
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing fusion in tetrathienylethene cored perylene diimide tetramers affords acceptors with strong and broad absorption in the near-UV to visible region†
Qiao
He‡
 a,
Flurin D.
Eisner‡
b,
Drew
Pearce
b,
Thomas
Hodsden
a,
Elham
Rezasoltani
b,
Daniel
Medranda
b,
Zhuping
Fei
c,
Jenny
Nelson
*b and
Martin
Heeney
*a
a,
Flurin D.
Eisner‡
b,
Drew
Pearce
b,
Thomas
Hodsden
a,
Elham
Rezasoltani
b,
Daniel
Medranda
b,
Zhuping
Fei
c,
Jenny
Nelson
*b and
Martin
Heeney
*a
aDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, London W12 0BZ, UK. E-mail: m.heeney@imperial.ac.uk
bDepartment of Physics and Centre for Processable Electronics, Imperial College London, London SW7 2AZ, UK. E-mail: jenny.nelson@imperial.ac.uk
cInstitute of Molecular Plus, Tianjin Key Laboratory of Molecular Optoelectronic Science, Tianjin University, Tianjin 300072, P. R. China
First published on 17th November 2020
Abstract
In this work, we designed and synthesized two novel perylene diimide (PDI) tetramers based on a tetrathienylethene core, named TTE-PDI4 and FTTE-PDI4, and investigated their application as non-fullerene acceptors for organic photovoltaics. The free rotation of PDIs and adjacent thiophene units renders TTE-PDI4 with a highly twisted molecular geometry. The ring fusion of TTE-PDI4 yields FTTE-PDI4, a more rigid molecule with increased intramolecular stacking. Interestingly, TTE-PDI4 and FTTE-PDI4 possess similar energy levels but very different UV-Vis absorptions, with the latter showing strong broad-band absorption with multiple sharp peaks in the 300–600 nm region. Through time-dependent density functional theory (TD-DFT) calculations, we show that this broad absorption spectrum in FTTE-PDI4 arises from the combination of multiple bright transitions in the visible region with a strong vibronic progression, tentatively assigned to the dominant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching mode. TTE-PDI4, despite having a lower energy absorption onset, shows weaker absorption at long wavelengths. Due to its higher absorption as well as its increased rigidity, FTTE-PDI4 shows a higher photocurrent and hence a higher power conversion efficiency (PCE), of 6.6%, when blended with the polymer donor PFBDB-T than TTE-PDI4 based blends (PCE of 3.8%). The greater rigidity of FTTE-PDI4 is likely to contribute to the good fill factor of the blend devices. Potential for further improvement through reducing voltage losses is identified.
C stretching mode. TTE-PDI4, despite having a lower energy absorption onset, shows weaker absorption at long wavelengths. Due to its higher absorption as well as its increased rigidity, FTTE-PDI4 shows a higher photocurrent and hence a higher power conversion efficiency (PCE), of 6.6%, when blended with the polymer donor PFBDB-T than TTE-PDI4 based blends (PCE of 3.8%). The greater rigidity of FTTE-PDI4 is likely to contribute to the good fill factor of the blend devices. Potential for further improvement through reducing voltage losses is identified.
Introduction
Among various photovoltaic energy-conversion technologies, organic photovoltaics (OPVs) have become one of the fastest-growing areas over the past few years, attributed to the successful design and development of non-fullerene acceptors (NFAs).1–6 Such materials have helped to overcome some of the limitations of fullerene based acceptors (PC61BM and PC71BM) including weak absorption in the visible region, elevated synthetic difficulty in tuning the electronic/optical properties, and poor morphology stability.7–9 These issues have severely retarded the further development of fullerene based OPVs, and thus have accelerated the development of NFAs as an alternative strategy. Compared to their fullerene counterparts, there are more available building blocks to prepare NFAs and this synthetic flexibility affords more readily tunable optoelectronic properties.With regards to the various types of electron-withdrawing units to construct NFAs, perylene diimide (PDI) has been among the earliest studied and most promising.10–15 Pioneering studies on the PDI monomer and its derivatives found that the tendency of the coplanar PDI monomers to strongly aggregate resulted in micrometer-sized phase-separated domains in bulk-heterojunction blends, leading to low OPV device efficiencies.16,17 Disrupting molecular planarity by intramolecular twisting has been found to be a useful approach to suppress the formation of large aggregates and improve performance.18–21 By connecting two PDI monomers via a spacer or linking more PDIs with a central core, PDI dimers, trimers and tetramers have been prepared with a twisted 3D or quasi-3D geometry, resulting in improved OPV device performance. The spacers can be simply a single or double bond, an aromatic ring or a larger conjugated central core.22–27 Although the disruption of planarity is generally beneficial, excessive molecular twisting can be detrimental to charge transport and, consequently, high structural non-planarity of PDI-based acceptors can result in reduced device performance.27–29 To tackle this issue, ring fusion has been proposed in several studies to finely tune the intermolecular π–π stacking.30–34 Concomitantly, evidence also suggests that ring fusion blue-shifts absorption onset, enhances absorption strength, and up-shifts the lowest unoccupied molecular orbital (LUMO) energy level, which can be beneficial for overall OPV device performance.30,35 Through the employment of geometry twisting and aromatic ring fusion, currently the power conversion efficiency (PCE) of the best performing PDI-based NFAs have been enhanced to over 10%.35,36
We were particularly interested by the observations that ring fusion can lead a blue-shifted absorption, since wide band gap NFA absorbers with a strong absorption in the green to near-UV region are of interest for the front cell in tandem devices or for smart window applications.37,38 Ring fusion in the bay region of the PDI has typically been achieved by an oxidative ring closure with an electron rich heterocycle like thiophene or selenophene,30,35 and in order to prevent excessive aggregation it should be combined with the twisting approach. As such, we identified tetrathienylethene (TTE) as a potentially interesting central core for the attachment of four PDI arms, combining the requisite thienyl groups for ring fusion with a non-planar geometry. TTE has previously been primarily investigated in the field of aggregation-induced emission (AIE).39,40 Although the emissive properties are not the focus of this paper, we note that other AIE cores like tetraphenylethene (TPE) have been used for the attachment of PDI arms, although the resulting TPE-PDI4 was not able to undergo ring fusion with the PDI.25 Here we report the synthesis of two new non-fused and fused PDI tetramers (TTE-PDI4 and FTTE-PDI4) based on the tetrathienylethene core and investigate their performance in organic photovoltaic devices. For the non-fused TTE-PDI4, freely rotating single bonds between thiophene and the central ethene, as well as thiophene and the PDI, induces strong steric hindrance leading to a highly twisted molecular structure. In comparison, DFT calculations suggest the fused tetramer, FTTE-PDI4, displays a more ordered geometry with the interlocking of thiophenes and PDI units. The ring fusion leads to an up-shifted LUMO level, and an intense and extremely broad absorption spectrum at visible to near-UV wavelengths. These are found by time-dependent DFT to arise from multiple bright electronic transitions. These beneficial changes contribute to higher short-circuit current density (JSC) and improved power conversion efficiency from 3.8% (TTE-PDI4) to 6.6% (FTTE-PDI4) in solar cell devices, showing that careful design of the central core on PDI tetramers can lead to promising wide band gap acceptors.
Results and discussion
Synthesis of TTE-PDI4 and FTTE-PDI4
The synthetic route to TTE-PDI4 and FTTE-PDI4 is presented in Scheme 1. The intermediates DTK and TTE were synthesized according to a previous report.41 The TTE-Bpin4 was prepared by tetraborylation of TTE via an Ir-catalyzed reaction. Subsequent fourfold Suzuki coupling reaction between TTE-Bpin4 and C6-PDI-Br afforded the first product TTE-PDI4 in 40% yield. By subsequent treatment with FeCl3/CH3NO2, an oxidative cyclization reaction occurred between the PDI subunits and their neighboring thiophene spacers, affording the fused product FTTE-PDI4 in 62.6% yield.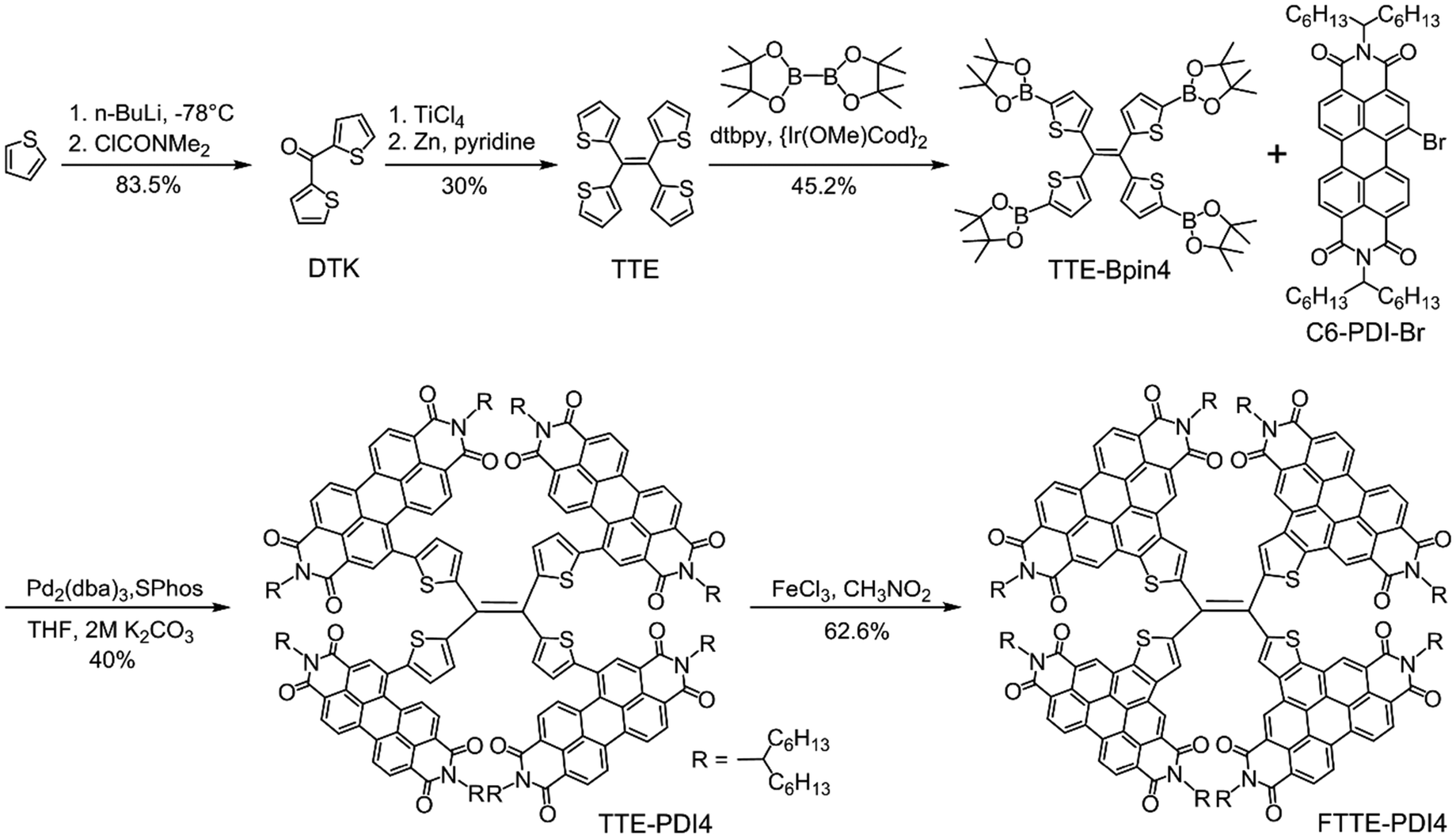 | ||
| Scheme 1 Synthetic route to TTE-PDI4 and FTTE-PDI4. | ||
TTE-PDI4 and FTTE-PDI4 were purified by a combination of column chromatography followed by preparative recycling gel-permeation chromatography (GPC) to remove non-tetrameric by-products (see ESI†). The identities of TTE-PDI4 and FTTE-PDI4 were confirmed by MALDI-TOF mass spectrometry (Fig. S25 and S27, ESI†). Room temperature 1H NMR spectra were broad and poorly resolved, similar to other PDI tetramers,15,34,35 as a result of the restricted rotation of the PDI subunits and presence of multiple conformers. Heating FTTE-PDI4 solutions at 40 °C, 80 °C and 120 °C resulted in improved resolution (Fig. S24, ESI†), particularly of the protons on the outer shell of the molecule which were clearly resolved as sets of doublets. The inner protons nearer the TTE core were less well resolved, possibly due to more restricted movement.
As such, it is difficult to completely rule out the presence of some incompletely fused material, although the significant change in optical properties (vide infra) suggests that this would only be a minor impurity if present.
Both compounds are readily soluble in common organic solvents such as chloroform and chlorobenzene at room temperature. The structures of the two PDI tetramers were characterized and confirmed by MALDI-TOF MS, 1H NMR, and 13C NMR. Both TTE-PDI4 and FTTE-PDI4 exhibited good thermal stability, with thermal decomposition temperatures (Td) of 320 °C and 375 °C, respectively, measured by thermogravimetric analysis (TGA, at 5% weight loss, Fig. S1, ESI†). In addition, in the range of 20 °C to 280 °C, no obvious crystallization or melting transitions could be observed for both PDI tetramers, indicating their weak crystallization tendency (Fig. S2, ESI†).
DFT and TD-DFT calculations
To investigate the molecular structural geometries and frontier molecular orbitals of TTE-PDI4 and FTTE-PDI4, DFT calculations were performed using the B3LYP functional and 6-31G basis set, with all alkyl chains replaced by methyl groups for computational simplicity. Both TTE-PDI4 and FTTE-PDI4 are predicted to exhibit highly twisted molecular geometries, with the former exhibiting a propeller type arrangement in which each arm is twisted with respect to the central ethene, with dihedral angles between the ethene and thiophene of 43°, and 57° between the thiophene and PDIs. As expected the ring fusion in FTTE-PDI4 eliminates the rotational freedom between the thiophenes and PDI units (Fig. 1A and Fig. S3, ESI†), and the predicted structure becomes more saddle shaped, with two PDI-Th planes at each end of the central ethene. There remains a large twist between the PDI-Th and the central ethene of 48°, with the two PDI-Th planes rigidly held in space, with the plane of one rotated by an angle of 59° with respect to the other. With regards to the frontier molecular orbitals, though the HOMOs are localised on the TTE central core for both fused and non-fused NFAs, the delocalization of LUMOs shows significant difference. In TTE-PDI4, the near degenerate LUMO and LUMO+1 orbitals localize on different PDI units. In contrast, the LUMO of FTTE-PDI4 delocalizes through the entire molecular backbone as the fused molecule becomes more planar and rigid. The internal reorganization energies upon positive polaron formation were calculated to be 0.105 eV and 0.116 eV for FTTE-PDI4 and TTE-PDI4, respectively. The lower reorganization energy for the FTTE-PDI4 is consistent with its increased rigidity.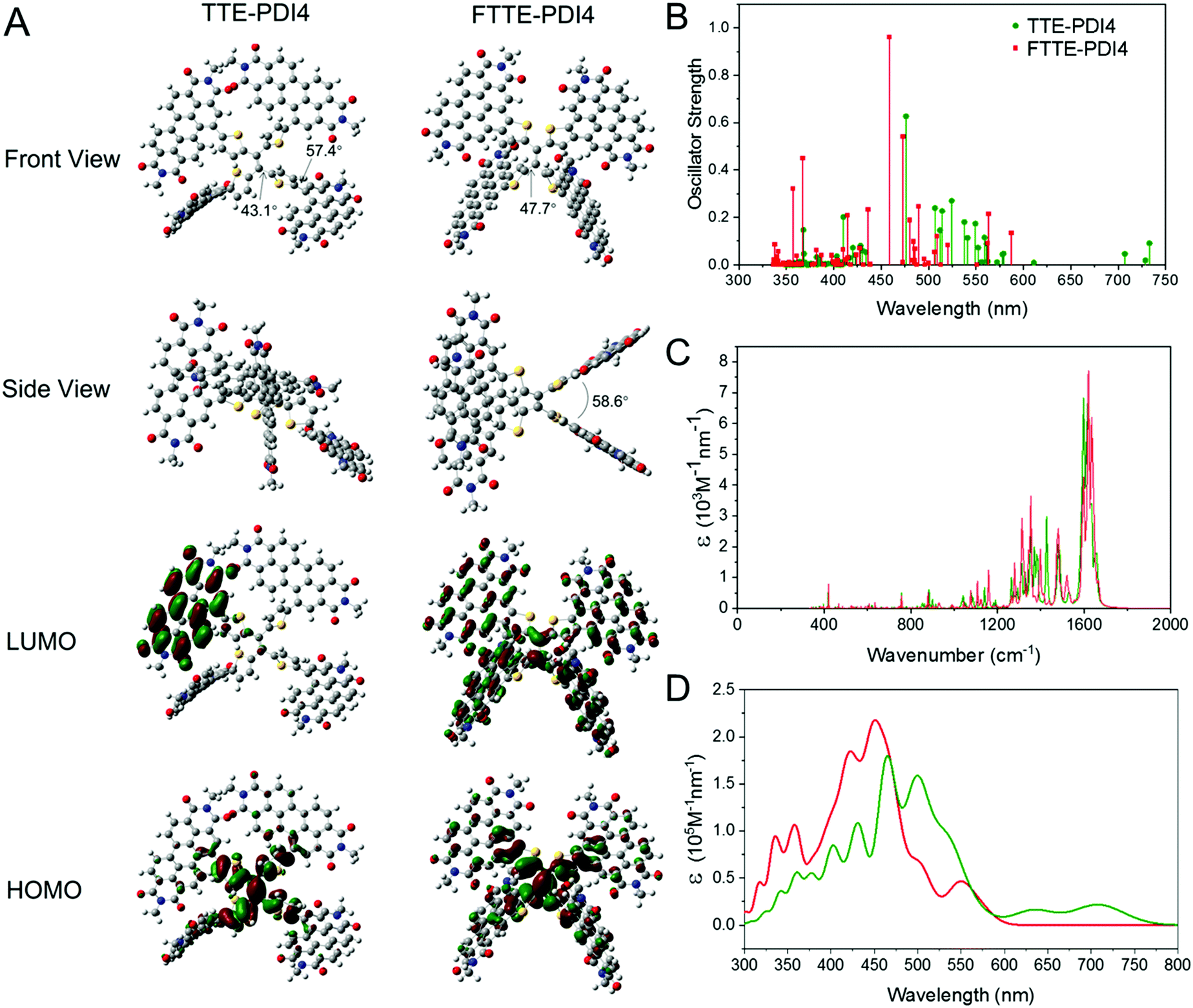 | ||
| Fig. 1 (A) Optimized geometry (front and side views) and frontier molecular orbitals of TTE-PDI4 and FTTE-PDI4 using DFT calculation at the B3LYP/6-31G level. (B) Oscillator strength (ε) calculations of the excited state transitions to the ground state and (C) extinction coefficients (ε) from vibrational transitions calculated using TD-DFT, with the B3LYP functional and 6-311g basis set; 100 states were calculated and broadened with a half-width at half height of 0.2 eV. Frequency calculations were also performed to obtain the vibrational transitions. (D) Extinction coefficient calculations based on a Franck–Condon lineshape applied to the electronic transitions. | ||
In order to investigate the effect of the different structural geometries of the two molecules on their absorption profile, we additionally carried out time-dependent DFT (TD-DFT) calculations. Fig. 1B and C thus show the electronic and vibrational structure calculations, respectively. The results firstly show that we expect TTE-PDI4 to have a smaller band gap than FTTE-PDI4 by around 0.1 eV due to a weak low energy transition that appears in the extinction spectrum as a long and shallow tail. Secondly, FTTE-PDI4 shows strong absorption at low wavelengths due to several bright electronic transitions in the visible, in agreement with previous studies,42 while lacking the weak low energy transition that creates the absorption tail in TTE-PDI4. Thirdly, vibrational structure calculations show that there is a strong vibrational band at 1600 cm−1 that is present in both molecules. The mode is assigned to the CC bond stretch within the PDI units, as shown in Fig. S5 (ESI†). When a mode of this frequency is used to model the vibronic progression of the extinction spectra of the two molecules (Fig. 1D) a spectrum with multiple peaks across the visible results. We note that broad absorption features at low wavelengths have often been reported in the absorption spectra of PDI-based molecules, in particular in fused PDIs;35,43 our calculations suggest that these features may result from the combination of multiple close lying electronic transitions with a strong vibronic progression.
Optical and electronic properties
The measured optical and electronic properties of TTE-PDI4 and FTTE-PDI4 are summarized in Table 1. As presented in Fig. 2A and B, the UV-Vis absorption spectra of TTE-PDI4 and FTTE-PDI4 films also exhibit significant differences. Both the solution and film absorptions of TTE-PDI4 exhibit an absorption peak in the region of 400–650 nm, similar to the PDI monomer, while those of FTTE-PDI4 show multiple distinctive sharp peaks in the region of 350–600 nm, and are in good agreement with the calculated absorption spectra (Fig. 1D). This comparison is also in agreement with the increased number of bright electronic transitions in the visible region in FTTE-PDI4 compared with TTE-PDI4, as identified from TD-DFT calculations. The relative intensities of the 0–1 and 0–0 transitions (I0–1/I0–0) have been previously identified as indicative of the H-aggregation of PDI chromophores.43–45 Since aggregation is inhibited by the non-planar structure of the molecules, the high I0–1 (450 nm)/I0–0 (500 nm) = 1.15 of FTTE-PDI4 in films thus suggests a degree of long-range coupling of the PDI chromophores possibly related to the rigid arrangement of the PDI units leading to H-aggregation of the PDI chromophores.46 Notably, fused FTTE-PDI4 also shows higher absorptivity in the 300–450 region nm and a blue-shifted absorption onset compared to TTE-PDI4, resulting in a higher band gap of 2.04 compared to 1.90 eV (calculated by the crossing point between absorption and emission, Fig. S6, ESI†), in agreement with the theoretical calculations.| λ max,sol (nm) | α max,sol (M−1 cm−1) | λ max,film (nm) | α max,film (cm−1) | E g,opt (eV) | E g,electro (eV) | LUMOc (eV) | HOMOc (eV) | |
|---|---|---|---|---|---|---|---|---|
| a Determined by the absorption onset of the films. b Electrochemical band gap, determined by (LUMO–HOMO). c Estimated from the reduction and oxidation onsets of the CV curves. | ||||||||
| TTE-PDI4 | 525 | 1.19 × 105 | 530 | 3.55 × 104 | 1.90 | 2.20 | −3.74 | −5.94 |
| FTTE-PDI4 | 429 | 1.41 × 105 | 424 | 3.77 × 104 | 2.00 | 2.29 | −3.68 | −5.97 |
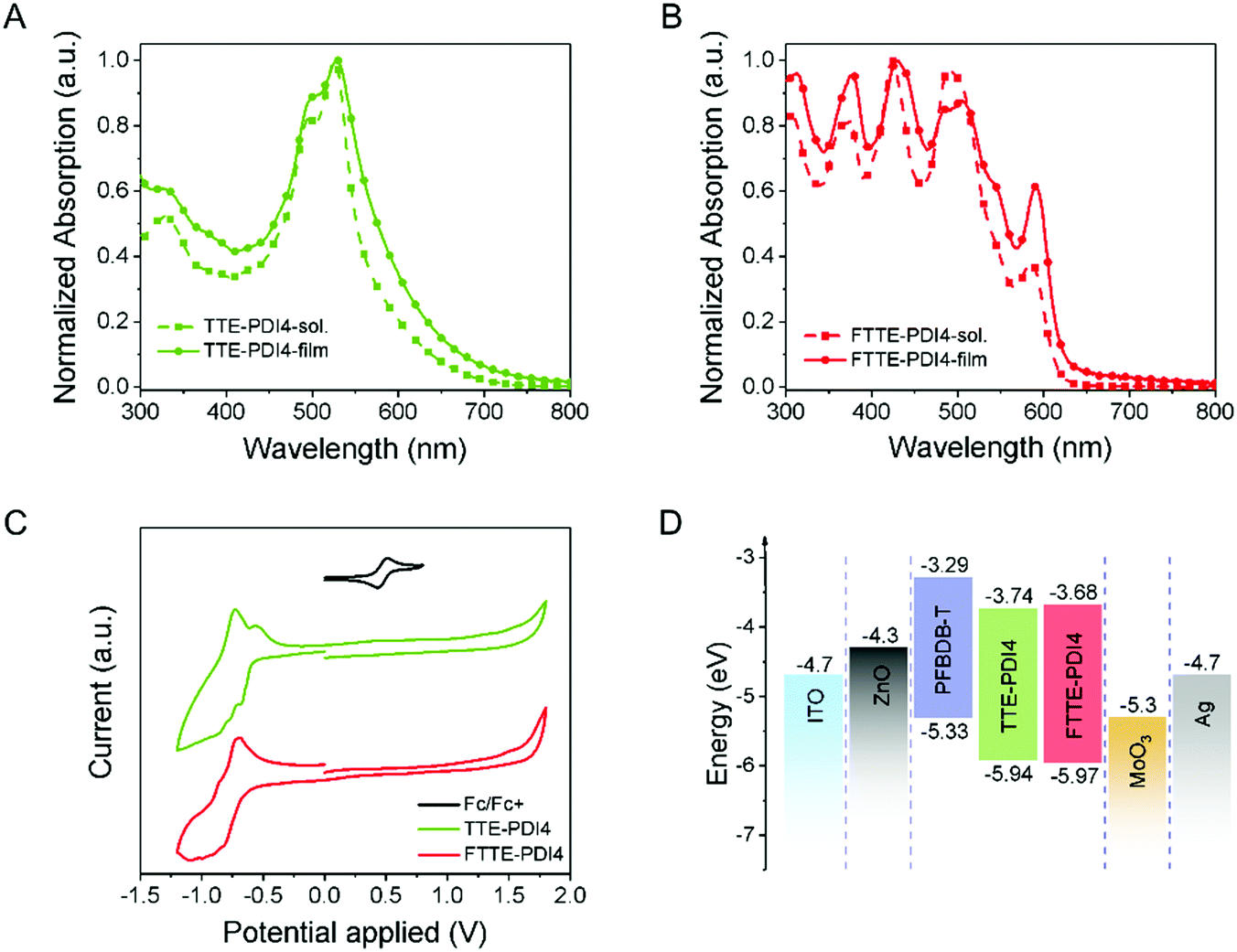 | ||
| Fig. 2 Normalized absorption spectra of (A) TTE-PDI4 and (B) FTTE-PDI4 in chloroform solution (10−5 M) and as pristine films. (C) Cyclic voltammetry curves of TTE-PDI4 and FTTE-PDI4 films. (D) Energy level diagram of the OPV device. | ||
In addition, the maximum extinction coefficients of FTTE-PDI4 in solution and in film were calculated to be 1.41 × 105 M−1 cm−1 and 3.77 × 104 cm−1, respectively, higher than those of TTE-PDI4 (1.19 × 105 M−1 cm−1 and 3.55 × 104 cm−1). The minor differences in the absorption spectra between solution and film further suggest that intermolecular packing is weak for both molecules. This is further confirmed by absence of any change in solution absorption measurements as a function of concentration (Fig. S10, ESI†). This additionally suggests that the breadth of the absorption of FTTE-PDI4, at over 50% of maximum intensity from 300 to 600 nm, must be due to the multiple bright electronic states of the molecules identified by TD-DFT rather than due to intermolecular interactions.
The electrochemical properties of these two compounds were investigated by cyclic voltammetry of thin films, with the LUMO/HOMO levels of TTE-PDI4 and FTTE-PDI4 (Fig. 2C) estimated to be similar at −3.74/−5.94 eV and −3.68/5.97 eV, respectively from the onset of the reduction and oxidation peaks. The slight upshift of the LUMO level upon ring fusion is in agreement with the DFT calculations and with other studies on fused PDIs.30,34,35
OPV performance
To evaluate the photovoltaic performance of these two compounds, OPV devices were fabricated with an inverted structure of ITO/ZnO/polymer donor:TTE-PDI4 or FTTE-PDI4/MoO3/Ag (Fig. 2D). To begin with, several high performance polymers (PTB7-th,47 PffBT4T-2OD,48 PBDB-T,49 PFBDB-T,6 and P4FBDB-T50) were used to fabricated devices with FTTE-PDI4 to determine the most suitable donor.Reasonably high efficiencies (∼5%) were achieved with PTB7-Th, PBDB-T and PFBDB-T as the donor (Table S1, ESI†), with devices with PFBDB-T showing the highest efficiency; we therefore selected this polymer to optimize fabrication conditions with both TTE-PDI4 and FTTE-PDI4 blends. The optimized photovoltaic parameters and typical J–V characteristics of the PFBDB-T:TTE-PDI4 and PFBDB-T:FTTE-PDI4 devices are presented in Fig. 3A and Table 2. The best performance with PFBDB-T:FTTE-PDI4 devices was achieved with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 blend ratio, 2% 1-chloronaphthalene (CN) as solvent additive and annealing at 140 °C, showing an average PCE of 6.4% (best 6.6%) with a JSC of 10.9 mA cm−2, an open-circuit voltage (VOC) of 1.0 V, and a FF of 0.59. The relatively large acceptor fraction required for maximum device performance is in agreement with weak intermolecular packing of the molecules (evidenced by the absorption data above) and the low crystallinity (evidenced by featureless DSC spectra). Non-crystalline acceptor molecules generally need to be added in greater excess than crystalline acceptors to achieve percolating networks and phase-pure domains for charge separation. Interestingly, a high PCE was also maintained in as-cast cells without additive and thermal annealing, with such devices showing a lower JSC but higher VOC and FF of 1.02 V and 0.63, respectively. We note, also, that the highest FF (0.64) is achieved in a 1:1.5 blend ratio, which is amongst the highest reported for PDI-based acceptors. The PFBDB-T:TTE-PDI4 based active layers were also systematically optimized under various conditions, with a donor:acceptor ratio of 1:1.5, no additive and thermal annealing at 140 °C leading to the optimized average PCE of the TTE-PDI4 based OPVs of 3.8%, with inferior current density and FF to the FTTE-PDI4 based OPV devices. A similar VOC was observed for TTE-PDI4 and FTTE-PDI4 based OPVs, in spite of the slightly higher-lying LUMO levels for the latter. Moreover, external quantum efficiencies (EQEs) of the optimal devices were measured to verify the JSC values obtained from the J–V measurement (Fig. 3B). The JSC values integrated from EQE are 7.4 and 11.9 mA cm−2, both in less than 10% deviation from JSC measured from the TTE-PDI4 and FTTE-PDI4 based OPV devices. The PFBDB-T:FTTE-PDI4 devices exhibit stronger photo-response in the spectral range between 350 and 600 nm, with a higher maximum EQE of 72% than the PBDB-T:TTE-PDI4 devices (EQEmax = 56%), in agreement with the stronger absorption of FTTE-PDI4 compared to TTE-PDI4 in that region. This indicates FTTE-PDI4 contributes more to the charge generation and JSC than TTE-PDI4 as electron acceptor in their corresponding blends.
:2 blend ratio, 2% 1-chloronaphthalene (CN) as solvent additive and annealing at 140 °C, showing an average PCE of 6.4% (best 6.6%) with a JSC of 10.9 mA cm−2, an open-circuit voltage (VOC) of 1.0 V, and a FF of 0.59. The relatively large acceptor fraction required for maximum device performance is in agreement with weak intermolecular packing of the molecules (evidenced by the absorption data above) and the low crystallinity (evidenced by featureless DSC spectra). Non-crystalline acceptor molecules generally need to be added in greater excess than crystalline acceptors to achieve percolating networks and phase-pure domains for charge separation. Interestingly, a high PCE was also maintained in as-cast cells without additive and thermal annealing, with such devices showing a lower JSC but higher VOC and FF of 1.02 V and 0.63, respectively. We note, also, that the highest FF (0.64) is achieved in a 1:1.5 blend ratio, which is amongst the highest reported for PDI-based acceptors. The PFBDB-T:TTE-PDI4 based active layers were also systematically optimized under various conditions, with a donor:acceptor ratio of 1:1.5, no additive and thermal annealing at 140 °C leading to the optimized average PCE of the TTE-PDI4 based OPVs of 3.8%, with inferior current density and FF to the FTTE-PDI4 based OPV devices. A similar VOC was observed for TTE-PDI4 and FTTE-PDI4 based OPVs, in spite of the slightly higher-lying LUMO levels for the latter. Moreover, external quantum efficiencies (EQEs) of the optimal devices were measured to verify the JSC values obtained from the J–V measurement (Fig. 3B). The JSC values integrated from EQE are 7.4 and 11.9 mA cm−2, both in less than 10% deviation from JSC measured from the TTE-PDI4 and FTTE-PDI4 based OPV devices. The PFBDB-T:FTTE-PDI4 devices exhibit stronger photo-response in the spectral range between 350 and 600 nm, with a higher maximum EQE of 72% than the PBDB-T:TTE-PDI4 devices (EQEmax = 56%), in agreement with the stronger absorption of FTTE-PDI4 compared to TTE-PDI4 in that region. This indicates FTTE-PDI4 contributes more to the charge generation and JSC than TTE-PDI4 as electron acceptor in their corresponding blends.
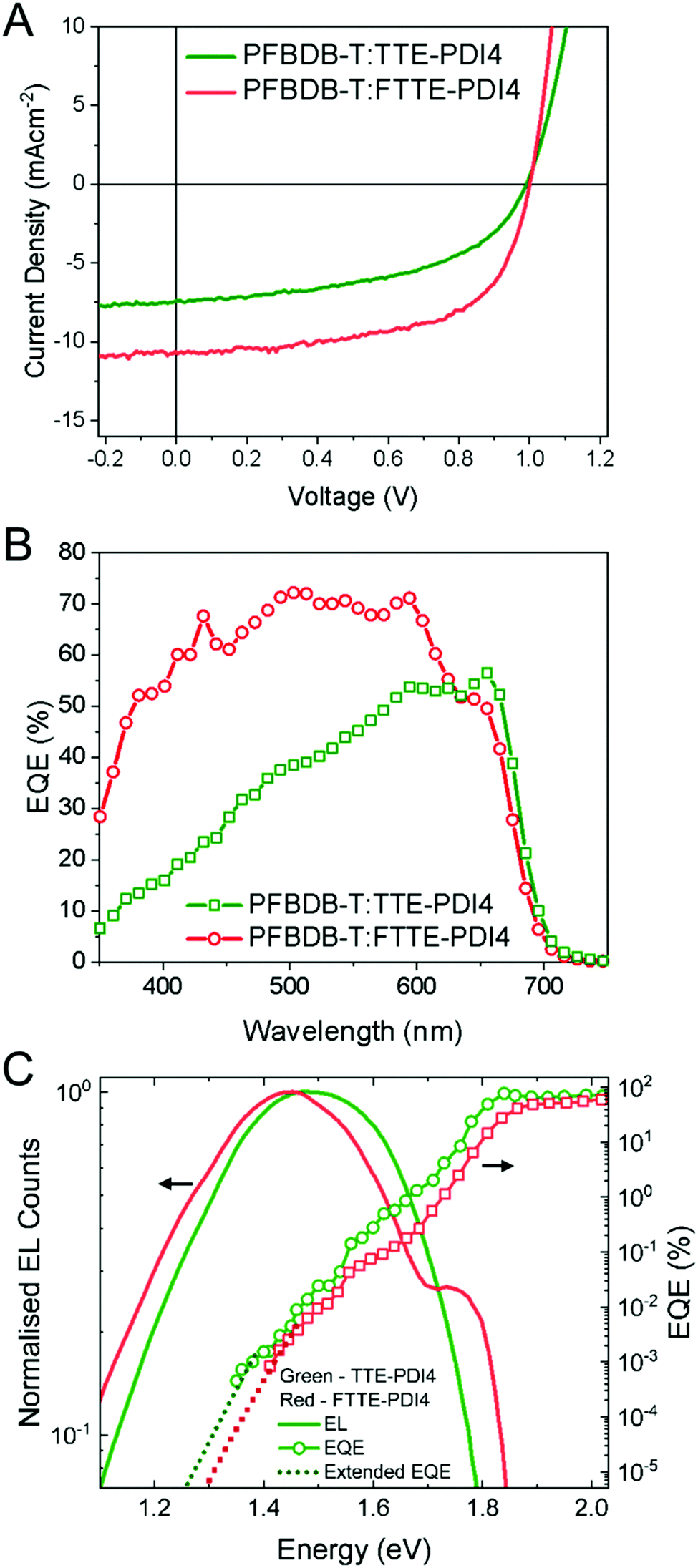 | ||
| Fig. 3 J–V curves (A) and EQEs (B) for the optimized devices based on the PFBDB-T:TTE-PDI4 and PFBDB-T:FTTE-PDI4 blends. (C) Electroluminescence (solid lines), external quantum efficiency (line and symbols), and extended external quantum efficiency (symbols) calculated using reciprocity from the EL spectra of PFBDB-T:TTE-PDI4 (green) and PFBDB-T:FTTE-PDI4 (red) blend devices. | ||
| Acceptor | V OC (V) | J SC (mA cm−2) | FF (%) | PCEa (%) | Calc. JSC (mA cm−2) | V OC,rad (V) | ΔVOC,nrad (V) |
|---|---|---|---|---|---|---|---|
| a The values in parentheses are the average PCEs. The average values are collected from >12 independent devices. | |||||||
| TTE-PDI4 | 0.99(0.99) | 7.5(7.3) | 51(50) | 3.8(3.6) | 8.2 | 1.35 | 0.36 |
| FTTE-PDI4 | 1.0(1.0) | 10.9(10.9) | 60(59) | 6.6(6.4) | 11.9 | 1.38 | 0.38 |
Using the measured EQE edge (Fig. 3C), we then calculated the photovoltaic band gap energy (EPVg), using the method proposed by Rau,51 for both blends to be 1.84 eV. These values suggested high voltage losses (EPVg/q − VOC) of 0.85 and 0.84 V for PFBDB-T:TTE-PDI4 and PFBDB-T:FTTE-PDI4 devices, respectively, where q is electronic charge. In order to better understand the origin of these high losses, we therefore performed a voltage-loss analysis of the optimized blends by employing electroluminescence (EL) spectra to extend the measured sub-bandgap EQE spectra (Fig. 3C), using the reciprocity relation between light absorption and light emission,52,53 to calculate the open-circuit voltage in the radiative limit (VOC,rad). VOC,rad is the ideal VOC that a device can achieve when there is only radiative recombination, with the difference between VOC,rad and the measured VOC of the device then being defined as the voltage loss due to non-radiative recombination, ΔVOC,nrad. The VOC,rad and ΔVOC,nrad values for the optimized devices are shown in Table 2 and we also note that the maximum ideal VOC in the Shockley–Queisser limit (VOC,sq) achievable by a device with the measured band gap of 1.84 eV is 1.54 V.
We firstly draw attention to the fact, then, that the difference between VOC,sq and VOC,rad is large for both blends, which suggests that losses due a broad absorption edge are large; this is expected given the large offset between the LUMOs of the donor (−3.29 eV) and the acceptors (−3.74 eV and −3.68 eV), and suggests that the VOC could be significantly improved by using a donor with a lower lying LUMO level. Secondly, we note that both blends show similarly high ΔVOC,nrad of 0.36 V and 0.38 V for PFBDB-T:TTE-PDI4 and PFBDB-T:FTTE-PDI4, respectively, with the higher value for the FTTE-PDI4 blend meaning that the VOC is not larger than that of the TTE-PDI4 blend despite the larger optical gap. An earlier study identified several molecular factors that could reduce the nonradiative voltage loss in solar cells,54 among which increased molecular rigidity, expressed as reduced reorganization energy, is one. Interestingly, whilst the higher molecular rigidity leading to the smaller reorganization energy of FTTE-PDI4 appears to facilitate charge transport in the blend (manifested through the higher FF), non-radiative recombination does not appear to be suppressed. It is therefore likely that the voltage losses are influenced also by microstructural properties of the blend films and not only by molecular parameters.
To confirm this hypothesis, atomic-force-microscopy (AFM) images were measured for both blends, as shown in Fig. 4. Both films display nanoscale phase separation with fine and small domain size, suggesting their twisted molecular geometry lead to the formation of favorable morphology for OPV devices. The root-mean-square (RMS) roughness are 3.0 nm and 7.6 nm for the PFBDB-T:TTE-PDI4 and PFBDB-T:FTTBPDI4 blends, respectively. As expected, the PFBDB-T:TTE-PDI4 blend shows strong intermixing and a smoother surface morphology, with fiber-like aggregates observed in the film, whilst the PFBDB-T:FTTEPDI4 blend shows more aggregated domains of roughly 25–30 nm in length, possibly due to the larger excess of acceptor in the optimized blend compared to in PFBDB-T:TTE-PDI4. This is likely to lead to a larger degree of disorder in the blend, which is consistent with the slightly larger ΔVOC,nrad observed in this blend, although further measurements would be required to confirm this.
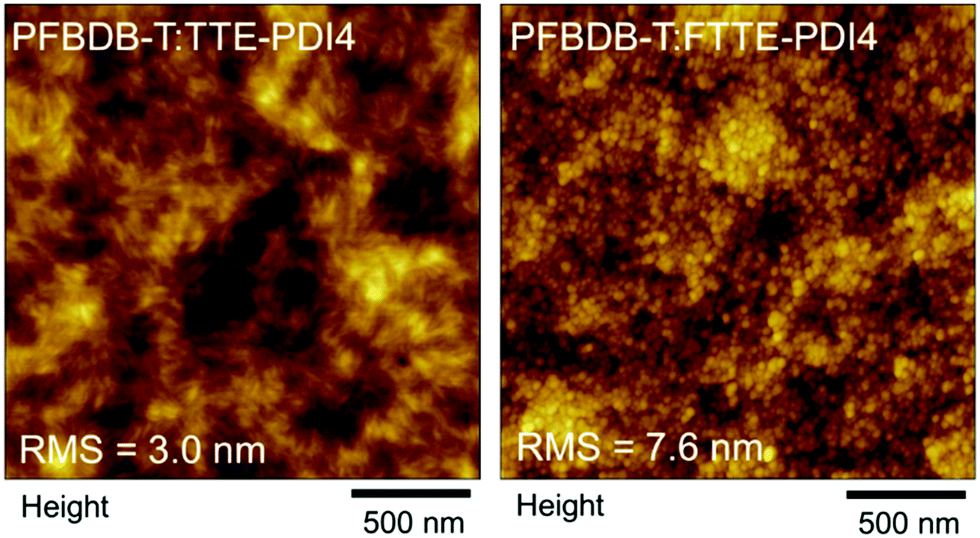 | ||
| Fig. 4 AFM height images (2 μm × 2 μm) of the PFBDB-T:TTE-PDI4 (left) and PFBDB-T:FTTBPDI4 (right) blend films. The topographical height difference (Δz) are 21 nm and 51 nm, respectively. | ||
The molecular packing behaviour of compounds TTE-PDI4, FTTE-PDI4, PFBDB-T and their blend films were also investigated by X-ray diffraction on drop-cast films. As shown in Fig. S12 (ESI†), a broad π–π stacking peak was observed for neat PFBDB-T film, with a maxima at 2θ = 24.26°. After annealing at 160 °C for 10 min, the film not only exhibited a π–π stacking at a similar position, but also weak diffraction peaks at 2θ = 12.72°, 14.14°, 18.60° and 21.68°, suggestive of enhanced order. The enhanced order after thermal annealing is in agreement with our previous GIWAXS results on thin-films.6 TTE-PDI4 and FTTE-PDI4 exhibited no diffraction peaks, indicating their low crystallinity in thin films, which was also consistent with DSC data. The optimal blend films of PFBDB-T:TTE-PDI4 and PFBDB-T:FTTE-PDI4 also showed no diffraction peaks from their drop cast blends after annealing at 140 °C for 10 min, suggesting blending may inhibit polymer crystallisation somewhat, with the caveat that any crystallites present may not be readily observed with the lab based diffractometer used in these measurements.
Conclusions
In conclusion, two PDI tetramers with non-fused and fused structures, TTE-PDI4 and FTTE-PDI4, were designed and synthesized. DFT calculations reveal that the fused FTTE-PDI4 displays reduced intramolecular twisting compared to non-fused TTE-PDI4 with a lower internal reorganization energy. FTTE-PDI4 exhibits a smaller band gap than TTE-PDI4, with a much broader and stronger absorption in the region of 300–600 nm, together with an up-shifted LUMO level. TD-DFT calculations confirm that the observed strong absorption features at low wavelengths, also reported in other fused-PDI molecules in the literature, are due to multiple bright electronic states coupled to a strong vibrational band at 1600 cm−1. Consequently, the PFBDB-T:FTTE-PDI4 devices achieved a highest PCE of 6.6% with a higher current density of 10.9 mA cm−2 and FF of 60%. This work demonstrates that ring fusion is an effective strategy to broaden the absorption spectra of PDI-type OPVs by induced bright low-lying bright electronic states, as well as finely tune the molecular shape, both of which work to boost device efficiency.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the China Scholarship Council (CSC) via the CSC Imperial Scholarship, Engineering and Physical Sciences Research Council (EPSRC) (Grant EP/L016702/1) and the Royal Society and the Wolfson Foundation (Royal Society Wolfson Fellowship) for funding. F. E. thanks the Engineering and Physical Sciences Research Council (EPSRC) for support via the Post-Doctoral Prize Fellowship, and J. N. and F. E. are grateful for funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 742708). The authors also thank Filip Aniés, Zhongyao Jiang and Richard Sweeney for valuable help.Notes and references
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Nat. Energy, 2018, 3, 720–731 CrossRef CAS.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2018, 48, 1596–1625 RSC.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- Z. Fei, F. D. Eisner, X. Jiao, M. Azzouzi, J. A. Röhr, Y. Han, M. Shahid, A. S. R. Chesman, C. D. Easton, C. R. McNeill, T. D. Anthopoulos, J. Nelson and M. Heeney, Adv. Mater., 2018, 30, 1705209 CrossRef.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS.
- Z. Liu, Y. Wu, Q. Zhang and X. Gao, J. Mater. Chem. A, 2016, 4, 17604–17622 RSC.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS.
- J. Wang and X. Zhan, Trends Chem., 2019, 1, 869–881 CrossRef CAS.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- W. Zhu, J. M. Alzola, T. J. Aldrich, K. L. Kohlstedt, D. Zheng, P. E. Hartnett, N. D. Eastham, W. Huang, G. Wang, R. M. Young, G. C. Schatz, M. R. Wasielewski, A. Facchetti, F. S. Melkonyan and T. J. Marks, ACS Energy Lett., 2019, 4, 2695–2702 CAS.
- Q. Wu, D. Zhao, A. M. Schneider, W. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7248–7251 CrossRef CAS.
- H. Yu, L. Arunagiri, L. Zhang, J. Huang, W. Ma, J. Zhang and H. Yan, J. Mater. Chem. A, 2020, 8, 6501–6509 RSC.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS.
- S. Li, W. Liu, C.-Z. Li, M. Shi and H. Chen, Small, 2017, 13, 1701120 CrossRef.
- Q. He, T. Li, C. Yan, Y. Liu, J. Wang, M. Wang, Y. Lin and X. Zhan, Dyes Pigm., 2016, 128, 226–234 CrossRef CAS.
- C. Zhan and J. Yao, Chem. Mater., 2016, 28, 1948–1964 CrossRef CAS.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS.
- Y. Zhong, M. T. Trinh, R. Chen, W. Wang, P. P. Khlyabich, B. Kumar, Q. Xu, C.-Y. Nam, M. Y. Sfeir, C. Black, M. L. Steigerwald, Y.-L. Loo, S. Xiao, F. Ng, X. Y. Zhu and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 15215–15221 CrossRef CAS.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375–380 CrossRef CAS.
- Q. Yan, Y. Zhou, Y.-Q. Zheng, J. Pei and D. Zhao, Chem. Sci., 2013, 4, 4389–4394 RSC.
- Y. Liu, C. Mu, K. Jiang, J. Zhao, Y. Li, L. Zhang, Z. Li, J. Y. L. Lai, H. Hu, T. Ma, R. Hu, D. Yu, X. Huang, B. Z. Tang and H. Yan, Adv. Mater., 2015, 27, 1015–1020 CrossRef CAS.
- S. Li, W. Liu, C.-Z. Li, F. Liu, Y. Zhang, M. Shi, H. Chen and T. P. Russell, J. Mater. Chem. A, 2016, 4, 10659–10665 RSC.
- Y. Duan, X. Xu, H. Yan, W. Wu, Z. Li and Q. Peng, Adv. Mater., 2017, 29, 1605115 CrossRef.
- H. Lin, S. Chen, H. Hu, L. Zhang, T. Ma, J. Y. L. Lai, Z. Li, A. Qin, X. Huang, B. Tang and H. Yan, Adv. Mater., 2016, 28, 8546–8551 CrossRef CAS.
- A. Kuzmich, D. Padula, H. Ma and A. Troisi, Energy Environ. Sci., 2017, 10, 395–401 RSC.
- H. Zhong, C.-H. Wu, C.-Z. Li, J. Carpenter, C.-C. Chueh, J.-Y. Chen, H. Ade and A. K. Y. Jen, Adv. Mater., 2016, 28, 951–958 CrossRef CAS.
- H. Qian, Z. Wang, W. Yue and D. Zhu, J. Am. Chem. Soc., 2007, 129, 10664–10665 CrossRef CAS.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef CAS.
- D. Meng, H. Fu, C. Xiao, X. Meng, T. Winands, W. Ma, W. Wei, B. Fan, L. Huo, N. L. Doltsinis, Y. Li, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2016, 138, 10184–10190 CrossRef CAS.
- Q. Wu, D. Zhao, J. Yang, V. Sharapov, Z. Cai, L. Li, N. Zhang, A. Neshchadin, W. Chen and L. Yu, Chem. Mater., 2017, 29, 1127–1133 CrossRef CAS.
- J. Zhang, Y. Li, J. Huang, H. Hu, G. Zhang, T. Ma, P. C. Y. Chow, H. Ade, D. Pan and H. Yan, J. Am. Chem. Soc., 2017, 139, 16092–16095 CrossRef CAS.
- G. Zhang, J. Feng, X. Xu, W. Ma, Y. Li and Q. Peng, Adv. Funct. Mater., 2019, 29, 1906587 CrossRef CAS.
- Y. Firdaus, V. M. Le Corre, J. I. Khan, Z. Kan, F. Laquai, P. M. Beaujuge and T. D. Anthopoulos, Adv. Sci., 2019, 6, 1802028 CrossRef.
- N. C. Davy, M. Sezen-Edmonds, J. Gao, X. Lin, A. Liu, N. Yao, A. Kahn and Y.-L. Loo, Nat. Energy, 2017, 2, 17104 CrossRef CAS.
- L. Viglianti, N. L. C. Leung, N. Xie, X. Gu, H. H. Y. Sung, Q. Miao, I. D. Williams, E. Licandro and B. Z. Tang, Chem. Sci., 2017, 8, 2629–2639 RSC.
- J.-J. Liu, J. Yang, J.-L. Wang, Z.-F. Chang, B. Li, W.-T. Song, Z. Zhao, X. Lou, J. Dai and F. Xia, Mater. Chem. Front., 2018, 2, 1126–1136 RSC.
- A. Yamamoto, Y. Matsui, T. Asada, M. Kumeda, K. Takagi, Y. Suenaga, K. Nagae, E. Ohta, H. Sato, S. Koseki, H. Naito and H. Ikeda, J. Org. Chem., 2016, 81, 3168–3176 CrossRef CAS.
- Y. Zhong, B. Kumar, S. Oh, M. T. Trinh, Y. Wu, K. Elbert, P. Li, X. Zhu, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 8122–8130 CrossRef CAS.
- J. Zhang, F. Bai, Y. Li, H. Hu, B. Liu, X. Zou, H. Yu, J. Huang, D. Pan, H. Ade and H. Yan, J. Mater. Chem. A, 2019, 7, 8136–8143 RSC.
- J. Seibt, P. Marquetand, V. Engel, Z. Chen, V. Dehm and F. Würthner, Chem. Phys., 2006, 328, 354–362 CrossRef CAS.
- N. J. Schuster, D. W. Paley, S. Jockusch, F. Ng, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2016, 55, 13519–13523 CrossRef CAS.
- A. Oleson, T. Zhu, I. S. Dunn, D. Bialas, Y. Bai, W. Zhang, M. Dai, D. R. Reichman, R. Tempelaar, L. Huang and F. C. Spano, J. Phys. Chem. C, 2019, 123, 20567–20578 CrossRef CAS.
- L. Ye, S. Zhang, W. Zhao, H. Yao and J. Hou, Chem. Mater., 2014, 26, 3603–3605 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. A. Tan and J. Hou, Macromolecules, 2012, 45, 9611–9617 CrossRef CAS.
- F. D. Eisner, M. Azzouzi, Z. Fei, X. Hou, T. D. Anthopoulos, T. J. S. Dennis, M. Heeney and J. Nelson, J. Am. Chem. Soc., 2019, 141, 6362–6374 CrossRef CAS.
- U. Rau, B. Blank, T. C. M. Müller and T. Kirchartz, Phys. Rev. Appl., 2017, 7, 044016 CrossRef.
- J. Yao, T. Kirchartz, M. S. Vezie, M. A. Faist, W. Gong, Z. He, H. Wu, J. Troughton, T. Watson, D. Bryant and J. Nelson, Phys. Rev. Appl., 2015, 4, 014020 CrossRef.
- U. Rau, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 085303 CrossRef.
- M. Azzouzi, J. Yan, T. Kirchartz, K. Liu, J. Wang, H. Wu and J. Nelson, Phys. Rev. X, 2018, 8, 031055 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc04110f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |