation.62 To be able to optimise this reaction towards the desired trans-product it was first investigated in silico. DFT calculations showed that the stereoselectivity is heavily influenced by conjugative effects between the diene and the aromatic ring substituents. The method was tested using aluminium tris(2,6-diphenylphenoxide) (ATPH) as Lewis acid catalyst on olefins 66 and 67.63 In contrast to earlier DFT calculations, ATPH dramatically inversed stereoselectivity to afford 68 and 69 in moderate trans-diastereoselectivity (Scheme 15). 69 was finally treated with methylmagnesium chloride and cyclised using ZnBr2 to obtain (−)-trans-Δ9-THC (7).47,48
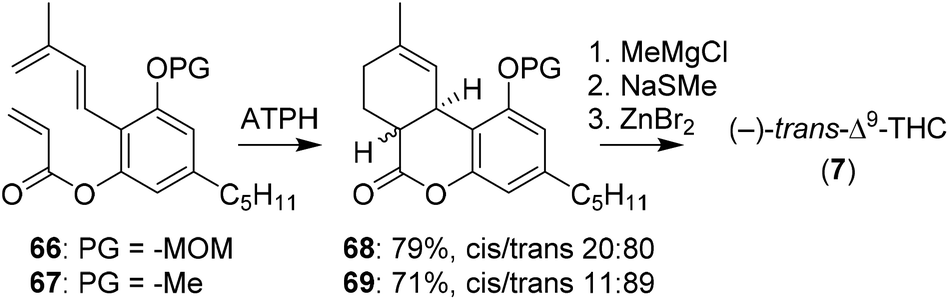 | ||
| Scheme 15 Sherburn and co-workers used a trans-selective Diels–Alder cyclisation for the preparation of (−)-trans-Δ9-THC (7). | ||
Minuti and co-workers applied late stage hyperbaric Diels–Alder reactions giving rise to a variety of Δ8-THC derivatives.64 The authors performed this reaction with six benzylidene acetone dienophiles (70) and two diene coupling partners (71a, 71b) promoted by mildly Lewis acidic conditions. This resulted in excellent yields, full trans-stereoselectivity, however, with additional formation of a regioisomeric product. The hyperbaric Diels–Alder reaction was applied to 1-olivetolic(alkylidene)acetone and afforded inseparable (+)- and (−)-trans-configured bicyclic products 72 (Scheme 16, top route). The bicyclic intermediate was subsequently treated with MeMgBr, selectively deprotected with NaSMe, cyclised using ZnBr2 and finally deprotected with additional NaSMe to afford rac-Δ8-THC. The authors resolved intermediate (±)-72via (S)-(−)-1-amino-2-(methoxymethyl)pyrrolidine (SAMP) resolution, separated the diastereoisomers and followed with a subsequent Brønsted acid-mediated hydrazone hydrolysis. After obtaining both (R,R)-(−)-72 and (S,S)-(+)-72 the intermediate products were transformed into the corresponding diastereomerically pure (−)- and (+)-trans-Δ8-THCs (9 and 76, respectively).
 | ||
| Scheme 16 Minuti's Diels–Alder approaches in the preparation of trans-Δ8-and Δ9-THC and derivatives. | ||
The influence of solvents in the Diels–Alder reaction and the preparation of Δ9-THC and derivatives was also demonstrated later. The previously used aryl alkene substituent 70 was transformed into dienophile 73 using a Wittig olefination, and reacted with alkene65 and alkyne66 coupling partners in a hyperbaric Diels–Alder cyclisation reaction (Scheme 16, bottom route). The optimal results with methyl vinyl ketone (74a) or methyl acrylate (74b) were obtained in ethanol at 9 kbar providing olefin 75 in high cis-selectivity. This result is in strong contrast with the previous trans-selective Diels–Alder reaction (70 to 72). Therefore, the authors investigated a small variety of dienophiles, which were even less stereoselective. To demonstrate the applicability of the system, both (±)-cis- and (±)-trans-Δ9-THC were prepared via the Diels–Alder approach using the endo- and exo-cycloadduct of 75, respectively. Additionally, the single (+)- and (−)-enantiomers of trans-Δ9-THC were obtained via the SAMP-hydrazone route (59 and 7, respectively). Despite the laborious and low yielding approach, all stereoisomers of Δ8- and Δ9-THCs were obtained, thereby widening the scope to prepare synthetic cannabinoid derivatives.
4. Other approaches
A different approach to create the BC-ring system of cannabinoids proceeds via pericyclic reactions. The first intramolecular cyclisation reaction was shown by Childers and Pinnick. Using a Claisen rearrangement at room temperature, the authors obtained mixtures of (±)-trans-Δ9-THCs. Another approach was reported by Kirschleger and co-workers who published a one-step Brønsted acid-mediated double cyclisation giving rise to (±)-cis/trans-Δ9-THCs.67 Alternatively, the synthesis of (±)-cis-11-nor-Δ9-THC-9-carboxylic acid was reported by Tius and Gu, using a similar key-step cationic cyclisation reaction.68 Despite the low stereoselectivity of these concise synthesis routes, they offer fast access to new pharmacologically relevant cannabinoids and aid in the development of new potential drugs.11,69One of the most recent stereoselective cyclisations was the preparation of (−)-perrottetinene (86), a phenethyl-substituted (−)-cis-Δ9-THC analogue. Kim and co-workers synthesised the cannabinoid using a key-step Ireland–Claisen rearrangement (Scheme 17A).70 Dihydropinosylvin (77) was regioselectively iodinated, and phenol protected to afford aryl iodide 78. The following Stille cross-coupling with tributylstannyl (S)-methyl vinyl carbinol (79) led to the isolation of 80, which was subsequently coupled with 5-methylhex-5-enoic acid (81) to obtain olefin 82. The following Ireland–Claisen rearrangement was conducted using LDA and TBSCl resulting in a silyl enol ether intermediate. The addition of HMPA enhances formation of the (Z)-configured silyl enol ether, which then rearranges through the chair-like transition state 83 to form the (S,R)-configured product 84 with high diastereoselectivity (>20:1). Precursor 85 was formed after esterification, treatment with MeMgBr and Grubbs olefin metathesis. (−)-Perrottetinene (86) was obtained after p-TSA-mediated cyclisation.
 | ||
| Scheme 17 Ireland–Claisen rearrangement by Kim and co-workers to afford (−)-perrottetinene (86), and the inversed Ireland–Claisen rearrangement by Leahy and co-workers to afford (−)-trans-Δ9-THC (7). | ||
The Ireland–Claisen rearrangement by Kim70 and the molybdenum-catalysed allylic alkylation reaction by Trost and Dogra,71 inspired Leahy and co-workers to design a chemoenzymatic synthesis of (−)-trans-Δ9-THC (7, Scheme 17B).72 The synthesis followed the methylation and formylation of olivetol 11 described by Trost and Dogra,71 but was then followed by an acetone aldol condensation reaction to afford the enone-substituted product 87. The enone was reduced using the Corey–Bakshi–Shibata oxazaborolidine, or via a sequence of NaBH4 reduction, vinyl butyrate acylation in presence of Savinase 12T and basic hydrolysis to obtain 88. Ester 89 was produced by coupling 88 with 5-methylhex-5-enoic acid (83) in a Steglich DCC coupling. Critically, as opposed to Kim's approach, the absence of HMPA resulted in formation of the (E)-configured silyl enol ether leading to the formation of chair-like intermediate 90. Ireland–Claisen rearrangement then afforded the (R,R)-configured product 91. Leahy demonstrated the versatility of 91 by transforming it into CBD or (−)-trans-Δ9-THC. CBD was prepared using a sequence of Grubbs olefin metathesis, Wittig olefination with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 and final deprotection and methylation using MeMgI (not shown). (−)-trans-Δ9-THC (7) was formed using an esterification, ring-closing metathesis, deprotection using MeMgI and a final Lewis acid-based cyclisation with ZnBr2.47,48,59
CH2 and final deprotection and methylation using MeMgI (not shown). (−)-trans-Δ9-THC (7) was formed using an esterification, ring-closing metathesis, deprotection using MeMgI and a final Lewis acid-based cyclisation with ZnBr2.47,48,59
Previously, the SN2 arylation of α-iodocyclohexanone was demonstrated by Kobayashi, in the preparation of (−)-trans-Δ9-THC and -CBD. Alternatively, Zhou and co-workers later found that α-iodocyclohexanones are effective candidates in Suzuki–Miyaura cross-coupling reactions (Scheme 18).73 The key step in the publication is a high-pressure asymmetric ketone reduction with ruthenium catalyst (Sa,R,R)-C2. Following one of their earlier publications,74 the authors set out to investigate the reduction of rac-2,6-dimethoxyphenylcyclohexanone, which appeared synthetically challenging due to the steric hindrance caused by the two ortho-methoxy groups. Fortunately, by changing one methoxy into a fluoride atom, the reaction proceeded more efficiently. The chemistry was applied in the synthesis of both (−)-trans-Δ9- and Δ8-THC isomers. Initially, the Suzuki–Miyaura cross-coupling of α-iodocyclohexenone 94 with boronic acid 93 and hydrogenation on Pd/C provided racemic 95. The crucial high-pressure asymmetric ketone reduction with (Sa,R,R)-C2 and concomitant racemisation were applied to substrate 95 to afford (−)-cis-configured alcohol 96. To obtain the target trans-configured cannabinoid scaffolds, a sequence of saponification, Wittig olefination and basic olefination was employed. From product 96 still 10 steps were required to produce both the (−)-trans-Δ9- and Δ8-THC isomers, in a total yield of 43 and 51%, respectively.
