
Stereoselective polymerization of rac-lactide catalyzed by zwitterionic calcium complexes†
Na
Liu‡
b,
Doudou
Liu‡
c,
Bo
Liu
 *a,
Hao
Zhang
*c and
Dongmei
Cui
a
*a,
Hao
Zhang
*c and
Dongmei
Cui
a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Science, 5625 Renmin Street, Changchun, 130022, China. E-mail: liubo@ciac.ac.cn; Fax: +86 431 85262774; Tel: +86 431 85262774
bKey Laboratory of Rare and Scattered Elements, Liaoning University, Shenyang, 110036, China
cCollege of Chemistry & Chemical Engineering, Inner Mongolia University, Hohhot, 010021, China
First published on 15th December 2020
Abstract
A reaction of heteroscorpionated ligands HL1 and HL2 (L1 = (3,5-Me2Pz)2CHP(Ph)2NPh, L2 = (3,5-Me2Pz)2CHP(Ph)2NPh(2-OMe), Pz = pyrazole) with homoleptic calcium amide Ca[N(SiMe3)2]2(THF)2, respectively, afforded novel zwitterionic calcium complexes L1CaN(SiMe3)2(THF) (1) and L2CaN(SiMe3)2 (2) through protolysis. 1 and 2 were characterized by NMR spectroscopy. The chemical shifts of the protons of pyrazole and phosphino phenyl within 1 are similar to those in complex 2, while the chemical shift of phosphine in complex 2 (28.66 ppm) is a little higher than that in complex 1 (11.43 ppm). The structures of complexes 1 and 2 were determined by X-ray diffraction analysis. Both 1 and 2 adopt a distorted trigonal bipyramidal geometry. L1 and L2 chelate to the calcium ion through a κ3 and κ4 fashion, respectively. 1 and 2 were assayed towards the ROP of rac-lactide. Both of them catalyzed the polymerization in a controlled manner. With the increase of the feeding ratio of the monomer to catalyst, the molecular weights increased linearly. At 25 °C, these two calcium complexes catalyzed the ROP of rac-lactide to afford atactic polylactides. Surprisingly, at −75 °C, complex 1 gave a heterotactic sequence enriched polylactide (Pr = 0.84); in contrast, complex 2 produced an isotactic sequence enriched polymer. The reason behind was elucidated.
Introduction
Polylactide (PLA) has a wide range of applications in packaging, agriculture, medicine, pharmaceuticals, and the tissue engineering field due in part to its relatively facile production from renewable agricultural sources, and its biodegradability and biocompatibility.1 Ring-opening polymerization (ROP) of lactide (LA) is a widely adopted manner to synthesize PLA. LA has three enantiomers L-LA, D-LA and meso-LA, thus producing PLA with various stereoisomers, such as poly(L-lactide) (PLLA), and poly(D-lactide) (PDLA).2 Both PLLA and PDLA have a melting point at 170 °C, but they start to decompose at 200 °C,3 leading to a narrow melt processing window. An interesting thermal property of PLA is that the melting temperature is raised to 230 °C upon stereo-complex formation of PLLA and PDLA.4 This stereo-complex formation is an attractive technique to improve its thermal stability. But it has not been widely industrialized because of the low efficiency and high cost of D-LA manufacturing. Isoselective ROP of rac-LA (a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of L-LA and D-LA) is considered to be another attractive approach to obtain a PLA stereo-complex. Much effort has been devoted to exploring efficient catalysts. Aluminum complexes ligated by salen ligands and their derivatives show impressive degrees of isoselective control, but suffer from low activities (taking days to reach completion) even at elevated temperatures (70–110 °C) as well as they need high initiator loadings (typically ∼1 mol%).5 Initiators based on other metals, such as Zn,6 Mg,7 In,8 K,9 group IV metals,10 and rare-earth metals,11 have been extensively studied, and some of them have displayed excellent stereo-control in the catalytic ROP of rac-LA. Nevertheless, only a few discrete isoselective catalysts with moderate to high activities have emerged recently.8e,9d,11m,12 Thus, exploring new initiators which integrate excellent isoselectivity with high activity as well as high productivity toward the ROP of rac-LA is still a challenge.
:1 mixture of L-LA and D-LA) is considered to be another attractive approach to obtain a PLA stereo-complex. Much effort has been devoted to exploring efficient catalysts. Aluminum complexes ligated by salen ligands and their derivatives show impressive degrees of isoselective control, but suffer from low activities (taking days to reach completion) even at elevated temperatures (70–110 °C) as well as they need high initiator loadings (typically ∼1 mol%).5 Initiators based on other metals, such as Zn,6 Mg,7 In,8 K,9 group IV metals,10 and rare-earth metals,11 have been extensively studied, and some of them have displayed excellent stereo-control in the catalytic ROP of rac-LA. Nevertheless, only a few discrete isoselective catalysts with moderate to high activities have emerged recently.8e,9d,11m,12 Thus, exploring new initiators which integrate excellent isoselectivity with high activity as well as high productivity toward the ROP of rac-LA is still a challenge.
Calcium is non-toxic, biocompatible and inexpensive. However, the development of calcium chemistry was impeded by the fact that the heteroleptic complexes Ca–Nu (Nu = nucleophilic group) readily decompose during deleterious Schlenk-type equilibria to generate the poorly reactive and ill-defined homoleptic species. This issue has been preliminarily addressed by employing sterically demanding multi-dentate ligands, such as tris(pyrazolyl)borates,13 aminotrop(on)iminates,14 β-diketiminates,15 bis- or tris-(imidazolin-2-ylidene-1-yl)borate,16 and phenolate.17 Well-defined heteroleptic calcium complexes have displayed remarkable catalysis in the ROP of bio-resourced cyclic esters, but except for a report by Panda et al., who reported that bis-phosphinimino-chalcogen amide and aminophosphine borane ligated homoleptic calcium complexes exhibited high iso-selectivity, with Pm values of 0.78–0.87;18 there are rare examples of calcium complexes being used in the isoselective ROP of rac-lactide in the literature.
Herein, we prepared two heteroleptic calcium complexes 1 and 2via a reaction of iminophosphine heteroscorpionate ligands (3,5-Me2Pz)2CHP(Ph)2NPh (HL1), (3,5-Me2Pz)2CHP(Ph)2NPh(2-OMe) (HL2) with homoleptic calcium amide, Ca[N(SiMe3)2]2(THF)2, respectively (Scheme 1). Their catalytic behaviors towards the ROP of rac-LA were assayed. Both of them showed high activity toward the ROP of rac-LA, but no stereoselectivity at 25 °C. Surprisingly, at low temperature, complexes 1 and 2 respectively showed heteroselectivity (Pr = 0.84) and isoselectivity (Pm = 0.78). The reason behind was elucidated.
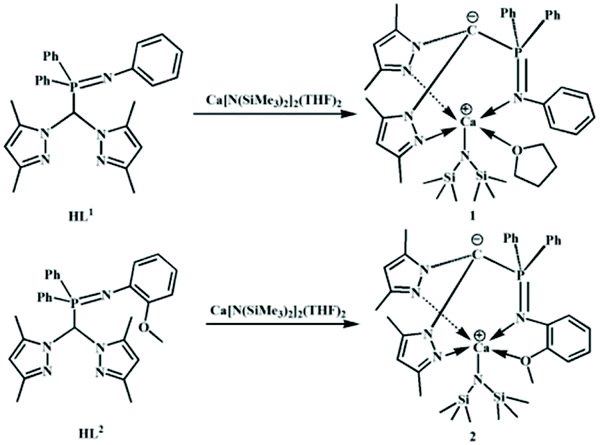 | ||
| Scheme 1 Synthesis of heteroscorpionate calcium complexes 1–2. | ||
Results and discussion
Synthesis and characterization of calcium complexes 1–2
Recently, our group demonstrated that iminophosphine ligated achiral heteroscorpionate zwitterionic rare-earth metal bisalkyl complexes showed high heteroselectivity (Pr = 0.89) towards the ROP of rac-lactide,19 whilst the corresponding zinc silylamido and benzyloxy complexes exhibited isoselectivity (Pm = 0.85).20 These diametrical results intrigued us to employ the iminophosphine heteroscorpionate ligand (3,5-Me2Pz)2CHP(Ph)2NPh (HL1) to prepare a calcium complex. Treatment of HL1 with 1.05 equiv. of Ca[N(SiMe3)2]2(THF)2 in THF at 25 °C for 5 h afforded a pale yellow solution. Most of the solvent was removed under vacuum. Then several drops of hexane were added. The mixture was stored at −30 °C to give colorless crystals. The product was firstly analyzed by 1H NMR spectroscopy. The disappearance of the singlet at 6.84 ppm assigned to the methine proton of HL1 suggested that the protolysis reaction occurred to generate a carbanion which gave a doublet resonance at 58.83 ppm with J = 154.2 Hz in the 13C NMR spectrum (Fig. S1†).20 The ratio of the integral intensity of resonance at 5.40 ppm to that at 0.55 ppm, respectively, derivated from the protons of 4-H of pyrazole and methyl protons of the amide groups, is 1:9, indicating the formation of the heteroleptic calcium amide 1 (Fig. S2†). The resonances at 3.71 ppm and 1.43 ppm are attributed to the protons of THF, suggesting the coordination of THF which is unwanted. HL2 (3,5-Me2Pz)2CHP(Ph)2NPh(2-OMe) containing an additional dentate was selected. By following a similar procedure to synthesize complex 1, complex 2 was isolated. As expected, there are no resonances assigned to THF. Compared to that in HL2 (3.12 ppm), the resonance of methoxyl protons shifts downfield (3.98 ppm), suggesting that the methoxyl group coordinates to the metal center (Fig. S3†). The chemical shifts of the protons of pyrazole and phosphino phenyl are similar to those in complex 1. While the chemical shift of phosphine in complex 2 (28.66 ppm) (Fig. S4†) is a little higher than that in complex 1 (11.43 ppm) (Fig. S5†).
The molecular structures of complexes 1 and 2 were determined by X-ray diffraction analysis (Fig. 1). The details of the structural and refinement parameters are given in Table S1.† The heteroscorpionate ligand HL1 facially chelates to the calcium ion through one nitrogen atom of phosphino imine and the two nitrogen atoms of the pyrazole rings in a κ3 mode. The atoms of C(1), P(1), N(5) and Ca(1) are almost coplanar with the amino phenyl. The THF molecule and amido group locate in the two sides of the above plane. In contrast, in complex 2, the heteroscorpionate ligand HL2 chelates to the calcium ion through N(2), N(4), N(5) and O(1) in a κ4 fashion. The dihedral angle of the four member ring composed by Ca(1), N(5), C(24) and C(29) is 13.31°, suggesting that the amino phenyl deviates from the plane composed by C(1), P(1), N(5) and Ca(1) owing to the coordination of the methoxyl group. In addition, the rigidity derived from the methoxyl coordination results in the angle of O(1)–Ca(1)–N(5) (65.49°) being much smaller than that in complex 1 (89.73°) and the distance between Ca(1) and C(1) (3.230 Å) being longer than that in complex 1 (3.160 Å). Apparently, the distance between the carbanion atom C(1) and the central calcium ion Ca(1) is beyond the normal range of the Ca–C bond, indicating that the complex is a zwitterion with one negative charge that resides on C(1) and a positive charge on Ca(1).
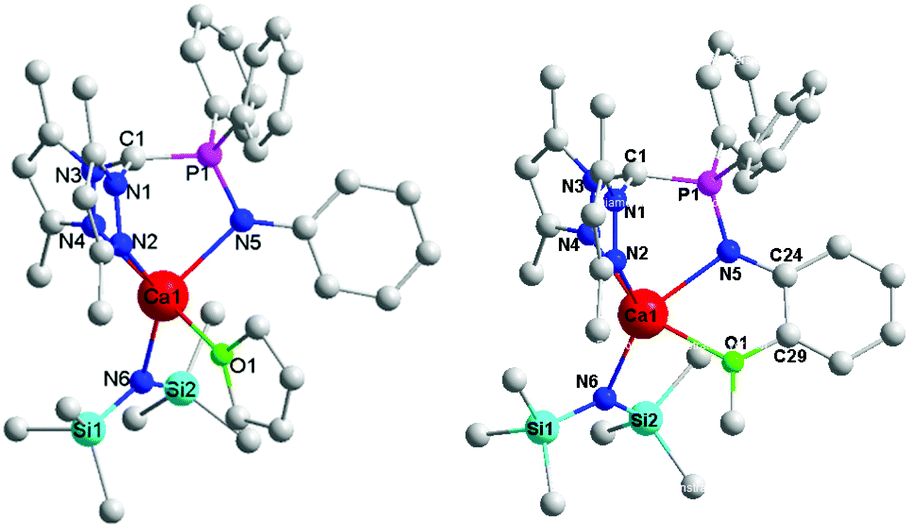 | ||
| Fig. 1 The crystal structures of complexes 1 (left) and 2 (right). All the hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (deg): complex 1: Ca(1)–N(2) 2.494, Ca(1)–N(4) 2.498, Ca(1)–N(5) 2.421, Ca(1)–N(6) 2.331, Ca(1)–O(1) 2.438, Ca(1)⋯C(1) 3.160, N(5)–P(1) 1.635, C(1)–N(1) 1.432, C(1)–N(3) 1.423, C(1)–P(1) 1.726, N(4)–Ca(1)–N(5) 90.19, N(5)–Ca(1)–O(1) 89.73, N(2)–Ca(1)–N(5) 88.33, N(5)–Ca(1)–N(6) 134.53, N(2)–Ca(1)–N(6) 137.07, P(1)–N(5)–Ca(1) 107.14, N(1)–C(1)–N(3) 114.99, N(1)–C(1)–P(1) 117.29, N(3)–C(1)–P(1) 118.05; complex 2: Ca(1)–N(2) 2.479, Ca(1)–N(4) 2.445, Ca(1)–N(5) 2.391, Ca(1)–N(6) 2.313, Ca(1)–O(1) 2.450, Ca(1)⋯C(1) 3.230, N(5)–P(1) 1.608, C(1)–N(1) 1.439, C(1)–N(3) 1.425, C(1)–P(1) 1.734, N(2)–Ca(1)–N(4) 83.89, N(2)–Ca(1)–N(5) 84.20, O(1)–Ca(1)–N(5) 65.49, N(5)–Ca(1)–N(4) 88.74, N(5)–Ca(1)–N(6) 142.82, N(2)–Ca(1)–N(6) 131.55, P(1)–N(5)–Ca(1) 110.44, N(1)–C(1)–N(3) 115.10, N(1)–C(1)–P(1) 114.83, N(3)–C(1)–P(1) 116.50. | ||
Ring-opening polymerization of rac-LA initiated by calcium complexes 1 and 2
Complex 1 as a single-component catalyst was initially assayed towards the ROP of rac-lactide at 25 °C. At a feeding ratio of [LA]0:[Ca]0 = 200:1, the monomer conversion reached 89% within 1 min (Table 1, entry 1). The molecular weight of the afforded polymer is 3.10 × 104, a little higher than the theory value, and the molecular weight distribution is 1.5. According to the homonuclear decoupled 1H NMR spectrum, the Pr value is 0.40, suggesting the formation of atactic PLA. Doubling the monomer feeding but keeping the monomer concentration as a constant, 97% conversion of the monomer was achieved within 5 min (Table 1, entry 2). The molecular weight of the corresponding polymer is 5.93 × 104, nearly twice higher than that of the former one, while the molecular weight distribution is still relatively narrow. A further increase in the feeding of rac-LA is necessary for achieving high conversion to prolong the polymerization time (Table 1, entries 3–5). The molecular weights of the afforded polymers increase linearly with the feeding ratio, suggesting that the polymerization proceeds in a controlled manner.21 Noteworthy is that the heteroselectivity slightly increases. This might be attributed to the decreasing polymerization rate. To confirm this hypothesis, the polymerization was carried out at 0 °C (Table 1, entry 6). The Pr value of the afforded polymer is 45%, 5% higher than that afforded at 25 °C. This inspired us to further lower the polymerization temperature. With the temperature decreasing from −20 °C to −75 °C, the Pr value increases from 0.56 to 0.84 (Table 1, entries 7–9). The molecular weights of polymers afforded at various temperatures are close to their corresponding theory values. Complex 1 was one of few calcium complexes that exhibited high hetero-selectivity.13
a
| Entry | [LA]0/[1]0 | T p (°C) | T (min) | Conv.b (%) | M n,calcd (104)c | M n,exp (104)d | M w/Mnd | P r |
|---|---|---|---|---|---|---|---|---|
| a Conditions: 1: 10 μmol, [LA]0 = 0.8 M, solvent: THF. b Determined by 1H NMR spectroscopy. c M n,calcd = [LA]0/[Cat]0 × 144.13 × conv. (%). d Determined by GPC in THF at 40 °C against the polystyrene standard, Mn using a correcting factor for polylactides (0.58).22 e Determined from the relative intensity of the tetrad signals of the methine region in the homonuclear decoupled 1H NMR spectra. Pr = 2I1/(I2 + I1), while I1 = δ 5.20–5.23 ppm (rmr, rmm) and I2 = δ 5.13–5.20 ppm (mmr, mmm, mrm).23 | ||||||||
| 1 | 200/1 | 25 | 1 | 89 | 2.56 | 3.10 | 1.5 | 0.40 |
| 2 | 400/1 | 25 | 5 | 97 | 5.59 | 5.93 | 1.4 | 0.42 |
| 3 | 800/1 | 25 | 5 | 63 | 7.26 | 6.32 | 1.6 | 0.43 |
| 4 | 800/1 | 25 | 20 | 96 | 11.07 | 9.17 | 1.6 | 0.43 |
| 5 | 1600/1 | 25 | 60 | 96 | 22.14 | 16.20 | 1.5 | 0.45 |
| 6 | 200/1 | 0 | 8 | 89 | 2.56 | 2.81 | 1.5 | 0.45 |
| 7 | 200/1 | −20 | 20 | 83 | 2.36 | 2.62 | 1.4 | 0.56 |
| 8 | 200/1 | −40 | 120 | 98 | 2.82 | 3.80 | 1.6 | 0.67 |
| 9 | 200/1 | −75 | 540 | 79 | 2.28 | 2.89 | 1.4 | 0.84 |
The catalytic behavior of complex 2 was also examined at various feeding ratios of monomer to catalyst and polymerization temperatures. The results are summarized in Table 2. With the increase of the feeding ratio, the molecular weights of the afforded polymers increase linearly, and the molecular weight distributions are relatively narrow (Table 2, entries 1–4), indicating that 2 also catalyzed the ROP of rac-lactide in a controlled manner. At 25 °C, 2 catalyzed the ROP of rac-lactide to generate atactic polylactide with a Pm value of ca. 0.55. The polymerization was carried out at 0 °C to produce polylactide with a Pm value of 0.58 (Table 2, entry 5). Surprisingly, on further decreasing the polymerization temperature from −20 °C to −40 °C, the Pm value gradually enhanced from 0.65 to 0.72 (Table 2, entries 6 and 7). The Pm value could reach as high as 0.78 at −75 °C (Table 2, entry 8), however, no melting temperature was detected in the second run through DSC analysis.24 Although Panda et al. recently reported calcium complexes stabilized by the bis-phosphinimino-chalcogen amide ligand and aminophosphine borane ligand18 catalyzed ROP of rac-lactide to give polylactide with the highest Pm value of 0.87, this is the first heteroleptic calcium complex that exhibited high isotactic selectivity. Thus, the reason behind was investigated.
| Entry | [LA]0/[2]0 | T p (°C) | T (min) | Conv.b (%) | M n,calcd (104)c | M n,exp (104)d | M w/Mnd | P m |
|---|---|---|---|---|---|---|---|---|
| a Conditions: 2: 10 μmol, [LA]0 = 0.8 M, solvent: THF. b Determined by 1H NMR spectroscopy. c M n,calcd = [LA]0/[Cat]0 × 144.13 × conv. (%). d Determined by GPC in THF at 40 °C against the polystyrene standard, Mn using a correcting factor for polylactides (0.58).22 e Determined from the relative intensity of the tetrad signals of the methine region in the homonuclear decoupled 1H NMR spectra. Pm = (I2 − I1)/(I2 + I1), while I1 = δ 5.20–5.23 ppm (rmr, rmm) and I2 = δ 5.13–5.20 ppm (mmr, mmm, mrm).23 | ||||||||
| 1 | 200/1 | 25 | 2 | 77 | 2.22 | 1.35 | 2.2 | 0.56 |
| 2 | 400/1 | 25 | 5 | 97 | 5.59 | 5.58 | 1.3 | 0.55 |
| 3 | 1200/1 | 25 | 40 | 97 | 16.78 | 13.23 | 1.6 | 0.54 |
| 4 | 1600/1 | 25 | 60 | 96 | 22.14 | 18.15 | 1.6 | 0.55 |
| 5 | 200/1 | 0 | 8 | 98 | 2.82 | 1.76 | 2.1 | 0.58 |
| 6 | 200/1 | −20 | 20 | 88 | 2.54 | 1.75 | 2.0 | 0.65 |
| 7 | 200/1 | −40 | 120 | 86 | 2.48 | 2.09 | 1.8 | 0.72 |
| 8 | 200/1 | −75 | 540 | 62 | 1.79 | 1.69 | 1.8 | 0.78 |
The difference between 1 and 2 is that there is an additional coordination side arm in 2, leading to the asymmetry of the molecular structure of 2 (Fig. 1), suggesting that the coordination of the side arm should be the key factor to determine the iso-selectivity. As the iso-selectivity was significantly affected by the temperature, complex 2 was investigated via the variable-temperature 1H NMR technique (Fig. 2). The protons of the methoxyl group give a sharp singlet at 4.06 ppm at 25 °C. This signal shifts to 4.00 ppm but still remains narrow at 0 °C. From −20 °C to −80 °C, the singlet attributed to methoxyl protons continues to shift upfield, but becomes broad gradually. Meanwhile, the multiplet assigned to the ortho protons of diphenylphosphine also becomes wide even flat. In contrast, the resonances derived from amino phenyl protons have no obvious change. This could be explained by the fluxional behavior of complex 2 due to the swing of the amino phenyl group as the Gibbs free energy of methoxyl disassociation is 9 kcal mol−1 (Fig. S6 and 7†).25 With the decrease of the temperature, the rate of the swing becomes slow. 2 could stay at a certain state long enough for the monomers with the same chirality continuous insertion, giving isotactic sequence enriched polylactide.
 | ||
| Fig. 2 Variable-temperature 1H-NMR spectra (toluene-d8, 400 MHz) showing the phenyl and OCH3 regions of the complex 2. | ||
To further investigate the microstructures of isotactic PLAs obtained in this work, the tetrad signal of a typical PLA sample obtained by 2 was analyzed via homonuclear decoupled 1H NMR spectroscopy. As depicted in Fig. 3, the resonance assigned to mmm tetrad splits, suggesting that the length of the isotactic sequence is not long, thus leading to no Tm being detected. The absence of resonance assigned to rmr tetrad excludes the existence of –RRRRSSRRRR–/–SSSSRRSSSS–sequences derived from single insertion stereo errors. In contrast, the intensity ratio of rmm:mmr:mrm is near 1:1:1, indicating that the polymer main chain is essentially stereo blocky (e.g. –RRRRRRSSSSSS–). Hence, it is conceivable that the mechanism of isoselectivity achieved by complex 2 is the chain end control.
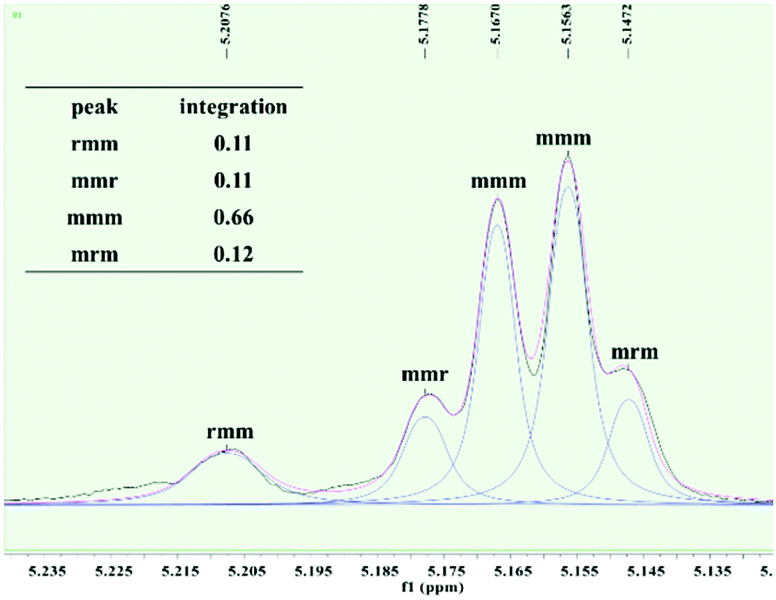 | ||
| Fig. 3 Methine region of the homonuclear-decoupled 1H NMR spectrum of the resulting PLA (Table 2, entry 8). | ||
To provide further insight into the mechanism, an oligomer Mn = 2900 g mol−1 was synthesized and characterized by 1H NMR spectroscopy and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). The MALDI-TOF mass spectrum (Fig. 4) consisted of two series of molecular ion peaks. One series is corresponding to linear PLA with -N(SiMe3)2 as chain ends which were corroborated from the 1H NMR spectrum of the oligomer, where the resonance at 0.16 ppm was assigned to the methyl protons of the N(SiMe3)2 group (Fig. S8†). The other series is corresponding to [LA]n, suggesting the existence of cyclic PLA with no chain ends. According to the literature,26 the formation of the cyclic polymers might be via intramolecular backbiting.
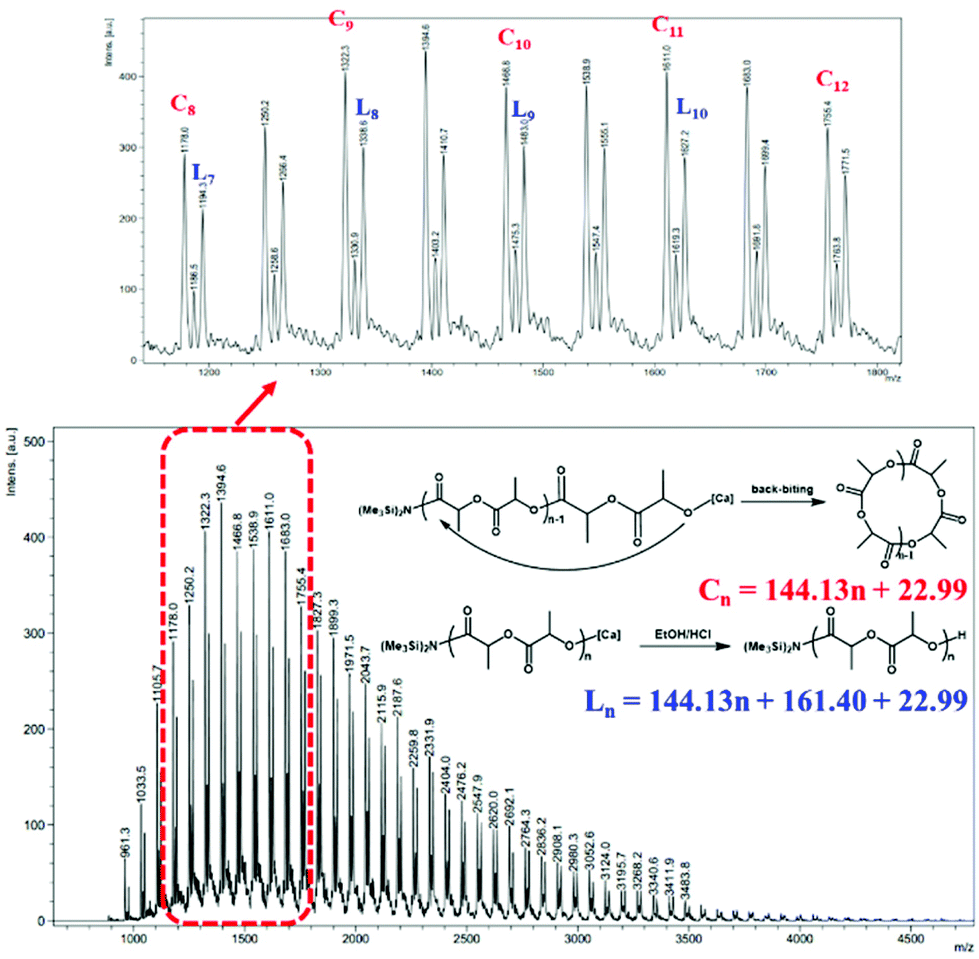 | ||
| Fig. 4 MALDI-TOF mass spectrum (major population: Na+) of the PLA sample (Mn,GPC = 2900 g mol−1, Mw/Mn = 1.88) prepared with catalyst 1 (C(cyclic) = 144.13n + 22.99, L(linear) = 144.13n + 161.40 + 22.99, where n is the degree of polymerization, MLA = 144.13 g mol−1, MNH(SiMe3)2 = 161.40 g mol−1, MNa = 22.99 g mol−1). | ||
Conclusions
Novel zwitterionic calcium complexes 1 and 2 were synthesized via treatment of heteroscorpionated iminophosphine ligands HL1 and HL2 with calcium amide, respectively. Compared to L1, L2 has an additional methoxyl group at the ortho position of the imino phenyl ring. Thus, L1 coordinates to the calcium center in a κ3 fashion, while L2 chelates to the metal center in a κ4 mode. The additional coordination side arm leads to the asymmetry of the structure of 2. Both 1 and 2 could catalyze the ring-opening polymerization of rac-lactide in a controlled manner to give atactic polylactide at 25 °C. While with the temperature decrease of polymerization catalyzed by 1, the content of the heterotactic sequence with the polylactide increased. At −75 °C, the Pr value reached 0.84. In contrast, the coordination side arm in 2 resulted in iso-selectivity towards the ROP of rac-lactide. The iso-selectivity of 2 increased with the decrease of the swing rate of the side arm. At −75 °C, the Pm value reached 0.78. The mechanism of isoselectivity achieved by complex 2 is the chain end control. The existence of a cyclic polymer indicated the occurrence of intramolecular backbiting during the polymerization. These interesting results will shed new light on designing catalyst precursors for specifically selective polymerization.Experimental section
General procedures
All reactions were carried out under a dry nitrogen atmosphere using Schlenk techniques or in a glovebox filled with dry nitrogen. Hexane was purified using an SPS Braun system. THF was dried by distillation over sodium with benzophenone as the indicator under a nitrogen atmosphere and was stored over freshly cut sodium in a glovebox. Calcium iodide was purchased from Aldrich and stored in a glove box. Ca[N(SiMe3)2]2(THF)2 can be prepared via salt metathesis involving treatment of metal halides with alkali metal amides.27 HL1 and HL2 were synthesized according to previous literatures.19,28rac-LA was recrystallized with dry ethyl acetate three times. Glassware and flasks used in the polymerization were dried in an oven at 115 °C for 14 h and exposed to a vacuum-nitrogen cycle three times. The molecular weights and the molecular weight distributions of the polymers were measured using a TOSOH HLC 8220 GPC instrument at 40 °C with THF as the eluent against the polystyrene standard. Complexes for NMR spectroscopy measurements were prepared in a glovebox by the use of a NMR tube sealed with paraffin film. 1H, 31P and 13C NMR spectra were recorded on a Bruker AV400 spectrometer. Elemental analyses were performed at National Analytical Research Centre of Changchun Institute of Applied Chemistry (CIAC).X-ray crystallographic study
A crystal for X-ray analysis was obtained as described in the following preparations. The crystal was manipulated in a glovebox. Data collection was performed at −86.5 °C using a Bruker SMART APEX diffractometer with a CCD area detector and graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The determination of the crystal class and unit-cell parameters was carried out by the SMART program package. The raw frame data were processed using SAINT and SADABS to yield the reflection data file. The structures were solved by using the SHELXTL program.ROP of rac-LA
Polymerizations of rac-LA were carried out in a 25 mL flask under a nitrogen atmosphere. A typical procedure was described as follows: a solution of complex 1 (10 μmol) in THF (2 mL) was added to a stirred solution of rac-LA (4 mmol) in THF (3 mL). The polymerization took place immediately at 25 °C. The system became viscous in 5 min. Then it was quenched by adding several drops of acidified ethanol, and a small sample of the viscous solution was separated for the measurement of conversion by 1H NMR. Then polymers were precipitated with abundant ethanol, collected and dried at 40 °C for 24 h in vacuo. The molecular weights and molecular weight distributions of the resulting polymers were determined by GPC. The tacticity of the PLA was calculated according to the methine region homonuclear decoupled 1H NMR spectrum.Synthesis of the calcium complexes
Author contributions
N. Liu and D. Liu performed the experiments. B. Liu and D. Cui conceived and designed the experiments. All authors discussed the results and commented on the manuscript. N. Liu and D. Liu contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from the National Natural Science Foundation of China for the project no. 21274143.Notes and references
- (a) S. Slomkowski, S. Penczek and A. Duda, Polym. Adv. Technol., 2014, 25, 436–447 CrossRef CAS; (b) M. Singhvi and D. Gokhale, RSC Adv., 2013, 3, 13558–13568 RSC; (c) T. A. Hottle, M. M. Bilec and A. E. Landis, Polym. Degrad. Stab., 2013, 98, 1898–1907 CrossRef CAS; (d) S. Inkinen, M. Hakkarainen, A. C. Albertsson and A. Sodergard, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS; (e) J. Ahmed and S. K. Varshney, Int. J. Food Prop., 2011, 14, 37–58 CrossRef CAS.
- (a) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS; (b) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- O. Wachsen, K. H. Reichert, R. P. Krüger, H. Much and G. Schulz, Polym. Degrad. Stab., 1997, 55, 225–231 CrossRef CAS.
- (a) Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS; (b) H. Tsuji, F. Horii, S. H. Hyon and Y. Ikada, Macromolecules, 1991, 24, 2719–2724 CrossRef CAS.
- (a) N. Spassky, M. Wisniewski, C. Pluta and A. LeBorgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS; (b) C. P. Radano, G. L. Baker and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 1552–1553 CrossRef CAS; (c) T. M. Ovitt and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4686–4692 CrossRef CAS; (d) N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS; (e) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS; (f) Z. Y. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS; (g) Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS; (h) Z. H. Tang, X. S. Chen, X. Pang, Y. K. Yang, X. F. Zhang and X. B. Jing, Biomacromolecules, 2004, 5, 965–970 CrossRef CAS; (i) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS; (j) M. H. Chisholm, N. J. Patmore and Z. P. Zhou, Chem. Commun., 2005, 127–129, 10.1039/b413266a; (k) N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433–4451 CrossRef CAS; (l) H. L. Chen, S. Dutta, P. Y. Huang and C. C. Lin, Organometallics, 2012, 31, 2016–2025 CrossRef CAS; (m) H. Du, X. Pang, H. Yu, X. Zhuang, X. Chen, D. Cui, X. Wang and X. Jing, Macromolecules, 2007, 40, 1904–1913 CrossRef CAS; (n) N. Maudoux, T. Roisnel, V. Dorcet, J. F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2014, 20, 6131–6147 CrossRef CAS; (o) A. Pilone, K. Press, I. Goldberg, M. Kol, M. Mazzeo and M. Lamberti, J. Am. Chem. Soc., 2014, 136, 2940–2943 CrossRef CAS; (p) K. Press, I. Goldberg and M. Kol, Angew. Chem., Int. Ed., 2015, 54, 14858–14861 CrossRef CAS.
- (a) M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS; (b) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS; (c) M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785–2794 CrossRef CAS; (d) C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS; (e) J.-C. Wu, B.-H. Huang, M.-L. Hsueh, S.-L. Lai and C.-C. Lin, Polymer, 2005, 46, 9784–9792 CrossRef CAS; (f) H.-Y. Chen, H.-Y. Tang and C.-C. Lin, Macromolecules, 2006, 39, 3745–3752 CrossRef CAS; (g) Y. Huang, W.-C. Hung, M.-Y. Liao, T.-E. Tsai, Y.-L. Peng and C.-C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2318–2329 CrossRef CAS; (h) H. J. Chuang, H. L. Chen, B. H. Huang, T. E. Tsai, P. L. Huang, T. T. Liao and C. C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1185–1196 CrossRef CAS; (i) S. D. Song, X. Y. Zhang, H. Y. Ma and Y. Yang, Dalton Trans., 2012, 41, 3266–3277 RSC; (j) M. Fuchs, S. Schmitz, P. M. Schafer, T. Secker, A. Metz, A. N. Ksiazkiewicz, A. Pich, P. Kogerler, K. Y. Monakhov and S. Herres-Pawlis, Eur. Polym. J., 2020, 122, 109302 CrossRef CAS; (k) P. P. Rade and B. Garnaik, Int. J. Polym. Anal. Charact., 2020, 25, 283–299 CrossRef CAS; (l) P. Marin, M. J. L. Tschan, P. Haquette, T. Roisnel, I. del Rosal, L. Maron and C. M. Thomas, Eur. Polym. J., 2019, 120, 109208 CrossRef CAS; (m) D. M. Gonzalez, J. Cisterna, I. Brito, T. Roisnel, J. R. Hamon and C. Manzur, Polyhedron, 2019, 162, 91–99 CrossRef CAS.
- (a) H. B. Wang, Y. Yang and H. Y. Ma, Macromolecules, 2014, 47, 7750–7764 CrossRef CAS; (b) H. Xie, Z. Mou, B. Liu, P. Li, W. Rong, S. Li and D. Cui, Organometallics, 2014, 33, 722–730 CrossRef CAS; (c) L. Wang and H. Ma, Macromolecules, 2010, 43, 6535–6537 CrossRef CAS; (d) S. D. Song, H. Y. Ma and Y. Yang, Dalton Trans., 2013, 42, 14200–14211 RSC; (e) Y. Wang, W. Zhao, X. L. Liu, D. M. Cui and E. Y. X. Chen, Macromolecules, 2012, 45, 6957–6965 CrossRef CAS; (f) Y. Yang, H. B. Wang and H. Y. Ma, Polyhedron, 2016, 117, 569–578 CrossRef CAS; (g) H. Wang, J. Guo, Y. Yang and H. Ma, Dalton Trans., 2016, 45, 10942–10953 RSC; (h) H. C. Quilter, R. H. Drewitt, M. F. Mahon, G. Kociok-Kohn and M. D. Jones, J. Organomet. Chem., 2017, 848, 325–331 CrossRef CAS.
- (a) A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS; (b) I. Yu, A. Acosta-Ramirez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758–12773 CrossRef CAS; (c) D. C. Aluthge, B. O. Patrick and P. Mehrkhodavandi, Chem. Commun., 2013, 49, 4295–4297 RSC; (d) D. C. Aluthge, J. M. Ahn and P. Mehrkhodavandi, Chem. Sci., 2015, 6, 5284–5292 RSC; (e) D. Myers, A. J. P. White, C. M. Forsyth, M. Bown and C. K. Williams, Angew. Chem., Int. Ed., 2017, 56, 5277–5282 CrossRef CAS; (f) J. Beament, M. F. Mahon, A. Buchard and M. D. Jones, New J. Chem., 2017, 41, 2198–2203 RSC.
- (a) J. J. Zhang, J. Xiong, Y. Y. Sun, N. Tang and J. C. Wu, Macromolecules, 2014, 47, 7789–7796 CrossRef CAS; (b) Z. R. Dai, Y. Y. Sun, J. Xiong, X. B. Pan and J. C. Wu, ACS Macro Lett., 2015, 4, 556–560 CrossRef CAS; (c) C. J. Chen, Y. Q. Cui, X. Y. Mao, X. B. Pan and J. C. Wu, Macromolecules, 2017, 50, 83–96 CrossRef CAS; (d) Y. Y. Sun, J. Xiong, Z. R. Dai, X. B. Pan, N. Tang and J. C. Wu, Inorg. Chem., 2016, 55, 136–143 CrossRef CAS; (e) M. Fernandez-Millan, P. Ortega, T. Cuenca, J. Cano and M. E. G. Mosquera, Organometallics, 2020, 39, 2278–2286 CrossRef CAS; (f) C. J. Chen, J. X. Jiang, X. Y. Mao, Y. Cong, Y. Q. Cui, X. B. Pan and J. C. Wu, Inorg. Chem., 2018, 57, 3158–3168 CrossRef CAS.
- (a) A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295, 10.1039/b718678a; (b) A. L. Zelikoff, J. Kopilov, I. Goldberg, G. W. Coates and M. Kol, Chem. Commun., 2009, 6804–6806, 10.1039/b915211c; (c) E. L. Whitelaw, M. D. Jones and M. F. Mahon, Inorg. Chem., 2010, 49, 7176–7181 CrossRef CAS; (d) E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004–10006 RSC; (e) A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS; (f) M. D. Jones, S. L. Hancock, P. McKeown, P. M. Schafer, A. Buchard, L. H. Thomas, M. F. Mahon and J. P. Lowe, Chem. Commun., 2014, 50, 15967–15970 RSC; (g) M. D. Jones, L. Brady, P. McKeown, A. Buchard, P. M. Schafer, L. H. Thomas, M. F. Mahon, T. J. Woodman and J. P. Lowe, Chem. Sci., 2015, 6, 5034–5039 RSC; (h) M. D. Jones, X. J. Wu, J. Chaudhuri, M. G. Davidson and M. J. Ellis, Mater. Sci. Eng., C, 2017, 80, 69–74 CrossRef CAS.
- (a) H. Y. Ma, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2006, 45, 7818–7821 CrossRef; (b) C. X. Cai, A. Amgoune, C. W. Lehmann and J. F. Carpentier, Chem. Commun., 2004, 330–331, 10.1039/b314030j; (c) A. Amgoune, C. M. Thomas, T. Roisnel and J. F. Carpentier, Chem. – Eur. J., 2006, 12, 169–179 CrossRef CAS; (d) A. Amgoune, C. M. Thomas and J. F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693–697 CrossRef CAS; (e) X. L. Liu, X. M. Shang, T. Tang, N. H. Hu, F. K. Pei, D. M. Cui, X. S. Chen and X. B. Jing, Organometallics, 2007, 26, 2747–2757 CrossRef CAS; (f) L. Clark, M. G. Cushion, H. E. Dyer, A. D. Schwarz, R. Duchateau and P. Mountford, Chem. Commun., 2010, 46, 273–275 RSC; (g) S. Yang, Z. Du, Y. Zhang and Q. Shen, Chem. Commun., 2012, 48, 9780–9782 RSC; (h) P. L. Arnold, J. C. Buffet, R. P. Blaudeck, S. Sujecki, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed., 2008, 47, 6033–6036 CrossRef CAS; (i) T. P. A. Cao, A. Buchard, X. F. Le Goff, A. Auffrant and C. K. Williams, Inorg. Chem., 2012, 51, 2157–2169 CrossRef CAS; (j) N. Maudoux, T. Roisnel, J. F. Carpentier and Y. Sarazin, Organometallics, 2014, 33, 5740–5748 CrossRef CAS; (k) C. Bakewell, T. P. A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577–20580 CrossRef CAS; (l) C. Bakewell, A. J. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226–9230 CrossRef CAS; (m) T. Q. Xu, G. W. Yang, C. Liu and X. B. Lu, Macromolecules, 2017, 50, 515–522 CrossRef CAS; (n) N. Y. Rad'kova, T. A. Kovylina, A. S. Shavyrin, A. V. Cherkasov, G. K. Fukin, K. A. Lyssenko and A. A. Trifonov, New J. Chem., 2020, 44, 7811–7822 RSC.
- S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689–692 CrossRef CAS.
- (a) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48–49, 10.1039/B208679d; (b) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS; (c) M. H. Chisholm, J. C. Gallucci, G. Yaman and T. Young, Chem. Commun., 2009, 1828–1830, 10.1039/b818375a; (d) M. H. Chisholm, J. C. Gallucci and G. Yaman, Dalton Trans., 2009, 368–374, 10.1039/b812228h; (e) M. G. Cushion and P. Mountford, Chem. Commun., 2011, 47, 2276–2278 RSC.
- (a) S. Datta, P. W. Roesky and S. Blechert, Organometallics, 2007, 26, 4392–4394 CrossRef CAS; (b) S. Datta, M. T. Gamer and P. W. Roesky, Organometallics, 2008, 27, 1207–1213 CrossRef CAS.
- (a) A. G. M. Barrett, I. J. Casely, M. R. Crimmin, M. S. Hill, J. R. Lachs, M. F. Mahon and P. A. Procopiou, Inorg. Chem., 2009, 48, 4445–4453 CrossRef CAS; (b) M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042–2043 CrossRef CAS; (c) A. G. Avent, M. R. Crimmin, M. S. Hill and P. B. Hitchcock, Dalton Trans., 2005, 278–284, 10.1039/b415468a; (d) S. Harder and J. Brettar, Angew. Chem., Int. Ed., 2006, 45, 3474–3478 CrossRef CAS; (e) C. Ruspic, S. Nembenna, A. Hofmeister, J. Magull, S. Harder and H. W. Roesky, J. Am. Chem. Soc., 2006, 128, 15000–15004 CrossRef CAS; (f) S. P. Sarish, H. W. Roesky, M. John, A. Ringe and J. Magull, Chem. Commun., 2009, 2390–2392, 10.1039/b822148k.
- (a) M. Arrowsmith, M. S. Hill and G. Kociok-Kohn, Organometallics, 2009, 28, 1730–1738 CrossRef CAS; (b) M. Arrowsmith, A. Heath, M. S. Hill, P. B. Hitchcock and G. Kociok-Köhn, Organometallics, 2009, 28, 4550–4559 CrossRef CAS.
- (a) B. Liu, T. Roisnel, J. P. Guegan, J. F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 18, 6289–6301 CrossRef CAS; (b) D. J. Darensbourg, W. Choi, P. Ganguly and C. P. Richers, Macromolecules, 2006, 39, 4374–4379 CrossRef CAS; (c) D. J. Darensbourg, W. Choi and C. P. Richers, Macromolecules, 2007, 40, 3521–3523 CrossRef CAS; (d) D. J. Darensbourg, W. Choi, O. Karroonnirun and N. Bhuvanesh, Macromolecules, 2008, 41, 3493–3502 CrossRef CAS; (e) V. Poirier, T. Roisnel, J. F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820–9827, 10.1039/b917799j; (f) Y. Sarazin, D. Rosca, V. Poirier, T. Roisnel, A. Silvestru, L. Maron and J. F. Carpentier, Organometallics, 2010, 29, 6569–6577 CrossRef CAS.
- (a) J. Bhattacharjee, A. Harinath, H. P. Nayek, A. Sarkar and T. K. Panda, Chemistry, 2017, 23, 9319–9331 CrossRef CAS; (b) J. Bhattacharjee, A. Harinath, A. Sarkar and T. K. Panda, Chem.– Asian J., 2020, 15, 860–866 CrossRef CAS.
- Z. Zhang and D. Cui, Chem. – Eur. J., 2011, 17, 11520–11526 CrossRef CAS.
- Z. Mou, B. Liu, M. Wang, H. Xie, P. Li, L. Li, S. Li and D. Cui, Chem. Commun., 2014, 50, 11411–11414 RSC.
- (a) X. Xu, Y. Chen, G. Zou, Z. Ma and G. Li, J. Organomet. Chem., 2010, 695, 1155–1162 CrossRef CAS; (b) J. Guo, P. Haquette, J. Martin, K. Salim and C. M. Thomas, Angew. Chem., Int. Ed., 2013, 52, 13584–13587 CrossRef CAS.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889–899 CrossRef CAS.
- L. F. Sanchez-Barba, A. Garces, J. Fernandez-Baeza, A. Otero, C. Alonso-Moreno, A. Lara-Sanchez and A. M. Rodriguez, Organometallics, 2011, 30, 2775–2789 CrossRef CAS.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433–4451 CrossRef CAS.
- K. Beckerle, R. Manivannan, T. P. Spaniol and J. Okuda, Organometallics, 2006, 25, 3019–3026 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS.
- M. Westerhausen, Inorg. Chem., 1991, 30, 96–101 CrossRef CAS.
- Z. Mou, B. Liu, X. Liu, H. Xie, W. Rong, L. Li, S. Li and D. Cui, Macromolecules, 2014, 47, 2233–2241 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 31P, 13C NMR spectra of complexes 1–2; the GPC traces and DSC curves of the afforded polymers; the crystallographic data and structure refinement details for complexes 1–2; the optimized structures and the free energy of the O atom of the methoxy group coordinated/dissociated with the central metal. CCDC 2034134 (2) and 2034135 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0py01397h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |