
Catechol-functionalized sequence-defined glycomacromolecules as covalent inhibitors of bacterial adhesion†
Lukas
Fischer
,
Ricarda C.
Steffens
,
Tanja J.
Paul
and
Laura
Hartmann
 *
*
Institut für Organische und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf, Germany. E-mail: Laura.Hartmann@hhu.de
First published on 26th August 2020
Abstract
Herein, we present the synthesis of catechol functionalized sequence-defined glycomacromolecules that can covalently block the binding site of lectins and bacterial adhesins. These structures produced on a solid phase support combine two important features: the multivalent presentation of carbohydrates for specificity, and catechols as anchors to go from highly reversible interactions to covalent attachment and more efficient inhibition. In our study we demonstrate this on the lectin Concanavalin A (ConA) by showing an increase in clustering for catechol ligands and on the effective inhibition of bacterial adhesion of E. coli on mannan surfaces by our catechol functionalized glycomacromolecules. Furthermore, covalent attachment is studied via MALDI-TOF measurements and SDS-PAGE analysis. Importantly, by replacing binding sugars with non-binding sugars, no inhibitory effects or covalent attachment were observed.
Introduction
Carbohydrate binding proteins such as lectins play key roles in numerous biological pathways. They are part of cell–cell communication, immune processes, tumorigenesis and pathogen infection.1–5 For example, galectin-3 mediates the adhesion of cancer cells towards the extracellular membrane, an important step in the formation of tumour metastases.6 Another example is the adhesin FimH from E. coli which binds to the glycocalyx of host cells and promotes biofilm formation.7,8 Blocking these interactions between the carbohydrate and protein can stop or slow the pathogenic process and is therefore a promising therapeutic route e.g. as antiviral or antibacterial treatments.9 However, single carbohydrates are weak binders and multivalent interactions are required to achieve strong and potentially selective binding.10,11 Such multivalency can be achieved by the presentation of multiple carbohydrate ligands on a synthetic scaffold, a process that has been shown to provide access to high affinity binders and efficient inhibitors e.g. against E. coli or influenza.12,13 It is important to keep in mind though that the carbohydrate–lectin interactions are still highly reversible usually resulting in only temporary inhibition, followed by disassociation from the target receptor and subsequent renal clearance.14 Ligands with the potential to covalently couple to a protein have the ability to overcome this challenge. For example, Wagner et al. recently introduced an epoxide functionalized D-galactose derivative to mark biofilm formation for P. aeruginosa, a multiple drug resistant bacteria that is cause for a high amount of hospital infections.15 The galactose derivative binds to the lectin LecA via the carbohydrate recognition domain (CRD) but then covalently attaches to the protein via a reactive epoxide group. This process was also shown to be specific as it depends on a cysteine found in the CRD, which however limits this approach to LecA. Next generation covalent ligands as therapeutic inhibitors could address this issue by fulfilling the following requirements: limited reactivity during distribution in order to avoid side-effects, selective covalent binding to the target protein after accumulation to achieve high levels of inhibition and a platform that can be adjusted to different targets e.g. different pathogens. As a first proof-of-concept study, here we introduce sequence-defined glycomacromolecules in combination with a catechol moiety and evaluate their potential as carbohydrate-based non-reversible inhibitors of bacterial adhesion. The goal of this study is to provide first insights into using catechols in covalent ligand design.Catechol groups oxidize to benzoquinone under basic to neutral conditions and afterwards are capable of reacting in a Michael addition-like reaction. This process allows them to covalently bind to proteins via surface exposed nucleophiles.16 One advantage of this process is that the oxidation rate depends on pH and chemical environment, for example dopamine prodrugs reach plasma half-lives of several hours with intact catechol moiety.17,18 This makes catechols promising covalent linker candidates as they could combine plasma stability for distribution with sufficient reactivity after accumulation. However, this advantage can also be a limitation if the catechol is slow to react.
Previously, we have introduced a class of precision glycomacromolecules as sequence-defined glycooligo(amidoamines). These compounds are accessible through the stepwise addition of building blocks on solid support, which allows for the control of the number, position and type of carbohydrates attached to the macromolecular scaffold, and gives access to tailor-made multivalent glycomacromolecules for targeting different proteins such as bacterial adhesins,19 viral capsid proteins20 and galectins.21
In this work, we introduce for the first time catechol groups to our precision glycomacromolecules to combine both the high affinity and selective binding of the glycomacromolecule with covalent inhibition. We envision that catechols can interact with the targeted protein only after binding through the carbohydrate-functionalized part of the scaffold. Over time the catechol moiety oxidizes to its benzoquichone derivative and can attach covalently e.g. to available surface lysines or cysteines (Fig. 1).
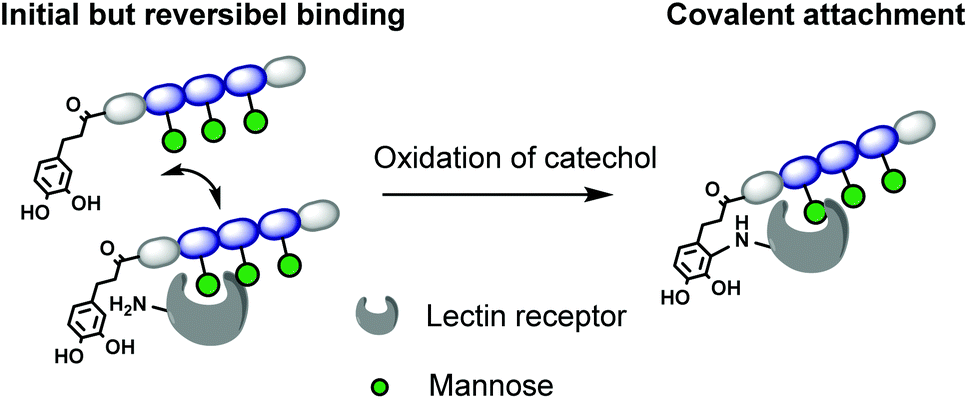 | ||
| Fig. 1 Model for irreversible inhibition by catechol-functionalized glycomacromolecules through specific binding via the carbohydrate ligands followed by irreversible attachment through the catechol group. | ||
Results and discussion
Synthesis of catechol-functionalized precision glycomacromolecules
Six different glycomacromolecules were synthesized presenting either three mannose (Man) or three galactose (Gal) residues and either zero, one or two catechol moieties positioned at the chain ends (Fig. 2). The synthesis of the glycofunctionalized scaffolds followed prior published protocols. In short, as previously introduced, tailor-made building blocks equipped with both an acid and an Fmoc-protected amine functionality, were assembled stepwise on solid support using standard Fmoc-peptide coupling chemistry.22 Site-specific introduction of alkyne side chains was achieved by using TDS (triple bond diethylenetriamine succinic acid) building blocks. Carbohydrate moieties were then coupled via Cu-mediated alkyne–azide-conjugation (CuAAC) using azidoethanol-functionalized Man and Gal derivatives. Catechol groups (Cat) were introduced via free primary amine groups and amide coupling using acetonide protected hydrocaffeic acid which was synthesized according to Wei et al.23 For the Cat containing glycomacromolecules 2, 3, 5 and 6, the N-terminal amine was used for functionalization after final Fmoc-deprotection. Additionally, for glycomacromolecules 3 and 6 with two catechol units, Boc-protected lysine was used during assembly on solid support to introduce a second primary amine after on-resin deprotection with HCl in dioxane. As last step, carbohydrate moieties were deprotected using sodium methanolate in methanol and the final glycomacromolecules were cleaved off the resin with trifluoroacetic acid, at the same time resulting in catechol deprotection.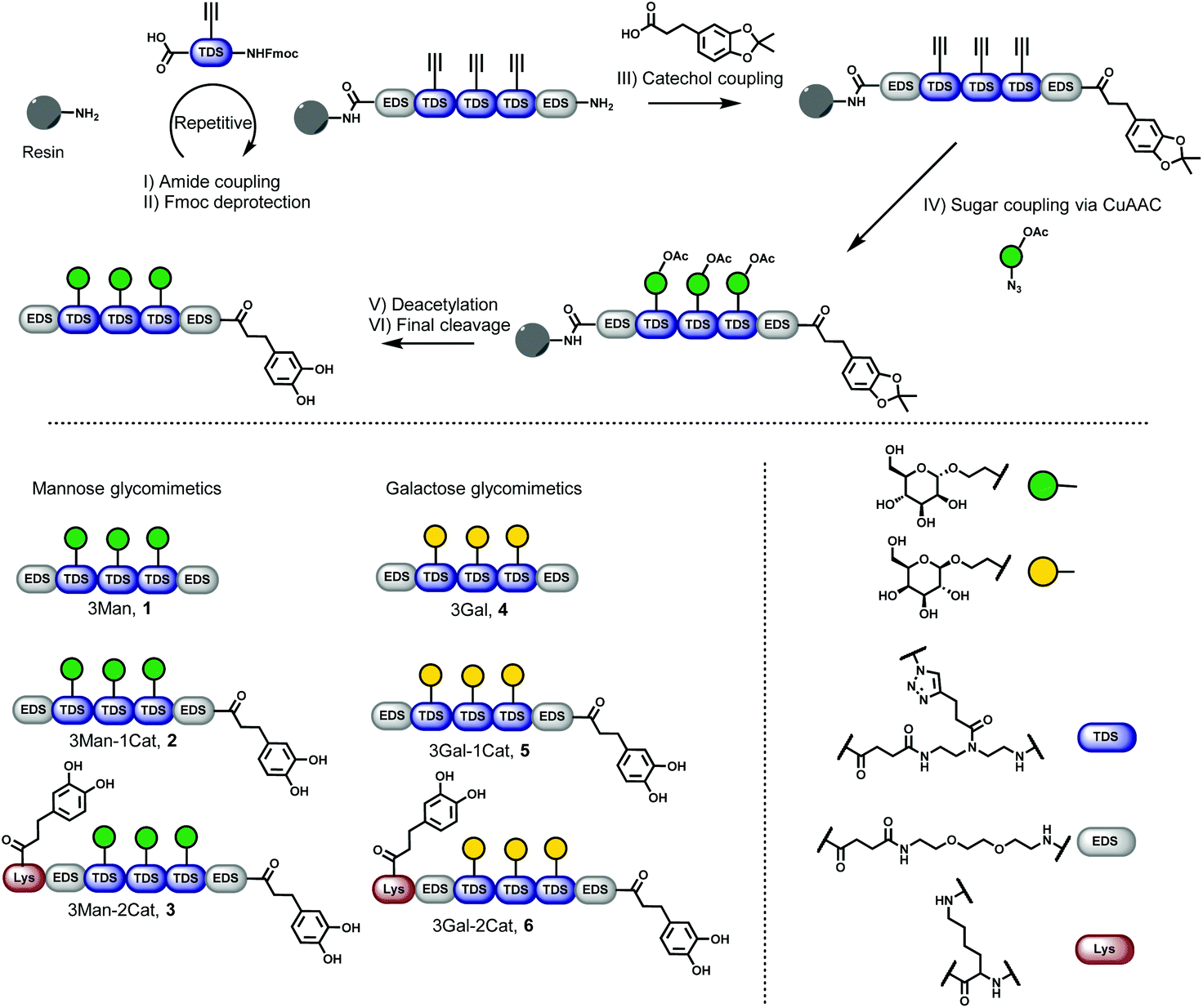 | ||
| Fig. 2 Solid phase synthesis of catechol containing glycomacromolecules (1–6). | ||
All structures were obtained with a relative purity of >90% after cleavage as determined by RP-HPLC and further characterized by 1H-NMR, 13C-NMR and HRMS (see ESI† for analytical data). Compounds were used in analytical and bioassays without further purification.
Catechol-functionalized precision glycomacromolecules binding to model lectin Concanavalin A (ConA)