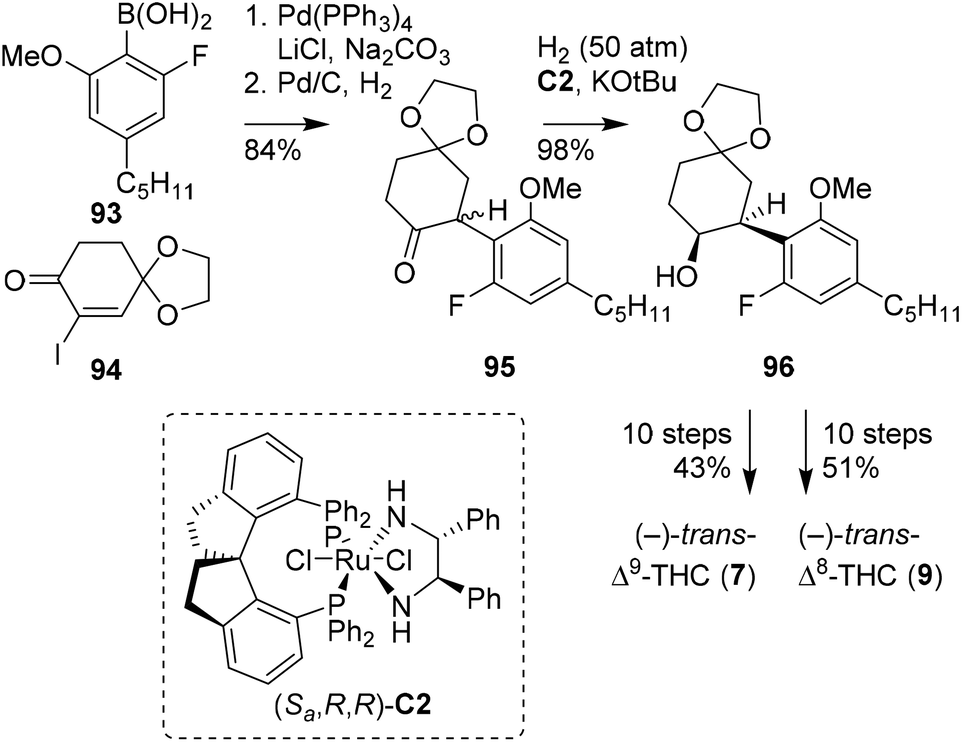 | ||
| Scheme 18 Suzuki–Miyaura cross-coupling and asymmetric hydrogenation reaction in the preparation of (−)-trans-THCs, reported by Zhou and co-workers. | ||
The group of Lupton has investigated an NHC-catalysed (4 + 2) annulation reaction for the enantioselective synthesis of β-lactones.75 Determined to extend the method to medicinal chemistry and natural product synthesis, Ametovski and Lupton investigated the applicability of the annulation reaction in the formation of THC scaffolds (Scheme 19).76 Starting from acyl fluoride 97, earlier reported cyclobutane 98 and NHC catalyst C3, the authors were delighted to directly obtain lactone 99 in excellent enantioselectivity (er 98:2). Compound 99 was saponified with KCN, oxidized with IBX, subjected to Krapcho decarboxylation and isomerised to afford (−)-trans-configured scaffold 100. Finally, by using known procedures, the authors were able to obtain both (−)-trans-Δ8- and Δ9-THC.
