
Thermo- and oxidation-sensitive poly(meth)acrylates based on alkyl sulfoxides: dual-responsive homopolymers from one functional group†
Doğuş
Işık
a,
Elisa
Quaas
b and
Daniel
Klinger
 *a
*a
aInstitute of Pharmacy, Freie Universität Berlin, Königin-Luise-Straße 2-4, 14195 Berlin, Germany. E-mail: daniel.klinger@fu-berlin.de
bInstitute of Chemistry, Freie Universität Berlin, Takustraße 3, 14195 Berlin, Germany
First published on 17th November 2020
Abstract
Ideal dual-responsive homopolymers are derived from monomers that unite two sensitive functionalities in one molecule. This design circumvents structural inhomogeneities that can occur during the preparation of similar copolymers from two different monomers. Among such “smart” homopolymers, materials that sense changes in temperature and redox activity are of special interest due to the ubiquitous availability and facile applicability of both stimuli. However, developing new monomers that combine both response mechanisms in one functional motif remains challenging. To address this challenge, we introduce a new class of (meth)acrylates bearing alkyl sulfoxide side groups to realize thermo- and oxidation-responsive homopolymers. The dual-responsive behavior follows specific design principles: first, the thermo-responsive behavior can be tuned by balancing sulfoxide-water hydrogen bonds and hydrophobic interactions of the respective alkyl side chains. Following this molecular design, we found that systematically varying the sulfoxides’ alkyl groups can be used to tune the thermo-responsive properties of the polymers. Second, an additional oxidation response is introduced by oxidizing the hydrophilic sulfoxides to their hydrophobic sulfone analogues. This decreases the polymer's overall hydrophilicity, thus reducing the cloud point temperature. As a result, the polymer exhibits a dual-responsive behavior. In addition, the gradual partial oxidation of sulfoxides gives the advantage to adjust the thermo-responsive profile though the second stimulus. We believe that this unique combination of thermal and oxidation response in one single functional unit, its facile synthesis, well-controlled polymerization, and biocompatibility is the starting point for the preparation of highly sophisticated materials for a wide variety of applications.
Introduction
Nature is able to control highly complex functions by precisely adapting macromolecular properties to multiple environmental changes. To mimic such outstanding biological versatility with “smart” synthetic polymers, different functional moieties need to be combined within one polymer chain.1–3 The resulting dual- or multi-responsive polymers enable new levels of materials’ variability and complexity, and are paving the way to new advanced applications.4–6Among such emerging dual-stimuli-responsive materials, (water soluble) polymers that sense changes in temperature and redox-activity are of special interest for applications in the biomedical field,7,8 information processing,9 and nanotechnology.10,11 This can be attributed to the ubiquitous availability and efficient applicability of both stimuli in different environments. In addition, the orthogonal sensitivity to temperature as physical trigger and redox active reagents, such as reactive oxygen species (ROS), as chemical oxidation trigger gives rise to innovative smart materials with non-interfering dual functions.12,13
While a plethora of polymers have been developed to exhibit either a thermo- or oxidation-response individually, examples that combine both properties in one dual-responsive-polymer are still limited. Existing multi-sensitive polymers have been realized mainly by (controlled) radical copolymerization of thermo- and oxidation-responsive monomers14,15 or by step growth polymerization i.e. of poly(ethylene glycol) diacrylate with bridging thioether groups.16 Even though such random copolymerization strategies are synthetically easy to realize, the concerted distribution of both stimuli-sensitive groups along the same backbone can significantly affect the individual response profiles.17–20 In case of step growth polymerizations, this approach often leads to broad molecular weight distributions restricts the access to well-defined (block)copolymer structures. Overall, the co-dependency between two monomers along one backbone as well as the distribution of functional moieties in the backbone and side chains can limit the synthetic flexibility and hinders the facile adjustment of accurate transition temperatures.14,20–22
To overcome such copolymerization disadvantages, new strategies aim to combine two different responsive groups in one multi-stimuli-responsive monomer.23–25 The resulting homopolymers allow accurate multi-stimuli-responsive profiles that do not depend on copolymer compositions.26,27 However, the synthetic introduction of multiple functional groups in one monomer is far from trivial and such strategies are, until now, mainly reported for combinations of temperature with light and/or pH.28–30 For example, monomers containing methacrylamides and aspartic acid groups were designed to exhibit temperature response due to the acrylamide groups and pH response due to the carboxylic acid groups.31 Difficulties of this approach stem from the large multi-functional monomers which can hinder efficient and controlled polymerization.14,23 In addition, the close spatial proximity of the sensitive groups can further influence the response behavior of each other.32,33
Hence, ideal multi-responsive polymers reduce the complexity of the macromolecular design even further: in such advantageous systems, monomers contain only one functional group that can respond to both stimuli.25 A prominent example of this approach is poly[N-[2-(diethylamino)ethyl] acrylamide] (pDEAEAm) where the dimethylamino group is responsive to both temperature and pH (and CO2) changes.34 For polymers that respond to temperature and oxidative species, such a design would also allow for the precise adjustment of a cooperative stimuli-response profile. Here, the response to one trigger (temperature) can be adjusted through the application of the second stimulus (oxidation). The resulting dual-responsive homopolymers enable to examine and apply structure–property-relations much more precisely than similar copolymer systems. While such multi-responsive functional groups are a promising concept, an actual simple molecular design and straightforward synthesis of thermo- and oxidation-responsive monomers is still very challenging.
To address this challenge, we have considered two important criteria for a respective molecular design: first, the polymers’ thermo-responsive behavior depends on a specific balance between polymer–water hydrogen bonds and intra-/inter-polymer hydrophobic interactions. Hence, the respective monomers are inherently amphiphilic and the balance between hydrophilic hydrogen bonds and hydrophobic groups determines the thermo-responsive profile.35 Second, introducing an additional oxidation response requires this thermo-responsive monomer to change its amphiphilicity upon oxidation. This can be realized if the hydrophilic group is oxidized to a more hydrophobic state with less hydrogen bonds. As a result, the hydrophilic-to-hydrophobic ratio of the thermo-responsive polymer is lowered and the polymer–water hydrogen bonds will be reduced. This will cause the cloud point temperature to decrease and the polymer will thus exhibit a multi-responsive behavior.
We found sulfoxide groups to meet these requirements perfectly. First, they are strong hydrogen bond acceptors and are therefore very hydrophilic.36,37 In polymers, side-chain sulfoxide poly(meth)acrylates are known for their very low cytotoxicity, their excellent biocompatibility, and their penetration enhancing features with potential applications in pharmaceutical and medical sciences.38–40 Due to this strong inherent hydrophilicity, polysulfoxides were also proposed to have active “stealth” properties similarly to polyethylene glycol (PEG).41 Second, when sulfoxides are oxidized to the corresponding sulfones, the ability to form hydrogen bonds with water is dramatically reduced. Since this oxidation of sulfoxide-containing polymers can occur effectively as response to reactive oxygen species (ROS), polymers containing sulfoxide side groups show great potential for realizing oxidation-responsive materials.41,42
Based on these considerations, we herein report the rational design of new alkyl sulfoxide containing (meth)acrylates as monomers for thermo- and oxidation-responsive polymers (see Scheme 1). In these monomers, the central role of the sulfoxides is twofold: on one hand, the strong hydrogen bond acceptors will be combined with hydrophobic alkyl side groups to realize an amphiphilic structure. This is the foundation for thermo-responsive polymer properties. Here, the cloud point temperature can easily be tuned by varying the alkyl side chain, i.e. the hydrophilic-to-hydrophobic balance of the monomer. On the other hand, oxidation of the sulfoxides to the respective sulfones will reduce the polymers’ hydrophilicity. Thus, a quantitative oxidation results in a dramatically reduced water solubility. In addition, the oxidation-sensitive behavior also offers the potential to gradually adjust the thermo-responsive profile. By controlling the conversion of sulfoxide to sulfone side groups, the cloud point temperature can be continuously decreased with an increasing number of sulfones per polymer.
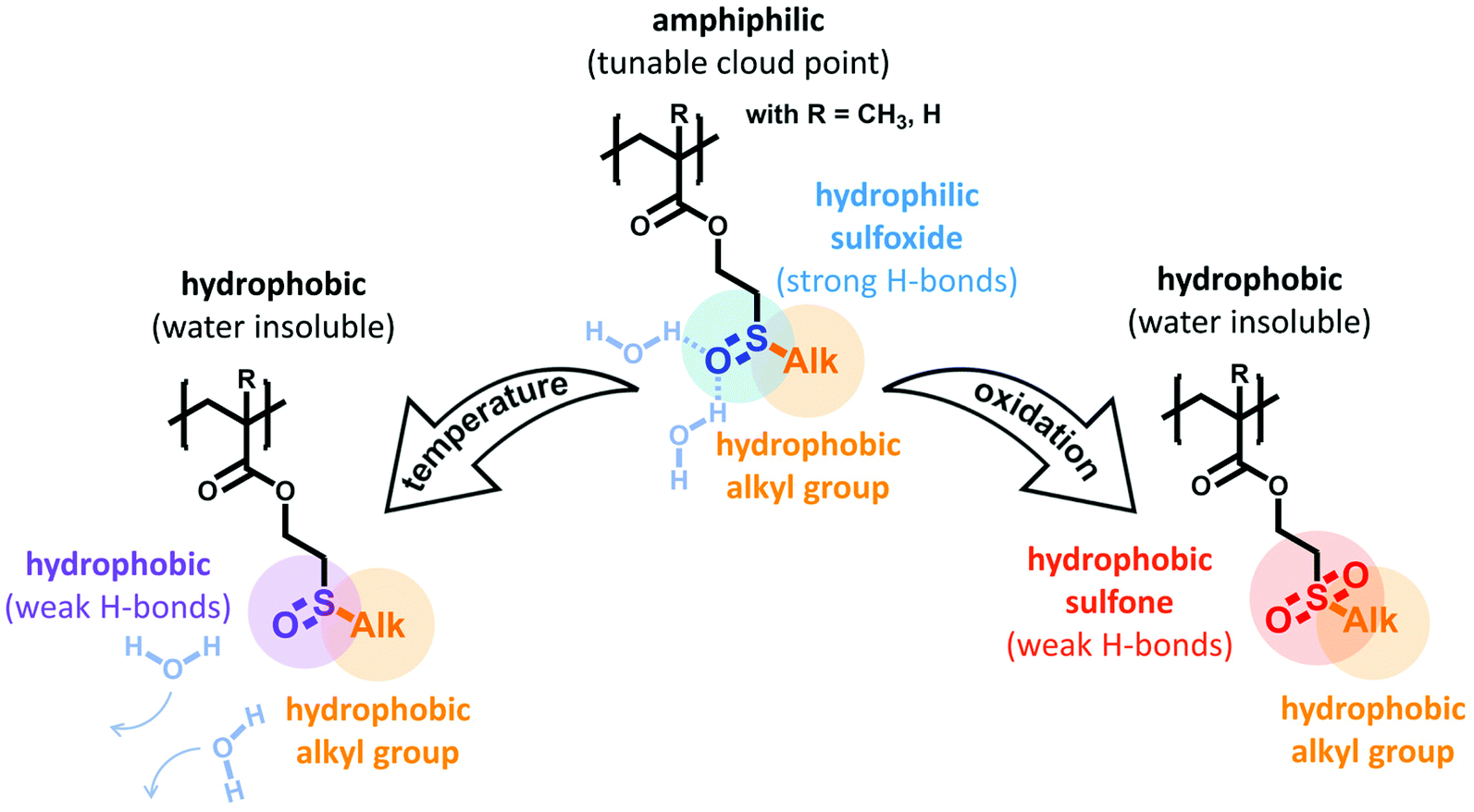 | ||
| Scheme 1 Sulfoxide side groups as thermo- and oxidation-responsive motifs. Temperature-sensitivity is based on reducing the amphiphilic balance between hydrophilic sulfoxide moieties and hydrophobic alkyl groups at elevated temperatures. A second oxidative trigger turns the sulfoxide moieties into the respective hydrophobic sulfone derivatives, thus reducing polymer solubility and realizing a dual-responsive system. | ||
We believe that this unique combination of the thermal and oxidation response in one single functional unit, it's facile synthesis, and well-controlled polymerization is the starting point for the preparation of highly sophisticated materials for a wide variety of applications.
Results and discussion
For the development of a sulfoxide-based thermo- and oxidation-responsive polymer, several steps need to be addressed. First, suitable functional groups have to be identified and incorporated into the monomer structure in a facile and efficient way. Second, a suitable polymerization method has to be chosen to give polymers with well-defined molecular weights and narrow molecular weight distributions. By doing so, a high degree of comparability is ensured that would enable to accurately determine the structure–property relationships. Third, the polymers should maintain their structural and functional integrity throughout the synthesis to preserve both the thermal und oxidation response. Addressing these challenges allows for an accurate and systematic investigation of a dual-stimuli-responsive polymer library.Preparation of an amphiphilic sulfoxide polymer library
Key to the rational macromolecular design in this work is the combination of hydrophilic and oxidation-responsive sulfoxide moieties with different hydrophobic alkyl groups (see Scheme 1). The resulting amphiphilic structure is generally regarded as a main requisite for the introduction of thermo-responsive polymer properties such as a lower critical solution temperature (LCST). Here, it is assumed that the cloud point temperature (Tcp) can easily be tuned by varying the hydrophobic contribution of the alkyl side group, i.e. larger alkyl side groups would decrease the Tcp. Additionally, the polymer backbone itself can alter the hydrophilic-to-hydrophobic balance of the polymer. In general, a polyacrylate backbone is less hydrophobic than its polymethacrylate analogue. This implies that the Tcp of polyacrylates would increase with respect to comparable polymethacrylates. To analyze these two structure–property-relations systematically, a library of sulfoxide-containing polymethacrylates and polyacrylates with varying alkyl side groups was investigated (Fig. 1a).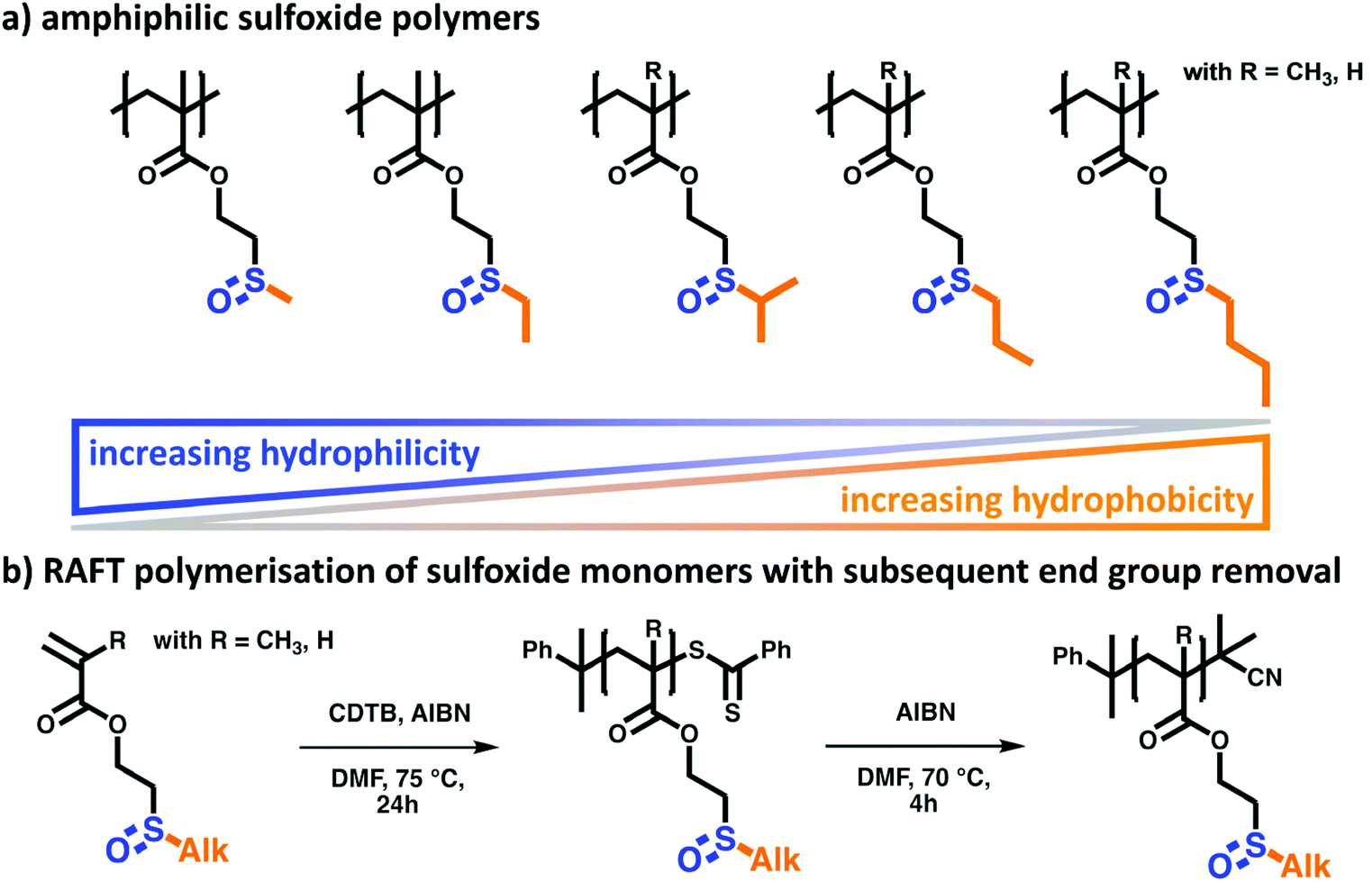 | ||
| Fig. 1 (a) The investigated library of alkyl sulfoxide (meth)acrylate polymers. Varying the polymer's amphiphilicity by changing the alkyl group and the polymer backbone gives access to analyzing the structure–property relations of the thermo-responsive polymers. (b) Polymer synthesis was achieved via RAFT polymerization with subsequent end group removal. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CDTB]0:[AIBN]0 = 150:1:0.125 (Table 1), thus targeting a DP of 150 for all polymers. As shown in Table 1, the RAFT method gave good control over the polymerization process for the methacrylate monomers. High conversions were reached and overall narrow molecular weight distributions (Đ ≤ 1.25) were obtained.
:[CDTB]:[I] = 150:1:0.125 at 75 °C for a reaction time of 24 h. Accounting for the lower conversion of the acrylate monomers iPr-SEA, nPr-SEA and nBu-SEA, the respective RAFT polymerizations were performed in dioxane ([M] = 1 M) with [M]:[BBDT]:[I] = 250:1:0.125 at 75 °C for a reaction time of 24 h
:[CDTB]0:[AIBN]0 = 150:1:0.125 (Table 1), thus targeting a DP of 150 for all polymers. As shown in Table 1, the RAFT method gave good control over the polymerization process for the methacrylate monomers. High conversions were reached and overall narrow molecular weight distributions (Đ ≤ 1.25) were obtained.
:[CDTB]:[I] = 150:1:0.125 at 75 °C for a reaction time of 24 h. Accounting for the lower conversion of the acrylate monomers iPr-SEA, nPr-SEA and nBu-SEA, the respective RAFT polymerizations were performed in dioxane ([M] = 1 M) with [M]:[BBDT]:[I] = 250:1:0.125 at 75 °C for a reaction time of 24 h
| Polymer | CTA | [M]/[CTA] | Conv.a (%) | M n,target (g mol−1) | M n,NMR (g mol−1) | DP | M n,GPC (g mol−1) | Đ |
|---|---|---|---|---|---|---|---|---|
| a Conversion measured by 1H NMR spectroscopy. b DMF LiBr (10 mM) eluent, linear PMMA standard. | ||||||||
| P(Me-SEMA) | CDTB | 150 | 84 | 26430 |
22100 |
124 | 20100 |
1.24 |
| P(Et-SEMA) | CDTB | 150 | 92 | 28540 |
26200 |
136 | 19900 |
1.24 |
| P(iPr-SEMA) | CDTB | 150 | 86 | 30640 |
26500 |
128 | 23200 |
1.20 |
| P(nPr-SEMA) | CDTB | 150 | 88 | 30640 |
26900 |
130 | 22400 |
1.21 |
| P(nBu-SEMA) | CDTB | 150 | 95 | 32750 |
31000 |
141 | 22400 |
1.19 |
| P(iPr-SEA) | BBDT | 250 | 51 | 47600 |
24400 |
127 | 19200 |
1.31 |
| P(nPr-SEA) | BBDT | 250 | 50 | 47600 |
23800 |
124 | 18900 |
1.33 |
| P(nBu-SEA) | BBDT | 250 | 48 | 51100 |
24600 |
119 | 15300 |
1.29 |
Furthermore, the RAFT solution homopolymerization of the alkyl sulfoxide acrylate monomers was initially performed using the same reaction conditions as for the methacrylates (CDTB, AIBN, DMF with [monomer]0:[CDTB]0:[AIBN]0 = 150:1:0.125). However, under these conditions only lower molecular weight polymers were obtained (see ESI section II, Table S1†). To address this observed lower conversion for the acrylate monomers different dithioester and trithiocarbonate CTA's, namely benzyl benzodithioate (BBDT), 4-cyano-4-(thiobenzoylthio)pentanoic acid, dibenzyltrithiocarbonate, 4-cyano-4-ethyl-trithiopentanoic acid, 4-cyano-4-dodecyl-trithiopentanoic acid and 2-(((butylthio)carbonothioyl)thio)-2-methylpropanoic acid were investigated. Unfortunately, for the majority of these CTAs either small molecular weights or high molecular weight dispersities were determined by GPC (data not shown). However, benzyl benzodithioate (BBDT) showed acceptable dispersities even though the molecular weight was lower than expected. This stems from the lower conversions in comparison to the polymethacrylates thus indicating a slower polymerization process.
Taking these finding into account, we aimed to prepare polyacrylates with higher molecular weights. For this, BBDT in dioxane was used with an initial [M]/[CTA] = 250 to account for the slow polymerization and the corresponding low conversion. Under these polymerization conditions, polyacrylates with degrees of polymerization comparable to the polymethacrylates with [M]/[CTA] = 150 were obtained with reasonable dispersities (Đ ≤ 1.33) (Table 1). The DP and molecular weights (Mn,NMR) of all RAFT polymers were calculated by 1H NMR spectroscopy (for details see ESI section III†).
After polymerization, the final alkyl sulfoxide-containing polymers were prepared by removal of the RAFT end group. This was necessary since the labile dithioester end groups might cause undesired side effects in upcoming polymer property measurements.45 To prevent this, the CTA of the macromolecules was cleaved via a radical substitution reaction with AIBN. This introduces a more robust dimethyl nitril group (Fig. 1b).
Once having obtained a library of sulfoxide-containing homopolymers, the respective polymer analogues with sulfone side groups were prepared as controls. In these polymers, the central role of the alkyl sulfones is as follows: regarding temperature-responsive properties, the sulfones are inherently hydrophobic with no ability to form strong hydrogen bonds. Thus, in combination with the alkyl side groups, this results in an overall hydrophobic polymer with no potential to show temperature-responsive solution properties. This is in direct contrast, to the amphiphilic sulfoxides where thermo-responsive polymer properties are easily tuned via changing the hydrophilic-to-hydrophobic balance. Regarding the oxidation-responsive mechanism, the quantitative oxidation of the sulfoxides to the respective sulfones is the endpoint of this reaction. Thus, the hydrophobic sulfone polymers act as the control group for the assessment of the special thermo- and oxidation-responsive properties of the sulfoxide polymers. To ensure comparability (same DP and Đ) between sulfoxide and sulfone polymers, a complementary library of sulfone (meth)acrylate polymers was prepared by simple oxidation of the previously prepared sulfoxide polymers (see ESI section IV† for further experimental details and characterization).
Screening of the thermo-responsive properties of the sulfoxide- and sulfone-containing polymers
Having established a straightforward synthetic route to prepare a functional library of comparable sulfoxide- and sulfone-containing (meth)acrylate polymers, their temperature-dependent solution properties were assessed. As mentioned in the previous section, it is assumed that the cloud point temperature (Tcp) decreases with longer alkyl groups. As shown in Fig. 2a, an initial screening of the solubility revealed that only polymers with alkyl side groups up to 3 carbons (Me to iPr or nPr) were water soluble at room temperature. The butyl sulfoxide-based polymethacrylate was completely insoluble in water and the polyacrylate analogue was only soluble upon cooling. As expected, all sulfone-based polymer analogues were non-soluble in water or alcohols as other polar, protic solvents (e.g. MeOH, EtOH). This demonstrates, that sulfone groups show a less pronounced ability to form strong hydrogen bonds and highlights the unique nature of the tunable amphiphilic sulfoxide polymers. In addition, these different solution properties of sulfoxides and sulfones support our assumption of using the sulfoxide oxidation as second trigger to change polymer solubility.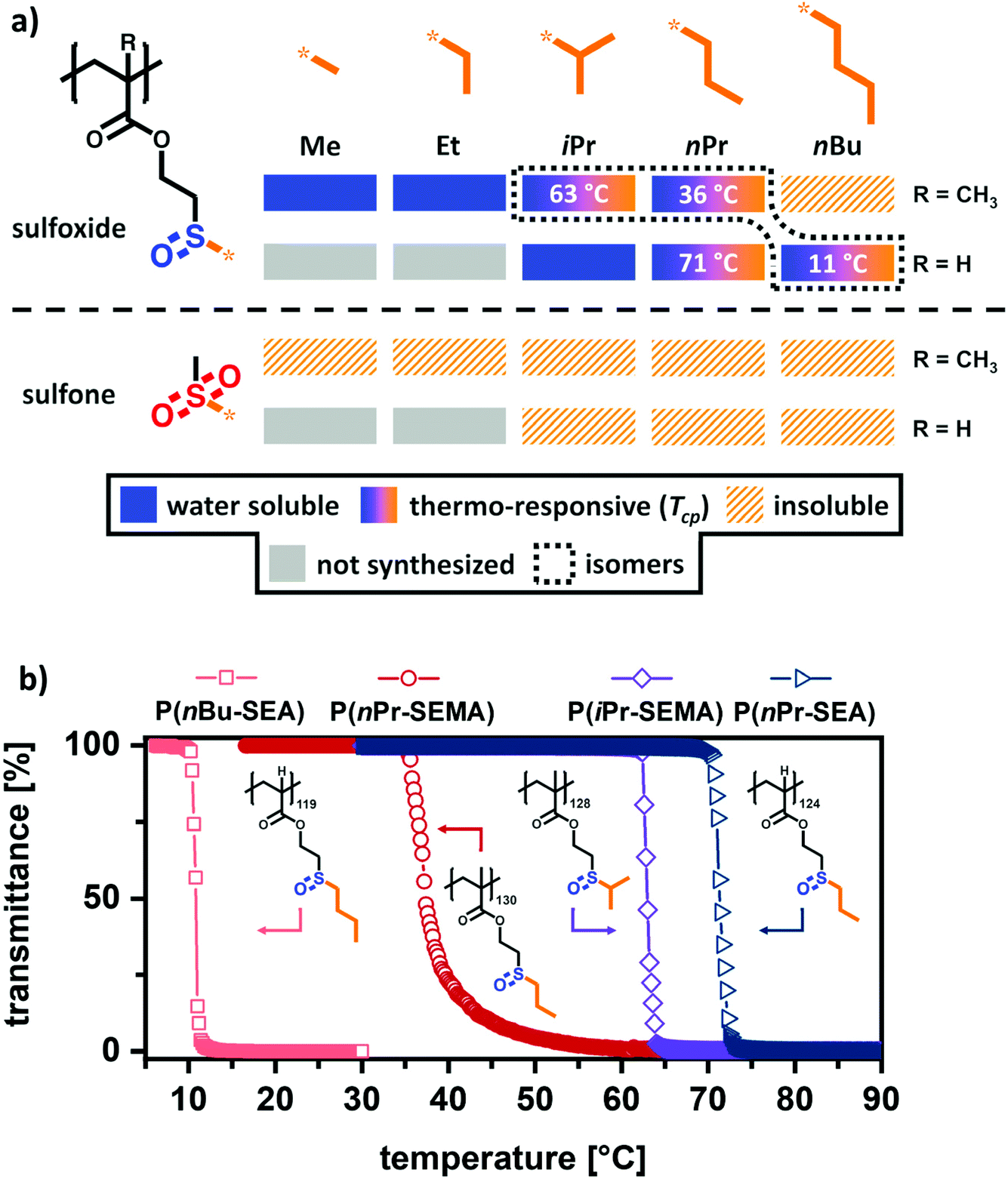 | ||
| Fig. 2 Overview of the temperature-dependent solution properties of sulfoxide and sulfone-based (meth)acrylate polymers. (a) Variation of alkyl length and backbone architecture induces a shift in amphiphilicity and thermo-responsive behavior, while the oxidized hydrophobic sulfone analogues are all water insoluble. (b) Of this library, the polymers P(nBu-SEA)119, P(nPr-SEMA)130, P(iPr-SEMA)128, and P(nPr-SEA)124 were observed to exhibit distinct cloud point temperatures of 11 °C, 36 °C, 63 °C and 71 °C, respectively. | ||
Looking into polymer solubility in more detail, temperature-dependent (5–90 °C) optical transmission measurements were performed to examine the temperature-responsiveness. As shown in Fig. 2a, a clear trend can be observed between an increasing alkyl group length and a reduced water solubility. While the sulfoxide polymers with a methyl P(Me-SEMA) or an ethyl group P(Et-SEMA) were completely water soluble over the whole investigated temperature range, the incorporation of an isopropyl P(iPr-SEMA) or n-propyl group P(nPr-SEMA) results in the occurrence of distinct cloud point temperatures. Adding one further –CH3 group (n-butyl) renders the methacrylate polymer P(nBu-SEMA) completely water insoluble in the examined temperature range. This trend can be further expanded to the acrylate polymers. As mentioned before, the tunability of the amphiphilic balance in our polymers is not only determined by the change in the alkyl groups’ length but also by changing the polymeric backbone itself. Here, the polyacrylate backbone is less hydrophobic and as consequence, the isopropyl polyacrylate derivative P(iPr-SEA) is completely water-soluble. This is in stark contrast to its polymethacrylate analogue P(iPr-SEMA). On the other hand, the n-propyl and n-butyl-sulfoxide polyacrylates P(nPr-SEA) and P(nBu-SEA) are observed to exhibit cloud point temperatures (Fig. 2a).
Interestingly, three sulfoxide polymers P(nBu-SEA)119, P(nPr-SEMA)130 and P(iPr-SEMA)128, that show a cloud point, are constitutional isomers. However, the polymer backbone and the different alkyl groups influence the temperature range in which the cloud point occurs very drastically. This can be seen in the respective transmittance versus temperature curves in Fig. 2b. From these curves, the cloud point temperatures were obtained as the minimum in plots of the first derivative (inflection point) against temperature (see ESI section V, Fig. S6†).46 From these measurements, it was determined that the polyacrylate P(nBu-SEA)119 exhibits a cloud point temperature at 11 °C, whereas the Tcp of the polymethacrylate isomer P(nPr-SEMA)130 is at 36 °C. Thus, the shorter n-propyl side group but additional methyl group in the backbone shift the Tcp close to human body temperature. In comparison, the Tcp of the isopropyl isomer P(iPr-SEMA)128 is increased significantly to 63 °C (Fig. 2b). Such a higher Tcp for the isopropyl polymer compared to the n-propyl-analogue has also been reported for poly(N-alkyl acrylamides). For example, the Tcp of poly(N-n-propyl acrylamide) (PNNPAM) (22 °C) is also decreased compared to its structural isomer poly(N-isopropyl acrylamide) (PNIPAM) (32 °C).47
In addition to Tcp changes between the constitutional isomers, the cloud point also changed with the polymer backbone. By comparing the polymethacrylate and the polyacrylate with the same n-propyl group, a 35 °C increase in cloud point temperature from P(nPr-SEMA)130 with Tcp = 36 °C to P(nPr-SEA)124 with Tcp = 71 °C (Fig. 2a) is observed. This shift of Tcp in dependence on the backbone is in accordance to literature where it was described for oligo(ethylene glycol) methacrylates and oligo(ethylene glycol) acrylates.48,49
As a consequence, the observed order of increasing cloud point temperature for the sulfoxide polymers P(nBu-SEMA) < P(nPr-SEMA) < P(iPr-SEMA) < P(nPr-SEA) agrees with established concepts.50
Detailed investigations on the RAFT polymerization of P(nPr-SEMA) and P(iPr-SEMA) as most promising functional polymers
Regarding potential applications, the polymers with cloud points above room temperature are of special interest. However, the utilization of such polymers in advanced materials requires a thorough understanding and control over the polymerization process itself (conversion, molecular weight, polymer dispersity, etc.). This would enable the synthesis of precisely tailored and complex polymer architectures such as functional block copolymers etc. To examine the level of control that can be achieved with the RAFT process, polymerization kinetics are assessed in detail for the nPr-SEMA and the iPr-SEMA monomers. Fig. 3a shows the conversion of nPr-SEMA during the polymerization. It can be seen that the monomer conversion increases rapidly to 64% in the first 8 hours and then reaches a plateau at around 82% with increasing polymerization time. As seen in the kinetic plot of ln([M]0/[M]) versus polymerization time in Fig. 3b, a linear correlation can be observed in the initial 8 h suggesting a constant number of active species. Taking the whole reaction time into account, however, the polymerization follows pseudo-first order kinetics. In a similar fashion, the RAFT polymerization of the isomer iPr-SEMA exhibits a conversion of 67% in the initial 8 hours with a plateau at around 75% (Fig. 3e) and similar polymerization kinetics (Fig. 3f). The slight deviation of monomer conversion from the ideal case of 100% may originate from the lower reactivity of the monomers. This could lead to increased chain termination processes which are in competition with the propagation of the polymer chain at the late stage of the polymerization.43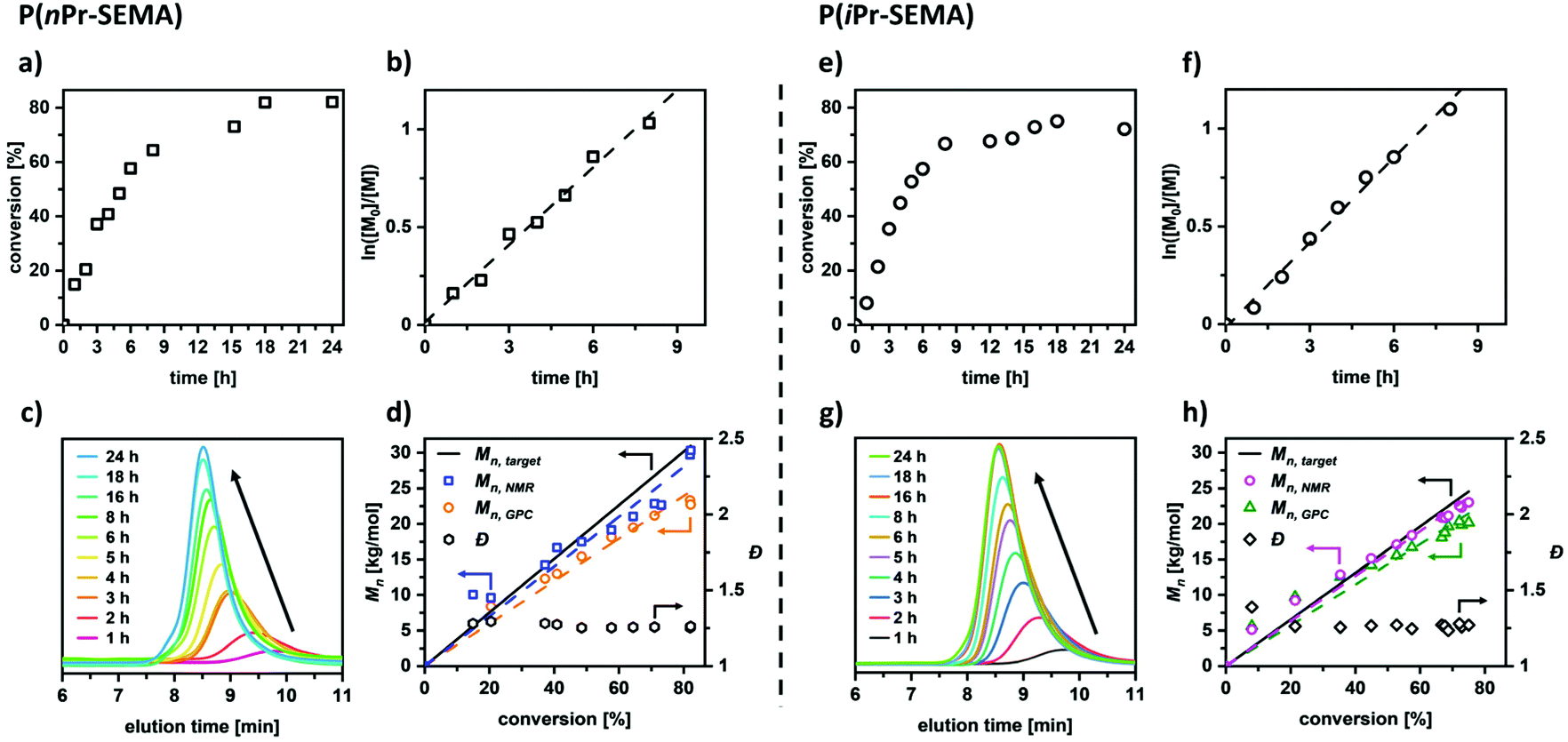 | ||
| Fig. 3 RAFT solution homopolymerization of 2-(n-propyl sulfoxide)ethyl methacrylate (nPr-SEMA) and 2-(isopropyl sulfoxide)ethyl methacrylate (iPr-SEMA) shows good control over the polymerization kinetics. (a and e) Monomer conversion and (b and f) kinetic plots of ln([M]0/[M]) versus polymerization time, (c and g) GPC traces as a function of elution time and (d and h) number-average molecular weights (Mn) and polymer dispersity (Đ) versus overall conversion under an initial feed ratio of [monomer]0:[CDTB]0:[AIBN]0 = 150:1:0.125. | ||
The respective GPC traces for the RAFT homopolymerizations confirm that the molecular weight grows faster in the initial 8 h, as shown by the decreasing shift of the GPC peaks with longer reaction times (see Fig. 3c for nPr-SEMA and Fig. 3g for iPr-SEMA). In addition to kinetic plots, the dependence of the molecular weight on the conversion serves as measure for the control over the system. For both investigated monomers (nPr-SEMA and iPr-SEMA), the number-average molecular weights Mn,GPC and Mn,NMR increase linearly with the monomer conversion. The molecular weight distributions remain relatively narrow throughout the whole polymerization process (Đ ≤ 1.25). As shown in Fig. 3d for nPr-SEMA and Fig. 3h for iPr-SEMA, the measured values for Mn, GPC are slightly lower than the calculated targeted molecular weight Mn, target. This might stem from the determination of the GPC values in comparison to PMMA calibration standards. In contrast, the values for Mn, NMR agree well with the theoretical molecular weights. Overall, the presented kinetic data demonstrate good control over the RAFT polymerization reaction, thus highlighting the potential of these versatile functional monomers for advanced polymeric materials.
Detailed studies on the thermo-responsive properties of P(nPr-SEMA) and P(iPr-SEMA)
Having demonstrated good control over the RAFT polymerization of P(nPr-SEMA) and P(iPr-SEMA), their thermo-responsive properties are assessed in more detail. It is known that the cloud point temperature (Tcp) of thermo-responsive polymers can depend on molecular weight, polymer concentration and salt concentration. Thus, it is of great importance to assess the influence of these parameters to gain a thorough understanding of the polymer properties.To examine the influence of polymer molecular weight, i.e. degree of polymerization (DP), additional polymers of varying chain lengths were synthesized (see ESI section VI, Table S3† for details). The respective DPs were 45, 130, and 213 for P(nPr-SEMA) and 48, 128, and 209 for P(iPr-SEMA). Comparing the temperature-dependent solubility of these polymers, it is observed that Tcp decreases with increasing polymer chain length for both polymers in solution (0.5 wt%). See Fig. 4a and d for temperature-dependent transmittance curves for P(nPr-SEMA) and P(iPr-SEMA), respectively. For P(nPr-SEMA), the Tcp decreases from 43 to 31 °C as the DP increases from 45 to 213. Thus, this polymer shows a moderate influence of the DP on the temperature response, which is similar to other typical thermo-responsive polymers like PNIPAM. Hence, the Tcp is robust against changes in polymer chain length. This is also the case for P(iPr-SEMA). Here, Tcp decreases from 71 °C to 61 °C as the DP increases from 48 to 209 (Fig. 4d). As known from other thermo-responsive polymers, this behavior is attributed to an increase in functional groups (alkyl sulfoxides) per polymer chain with increasing molecular weight. As a consequence, the net gain of free energy increases with an increased number of temperature-induced hydrophobic associations and released water molecules.50
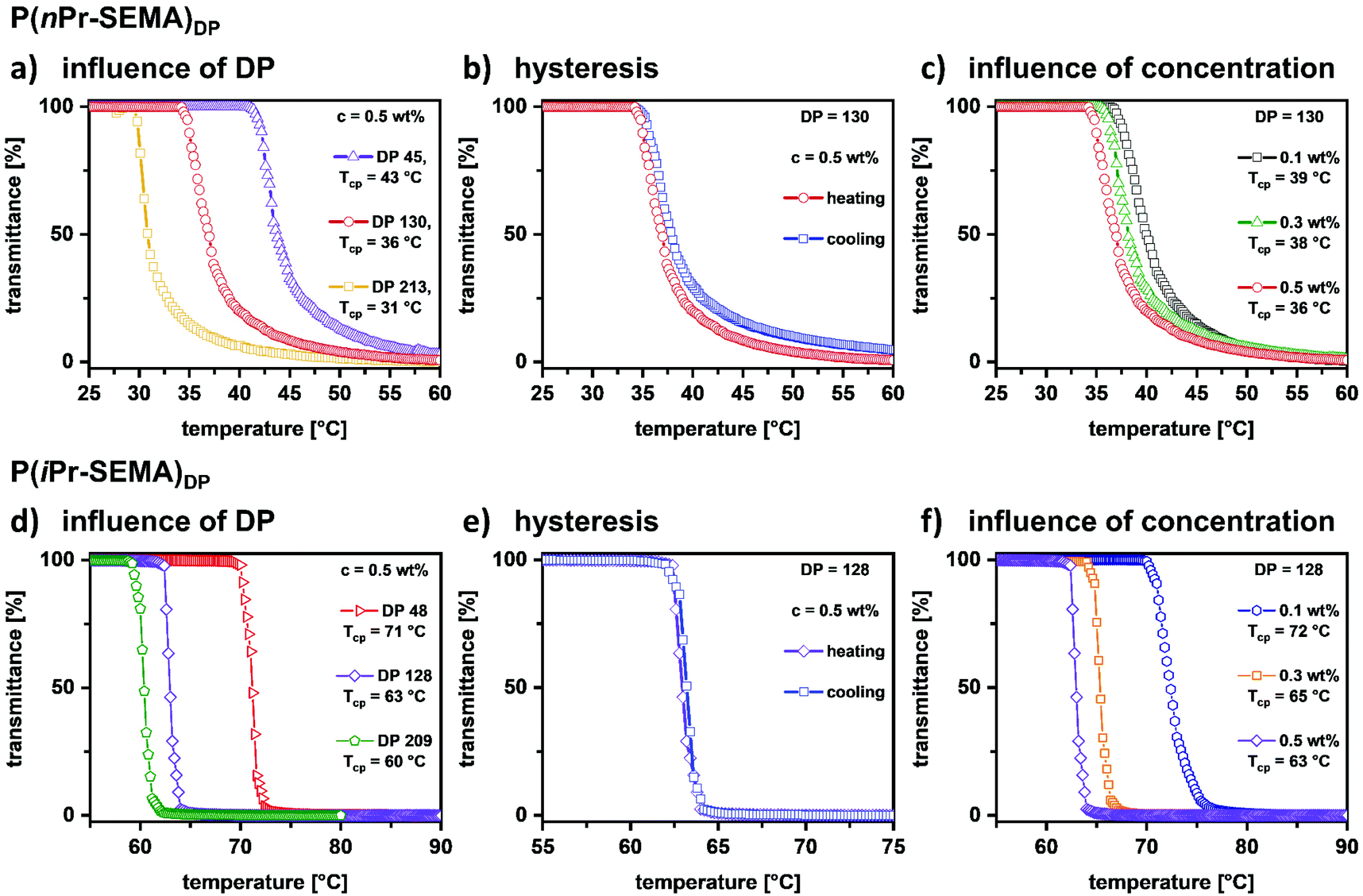 | ||
| Fig. 4 Detailed temperature-responsive examinations on P(iPr-SEMA) and P(nPr-SEMA) in aqueous solution to study the effect of: (a and d) changing the polymer molecular weight; (b and e) the heating or cooling cycle (hysteresis); and (c and f) the polymer concentration. | ||
Based on these findings, we assessed the influence of polymer dispersity, i.e. the width of the molecular weight distribution, on the Tcp. For this, free radical polymerizations were conducted to obtain polymers with comparable molecular weights but drastically higher dispersity values between Đ = 2.9 and 3.3 (see ESI section VII, Fig. S7 and S8†). For these free radical polymers, the observed effect of Mn on the Tcp is only marginal. This indicates that for such broad molecular weight distributions, the difference between a Mn of 14000 g mol−1 and 28900 g mol−1 is not as significant as for the corresponding RAFT polymers with much narrower dispersities. For these RAFT polymers with dispersities of around Đ = 1.2 to 1.3, the difference in Tcp is much more evident. This indicates the importance of low dispersity values for tunability of the thermo-responsive properties by the adjustment of the molecular weight (for a detailed discussion see ESI section VII†).
Furthermore, the influence of heating and cooling on the temperature-dependent phase transition was assessed for both polymers. This serves as a measure for the reversibility and robustness of the observed cloud point temperatures. Fig. 4b and e show the heating and cooling curves for P(nPr-SEMA)130 and P(iPr-SEMA)128, respectively (see ESI section VIII† for additional hysteresis curves). For all polymers these curves are nearly overlapping, thus not showing a pronounced hysteresis. This can be attributed to the lack of H-bond donors in their molecular structures. In general, during the cooling process, polymers that contain both (H-acceptors and H-donors) will form polymer–water H-bonds while also replacing the additional polymer–polymer H-bonds.35 Hence, such polymers will exhibit a thermo-responsive hysteresis.51 However, polymers without such proton donors were found to exhibit no hysteresis since no polymer–polymer H-bonds need to be overcome.52 As example for the latter, the new sulfoxide-based thermo-responsive polymers show distinct cloud point temperatures independent from the direction of the temperature change.
Next, the concentration-dependency of the transition temperatures was investigated as shown in Fig. 4c and f. With increasing polymer concentration from 0.1 to 0.5 wt%, the Tcp decreases from 39 °C to 36 °C for P(nPr-SEMA)130 and from 72 °C to 63 °C for P(iPr-SEMA)128, respectively. Also, the transition temperature range becomes slightly broader upon dilution. This well-documented effect is based on preferential intermolecular hydrophobic aggregation of the polymers at higher concentrations. Thus, a more concentrated polymer solution results in a lower Tcp and a narrower transition zone. Overall, a marginal deviation of 3 °C for the P(nPr-SEMA)130 highlights the robustness of the phase transition temperature against concentration effects, thus increasing its potential for applications.50
Ultimately, the influence of the salt concentration is examined since it is known that salt can reduce the transition temperature. Thus, determining this influence is important to assess the potential of the polymers for different applications where salts are present, e.g. in biological environments such as blood, mucus, etc. To analyze this behavior, sodium chloride was used as a typical example to influence the ionic strength of the polymer solutions. The temperature-dependent transmittance curves for both polymers were determined in dependence on c(NaCl) (Fig. 5a for P(nPr-SEMA)130 and Fig. 5c for P(iPr-SEMA)128). As expected, the cloud point temperature of both polymers decreases with increasing the sodium chloride concentration from 0 to 1 M. For P(nPr-SEMA)130Tcp decreases from 40 °C to 27 °C and for P(iPr-SEMA)128 from 71 °C to 47 °C (Fig. 5a and c). This trend becomes more evident if the Tcp values are plotted against the sodium chloride concentration. As shown in Fig. 5b for P(nPr-SEMA)130 and Fig. 5d for P(iPr-SEMA)128, both polymers exhibit a linear dependence of Tcp on the salt concentration. In addition, the thermal response in phosphate buffered saline (PBS) solution as a biologically more relevant environment was analyzed. Here, a Tcp of 39 °C for P(nPr-SEMA)130 and 64 °C P(iPr-SEMA)128 was observed which is in good agreement with the total sodium chloride concentration of 137 mM in PBS (Fig. 5b for P(nPr-SEMA)130, Fig. 5d for P(iPr-SEMA)128, see ESI section IX, Fig. S10† for respective transmittance curves). Overall, these results agree with the literature where the so-called “salting-out”-effect is often observed for thermo-responsive polymers. Salt ions disrupt the hydrogen-bonds between water molecules and polymer chains, thus promoting the hydrophobic polymer–polymer interactions. As a result, the transition temperatures decrease.
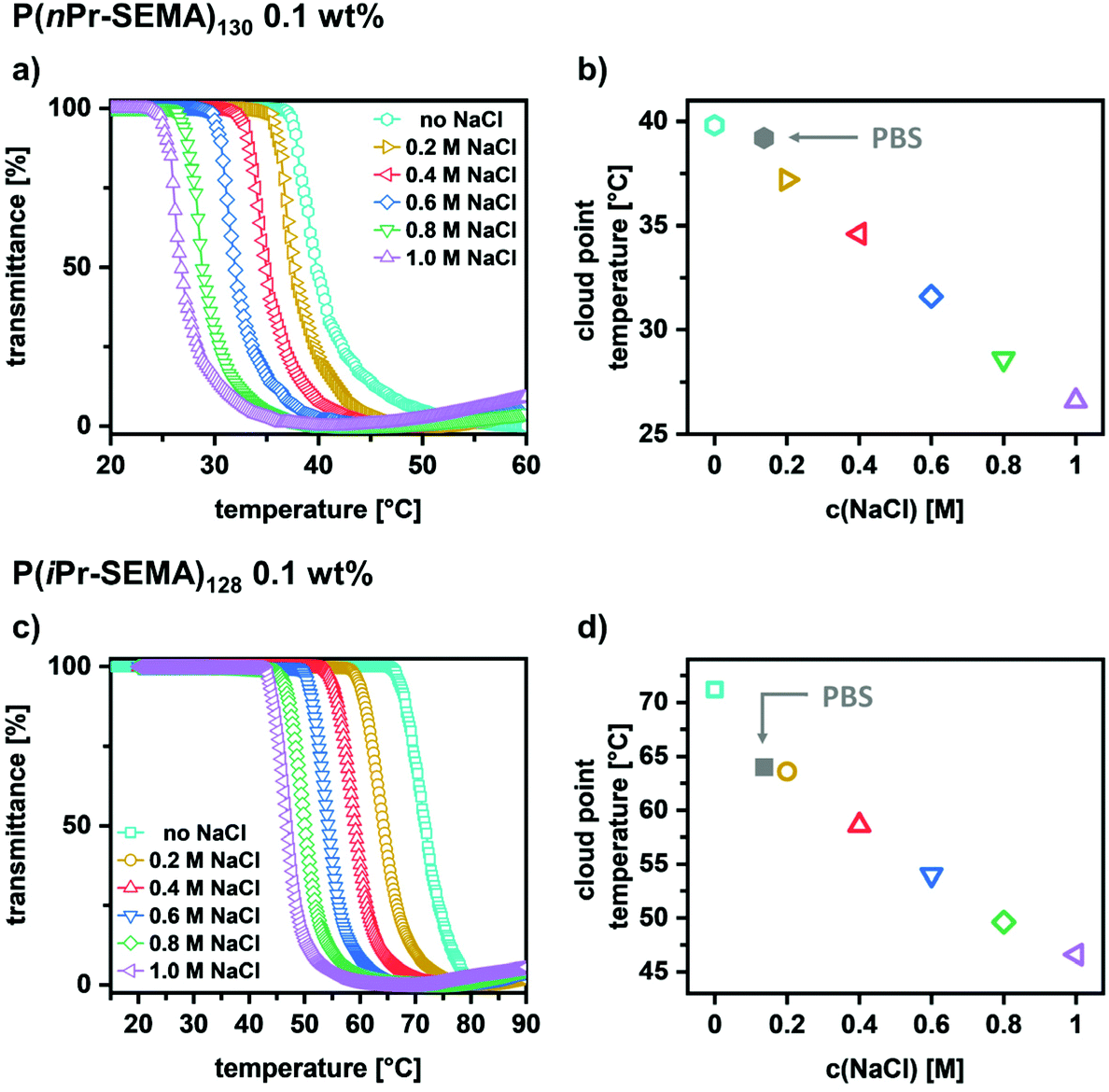 | ||
| Fig. 5 Influence of the sodium chloride concentration on the cloud point temperature of aqueous (a) P(nPr-SEMA)130 and (c) P(iPr-SEMA)128 solutions (0.1 wt%) showing the common observed “salting-out”-effect for thermo-responsive polymers. A linear dependency of the Tcp values on the salt concentration was observed for both polymers (b and d). | ||
Oxidation-responsive properties of P(nPr-SEMA) and P(iPr-SEMA)
Due to the sulfoxide groups, the thermo-responsive phase transition of P(nPr-SEMA) and P(iPr-SEMA) is expected to change upon oxidation to the respective sulfone moieties. It is assumed that an increasing degree of oxidation, i.e. increasing sulfone content, per polymer decreases the homopolymer hydrophilicity, thus shifting Tcp to lower temperatures.These anticipated shifts in thermal response were investigated for P(nPr-SEMA)130 and P(iPr-SEMA)128. For this, the respective aqueous polymer solutions (0.1 wt%) were first subjected to different hydrogen peroxide concentrations for 6 hours at 37 °C. Hydrogen peroxide was used here as a more biological relevant oxidizing agent. 1H-NMR investigations on the resulting polymers revealed partial oxidation of the sulfoxides to sulfone side groups (see ESI section X† for details). Thus, as a response to the reactive oxygen species in H2O2, the sulfoxide homopolymers are transformed into copolymers containing sulfoxide and sulfone side groups (Fig. 6a). Here, the content of sulfone groups increases with the H2O2 concentration and dramatically influences the transition temperatures. This can be seen from the temperature-dependent transmittance measurements shown in Fig. 6b for P(nPr-SEMA)130 and Fig. 6e for P(iPr-SEMA)128 at 0.1 wt% polymer concentration.
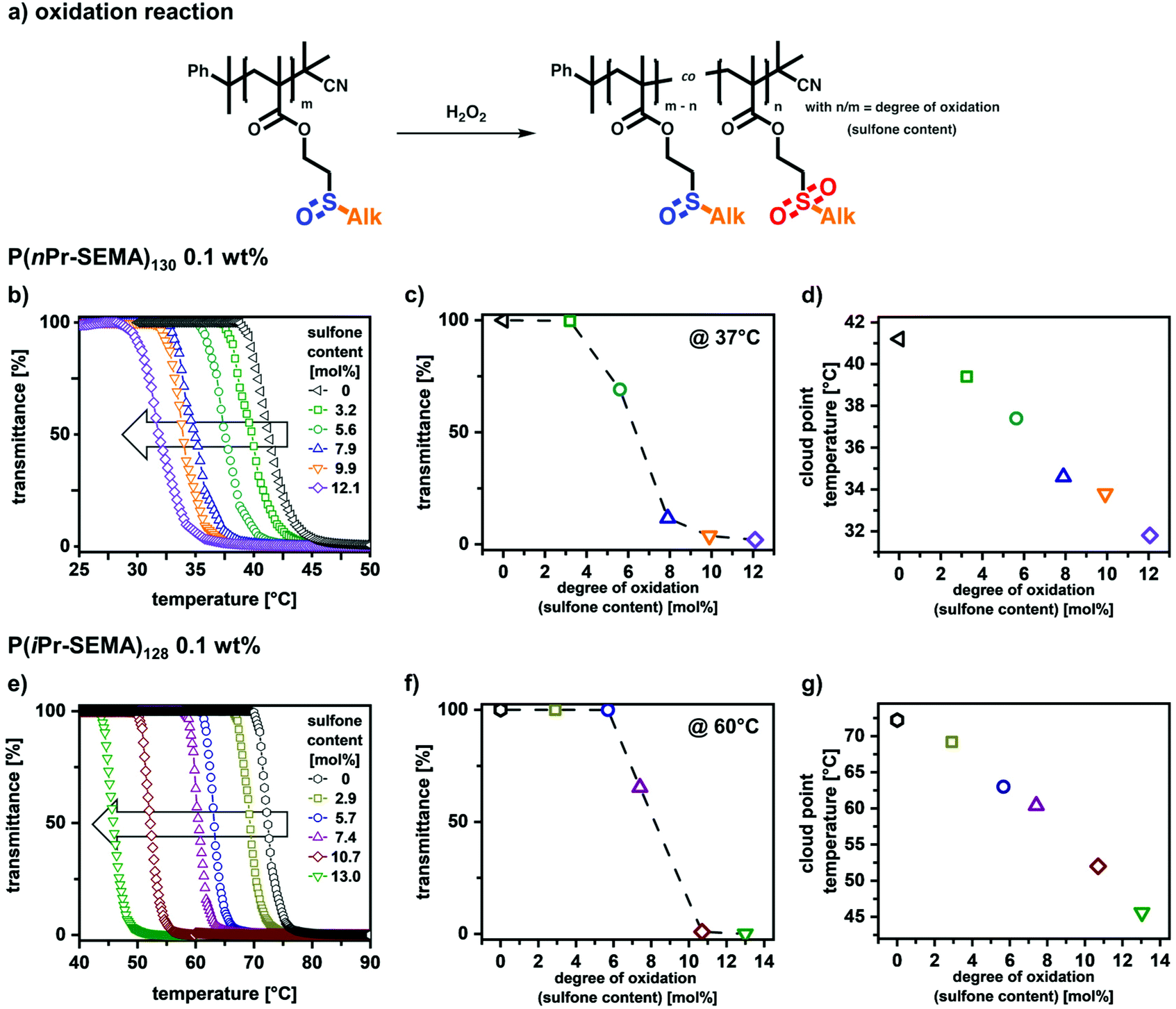 | ||
| Fig. 6 Demonstration of the oxidation-responsive behavior of P(nPr-SEMA)130 and P(iPr-SEMA)128. (a) Transformation of sulfoxide homopolymers into copolymers containing sulfoxide and sulfone side groups occurs via a facile oxidation reaction with H2O2. (b and e) The oxidation-induced shifts in cloud point temperature are presented as temperature-dependent transmittance curves for the partially oxidized P(nPr-SEMA)130 and P(iPr-SEMA)128 (0.1 wt% solutions). (c and f) At a fixed temperature, e.g. 37 °C for P(nPr-SEMA)130 and 60 °C for P(iPr-SEMA)128, the solubility depends drastically on the degree of oxidation. (d and g) The Tcp decrease linearly with the degree of oxidation, thus demonstrating the ability to gradually adjust the thermo-responsive profile via application of the second oxidation stimulus. | ||
For P(nPr-SEMA)130, the native non-oxidized polymer exhibits a Tcp at 41 °C. With increasing oxidation of hydrophilic sulfoxide groups to more hydrophobic sulfones, this Tcp is decreasing to 32 °C for a sulfone content of 12 mol% (Fig. 6d). Regarding the structural isomer P(iPr-SEMA)128, the decrease of Tcp from 72 °C (non-oxidized) to 46 °C (sulfone content of around 13 mol%) is much more significant (Fig. 6g). For both polymers, this partial oxidation leads to random amphiphilic copolymers which assemble into colloidal aggregates with different sizes in aqueous dispersion above the respective Tcp (see ESI section XI† for further information). In contrast, for sulfone contents over 20 mol% both polymers become completely water insoluble.
These results demonstrate two important aspects; first, a clear oxidation-responsive behavior was shown for P(nPr-SEMA) and P(iPr-SEMA). At a specific constant temperature, the polymer solubility clearly depends on the oxidation state of the sulfur-based side groups and the degree of oxidation. As shown in Fig. 6c for P(nPr-SEMA), this becomes interesting for the oxidation reaction at 37 °C. Here, a dramatic decrease in solubility occurs upon reaching a sulfone content of around 6.5 mol%. Similar effects are observed for P(iPr-SEMA). However, since here the Tcp is shifted to higher temperatures, its oxidation-dependent solubility was shown at 60 °C. At this exemplary temperature, a dramatic decrease in transmittance was observed upon reaching a sulfone content of around 8 mol% (Fig. 6f). Consequently, this oxidation sensitivity can be used to induce “smart” materials’ responses at fixed temperatures.
Second, in addition to this oxidation response, the partial oxidation of the monomeric units can also be used as synthetic concept to tune the Tcp of P(nPr-SEMA) and P(iPr-SEMA) very precisely. Upon oxidation, the sulfoxide-based homopolymers are transformed into copolymers containing both sulfoxide and sulfone groups randomly distributed along the same backbone. The balance between hydrophilicity (sulfoxide groups) and hydrophobicity (sulfone groups) can be varied accurately by the degree of oxidation. This results in a well-defined linear dependency between Tcp and the sulfone content (Fig. 6d for P(nPr-SEMA) and Fig. 6g for P(iPr-SEMA)) and allows for the precise tuning of the thermo-responsive polymer properties. A similar control is mostly realized via copolymer systems. However, copolymerization of two monomers always requires the synthesis of a new batch to vary the composition. Since this can also result in varying molecular weights and polymer dispersities, our oxidative post-polymerization modification approach drastically simplifies this tunability.
Demonstrating good biocompatibility via basic toxicological tests
Having demonstrated tunable thermo-responsive properties of the sulfoxide polymers, their biocompatibility was assessed via in vitro cell viability experiments. Since P(nPr-SEMA) exhibits a Tcp near body temperature, it is important to exclude potential polymer cytotoxicity in order to demonstrate the potential of our materials for applications in the biomedical field.For this, the cell counting kit-8 (CCK-8) colorimetric assay was conducted on cultured subcutaneous connective tissue cells of mice (L929). This cell type is used among others as standard biological material to assess the tolerability of polymeric materials in an in vitro environment.53–55 The tests were performed with four different polymer concentrations of 0.01, 0.05, 0.1, and 0.5 mg mL−1 and incubated for 24 hours at 37 °C. Non-cell treated medium was used as control.
The results of the CCK-8 assay are shown in Fig. 7 and indicate that all tested functional sulfoxide polymers are well tolerated by L929 cells after 24 hours. Overall, this supports good biocompatibility of the presented dual-functional sulfoxide polymers. These biological properties were already shown for methyl sulfoxide acrylates which is regarded as potential PEG alternative.36,41 However, it is of great importance to show this non-toxic behavior also for our sulfoxide polymers with increasing hydrophobic alkyl groups. While such amphiphilic structures may present undesired hydrophobic interactions with the cell membrane, no significant influence on the cell viability was observed. This highlights the potential of our multi-stimuli-responsive sulfoxide homopolymers to be utilized for a variety of biological applications.
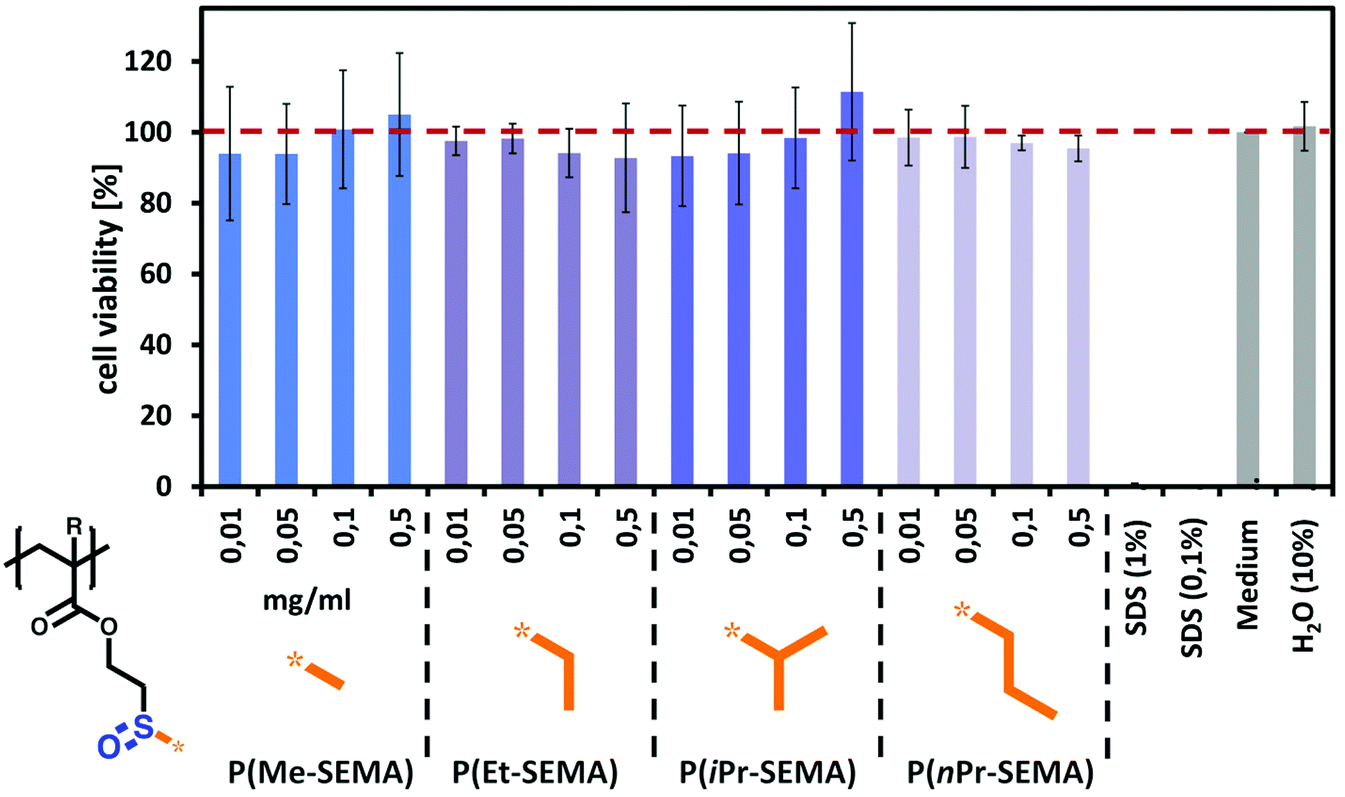 | ||
| Fig. 7 Cell viability tests on cultured subcutaneous connective tissue cells (L929) show no pronounced cytotoxic effects, thus suggesting good biocompatibility of the functional polymers. Cell viability is presented as percentage with respect to the medium and as mean ± SD (n = 3). The data were assessed via the CCK-8 assay after exposure of the cells to aqueous solutions of the respective sulfoxide methacrylates (0.01, 0.05, 0.1, and 0.5 mg mL−1) for 24 hours. | ||
Conclusion
We have demonstrated that a rational molecular design can be used to introduce thermo- and oxidation-responsive properties into one functional monomer. Key to this approach is the combination of highly hydrophilic sulfoxide moieties with varying hydrophobic alkyl groups.By systematically investigating the influence of different structural parameters, i.e. alkyl groups, polymer backbone, degree of polymerization (DP), this study lays the foundation for the preparation of new materials with well-defined and robust thermo-responsive profiles. As one promising candidate, P(nPr-SEMA) was found to exhibit a cloud point temperature close to human body temperature, thus making the thermal-response attractive for potential biomedical applications. Regarding the dual-stimuli-responsive properties, combining the thermal-response with an oxidation-sensitivity represents a unique way to realize more complex response profiles.
On one hand, this is achieved through the oxidation-dependent shift of the polymer's solubility at a fixed temperature. On the other hand, this allows to precisely tune the cloud point temperature by altering the balance between hydrophilic sulfoxide groups and hydrophobic sulfone groups. In contrast to this simple post-polymerization oxidation strategy, a similar control was mostly realized via copolymer systems. However, copolymerization of two monomers always requires the synthesis of a new batch to vary the composition which can influence molecular weights and polymer dispersities. By simplifying this tunability of thermo-responsiveness, our approach represents a simple yet powerful expansion of the functional polymer toolbox.
Overall, we believe that combining the thermal and oxidation response in one single functional unit, its facile synthesis, and well-controlled polymerization, presents the starting point for the preparation of highly sophisticated materials for a wide variety of applications.
Materials and methods
Materials
All starting materials and reagents were purchased from commercial sources and used without further purification, unless otherwise stated. Methacryloyl chloride (97%) and triethylamine (99%) were purchased from abcr GmbH. Anhydrous N,N-dimethylformamide (DMF, 99.8%, extra dry, stored over molecular sieve) was purchased from Acros Organics. 2-(Methylthio)ethan-1-ol, 2-(ethylthio)ethan-1-ol, 2-(i-propylthio)ethan-1-ol, 2-(n-propylthio)ethan-1-ol, 2-(n-butylthio)ethan-1-ol and p-toluenesulfonic acid monohydrate were purchased from Fluorochem Ltd. Acryloyl chloride (stabilized with phenothiazine) and α-methylstyrene (99%) were purchased from Merck KGaA. 2,2′-Azobis(2-methylpropionitrile) (AIBN, 98%), carbon disulfide (≥99.9%), 2,6-di-tert-butyl-p-cresol (>99%), meta-chloroperbenzoic acid (mCPBA, ≥77%) and phenyl magnesium bromide solution (1 M in THF) were purchased from Sigma-Aldrich. Cumyl dithiobenzoate (CDTB) was synthesized according to literature procedures.56 Ultrapure water was taken from a LaboStar UV 2 water system. Moisture and/or air sensitive reactions were carried out in dry glassware under nitrogen atmosphere.600000 g mol−1). Polymer sample solutions (1.5 mg ml−1) were prepared in the GPC eluent and filtered (PTFE, 450 μm) prior to injection. Chromatograms were processed using the PSS WinGPC UniChrom software.
Syntheses
Me-TEMA (chromatography: DCM/methanol 3 v/v%; yield 92%) 1H NMR (500 MHz, CDCl3): δ = 6.12 (s, 1H, Hcis), 5.57 (s, 1H, Htrans), 4.32 (t, J = 6.9 Hz, 2H, –O–CH2–), 2.76 (t, J = 6.9 Hz, 2H, –CH2–O–), 2.16 (s, 3H, –S–CH3), 1.94 (s, 3H, –CH3) ppm; 13C NMR (126 MHz, CDCl3): δ = 167.33, 136.28, 125.91, 63.55, 32.71, 18.42, 15.98 ppm; HRMS: calc. for C7H12O2S [M + H]+: 161.0636, found [M + H]+: 161.0641.
Et-TEMA (chromatography: DCM/methanol 3 v/v%; yield 88%) 1H NMR (500 MHz, CDCl3): δ = 6.11 (s, 1H, Hcis), 5.57 (s, 1H, Htrans), 4.29 (t, J = 7.0 Hz, 2H, –O–CH2–), 2.79 (t, J = 7.0 Hz, 2H, –CH2–S–), 2.60 (q, J = 7.5 Hz, 2H, –S–CH2–), 1.94 (s, 3H, –CH3), 1.26 (t, J = 7.4 Hz, 3H, –CH2–![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3) ppm; 13C NMR (126 MHz, CDCl3): δ = 167.32, 136.30, 125.89, 64.06, 30.10, 26.34, 18.42, 14.94 ppm; HRMS: calc. for C8H14O2S [M + K]+: 213.0352, found [M + K]+: 213.0554.
3) ppm; 13C NMR (126 MHz, CDCl3): δ = 167.32, 136.30, 125.89, 64.06, 30.10, 26.34, 18.42, 14.94 ppm; HRMS: calc. for C8H14O2S [M + K]+: 213.0352, found [M + K]+: 213.0554.
iPr-TEMA (distillation: 0.12 mbar, 39–42 °C; yield 71%) 1H NMR (500 MHz, CDCl3): δ = 6.12 (s, 1H, Hcis), 5.58 (t, J = 1.6 Hz, 1H, Htrans), 4.28 (t, J = 7.1 Hz, 2H, –O–CH2–), 3.00 (p, J = 6.7 Hz, 1H, –S–CH–), 2.80 (t, J = 7.1 Hz, 2H, –CH2–S–), 1.94 (s, 3H, –CH3), 1.28 (d, J = 6.7 Hz, 6H, (–CH3)2) ppm; 13C NMR (126 MHz, CDCl3): δ = 167.34, 136.32, 125.90, 64.34, 35.23, 29.00, 23.57, 18.44 ppm; HRMS: calc. for C9H16O2S [M + Na]+: 211.0769, found [M + Na]+: 211.0768.
nPr-TEMA (distillation: 0.12 mbar, 40–45 °C; yield 82%) 1H NMR (500 MHz, CDCl3): δ = 6.11 (s, 1H, Hcis), 5.57 (s, 1H, Htrans), 4.29 (t, J = 7.0 Hz, 2H, –O–CH2–), 2.77 (t, J = 7.0 Hz, 2H, –CH2–S–), 2.55 (t, 2H, –S–CH2–), 1.94 (s, 3H, –CH3), 1.67–1.57 (m, 2H, –2–CH3), 0.98 (t, J = 7.3 Hz, 3H, –CH2–3) ppm; 13C NMR (126 MHz, CDCl3): δ = 167.33, 136.31, 125.89, 64.09, 34.56, 30.48, 23.14, 18.43, 13.52 ppm; HRMS: calc. for C9H16O2S [M + Na]+: 211.0769, found [M + Na]+: 211.0754.
nBu-TEMA (chromatography: DCM/methanol 3 v/v%; yield 58%) 1H NMR (500 MHz, CDCl3): δ = 6.11 (s, 1H, Hcis), 5.57 (s, 1H, Htrans), 4.29 (t, J = 7.0 Hz, 2H, –O–CH2–), 2.78 (t, J = 7.0 Hz, 2H, –CH2–S–), 2.60–2.54 (m, 2H, –S–CH2–), 1.94 (s, 3H, –CH3), 1.58 (dt, J = 15.0, 6.8 Hz, 2H, –S–CH2–2–), 1.40 (dq, J = 14.6, 7.4 Hz, 2H, –2–CH3), 0.91 (t, J = 7.4 Hz, 3H, –CH2–3) ppm. 13C NMR (126 MHz, CDCl3): δ = 167.33, 136.31, 125.88, 64.10, 32.24, 31.92, 30.57, 22.06, 18.43, 13.78 ppm. HRMS: calc. for C10H18O2S [M + Na]+: 225.0925, found [M + Na]+: 225.0913.
iPr-TEA (chromatography: DCM/methanol 3 v/v%; yield 48%) 1H NMR (500 MHz, CDCl3): δ = 6.42 (dd, J = 17.3, 1.5 Hz, 1H, Hcis), 6.12 (dd, J = 17.3, 10.5 Hz, 1H, –CH–), 5.84 (dd, J = 10.4, 1.5 Hz, 1H, Htrans), 4.30 (t, J = 7.1 Hz, 2H, –O–CH2–), 2.99 (p, J = 6.7 Hz, 1H, –S–CH–), 2.80 (t, J = 7.1 Hz, 2H, –CH2–S–), 1.28 (d, J = 6.7 Hz, 6H, (–CH3)2) ppm. 13C NMR (126 MHz, CDCl3): δ = 166.10, 131.21, 128.39, 64.11, 35.26, 28.99, 23.55 ppm. HRMS: calc. for C8H14O2S [M + Na]+: 197.0612, found [M + Na]+: 197.0602.
nPr-TEA (chromatography: DCM/methanol 3 v/v%; yield 53%) 1H NMR (500 MHz, CDCl3): δ = 6.42 (dd, J = 17.3, 1.4 Hz, 1H, Hcis), 6.12 (dd, J = 17.3, 10.5 Hz, 1H, –CH–), 5.84 (dd, J = 10.5, 1.4 Hz, 1H, Htrans), 4.30 (t, J = 7.0 Hz, 2H, –O–CH2–), 2.77 (t, J = 7.0 Hz, 2H, –CH2–S–), 2.55 (t, J = 7.3 Hz, 2H, –S–CH2–), 1.68–1.54 (m, 2H, –2–CH3), 0.99 (t, J = 7.3 Hz, 3H, –CH3) ppm. 13C NMR (126 MHz, CDCl3): δ = 166.09, 131.20, 128.38, 63.88, 34.58, 30.49, 23.13, 13.53 ppm. HRMS: calc. for C8H14O2S [M + Na]+: 197.0612, found [M + Na]+: 197.0582.
nBu-TEA (chromatography: DCM/methanol 3 v/v%; yield 62%) 1H NMR (500 MHz, CDCl3): δ = 6.42 (dd, J = 17.3, 1.4 Hz, 1H, Hcis), 6.13 (dd, J = 17.3, 10.4 Hz, 1H, –CH–), 5.84 (dd, J = 10.5, 1.4 Hz, 1H, Htrans), 4.30 (t, J = 7.0 Hz, 2H, –O–CH2–), 2.78 (sbr, 2H, –CH2–S–), 2.58 (sbr, 2H, –S–CH2–), 1.58 (p, J = 7.4 Hz, 2H, –S–CH2–2–), 1.41 (h, J = 7.4 Hz, 2H, –2–CH3), 0.91 (t, J = 7.4 Hz, 3H, –CH3) ppm. 13C NMR (126 MHz, CDCl3): δ = 166.10, 131.21, 128.39, 63.90, 32.23, 31.92, 30.53, 22.06, 13.79 ppm. HRMS: calc. for C9H16O2S [M + Na]+: 211.0769, found [M + Na]+: 211.0759.
Me-SEMA (solid; yield 78%) 1H NMR (500 MHz, CDCl3): δ = 6.12 (s, 1H, Hcis), 5.58 (s, 1H, Htrans), 4.32 (t, J = 6.8 Hz, 2H, –O–CH2–), 2.80 (sbr, 2H, –CH2–SO–), 2.20 (sbr, 3H, –SO–CH3), 1.94 (s, 3H, –CH3) ppm. 13C NMR (126 MHz, CDCl3): δ = 167.32, 136.28, 129.10, 125.91, 32.63, 18.42, 18.05 ppm. HRMS: calc. for C7H13O3S [M + Na]+: 199.0405, found [M + Na]+: 199.0414.
Et-SEMA (liquid; yield 82%) 1H NMR (400 MHz, CDCl3): δ = 6.14 (s, 1H, Hcis), 5.61 (s, 1H, Htrans), 4.69–4.48 (m, 2H, –O–CH2–), 3.10–2.92 (m, 2H, –CH2–SO–), 2.79 (qd, J = 7.5, 3.1 Hz, 2H, –SO–CH2–), 1.94 (s, 3H, –CH3), 1.36 (t, J = 7.5 Hz, 3H, –CH2–3) ppm. 13C NMR (101 MHz, CDCl3): δ = 167.06, 135.82, 126.62, 57.60, 50.79, 46.24, 18.35, 6.89 ppm. HRMS: calc. for C8H14O3S [M + Na]+: 213.0561, found [M + Na]+: 213.0576.
iPr-SEMA (liquid; yield 75%) 1H NMR (500 MHz, CDCl3): δ = 6.14 (s, 1H, Hcis), 5.61 (s, 1H, Htrans), 4.69–4.51 (m, 2H, –O–CH2–), 3.06–2.94 (m, 1H, –SO–CH–), 2.92–2.81 (m, 2H, –CH2–SO–), 1.94 (s, 3H, –CH3), 1.31 (dd, J = 25.0, 6.9 Hz, 6H, (–CH3)2) ppm. 13C NMR (126 MHz, CDCl3): δ = 167.10, 135.88, 126.55, 57.90, 50.87, 48.06, 18.35, 16.01, 14.82 ppm. HRMS: calc. for C9H16O3S [M + Na]+: 227.0718, found [M + Na]+: 227.0712.
nPr-SEMA (liquid; yield 85%) 1H NMR (600 MHz, CDCl3): δ = 6.13 (s, 1H, Hcis), 5.61 (s, 1H, Htrans), 4.69–4.48 (m, 2H, –O–CH2–), 3.09–2.92 (m, 2H, –CH2–SO–), 2.82–2.64 (m, 2H, –SO–CH2–), 1.94 (s, 3H, –CH3), 1.88–1.79 (m, 2H, –2–CH3), 1.09 (t, J = 7.4 Hz, 3H, –CH2–3) ppm. 13C NMR (151 MHz, CDCl3): δ = 167.05, 135.84, 126.60, 126.59, 57.57, 55.08, 51.62, 51.60, 18.36, 16.44, 13.50 ppm. HRMS: calc. for C9H16O3S [M + Na]+: 227.0718, found [M + Na]+: 227.0730.
nBu-SEMA (liquid; yield 69%) 1H NMR (600 MHz, CDCl3): δ = 6.13 (s, 1H, Hcis), 5.60 (s, 1H, Htrans), 4.67–4.50 (m, 2H, –O–CH2–), 3.11–2.93 (m, 2H, –CH2–SO–), 2.85–2.68 (m, 2H, –SO–CH2–), 1.94 (s, 3H, –CH3), 1.82–1.71 (m, 2H, –SO–CH2–2–), 1.58–1.40 (m, 2H, –2–CH3), 0.96 (t, J = 7.3 Hz, 3H, –CH2–3) ppm. 13C NMR (151 MHz, CDCl3): δ = 167.03, 135.82, 126.59, 57.58, 52.74, 51.46, 24.71, 22.15, 18.35, 13.78, 13.78 ppm. HRMS: calc. for C10H18O3S [M + Na]+: 241.0874, found [M + Na]+: 241.0862.
nPr-SEA (liquid; yield 85%) 1H NMR (500 MHz, CDCl3): δ = 6.44 (dd, J = 17.3, 1.3 Hz, 1H, Hcis), 6.13 (dd, J = 17.4, 10.5 Hz, 1H, –CH–), 5.88 (dd, J = 10.5, 1.3 Hz, 1H, Htrans), 4.68–4.51 (m, 2H, –O–CH2–), 3.08–2.91 (m, 2H, –CH2–SO–), 2.83–2.62 (m, 2H, –SO–CH2–), 1.88–1.77 (m, 2H, –2–CH3), 1.09 (t, J = 7.4 Hz, 3H, –CH3) ppm. 13C NMR (126 MHz, CDCl3): δ = 131.92, 127.86, 120.04, 57.46, 54.99, 51.46, 16.42, 13.49 ppm. HRMS: calc. for C8H14O3S [M + Na]+: 213.0561, found [M + Na]+: 213.0563.
iPr-SEA (liquid; yield 73%) 1H NMR (500 MHz, CDCl3): δ = 6.44 (dd, J = 17.3, 1.3 Hz, 1H, Hcis), 6.13 (dd, J = 17.3, 10.5 Hz, 1H, –CH–), 5.87 (dd, J = 10.5, 1.3 Hz, 1H, Htrans), 4.69–4.52 (m, 2H, –O–CH2–), 3.04–2.95 (m, 1H, –SO–CH–), 2.91–2.82 (m, 2H, –CH2–SO–), 1.31 (dd, J = 24.6, 6.9 Hz, 6H, (–CH3)2) ppm. 13C NMR (126 MHz, CDCl3): δ = 165.85, 131.85, 127.91, 57.78, 50.93, 47.96, 15.98, 14.82 ppm. HRMS: calc. for C8H14O3S [M + Na]+: 213.0561, found [M + Na]+: 213.0571.
nBu-SEA (liquid; yield 77%) 1H NMR (600 MHz, CDCl3): δ = 6.43 (dd, J = 17.4, 1.2 Hz, 1H, Hcis), 6.12 (dd, J = 17.3, 10.5 Hz, 1H, –CH–), 5.87 (dd, J = 10.4, 1.2 Hz, 1H, Htrans), 4.70–4.49 (m, 2H, –O–CH2–), 3.12–2.91 (m, 2H, –CH2–SO–), 2.85–2.66 (m, 2H, –SO–CH2–), 1.83–1.69 (m, –SO–CH2–2–), 1.58–1.40 (m, 2H, –2–CH3), 0.96 (t, J = 7.3 Hz, 3H, –CH3) ppm. 13C NMR (151 MHz, CDCl3): δ = 165.79, 131.92, 127.83, 57.46, 52.77, 51.39, 24.68, 22.13, 13.78 ppm. HRMS: calc. for C9H16O3S [M + Na]+: 227.0718, found [M + Na]+: 227.0702.
:1 v/v%). After a lyophilization step, the final sulfone homopolymers were yielded as colorless, fluffy solids.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. Işık and D. Klinger acknowledge financial support for consumables from Verband der Chemischen Industrie eV (VCI) and the Focus Area Nanoscale of the Freie Universität Berlin. We would like to acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG.References
- J. J. Green and J. H. Elisseeff, Nature, 2016, 540, 386–394 CrossRef CAS
.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858–875 CrossRef CAS
- P. Schattling, F. D. Jochum and P. Theato, Polym. Chem., 2014, 5, 25–36 RSC
- C. M. Wells, M. Harris, L. Choi, V. P. Murali, F. D. Guerra and J. A. Jennings, J. Funct. Biomater., 2019, 10, 34 CrossRef CAS
- L. Hu, Y. Wan, Q. Zhang and M. J. Serpe, Adv. Funct. Mater., 2020, 30, 1903471 CrossRef CAS
- J. Qu, Q.-y. Wang, K.-l. Chen, J.-b. Luo, Q.-h. Zhou and J. Lin, Colloids Surf., B, 2018, 170, 373–381 CrossRef CAS
- M. Tang, Q. Zheng, N. Tirelli, P. Hu, Q. Tang, J. Gu and Y. He, React. Funct. Polym., 2017, 110, 55–61 CrossRef CAS
- P. Schattling, F. D. Jochum and P. Theato, Chem. Commun., 2011, 47, 8859–8861 RSC
- Z. Cheng, S. Liu, P. W. Beines, N. Ding, P. Jakubowicz and W. Knoll, Chem. Mater., 2008, 20, 7215–7219 CrossRef CAS
- S. E. Herrera, M. L. Agazzi, M. L. Cortez, W. A. Marmisollé, M. Tagliazucchi and O. Azzaroni, Chem. Commun., 2019, 55, 14653–14656 RSC
- S. Wang, Z. Xu, T. Wang, T. Xiao, X.-Y. Hu, Y.-Z. Shen and L. Wang, Nat. Commun., 2018, 9, 1737 CrossRef
- K. Zhang, M. Zhang, X. Feng, M. A. Hempenius and G. J. Vancso, Adv. Funct. Mater., 2017, 27, 1702784 CrossRef
- H. Zhang, W. Wu, X. Zhao and Y. Zhao, Macromolecules, 2017, 50, 3411–3423 CrossRef CAS
- T. Uemukai, T. Hioki and M. Ishifune, Int. J. Polym. Sci., 2013, 2013, 196145 Search PubMed
- C. Xiao, J. Ding, L. Ma, C. Yang, X. Zhuang and X. Chen, Polym. Chem., 2015, 6, 738–747 RSC
- H. Fu, D. M. Policarpio, J. D. Batteas and D. E. Bergbreiter, Polym. Chem., 2010, 1, 631–633 RSC
- N. Kuramoto, Y. Shishido and K. Nagai, Macromol. Rapid Commun., 1994, 15, 441–444 CrossRef CAS
- N. Kuramoto, Y. Shishido and K. Nagai, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1967–1972 CrossRef CAS
- N. Kuramoto and Y. Shishido, Polymer, 1998, 39, 669–673 CrossRef CAS
- C. Tonhauser, A. Alkan, M. Schömer, C. Dingels, S. Ritz, V. Mailänder, H. Frey and F. R. Wurm, Macromolecules, 2013, 46, 647–655 CrossRef CAS
- S. Matsumoto, A. Kanazawa, S. Kanaoka and S. Aoshima, Polym. Chem., 2019, 10, 4134–4141 RSC
- J. Huang, H. Qin, B. Wang, Q. Tan and J. Lu, Polym. Chem., 2019, 10, 6379–6384 RSC
- R. Chen, Z. Xiang, Y. Xia, Z. Ma, Q. Shi, S.-C. Wong and J. Yin, Macromol. Rapid Commun., 2020, 41, 2000206 CrossRef CAS
- F. A. Plamper, M. Ruppel, A. Schmalz, O. Borisov, M. Ballauff and A. H. E. Müller, Macromolecules, 2007, 40, 8361–8366 CrossRef CAS
- C. Liang, X. Wang, R. Zhou, H. Shi, S. Yan, Y. Ling, S. Luan and H. Tang, Polym. Chem., 2019, 10, 2190–2202 RSC
- O. Bertrand, A. Vlad, R. Hoogenboom and J.-F. Gohy, Polym. Chem., 2016, 7, 1088–1095 RSC
- F. D. Jochum, L. zur Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854–7862 CrossRef CAS
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Polym. Chem., 2014, 5, 5403–5411 RSC
- J. Lee, A. J. McGrath, C. J. Hawker and B.-S. Kim, ACS Macro Lett., 2016, 5, 1391–1396 CrossRef CAS
- C. Luo, Y. Liu and Z. Li, Macromolecules, 2010, 43, 8101–8108 CrossRef CAS
- J. Huang, X. Chen, H. Qin, H. Liang and J. Lu, Polymer, 2019, 160, 99–106 CrossRef CAS
- K. Wang, S. Chen and W. Zhang, Macromolecules, 2017, 50, 4686–4698 CrossRef CAS
- Z. Song, K. Wang, C. Gao, S. Wang and W. Zhang, Macromolecules, 2016, 49, 162–171 CrossRef CAS
- C. Zhao, Z. Ma and X. X. Zhu, Prog. Polym. Sci., 2019, 90, 269–291 CrossRef CAS
- S. Li, H. S. Chung, A. Simakova, Z. Wang, S. Park, L. Fu, D. Cohen-Karni, S. Averick and K. Matyjaszewski, Biomacromolecules, 2017, 18, 475–482 CrossRef CAS
- F. H. Sobotta, F. Hausig, D. O. Harz, S. Hoeppener, U. S. Schubert and J. C. Brendel, Polym. Chem., 2018, 9, 1593–1602 RSC
- C. Fu, C. Zhang, H. Peng, F. Han, C. Baker, Y. Wu, H. Ta and A. K. Whittaker, Macromolecules, 2018, 51, 5875–5882 CrossRef CAS
- H. Batz, V. Hofmann and H. Ringsdorf, Macromol. Chem. Phys., 1973, 169, 323–325 CrossRef CAS
- S. Li, M. Omi, F. Cartieri, D. Konkolewicz, G. Mao, H. Gao, S. E. Averick, Y. Mishina and K. Matyjaszewski, Biomacromolecules, 2018, 19, 3754–3765 CrossRef CAS
- F. El Mohtadi, R. d'Arcy, X. Yang, Z. Y. Turhan, A. Alshamsan and N. Tirelli, Int. J. Mol. Sci., 2019, 20, 4583 CrossRef CAS
- F. El Mohtadi, R. d'Arcy and N. Tirelli, Macromol. Rapid Commun., 2019, 40, 1800699 CrossRef
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS
- Y. Liu, J. He, J. Xu, D. Fan, W. Tang and Y. Yang, Macromolecules, 2005, 38, 10332–10335 CrossRef CAS
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC
- M. Kano and E. Kokufuta, Langmuir, 2009, 25, 8649–8655 CrossRef CAS
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS
-
V. Aseyev, H. Tenhu and F. M. Winnik, in Self Organized Nanostructures of Amphiphilic Block Copolymers II, Springer, 2010, pp. 29–89 Search PubMed
- L. D. Blackman, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 233–244 RSC
- K. Zhou, Y. Lu, J. Li, L. Shen, G. Zhang, Z. Xie and C. Wu, Macromolecules, 2008, 41, 8927–8931 CrossRef CAS
- M. Wadhwa, C. Bird, P. Dilger, P. Rigsby, H. Jia and M. E. B. Gross, J. Immunol. Methods, 2017, 447, 14–22 CrossRef CAS
- C. A. Chaves, C. E. Vergani, D. Thomas, A. Young, C. A. Costa, V. M. Salih and A. L. Machado, J. Tissue Eng., 2014, 5, 2041731414540911 Search PubMed
- M. A. Al Enezy-Ulbrich, H. Malyaran, R. D. de Lange, N. Labude, R. Plum, S. Rütten, N. Terefenko, S. Wein, S. Neuss and A. Pich, Adv. Funct. Mater., 2020, 2003528 CrossRef CAS
- H. Otsuka, T. Hagiwara and S. Yamamoto, J. Nanosci. Nanotechnol., 2014, 14, 6764–6773 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01321h |
| This journal is © The Royal Society of Chemistry 2020 |