To investigate the influence of the Cat moieties on the binding behaviour of glycomacromolecules towards lectins, binding to ConA was first studied via turbidity and precipitation assays. ConA is a Man-binding lectin extracted from the jack-bean and is widely used as a model system for sugar–lectin interactions. Forming a tetramer at pH greater than 7, it is capable of crosslinking multivalent glycomimetics, undergoing clustering and resulting in observable precipitation.24 This precipitation can be quantified via UV/Vis spectroscopy to measure the affinity of carbohydrates towards ConA. For the turbidity assay, a protocol adapted from Kiessling et al. was used25,26 to determine the reciprocal half-maximal turbidity (1/c1/2Tmax) as a value relative to the affinity towards ConA. Additional information was gathered by using a quantitative precipitation assay to derive the amount of ConA precipitated per glycomacromolecule (see ESI†).27The turbidity assay (Fig. 3, dark blue) clearly shows an increase in clustering of ConA in presence of a Cat moiety, with a five-fold increase for glycomacromolecule 2 with one Cat and a three-fold increase for glycomacromolecule 3 with two Cat in comparison to glycomacromolecule 1 having no Cat. Importantly, negative controls presenting Gal instead of Man but also including one or two Cat groups (5, 6) showed no turbidity and thus no binding to ConA (see ESI†). The catechol increases the clustering of the glycomacromolecule but only in combination with the binding carbohydrates, suggesting that Cat can only interact with ConA after the initial binding of the Man moieties. Thus, the overall specificity is retained. When looking at the quantitative precipitation assay (Fig. 3, light blue), more ConA per ligand is precipitated for the structure with two Cat (3), with 1.9 ConA bound per ligand vs. 0.7 ConA per ligand for structure 2. Both show a significant increase in comparison to 1 with 0.1 ConA per glycomacromolecule. Again, negative controls with Gal (4, 5, 6) showed no binding to ConA. There is an important difference between the two assays: in the turbidity assay, glycomacromolecule and ConA were incubated for 20 min before measurement, while for the precipitation assay glycomacromolecule and receptor were incubated for 24 h. This indicates that a second step of binding occurs – the bond formation via the catechol group, which probably takes time due to the oxidation and thus a fully non-reversible ligand-complex formation is only observed in the precipitation assay.
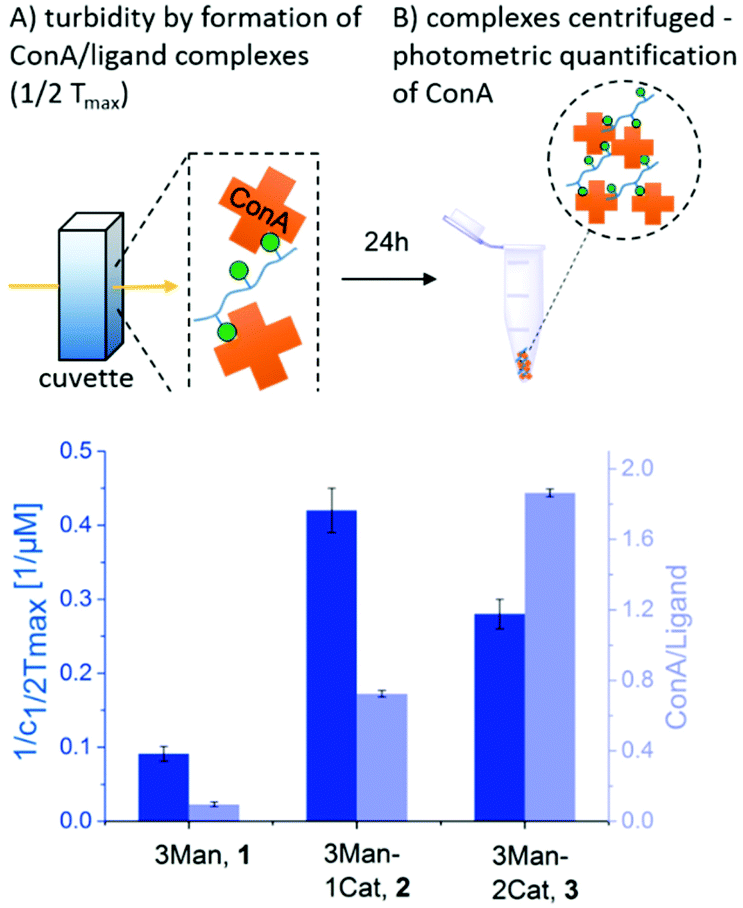 | ||
| Fig. 3 Results of the turbidity assay (dark blue) and quantitative precipitation assay (light blue) (schematic presentation of the assays are not to scale). Notably, none of the Gal-functionalized glycomacromolecules, with or without catechol, showed any binding in these assays (see ESI, Fig. S29†). | ||
Together, both assays show a clear increase in binding towards ConA upon the introduction of catechol groups. Binding of the ligands remains specific as the negative controls presenting a non-binding carbohydrate motif did not show any interaction. We have also seen that the second step, the covalent attachment, seems to be slower than the carbohydrate-mediated first step of the complex formation.
Bacterial adhesion-inhibition studies applying catechol-functionalized precision glycomacromolecules
To further show the applicability of catechol-functionalized precision glycomacromolecules as inhibitors in biological systems and to study the time-dependence of ligand binding, bacterial adhesion-inhibition studies were performed (Fig. 4). Here we used type 1-fimbriated E. coli binding to a mannan coated surfaces as model system. Adhesion of E. coli onto the mannan surface is promoted by mannose specific FimH receptors and can be reversed by addition of FimH binding molecules such as α-methyl D-mannopyranoside (MeMan).28 The assay was performed with GFP-tagged E. coli and adhesion was inhibited with increasing concentrations of glycomacromolecules giving half-maximum inhibitory concentration (IC50) values by detecting the fluorescence signal.19 Using MeMan as standard inhibitor, the IC50 value can be converted to the valency corrected relative inhibitory potential (RIPVC) by referencing the IC50 values and number of Man per glycomacromolecule to MeMan allowing for comparison of values across different experiments.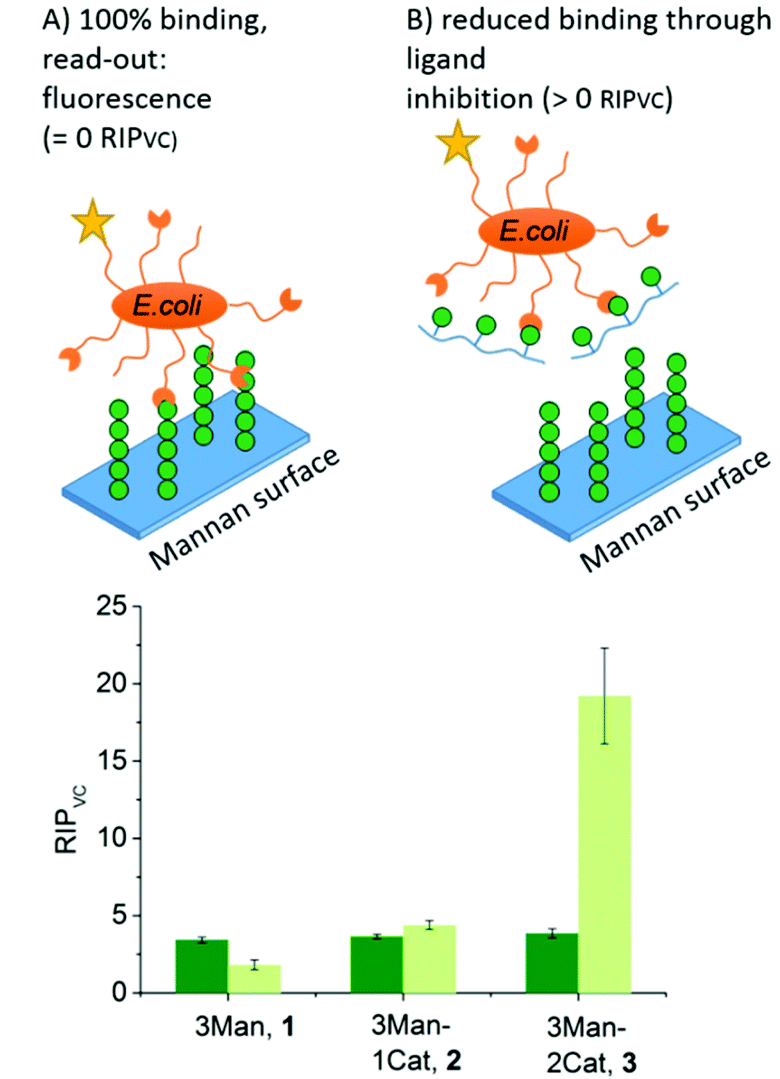 | ||
| Fig. 4 Results of the E. coli adhesion-inhibition assay after 1 h (green) and 24 h (light green) incubation (schematic presentation of the assays are not to scale). Notably, none of the Gal-functionalized glycomacromolecules, with or without catechol, showed any inhibition in these assays (see ESI, Fig. S39–S41†). | ||
Fig. 4 shows the RIPVC values of glycomacromolecules 1–3 after 1 h (green) and 24 h (light green) incubation with E. coli. After 1 h, all Man-containing glycomacromolecules showed similar inhibition potentials, which is about 3.5 more effective per sugar in comparison to MeMan. These values are in the same range as for similar trivalent glycomacromolecules without Cat units as previously studied by our group in bacterial adhesion-inhibition studies.19 Interestingly, incubating the structures with E. coli for 24 h significantly increases the efficacy of glycomacromolecule 3 by a factor of four while glycomacromolecules 1 and 2 remain at a similar inhibitory potential. This clear effect in longer incubation times for glycomacromolecule 3 again indicates the slow oxidation of the catechols and covalent binding over time. It also seems that binding with two Cat moieties leads to a more efficient blocking of the binding site than just one Cat. A possible explanation could be that if the glycomacromolecule is only attached via one chain end, the carbohydrate-containing scaffold can still unbind and release a free binding site, while a glycomacromolecule with Cat groups at both chain ends acts as a staple completely blocking the binding site.
Studying covalent attachment of catechol-functionalized precision glycomacromolecules
While the previous studies showed an effect of the introduction of Cat moieties on enhanced binding to ConA/FimH, we sought further support for a covalent attachment by examining glycomacromolecule-ConA complexes with MALDI-TOF. Equimolar amounts of ConA and glycomacromolecules were incubated for 24 h and then analysed by MALDI-TOF mass spectrometry.Fig. 5 shows the MALDI-TOF measurement for ConA alone and in presence of glycomacromolecules 2 (Man and Cat) and 6 (Gal and Cat). The main mass peak for ConA was found to be 25.6 kDa, which represents the mass of monomeric ConA as the tetramer is divided into its subunits during measurement.29 In presence of glycomacromolecules 2, additional mass peaks were found at 27.8 kDa, 30.0 kDa and 32.2 kDa. The mass difference between these peaks is 2.2 kDa, which corresponds to the mass of the glycomacromolecule and thus shows covalent attachment of the ligand to ConA. The MALDI-TOF samples were further investigated by polyacrylamide gel electrophoresis under denaturating conditions (SDS-PAGE) to show spots corresponding to the mass of ConA and glycomacromolecules. Indeed, here we also observed additional spots with higher molecular weight for structure 2 and 3, pointing towards the covalent attachment to the protein (see ESI†).
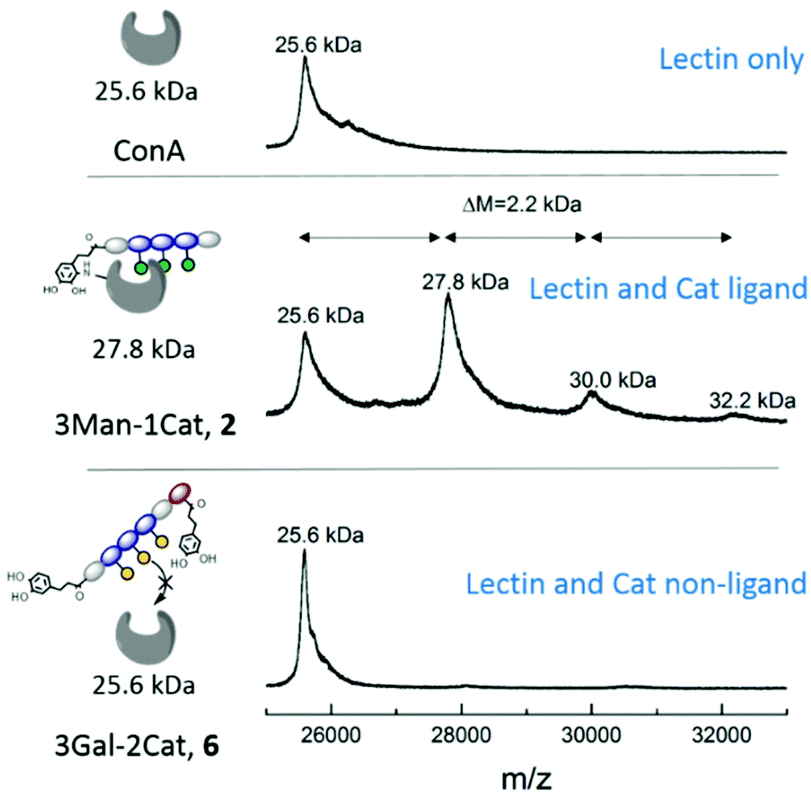 | ||
| Fig. 5 MALDI-TOF MS measurement after 24 h incubation of glycomacromolecules with ConA. | ||
Surprisingly, MS data shows that multiple glycomacromolecules bound to one protein, here a maximum of three ligands per protein were detected. This indeed supports our hypothesis, that for glycomacromolecules with just one Cat, the carbohydrate-containing scaffold can still diffuse out of the binding site, which enables the binding of a second glycomacromolecule and successive coupling to the protein. This could then take place repeatedly until the binding site is too sterically hindered by previously attached glycomacromolecules to allow for another structure to bind. Similar behaviour was observed for ligand 3 presenting two catechol units but only with up to two bound oligomers per protein (see ESI†) indicating that two catechols more effectively anchor around the binding site which is also in agreement with the quantitative precipitation study. Importantly, no covalent attachment to the protein was observed for glycomacromolecule 1 presenting Man but no Cat and for Gal glycomacromolecules 5 and 6 with Cat (see ESI†).
Overall, these findings support our model of binding for Cat-functionalized glycomacromolecules where the Cat unit can only interact with ConA after initial binding mediated by the carbohydrate-containing scaffold and it is the interplay of both binding units that enables effective inhibition.
Conclusions
In summary, this study demonstrates the synthesis of Cat-functionalized glycomacromolecules and their potential to covalently inhibit lectin receptors. By investigating the clustering of ConA we could show that the introduction of a Cat moiety increases the apparent binding affinity of glycomacromolecules. The interaction between Cat and receptor seems to occur after initial binding of the carbohydrate-containing scaffold and only if the carbohydrate moiety binds to the targeted receptor. The effect of Cat increases over time, likely due to slow oxidation and covalent bonding. Strong evidence for covalent binding was found in an MALDI-TOF MS assay and confirmed via SDS-PAGE. Finally, we show the possible application of these glycomacromolecules to inhibit bacterial adhesion of E. coli to mannan coated surfaces. In order to evaluate whether these systems can also be applied to more complex biological settings and might have a long-term perspective for clinical use, future studies will have to further explore the combination of precision glycomacromolecules and Cat moieties and their inhibition potential.Experimental
General procedure for solid phase synthesis
All glycomacromolecules were synthesized on solid support according to literature using the building blocks EDS (ethylene glycol diamine succinic acid), TDS and Fmoc-Lys(Boc).26 Tentagel® S RAM was used as a resin, and the structures were synthesized by repetitive Fmoc cleavage and amide coupling. For Fmoc cleavage, the resin was treated with 20% piperidine in DMF for 30 min. For the amide coupling, the resin was treated with a solution of 5 eq. building block, 5 eq. PyBOP and 10 eq. DIPEA in DMF for 1 h. After assembly of the full sequence the lysine was deprotected on solid support using 4 M HCl in dioxane for 30 min. The catechol moiety was introduced at the terminal amine or deprotected lysine or both. For this the resin was treated with 5 eq. 3-(2,2-dimethylbenzo[d][1,3]dioxol-5-yl)propionic acid, 5 eq. PyBOP and 10 eq. DIPEA in DMF for 1 h. For oligomers without catechol the terminal amine was capped with acetic anhydride for 5 min. After assembly of the scaffold and either catechol coupling or end capping, sugars were introduced via an established CuAAC protocol.26 Afterwards the sugars were deprotected on solid support using 0.1 M sodium methoxide in methanol. For final cleavage the resin was treated with 95% TFA, 2.5% TIPS and 2.5% DCM for 1 h. The glycomacromolecules were precipitated in diethyl ether and freeze dried.Concentration dependent turbidity assay
A solution of 5 μM ConA in LBB buffer (10 mM HEPES, 50 mM NaCl, 1 mM MnCl2, 1 mM CaCl2, pH 7.4) was prepared. The transmission of 1 ml of this solution was measured as 100% transmission baseline. Afterwards glycomacromolecules were stepwise titrated to the ConA solution and after 20 min incubation the transmission was measured. Every structure was measured three times.Quantitative precipitation assay
A solution of 15 μM ConA in LBB buffer (10 mM HEPES, 50 mM NaCl, 1 mM MnCl2, 1 mM CaCl2, pH 7.4) was prepared and the concentration was measured at 280 nm. Afterwards aliquots of this solution were mixed with different concentrations of glycomacromolecules, incubated for 24 h and centrifuged for 5 min at 4400 rpm. The precipitate was resuspended in LBB buffer with 50 mM α-methyl D-mannoside and the ConA concentration was determined at 280 nm. To calculate the amount of ConA precipitated per ligand the linear slope between 1 and 5 μM ligand was used.MALDI-TOF measurement
For the determination of a covalent bond between ConA and ligand, equimolar amounts of ConA (8 μM) and glycomacromolecule (8 μM) were incubated in LBB buffer (10 mM HEPES, 50 mM NaCl, 1 mM MnCl2, 1 mM CaCl2, pH 7.4) for 24 h. Afterwards the samples were filtrated and measured via MALDI-TOF in linear mode.Bacterial adhesion-inhibition assay
The E. coli strain PKL1162 was cultured from a stock in LB media (ampicillin 100 mg ml−1 and chloramphenicol 50 mg ml−1) at 37 °C overnight. The bacterial cells were centrifuged and washed twice and suspended in PBS buffer to a cell concentration of OD600 = 0.4. The adhesion-inhibition assay was conducted as described prior in this working group.19 Black 96-well microtiter plates (Nunc, MaxiScorp) were treated with mannan (1.2 mg ml−1 in carbonate buffer pH 9.6) for 12 h at 37 °C until full evaporation of water. The plates were washed three times with PBST buffer (PBS buffer + 0.05% v/v Tween®20) and blocked with PVA (1% in PBS) for 2 h. Afterwards the plates were washed with PBST twice and PBS once. For the measurement a serial dilution of glycomacromolecules on the mannan-coated microtiter plates was performed (50 μl). The bacterial suspension was added (50 μl) and the plates were incubated for either 1 h or 24 h at 37 °C. After incubation the microtiter plates were washed three times with PBS and refilled with PBS (100 μl) to measure the fluorescence intensity (excitation 485 nm, emission 535 nm).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Monir Tabatabai and Jun.-Prof. Dr Stephan Schmidt for their helpful discussions and the Heinrich Heine University (HHU) for its support.References
- P. R. Crocker and T. Feizi, Curr. Opin. Struct. Biol., 1996, 6, 679–691 CrossRef CAS.
- A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS.
- S. D. Rosen, Semin. Immunol., 1993, 5, 237–247 CrossRef CAS.
- H. Ghazarian, B. Idoni and S. B. Oppenheimer, Acta Histochem., 2011, 113, 236–247 CrossRef CAS.
- N. Sharon and H. Lis, Glycobiology, 2004, 14, 53R–62R CrossRef CAS.
- Y. Takenaka, T. Fukumori and A. Raz, Glycoconjugate J., 2002, 19, 543–549 CrossRef CAS.
- S. L. Chen, C. S. Hung, J. S. Pinkner, J. N. Walker, C. K. Cusumano, Z. Li, J. Bouckaert, J. I. Gordon and S. J. Hultgren, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22439–22444 CrossRef CAS.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740 CrossRef CAS.
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K.-E. Jaeger and M. Lahmann, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS.
- T. K. Dam, R. Roy, S. K. Das, S. Oscarson and C. F. Brewer, J. Biol. Chem., 2000, 275, 14223–14230 CrossRef CAS.
- M. Hartmann and T. K. Lindhorst, Eur. J. Org. Chem., 2011, 2011, 3583–3609 CrossRef CAS.
- X.-L. Sun, Curr. Med. Chem., 2007, 14, 2304–2313 CrossRef CAS.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677 CrossRef CAS.
- S. Wagner, D. Hauck, M. Hoffmann, R. Sommer, I. Joachim, R. Müller, A. Imberty, A. Varrot and A. Titz, Angew. Chem., Int. Ed., 2017, 56, 16559–16564 CrossRef CAS.
- S. Ito, T. Kato and K. Fujita, Biochem. Pharmacol., 1988, 37, 1707–1710 CrossRef CAS.
- L. Giannola, V. De Caro, G. Giandalia, M. Siragusa and L. Lamartina, Die Pharmazie – Int. J. Pharm. Sci., 2008, 63, 704–710 CAS.
- M. Carafa, C. Marianecci, L. D. Marzio, V. D. Caro, G. Giandalia, L. I. Giannola and E. Santucci, J. Liposome Res., 2010, 20, 250–257 CrossRef CAS.
- S. Boden, F. Reise, J. Kania, T. K. Lindhorst and L. Hartmann, Macromol. Biosci., 2019, 19, e1800425 CrossRef.
- M. Baier, N. H. Rustmeier, J. Harr, N. Cyrus, G. J. Reiss, A. Grafmüller, B. S. Blaum, T. Stehle and L. Hartmann, Macromol. Biosci., 2019, 19, 1800426 CrossRef.
- T. Freichel, D. Laaf, M. Hoffmann, P. B. Konietzny, V. Heine, R. Wawrzinek, C. Rademacher, N. L. Snyder, L. Elling and L. Hartmann, RSC Adv., 2019, 9, 23484–23497 RSC.
- D. Ponader, F. Wojcik, F. Beceren-Braun, J. Dernedde and L. Hartmann, Biomacromolecules, 2012, 13, 1845–1852 CrossRef CAS.
- Q. Wei, K. Achazi, H. Liebe, A. Schulz, P. L. M. Noeske, I. Grunwald and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 11650–11655 CrossRef CAS.
- V. Wittmann, Curr. Opin. Chem. Biol., 2013, 17, 982–989 CrossRef CAS.
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef CAS.
- C. Gerke, M. F. Ebbesen, D. Jansen, S. Boden, T. Freichel and L. Hartmann, Biomacromolecules, 2017, 18, 787–796 CrossRef CAS.
- Y. Gou, J. Geng, S. J. Richards, J. Burns, C. Remzi Becer and D. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2588–2597 CrossRef CAS.
- T. K. Lindhorst, S. Oscarson and J. Bouckaert, Glycoscience and Microbial Adhesion, Springer, 2009 Search PubMed.
- K. Sparbier, T. Wenzel and M. Kostrzewa, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 840, 29–36 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00975j |
| This journal is © The Royal Society of Chemistry 2020 |