 | ||
| Scheme 19 NHC-catalysed (4 + 2) annulation reaction to afford cannabinoid scaffold 99, demonstrated by Ametovski and Lupton. | ||
5. Outlook and conclusion
In conclusion, we have outlined several chiral pool and asymmetric catalytic approaches to THC and derivatives. Chiral pool approaches generally provide a fast entry into cannabinoids, but the final product mixtures are challenging to purify, have a limited substrate scope and may lack regioselectivity. Alternatively, asymmetric catalysis excels in substrate scope, high regio- and stereoselectivity, but requires more elaborate synthesis routes.The published approaches are generally well applicable and already provided several potential drug candidates. However, more research is required to afford straightforward and efficient access to new cannabinoid derivatives, as many examples are based on multistep organic transformations. A recent effective approach to obtain synthetic cannabinoids was published by Westphal and co-workers, who demonstrated the late stage derivatisation of a functionalised (−)-trans-Δ9-THC scaffold.77 Currently, this late-stage approach is attractive to efficiently prepare synthetic cannabinoid derivatives and is a hot topic in the scientific field.78–80 Alternatively, the groups of Carreira and Studer have disclosed stereodivergent syntheses of cannabinoids, affording a variety of cannabinoids by varying catalyst ligands.81–83 This latter approach may provide fast access into more stereodiverse cannabinoids.
Without question, organic synthesis is most suitable for the preparation of biologically relevant molecules, but there is still a need for more efficient preparation techniques. At this moment, only one continuous flow approach has been applied in the synthesis of ortho-substituted cannabinoids.84 Generally, the current preparation of cannabinoids and pharmaceuticals such as Nabilone occurs via batch chemistry,27,68,85 but may well be changed to flow processes because of the excellent scalability, safety and efficiency.
Besides the synthetic effort to obtain facile access to new cannabinoid derivatives, another goal is to elucidate the biological mechanisms of the endocannabinoid system. Despite numerous publications in literature, it is still unclear how the CB1, CB2 (and possibly CB3) receptors interact in various diseases.4–9 The receptors have shown to play a key role in various health conditions such as nausea during chemotherapy, (neuropathic) pain, multiple sclerosis and Tourette syndrome.
The preparation of new and specialised cannabinoids might help to further elucidate the biological mode of action of these receptors and enable treatment of these and other diseases. Clearly, the multidisciplinary scientific field of cannabinoids, with the continuous development of new synthetic methods, remains to hold considerable promise for the development of novel pharmaceuticals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We kindly acknowledge funding by the H2020-FETOPEN-2016-2017 programme of the European Commission (Grant agreement number: 737266-ONE FLOW).Notes and references
- J. L. Butrica, J. Cannabis Ther., 2002, 2, 51–70 CrossRef.
- D. T. Brown, Cannabis: the genus cannabis, CRC Press, 2003 Search PubMed.
- Y. Gaoni and R. Mechoulam, J. Am. Chem. Soc., 1964, 86, 1646–1647 CrossRef CAS.
- T. Hua, K. Vemuri, M. Pu, L. Qu, G. W. Han, Y. Wu, S. Zhao, W. Shui, S. Li and A. Korde, Cell, 2016, 167, 750–762 CrossRef CAS PubMed.
- X. Li, T. Hua, K. Vemuri, J.-H. Ho, Y. Wu, L. Wu, P. Popov, O. Benchama, N. Zvonok, K. a. Locke, L. Qu, G. W. Han, M. R. Iyer, R. Cinar, N. J. Coffey, J. Wang, M. Wu, V. Katritch, S. Zhao, G. Kunos, L. M. Bohn, A. Makriyannis, R. C. Stevens and Z.-J. Liu, Cell, 2019, 176, 459–467 CrossRef CAS PubMed.
- E. W. Bow and J. M. Rimoldi, Perspect. Med. Chem., 2016, 8, S32171 CrossRef PubMed.
- C. E. Turner, M. A. Elsohly and E. G. Boeren, J. Nat. Prod., 1980, 43, 169–234 CrossRef CAS PubMed.
- E. Ryberg, N. Larsson, S. Sjogren, S. Hjorth, N. O. Hermansson, J. Leonova, T. Elebring, K. Nilsson, T. Drmota and P. J. Geasley, Br. J. Pharmacol., 2007, 152, 1092–1101 CrossRef CAS PubMed.
- J. E. Lauckner, J. B. Jensen, H. Y. Chen, H. C. Lu, B. Hille and K. Mackie, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2699–2704 CrossRef CAS PubMed.
- S. J. Gagne, J. M. Stout, E. Liu, Z. Boubakir, S. M. Clark and J. E. Page, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12811–12816 CrossRef CAS PubMed.
- B. Yagen and R. Mechoulam, Tetrahedron Lett., 1969, 10, 5353–5356 CrossRef.
- M. Kajima and M. Piraux, Phytochemistry, 1982, 21, 67–69 CrossRef CAS.
- L. O. Hanuš, S. M. Meyer, E. Muñoz, O. Taglialatela-Scafati and G. Appendino, Nat. Prod. Rep., 2016, 33, 1357–1392 RSC.
- G. A. Thakur, R. I. Duclos Jr. and A. Makriyannis, Life Sci., 2005, 78, 454–466 CrossRef CAS PubMed.
- B. Jung, J. K. Lee, J. Kim, E. K. Kang, S. Y. Han, H.-Y. Lee and I. S. Choi, Chem.– Asian J., 2019, 14, 3749–3762 CrossRef CAS PubMed.
- K. Fahrenholtz, M. Lurie and R. Kierstead, J. Am. Chem. Soc., 1966, 88, 2079–2080 CrossRef CAS.
- K. E. Fahrenholtz, M. Lurie and R. W. Kierstead, J. Am. Chem. Soc., 1967, 89, 5934–5941 CrossRef CAS.
- R. Mechoulam, P. Braun and Y. Gaoni, J. Am. Chem. Soc., 1967, 89, 4552–4554 CrossRef CAS PubMed.
- R. Razdan, A. Puttick, B. Zitko and G. Handrick, Experientia, 1972, 28, 121–122 CrossRef CAS PubMed.
- R. Mechoulam, P. Braun and Y. Gaoni, J. Am. Chem. Soc., 1972, 94, 6159–6165 CrossRef CAS PubMed.
- D. H. Dethe, S. Das, B. D. Dherange and S. Mahapatra, Chem. – Eur. J., 2015, 21, 8347–8350 CrossRef CAS PubMed.
- T. Petrzilka and C. Sikemeier, Helv. Chim. Acta, 1967, 50, 1416–1419 CrossRef CAS PubMed.
- T. Petrzilka, W. Haefliger and C. Sikemeier, Helv. Chim. Acta, 1969, 52, 1102–1134 CrossRef CAS.
- R. K. Razdan, H. C. Dalzell and G. R. Handrick, J. Am. Chem. Soc., 1974, 96, 5860–5865 CrossRef CAS PubMed.
- R. Razdan, in Total Synthesis of Natural Products, Wiley, New York, 1981, vol. 4, pp. 185–262 Search PubMed.
- J. W. Huffman, X. Zhang, M. J. Wu and H. H. Joyner, J. Org. Chem., 1989, 54, 4741–4743 CrossRef CAS.
- M. A. Tius and G. K. Kannangara, J. Org. Chem., 1990, 55, 5711–5714 CrossRef CAS.
- V. Vaillancourt and K. F. Albizati, J. Org. Chem., 1992, 57, 3627–3631 CrossRef CAS.
- M. A. Tius, in Studies in Natural Products Chemistry, Elsevier, 1996, vol. 19, pp. 185–244 Search PubMed.
- R. W. Rickards and H. Rönneberg, J. Org. Chem., 1984, 49, 572–573 CrossRef CAS.
- G. Handrick, D. Uliss, H. Dalzell and R. Razdan, Tetrahedron Lett., 1979, 681–684 CrossRef CAS.
- P. Stoss and P. Merrath, Synlett, 1991, 553–554 CrossRef CAS.
- J. E. Cabaj, J. M. Lukesh, R. J. Pariza and P. M. Zizelman, Org. Process Res. Dev., 2009, 13, 358–361 CrossRef CAS.
- G. Ohloff and W. Giersch, Helv. Chim. Acta, 1968, 51, 1328–1342 CrossRef CAS.
- R. K. Razdan and G. R. Handrick, J. Am. Chem. Soc., 1970, 92, 6061–6062 CrossRef CAS PubMed.
- L. Crombie, W. M. L. Crombie, C. J. Palmer and S. V. Jamieson, Tetrahedron Lett., 1983, 24, 3129–3132 CrossRef CAS.
- D. Uliss, R. Razdan, H. Dalzell and G. Handrick, Tetrahedron, 1977, 33, 2055–2059 CrossRef CAS.
- L. Crombie, W. M. L. Crombie, S. V. Jamieson and C. J. Palmer, J. Chem. Soc., Perkin Trans. 1, 1988, 1243–1250 RSC.
- J. L. Montero and F. Winternitz, Tetrahedron, 1973, 29, 1243–1252 CrossRef CAS.
- W. F. Erman, J. Am. Chem. Soc., 1967, 89, 3828–3841 CrossRef CAS.
- J. Hurst and G. Whitham, J. Chem. Soc., 1960, 2864–2869 RSC.
- R. K. Razdan, G. R. Handrick and H. C. Dalzell, Experientia, 1975, 31, 16–17 CrossRef CAS PubMed.
- J. W. Huffman, X. Zhang, M. J. Wu, H. H. Joyner and W. T. Pennington, J. Org. Chem., 1991, 56, 1481–1489 CrossRef CAS.
- Q. Wang, Q. Huang, B. Chen, J. Lu, H. Wang, X. She and X. Pan, Angew. Chem., Int. Ed., 2006, 45, 3651–3653 CrossRef CAS.
- Q. Huang, Q. Wang, J. Zheng, J. Zhang, X. Pan and X. She, Tetrahedron, 2007, 63, 1014–1021 CrossRef CAS.
- Q. Huang, B. Ma, X. a. Li, X. Pan and X. She, Synthesis, 2010, 1766–1770 CAS.
- A. D. William and Y. Kobayashi, Org. Lett., 2001, 3, 2017–2020 CrossRef CAS PubMed.
- A. D. William and Y. Kobayashi, J. Org. Chem., 2002, 67, 8771–8782 CrossRef CAS PubMed.
- Y. Kobayashi, A. Takeuchi and Y.-G. Wang, Org. Lett., 2006, 8, 2699–2702 CrossRef CAS PubMed.
- H. Kawada, A. Ikoma, N. Ogawa and Y. Kobayashi, J. Org. Chem., 2015, 80, 9192–9199 CrossRef CAS PubMed.
- B. Lesch, J. Toraeng, M. Nieger and S. Braese, Synthesis, 2005, 1888–1900 CAS.
- F. Korte, E. Dlugosch and U. Claussen, Liebigs Ann., 1966, 693, 165–170 CrossRef CAS.
- H. J. Kurth, U. Kraatz and F. Korte, Chem. Ber., 1976, 109, 2164–2174 CrossRef CAS.
- U. Kraatz and F. Korte, Z. Naturforsch., B: J. Chem. Sci., 1976, 31, 1382–1386 Search PubMed.
- D. A. Evans, E. A. Shaughnessy and D. M. Barnes, Tetrahedron Lett., 1997, 38, 3193–3194 CrossRef CAS.
- J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325–335 CrossRef CAS PubMed.
- G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561–3651 CrossRef CAS PubMed.
- S. Reymond and J. Cossy, Chem. Rev., 2008, 108, 5359–5406 CrossRef CAS PubMed.
- D. A. Evans, D. M. Barnes, J. S. Johnson, T. Lectka, P. von Matt, S. J. Miller, J. A. Murry, R. D. Norcross, E. A. Shaughnessy and K. R. Campos, J. Am. Chem. Soc., 1999, 121, 7582–7594 CrossRef CAS.
- A. G. Chittiboyina, C. R. Reddy, E. B. Watkins and M. A. Avery, Tetrahedron Lett., 2004, 45, 1689–1691 CrossRef CAS.
- L. Xia and Y. R. Lee, Synlett, 2008, 1643–1646 CAS.
- E. L. Pearson, N. Kanizaj, A. C. Willis, M. N. Paddon-Row and M. S. Sherburn, Chem. – Eur. J., 2010, 16, 8280–8284 CrossRef CAS PubMed.
- K. Maruoka, H. Imoto and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 12115–12116 CrossRef CAS.
- E. Ballerini, L. Minuti and O. Piermatti, J. Org. Chem., 2010, 75, 4251–4260 CrossRef CAS PubMed.
- L. Minuti and E. Ballerini, J. Org. Chem., 2011, 76, 5392–5403 CrossRef CAS PubMed.
- L. Minuti, E. Ballerini, A. Barattucci, P. M. Bonaccorsi, M. L. Di Gioia, A. Leggio, C. Siciliano and A. Temperini, Tetrahedron, 2015, 71, 3253–3262 CrossRef CAS.
- J.-Y. Goujon, F. Zammattio and B. Kirschleger, J. Chem. Soc., Perkin Trans. 1, 2002, 1564–1567 RSC.
- M. A. Tius and X.-q. Gu, J. Chem. Soc., Chem. Commun., 1989, 62–63 RSC.
- F. Pollastro, D. Caprioglio, D. Del Prete, F. Rogati, A. Minassi, O. Taglialatela-Scafati, E. Munoz and G. Appendino, Nat. Prod. Commun., 2018, 13, 1189–1194 CrossRef.
- Y. Song, S. Hwang, P. Gong, D. Kim and S. Kim, Org. Lett., 2008, 10, 269–271 CrossRef CAS PubMed.
- B. M. Trost and K. Dogra, Org. Lett., 2007, 9, 861–863 CrossRef CAS PubMed.
- Z. P. Shultz, G. A. Lawrence, J. M. Jacobson, E. J. Cruz and J. W. Leahy, Org. Lett., 2018, 20, 381–384 CrossRef CAS PubMed.
- L.-J. Cheng, J.-H. Xie, Y. Chen, L.-X. Wang and Q.-L. Zhou, Org. Lett., 2013, 15, 764–767 CrossRef CAS.
- J.-H. Xie, L.-X. Wang, Y. Fu, S.-F. Zhu, B.-M. Fan, H.-F. Duan and Q.-L. Zhou, J. Am. Chem. Soc., 2003, 125, 4404–4405 CrossRef CAS.
- A. Levens, A. Ametovski and D. W. Lupton, Angew. Chem., 2016, 128, 16370–16374 CrossRef.
- A. Ametovski and D. W. Lupton, Org. Lett., 2019, 21, 1212–1215 CrossRef CAS PubMed.
- M. V. Westphal, M. A. Schafroth, R. C. Sarott, M. A. Imhof, C. P. Bold, P. Leippe, A. Dhopeshwarkar, J. M. Grandner, V. Katritch, K. Mackie, D. Trauner, E. M. Carreira and J. A. Frank, J. Am. Chem. Soc., 2017, 139, 18206–18212 CrossRef CAS PubMed.
- V. R. L. J. Bloemendal, D. Sondag, H. Elferink, T. J. Boltje, J. C. M. van Hest and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2019, 2289–2296 CrossRef CAS PubMed.
- G. Hoffmann, C. G. Daniliuc and A. Studer, Org. Lett., 2019, 21, 563–566 CrossRef CAS PubMed.
- M. Soethoudt, G. Alachouzos, E. J. van Rooden, M. D. Moya-Garzón, R. J. van den Berg, L. H. Heitman and M. van der Stelt, Cannabis Cannabinoid Res., 2018, 3, 136–151 CrossRef CAS PubMed.
- M. A. Schafroth, G. Zuccarello, S. Krautwald, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2014, 53, 13898–13901 CrossRef CAS PubMed.
- F. Klotter and A. Studer, Angew. Chem., Int. Ed., 2015, 54, 8547–8550 CrossRef CAS PubMed.
- A. Chicca, M. A. Schafroth, I. Reynoso-Moreno, R. Erni, V. Petrucci, E. M. Carreira and J. Gertsch, Sci. Adv., 2018, 4, eaat2166 CrossRef CAS PubMed.
- P. D. Giorgi, V. Liautard, M. Pucheault and S. Antoniotti, Eur. J. Org. Chem., 2018, 1307–1311 CrossRef CAS.
- R. A. Archer, W. Blanchard, W. A. Day, D. W. Johnson, E. Lavagnino, C. Ryan and J. E. Baldwin, J. Org. Chem., 1977, 42, 2277–2284 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |