
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCryptophane–xenon complexes for 129Xe MRI applications
Serge D. Zemerov and
Ivan J. Dmochowski *
*
Department of Chemistry, University of Pennsylvania, 231 South 34th St., Philadelphia, PA 19104-6323, USA. E-mail: ivandmo@sas.upenn.edu
First published on 17th February 2021
Abstract
The use of magnetic resonance imaging (MRI) and spectroscopy (MRS) in the clinical setting enables the acquisition of valuable anatomical information in a rapid, non-invasive fashion. However, MRI applications for identifying disease-related biomarkers are limited due to low sensitivity at clinical magnetic field strengths. The development of hyperpolarized (hp) 129Xe MRI/MRS techniques as complements to traditional 1H-based imaging has been a burgeoning area of research over the past two decades. Pioneering experiments have shown that hp 129Xe can be encapsulated within host molecules to generate ultrasensitive biosensors. In particular, xenon has high affinity for cryptophanes, which are small organic cages that can be functionalized with affinity tags, fluorophores, solubilizing groups, and other moieties to identify biomedically relevant analytes. Cryptophane sensors designed for proteins, metal ions, nucleic acids, pH, and temperature have achieved nanomolar-to-femtomolar limits of detection via a combination of 129Xe hyperpolarization and chemical exchange saturation transfer (CEST) techniques. This review aims to summarize the development of cryptophane biosensors for 129Xe MRI applications, while highlighting innovative biosensor designs and the consequent enhancements in detection sensitivity, which will be invaluable in expanding the scope of 129Xe MRI.
1. Introduction
Proton magnetic resonance imaging (1H MRI) is a commonly used imaging modality for the detection of disease pathology. Its high spatiotemporal resolution, penetration depth, and use of non-ionizing radiation make it an attractive technique for obtaining key anatomical and physiological information. However, due to the large background signal from water and fat molecules, it has limited applications in the identification of low-abundance molecular signatures of disease, which is often paramount in early disease detection. To address this sensitivity issue, MRI with heteronuclei lacking background signal has been investigated as a complement to standard 1H MRI techniques. Here, we will focus on the spin-1/2 noble gas isotope 129Xe, which has good solubility in aqueous and organic media,1 a relatively high natural abundance of 26%,2 low toxicity, and a large, polarizable electron cloud. Its high polarizability results in a 129Xe NMR chemical shift window of over 300 ppm,3,4 which facilitates well-resolved detection in different chemical environments. This makes 129Xe an attractive candidate for biomolecular imaging applications, including multiplexed detection. Additionally, 129Xe can be readily hyperpolarized (hp) via spin-exchange optical pumping,5 whereby electron spin polarization from Rb or Cs atoms is transferred to the nuclear spin of 129Xe. By this process, the signal-to-noise ratio of the MRI measurement can be increased by several orders of magnitude over the normal Boltzmann distribution.6,7 Additionally, the T1 of hp 129Xe is sufficiently long (99 h at 14.1 T8 in the gas phase and 66 s when dissolved in saline at 9.4 T9)such that high polarization levels can be maintained during transport. With respect to clinically relevant conditions, the T1 values of hp 129Xe at 1.5 T in the lungs10 and oxygenated blood11 are 20 and 8 s, respectively, which are acceptable for in vivo applications.The compatibility of 129Xe with in vivo imaging was first demonstrated by Albert and colleagues, who obtained a high-resolution image of mouse lungs after inhalation of hp 129Xe.12 This pioneering experiment has resulted in the development of hp 129Xe MRI techniques for the imaging of human lungs10,13–17 and brain,18–20 of which there are now numerous examples. Efforts in 129Xe MRI have also focused on using Xe host–guest chemistry, where the Xe guest interacts with a protein or small-molecule host, in biosensing applications. Genetically encoded Xe biosensors such as gas vesicles21 and monomeric proteins22–26 possessing hydrophobic cavities that allow for transient Xe interactions are being developed as MRI analogues of commonly used optical reporters such as green fluorescent protein. Additionally, small-molecule Xe hosts, which can be appended to an affinity tag for a biomarker of interest, can function as Xe biosensors via the modulation of the chemical shift or intensity of the 129Xe signal by interactions between the Xe host and the target. For example, encapsulated Xe can generate a unique “bound” 129Xe NMR chemical shift when the host binds to its target, well-resolved from the 129Xe signal of unbound biosensor, as well as from free 129Xe in solution. The diverse family of Xe biosensors has been thoroughly analyzed in recent reviews by our laboratory.27,28
The most investigated Xe biosensors involve organic cage-like host molecules known as cryptophanes. As seen in Fig. 1, these cages provide a unique chemical environment for Xe, and highlight the considerable sensitivity of the 129Xe NMR chemical shift to its environment: the chemical shifts of 129Xe bound inside compounds 1–4 span a range of ca. 40 ppm with only an 8% change in cavity volume,29,30 while that of compound 5 is shifted to 308 ppm due to its six electron-withdrawing [(η5-C5Me5)RuII]+ moieties.4 The prototypal cryptophane cage, cryptophane-A, was first synthesized in 1981, via the linkage of two cyclotriveratrylene (CTV) caps by three ethylene linkers.31 Over the past four decades, considerable progress has been made in the synthesis of novel cryptophane derivatives, characterization of their structural aspects, and exploration of their unique host–guest chemistry, in both organic and aqueous solutions. We refer the reader to excellent in-depth reviews of these subjects.32,33 This review focuses on the biosensing features of Xe–cryptophane interactions, with applications in 129Xe MRI. We seek to familiarize the reader with the fundamentals of cryptophane-based biosensing utilized in 129Xe magnetic resonance spectroscopy (MRS), and highlight recent advances in biomarker detection and imaging. We conclude with several cutting-edge methods to advance the frontiers of Xe–cryptophane host–guest chemistry and offer our perspectives on future research.
 | ||
| Fig. 1 Chemical structure and 129Xe NMR chemical shifts of cryptophanes with varying alkoxy linker length in 1,1,2,2-tetrachloroethane-d2 (1–4)30 and of a cryptophane functionalized with six [(η5-C5Me5)RuII]+ moieties (5)4 in D2O. | ||
2. Cryptophanes as biosensors in 129Xe MRS
2.1 Direct detection of cryptophane-encapsulated 129Xe
The Xe-binding properties of cryptophane-A were first discovered in 1998, with 1H NMR experiments revealing good Xe affinity (Ka ≈ 3000 M−1 in C2D2Cl4 at 278 K), and well-resolved 129Xe NMR signals from both solvated 129Xe and cryptophane-bound 129Xe.34 In 2001, Pines, Schultz, and colleagues exploited these features of cryptophane-A in designing the first cryptophane-based biosensor.35 After modifying the cryptophane-A molecule with a solubilizing peptide and a tether linking the cage to a biotin moiety, a 129Xe NMR peak corresponding to bound 129Xe was observed at ca. 70.2 ppm, referenced to gaseous 129Xe. Addition of 80 nmol avidin shifted this peak 2.3 ppm downfield, which was attributed to mechanical deformation of cryptophane and the corresponding distortion of xenon's highly polarizable electron cloud due to the biotin–avidin interaction (Fig. 2). | ||
| Fig. 2 Top: Chemical structure of a biosensor comprised of cryptophane-A, a solubilizing peptide and a tether linking the construct to biotin. Bottom: 129Xe NMR spectrum showing the 129Xe(aq) signal at ca. 193 ppm and the bound 129Xe signals at ca. 70 ppm. The peak corresponding to 129Xe encapsulated by this biosensor shifts from ca. 70.2 to 72.5 ppm upon addition of avidin to solution. Reproduced with permission from ref. 35. Copyright 2001 National Academy of Sciences. | ||
This innovative experiment demonstrated the feasibility of using functionalized cryptophanes and 129Xe NMR to detect analytes with high sensitivity and has prompted the use of this technique in the detection of numerous biomarkers of interest. Generally, this methodology employs the use of a targeting moiety which can bind to or otherwise interact with the biomarker, resulting in a “bound” 129Xe NMR signal that is distinct from “free” biosensor. Appending the targeting moiety to the cryptophane core can be accomplished via copper-catalyzed [3 + 2] azide–alkyne cycloaddition, whereby an affinity tag with an azido group can be appended onto a cryptophane-A derivative with propargyl moieties.36 The connection of a targeting moiety with an amine group to a carboxylated cryptophane via an amide linkage is also commonly utilized. For hydrophobic cryptophane scaffolds, one or more solubilizing groups possessing such ionizable moieties as carboxylic acids37–39 or amines40 can also be attached in order to increase the water solubility of the cage. Using this approach, the detection of such biomedically relevant targets as integrin receptor,41 carbonic anhydrase,42–44 matrix metalloproteinase-7 (MMP-7),45 and major histocompatibility complex class II protein46 was feasible with cryptophanes functionalized with affinity tags cyclic RGDyK peptide, benzenesulfonamide, MMP-7 specific peptide substrate, and hemagglutinin peptide, respectively.
In addition, the detection of metal ions via functionalized cryptophanes has become a valuable tool to supplement traditional analytical methods. In 2012, Kotera et al. demonstrated the first example of cryptophane-based metal ion detection by appending the zinc-chelating group nitrilotriacetic acid to cryptophane for the detection of Zn2+.47 A follow up study showed that this construct could also be used to detect Cd2+ and Pb2+, with 129Xe NMR chemical shifts migrating 1.5, 0.3, and 4.5 ppm downfield from free biosensor for Zn2+, Cd2+, and Pb2+, respectively.48 In recent years, efforts have focused on the design of cryptophane-based metal ion sensors with high selectivity and sensitivity. Zhang et al. designed a highly selective cryptophane functionalized with 2-(diphenylphosphino)benzenamine, which exhibited a sizable 6.4 ppm upfield 129Xe NMR chemical shift upon Zn2+ binding, while being unresponsive to Ag+, Cu+, Pd2+, Hg2+, Mg2+, and Ca2+.49 Additionally, Jeong et al. demonstrated how a cryptophane functionalized with the chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid was able to differentiate between Ca2+, Cu2+, Ce3+, Zn2+, Cd2+, Ni2+, Co2+, Cr2+, Fe3+, and Hg2+ by exhibiting distinct 129Xe NMR chemical shifts upon metal ion binding.50
Cryptophane-based biosensors have also been designed to report on more complex systems. For example, Roy et al. observed a 1.5 ppm upfield 129Xe NMR chemical shift change upon binding of a cryptophane functionalized with a 20-mer nucleotide to its complementary DNA strand, and no such change in the presence of a non-complementary strand.51 Berthault et al. demonstrated how the NMR chemical shift of 129Xe encapsulated by a hexa-carboxylate-functionalized cryptophane is highly pH dependent,52 and later used the chemical shift difference of encapsulated 129Xe in two different cryptophanes for more precise pH measurement.53 Finally, Yang et al. designed a cryptophane functionalized with a thiol-reactive acrylate moiety, which allowed for the detection of, and differentiation between biothiols such as cysteine, homocysteine, and glutathione.54 These innovative examples of cryptophane-based biosensor design highlight where recognition moieties can be tailored for highly specific and selective binding. Conversely, using a more universal approach to 129Xe NMR biosensing, Kotera et al. demonstrated how Cys4-tagged proteins can be detected using a cryptophane linked to a CrAsH probe (a carboxy derivative of the bisarsenical FlAsH probe).55,56 This method allows for the detection of wide families of proteins that contain the small tetracysteine tag, which is commonly used for analyzing protein function and location.
Despite this recent progress, there is still much optimization necessary in the design of cryptophane-based biosensors for direct detection 129Xe NMR. For example, there have been few examples of chemical shift changes >5 ppm upon biosensor-target binding, which can pose resolution issues at clinical magnetic field strengths. Additionally, due to chirality of the cryptophane (typically synthesized as a racemic mixture), and the chirality of many affinity tags, multiple peaks corresponding to different diastereomers can be observed in the 129Xe NMR spectrum.57 This phenomenon dilutes the signal-to-noise by the number of diastereomers, although this can be mitigated through enantiopure cryptophane synthesis.36,44,58 Xe access to the cryptophane interior can be limited by the energy necessary to displace confined water molecules inside the cage,59 and precluded entirely by collapsed conformations of the cryptophane cage in which one CTV unit folds inward into the cavity.60 Spectral analysis can be further complicated by the presence of residual peaks corresponding to unbound biosensor. The use of ‘turn-on’ biosensors, where 129Xe NMR signal is observed only in the presence of target, can simplify these analyses. Our laboratory recently reported the first turn-on cryptophane biosensor, which consisted of a FRRIAR peptide conjugated to cryptophane for the detection of calmodulin (CaM).61 Signal corresponding to bound 129Xe was only observed in the presence of Ca2+-bound CaM.
Additionally, we showed that cryptophane-based biosensors have a propensity to form water-soluble aggregates in solution,62 which can further complicate chemical shift assignments. Using a benzenesulfonamide-functionalized cryptophane (C8B) that was previously used42 for the detection of carbonic anhydrase (CA) as a model system, we observed that the biosensor formed aggregates that were hundreds of nanometers in diameter, but became monomeric upon binding to its protein target.62 We showed that as increasing amounts of CA were titrated into solutions of C8B, the chemical shift of encapsulated 129Xe progressively shifted downfield, which we attributed to mechanical perturbation of the encapsulated 129Xe electron cloud,35,57,63,64 and also the disaggregation phenomenon. Biosensor disaggregation can contribute significantly to the 129Xe NMR chemical shift change observed upon target binding. Indeed, we hypothesize that only biosensor–target interactions that cause a significant change in the cryptophane aggregation state will produce substantial chemical shift changes. This hypothesis is supported by recent studies from our laboratory with an adamantyl-functionalized cryptophane-A sensor, which remains monomeric in aqueous solution, and does not exhibit a 129Xe NMR chemical shift change upon binding the model receptor β-cyclodextrin.65 The modulation of the 129Xe NMR chemical shift as a function of such factors as stereochemistry and aggregation is a relatively unexplored area of cryptophane-based biosensing that is sure to receive additional attention as biosensing techniques are optimized.
2.2 Increasing 129Xe NMR detection sensitivity with hyper-CEST
Hyperpolarization of 129Xe offers a significant improvement in signal-to-noise relative to thermal polarization, but direct detection of encapsulated 129Xe NMR signal nevertheless requires micromolar concentrations of biosensor to avoid long acquisition times.66 Previous work showed that peptide-functionalized cryptophanes are relatively nontoxic to cells at concentrations of <100 μM,67 yet lower working concentrations of biosensor are still desired to maximize cell viability and to detect low-abundance molecular markers of disease. To increase detection sensitivity further, the hyper-CEST technique was developed, which combines 129Xe hyperpolarization with chemical exchange saturation transfer (CEST).68 Hyper-CEST takes advantage of the constant exchange of Xe into and out of the cryptophane, which typically occurs on the order of tens of Xe atoms per second in aqueous solution at rt.30,35 The encapsulated hp 129Xe can be selectively depolarized by applying radiofrequency (rf) pulses at the resonance frequency of the bound 129Xe atom. The depolarized 129Xe then exchanges into solution, causing depolarization of solvated 129Xe nuclei (and rapid thermal equilibration). The subsequent loss of 129Xe(aq) signal acts as a reporter for the encapsulated 129Xe pool, allowing for ultrasensitive detection of the cryptophane host (Fig. 3). | ||
| Fig. 3 Top: Mechanism of the hyper-CEST NMR experiment. Hp 129Xe atoms (green) bind to the interior of the cryptophane cage, and are depolarized (orange) by a selective rf pulse. The exchange of bound 129Xe out of the cage causes the depolarization of bulk 129Xe nuclei. This scheme was modified with permission from ref. 66. Copyright 2014 Wiley-VCH. Bottom: Representative 129Xe hyper-CEST NMR z-spectrum of a cryptophane host. The individual peaks represent the intensity of the 129Xe signal as a function of the frequency at which rf pulses are applied and the black trace represents the collective z-spectrum. | ||
In order to approximate the contrast that one would observe in a 129Xe MRI experiment, a time-dependent saturation profile of the Xe host can be obtained. First demonstrated by Meldrum et al.,69 this technique involves applying rf pulses at the bound 129Xe resonance frequency and monitoring the decrease in 129Xe(aq) NMR signal as a function of saturation time. This on-resonance decay of the 129Xe(aq) signal can be compared to an off-resonance decay, where rf pulses are applied at a resonance frequency where no hyper-CEST effect is observed, and the difference between the two decay curves, which is weighted by the saturation time, can be quantified as saturation contrast.70
Several recent examples highlight the increase in sensitivity afforded by the hyper-CEST technique. For instance, a peptide-functionalized cryptophane biosensor developed by Riggle et al. was utilized to label cells in acidic environments.71 A cryptophane-conjugated EALA-repeat peptide assumed a disordered form at neutral pH but became α-helical at pH 5.5, which resulted in insertion into HeLa cell membranes and a subsequent 13 ppm downfield chemical shift change of the encapsulated 129Xe (Fig. 4). Importantly, significant hyper-CEST contrast was observed with 34 pM biosensor, which improved detection sensitivity by 6 orders of magnitude relative to direct detection of the “bound” 129Xe NMR signal. In addition to allowing for the highly sensitive detection of specific Xe host molecules, hyper-CEST can also facilitate the differentiation between biosensor in the absence and presence of target. Zeng et al. designed a mitochondria-targeted biosensor for the detection of biothiols, comprising cryptophane-A, a disulfide linker, a naphthalimide fluorescent moiety, and a triphenylphosphonium mitochondrial targeting tag.72 Hyper-CEST revealed a 200 pM detection limit for this biosensor, but also allowed for the monitoring of intracellular biothiol levels. It was observed that the CEST effect for this biosensor incubated with lung cancer cells was significantly larger than for biosensor incubated with lung cancer cells and N-ethylmaleimide, a thiol scavenger. This was due to the biosensor becoming more water-soluble and accessible to Xe after thiol-induced cleavage of the disulfide bond, which highlights the importance of understanding solution properties in biosensor design. Finally, Milanole et al. used hyper-CEST to show that their cryptophane-based biosensor, functionalized with a fluorescein moiety and an antibody targeted toward epidermal growth factor receptor (EGFR), was selective toward cells overexpressing EGFR.73 Saturation contrast was only observed with biosensor incubated with the HCC827 cell line (high EGFR expression) and not with biosensor incubated with the A549 cell line (intermediate EGFR expression), with intracellular biosensor concentration in the low-nanomolar regime.
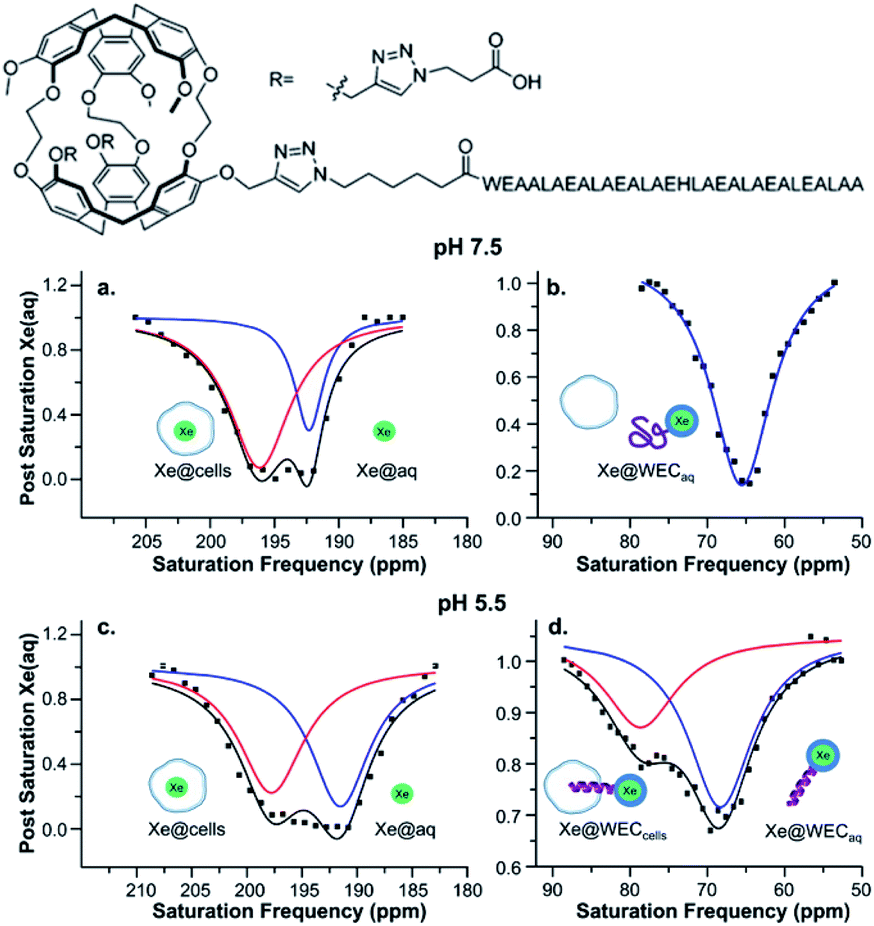 | ||
| Fig. 4 Top: Chemical structure of water-soluble EALA-cryptophane (WEC). Bottom: Hyper-CEST 129Xe NMR of 5–10 μM WEC with 1 × 107 HeLa cells per mL at pH 7.5 (a and b) and pH 5.5 (c and d). Reproduced with permission from ref. 71. Copyright 2015 American Chemical Society. | ||
3. From spectroscopy to imaging
3.1 In vitro 129Xe MRI studies
Heretofore, we have discussed the contributions made to cryptophane-based biosensing via spectroscopic studies. These experiments and analyses have not only advanced the growth of this field, but have also stimulated progression into the clinical realm, where obtaining high-contrast images of target is made feasible by designing cryptophane biosensors with optimal hyper-CEST properties. For more details on technical aspects related to the development of 129Xe MRI techniques, we refer the reader to prior reviews of this subject.74–76 We note that several of the biosensors discussed previously, as well as several of those that will be discussed in this section, are classified as “bimodal” contrast agents, containing both cryptophane and a fluorophore, and thus detectable via both 129Xe MRI and fluorescence imaging.55,56,73,77–82 The capacity for analysis via a secondary detection method such as fluorescence can be greatly beneficial in complementing the 129Xe MRI readout. Additionally, it has been shown that differences in membrane fluidity can affect the depolarization time of hp 129Xe. Studies by Schnurr et al. showed that high membrane fluidity resulted in short depolarization times and saturated hyper-CEST responses with short saturation pulses.83 Additional work by Booker and Sum provided valuable insight with respect to Xe–membrane interaction, showing that Xe localizes in the hydrophobic core of membranes, and by doing so increases bilayer thickness, membrane fluidity, and lipid head group spacing.84 This interaction of Xe with membranes has also been exploited in the design of liposome bilayers used for the delivery of the gas in rat models of stroke.85–87 Thus, having a secondary imaging modality unaffected by the dynamic interactions of Xe with the cellular environment becomes valuable when performing experiments.Kotera et al. first demonstrated the enhanced sensitivity of 129Xe MRI with their nitrilotriacetic acid-functionalized cryptophane for the detection of Zn2+ ions.47 They observed a significant increase in MRI contrast when Ca2+ ions were replaced with Zn2+ in a solution of biosensor, and showed that the limit of Zn2+ detection was as low as 100 nM, which was at the time an improvement of two orders of magnitude over conventional MRI methods. Efforts from the Zhou laboratory and collaborators have also focused on the development of 129Xe MRI contrast agents. For example, they designed a unique biosensor for Hg2+ detection comprised of two cryptophane-A cages linked by a dipyrrolylquinoxaline moiety.88 Hg2+ binding resulted in a clamp-like conformational change of the biosensor, in which the two cryptophanes converge and their electron clouds overlap, causing a 129Xe NMR chemical shift change. This change was enough to observe 129Xe MRI contrast in the presence of Hg2+, with no contrast observed with biosensor alone. They also demonstrated the use of 129Xe MRI in the detection of biothiols using the previously mentioned acrylate-functionalized cryptophane,54 where contrast was observed only when the biosensor was incubated with cysteine; and in the detection of H2S.82 This latter study featured a biosensor comprised of cryptophane-A functionalized with 4-azide-1,8-naphthalic anhydride, which was able to detect HS− due to the 129Xe NMR chemical shift change that occurred after the HS−-induced reduction of the azido group to an amine (Fig. 5). 129Xe MRI contrast was observed only after the biosensor was incubated with HS−. It should be noted that the design of this biosensor featured a fluorescent tag that could simultaneously function as an analyte probe, as the HS−-induced reduction of the azido group also caused an increase in the fluorescence intensity as well as a change in the 129Xe NMR chemical shift. Integration of two detection modalities into one sensor should simplify synthetic efforts and facilitate biocompatibility in future designs.
 | ||
| Fig. 5 Top: Scheme of biosensor for H2S, activatable via both fluorescence and 129Xe MRI modalities. Bottom: 129Xe MRI images of an NMR tube containing 100 μM biosensor (A) and 100 μM biosensor with 10 equiv. HS− (B). Reproduced with permission from ref. 82. Copyright 2017 Wiley-VCH. | ||
In addition to being used in the detection of biomedically relevant analytes, 129Xe MRI with cryptophanes has also found use in thermometry applications. Schilling et al. showed that the chemical shift of 129Xe encapsulated by cryptophane-A has a temperature dependence of 0.29 ppm °C−1, which is ca. 30-fold greater than water.89 This temperature dependence arises from the large electron cloud of Xe, which is able to interact with less shielded areas of the cryptophane interior, as well as from the greater conformational flexibility of the cryptophane at higher temperatures. Additionally, using chemical shift imaging with a cryptophane concentration of 150 μM, they were able to determine that the lower limit of the detectable temperature change was 0.1 °C, which suggests that highly sensitive and accurate temperature mapping is feasible via 129Xe hyper-CEST MRI.
3.2 In cellulo 129Xe MRI studies
Even though considerable efforts are still needed to successfully bridge the gap to clinical in vivo studies, there have been numerous pioneering experiments that illustrate the potential of cryptophanes for in vivo 129Xe MRI applications. In particular, the Schröder laboratory and their collaborators have made valuable contributions to developing 129Xe MRI with cryptophane-based biosensors by performing numerous in cellulo studies. In 2012, they demonstrated that using cryptophane-A in imaging applications requires only ca. 100 μM 129Xe and only 30 nM of NMR-active cryptophane with a CEST saturation pulse of 26 s, which can be reduced down to sub-second timescales if ca. 10 μM NMR-active cage is used.90 Their group was also the first to use 129Xe MRI with cryptophane to image live cells,80 which revealed valuable information about the in cellulo distribution of Xe. Using mouse fibroblasts, they observed the expected pools of Xe in bulk solution (192 ppm) and encapsulated by cryptophane-A (59 ppm), but also solvated Xe in cells (196 ppm) and Xe encapsulated by cell-internalized cryptophane (69 ppm). Due to the sizable chemical shift difference between Xe encapsulated in free and cell-internalized cryptophane, as well as the sensitivity of hyper-CEST, it was possible to perform selective 129Xe MRI of this latter pool. By using a construct comprised of cryptophane-A functionalized with fluorescein, they were able to use 129Xe MRI to clearly differentiate between labeled and non-labeled cells (Fig. 6), with an average intracellular cryptophane concentration of 15 μM, achieved after incubating cells with 50 μM cryptophane for 20 h. In a follow-up study, they showed that cryptophane-A functionalized with a different fluorophore, tetramethylrhodamine, was able to achieve similar 129Xe MRI contrast to that observed with the former construct.91 This illustrated the flexibility of cryptophane-based 129Xe imaging, and suggested the possibility of fluorescence multiplexing applications. | ||
| Fig. 6 Top: Chemical structure of fluorescein-conjugated cryptophane (CrA-FAM). Bottom: 129Xe hyper-CEST MRI with underlying 1H MRI images of mouse fibroblasts labeled with CrA-FAM (15 μM intracellular concentration) and control cells, encapsulated in alginate beads. Laser scanning microscopy images of each cell population are shown, with CrA-FAM accumulated in cells in green and dead cells stained with ethidium homodimer III (EthD-III) in red. Reproduced with permission from ref. 80. Copyright 2014 Wiley-VCH. | ||
After having succeeded in obtaining 129Xe MRI images of cells with nonspecific cryptophane uptake, they demonstrated the feasibility of performing cell-specific targeting using a functionalized cryptophane. In a key proof of principle study, they targeted cells expressing the cell surface protein and macrophage marker cluster of differentiation 14 (CD14).78 By functionalizing cryptophane-A and fluorescein with biotin, conjugating an anti-CD14 antibody to avidin, and incubating these constructs with high-CD14-expressing macrophages and a control fibroblast cell line, they were able to distinguish between the two types of cells using 129Xe MRI and fluorescence with only ca. 20 nM biosensor. Notably, these experiments demonstrated the versatility of their modular approach, as the biosensor can be adapted to detect any target for which there is a specific antibody or other binding element available. For example, they later showed how this general targeting scheme could be used for the detection of members of the claudin (Cldn) protein family.92 In this study, they conjugated Clostridium perfringens enterotoxin, which has high affinity for specific claudins, to avidin, and used the previously mentioned cryptophane–biotin and fluorescein–biotin constructs as 129Xe MRI and fluorescence readout modules, respectively. This biosensor was successfully used to image and distinguish Cldn4-expressing HEK cells (59% CEST effect) from control non-transfected HEK cells (11% CEST effect) via 129Xe MRI.
In their next study, they demonstrated a technique for delivering cryptophane to human brain microvascular endothelial cells (HBMECs) by using large unilamellar vesicles (LUVs).93 In their experimental design, they functionalized the lipid bilayer of cryptophane-containing LUVs (ca. 3800 cryptophane units per liposome) with arginine-rich peptides to facilitate uptake into HBMECs. The target cells and control human aortic endothelial cells (HAoECs) were then incubated with both functionalized and unfunctionalized LUVs, and cellular uptake was studied via fluorescence and 129Xe MRI modalities. Indeed, uptake of the biosensor into HBMECs resulted in an MRI CEST effect ca. 3-fold larger than that observed with HAoECs, allowing for the two cell types to be spatially distinguished. Because of the high loading capacity of LUVs, these images were able to be obtained with only 1 nM of biosensor, illustrating the high sensitivity of this approach. This technique was later improved by using a lipopeptide comprised of a cationic peptide, a cryptophane-A unit, and a palmitoyl chain.94 After micelle formation, it was estimated that the average cryptophane content per unit volume was ca. 7-fold higher in the micelles than in LUVs. This allowed for a significantly higher CEST effect to be observed with HBMECs relative to HAoECs, with the important difference being that the cells were incubated with a cryptophane concentration ca. 10-fold more dilute than that used in the LUV study. This was facilitated by the high local cryptophane concentration in this biosensor design, resulting in a high density of depolarization seed points. Finally, an additional study focused on targeting cell-surface glycans by functionalizing cryptophane-A with a fluorophore and bicyclo[6.1.0]nonyne for glycan labeling.79 Cells treated with the synthetic sugar Ac4ManNAz for the purpose of expressing an azido-functionalized sialic acid on their surface were able to be clearly imaged and differentiated from untreated cells using this biosensor. Notably, this study involved covalently linking the biosensor to its target via azide–alkyne cycloaddition, thus eliminating signal from unbound biosensor after washout.
4. New frontiers in cryptophane-based biosensing
Over the past decade, there has been considerable progress in the creative utilization of cryptophanes beyond their traditional roles as single-unit hosts for 129Xe biosensing. In this section, we highlight several inventive examples that push the boundaries of cryptophanes for 129Xe MRI.One of the critical goals in this area has been to enhance the detection sensitivity of cryptophane hosts. In order to achieve this, significant efforts have focused on grafting multiple cryptophane units onto a larger scaffold, notably by the laboratories of Pines, Wemmer, and collaborators. In 2006, they demonstrated the first example of this strategy by attaching a biotin moiety to a polyamidoamine dendrimer, which was able to encapsulate two cryptophane-A units when bound to avidin, and increased signal-to-noise by a factor of 8 relative to their original biotinylated cryptophane biosensor.35,95 They improved upon this concept four years later by using bacteriophage MS2 as a scaffold for attaching multiple cryptophane-A units via linker molecules.69 This allowed for the attachment of 125 cryptophane-A units per viral capsid, which resulted in a 700 fM detection limit for the cryptophane-capsid construct. Further improvements in detection sensitivity were achieved with the bacteriophage M13, which possesses 15 times as many coat proteins on its capsid as MS2, allowing for 1050 cryptophane-A host molecules per capsid to be attached, and a detection sensitivity of 230 fM.96 Biosensing capability with a construct comprising aminooxy-functionalized cryptophane-A ligated to filamentous bacteriophage fd expressing single-chain antibody variable fragments (scFvs) was then established.97 Significant hyper-CEST contrast in the presence of MDA-MB-231 cells expressing high levels of EGFR, recognized by the scFvs, and minimal contrast in the presence of Jurkat cells expressing low levels of EGFR was observed. Importantly, this demonstrated that constructs possessing multiple cryptophane units can avoid the nonspecific cell uptake previously observed with singular cryptophane-A.80 Finally, in the most recent demonstration of specific cell targeting, it was established that it was possible to use a cryptophane-bacteriophage construct modified on its exterior with DNA aptamers to target mIgM markers on the surfaces of lymphoma cells (Ramos line) (Fig. 7).98 Taken together, these experiments illustrate the potential of greatly enhancing cryptophane detection sensitivity by grafting individual units onto a larger scaffold. We note that it should also be possible to attach other recognition elements, such as antibodies or peptides, onto the viral capsids for the detection of a wide range of targets.
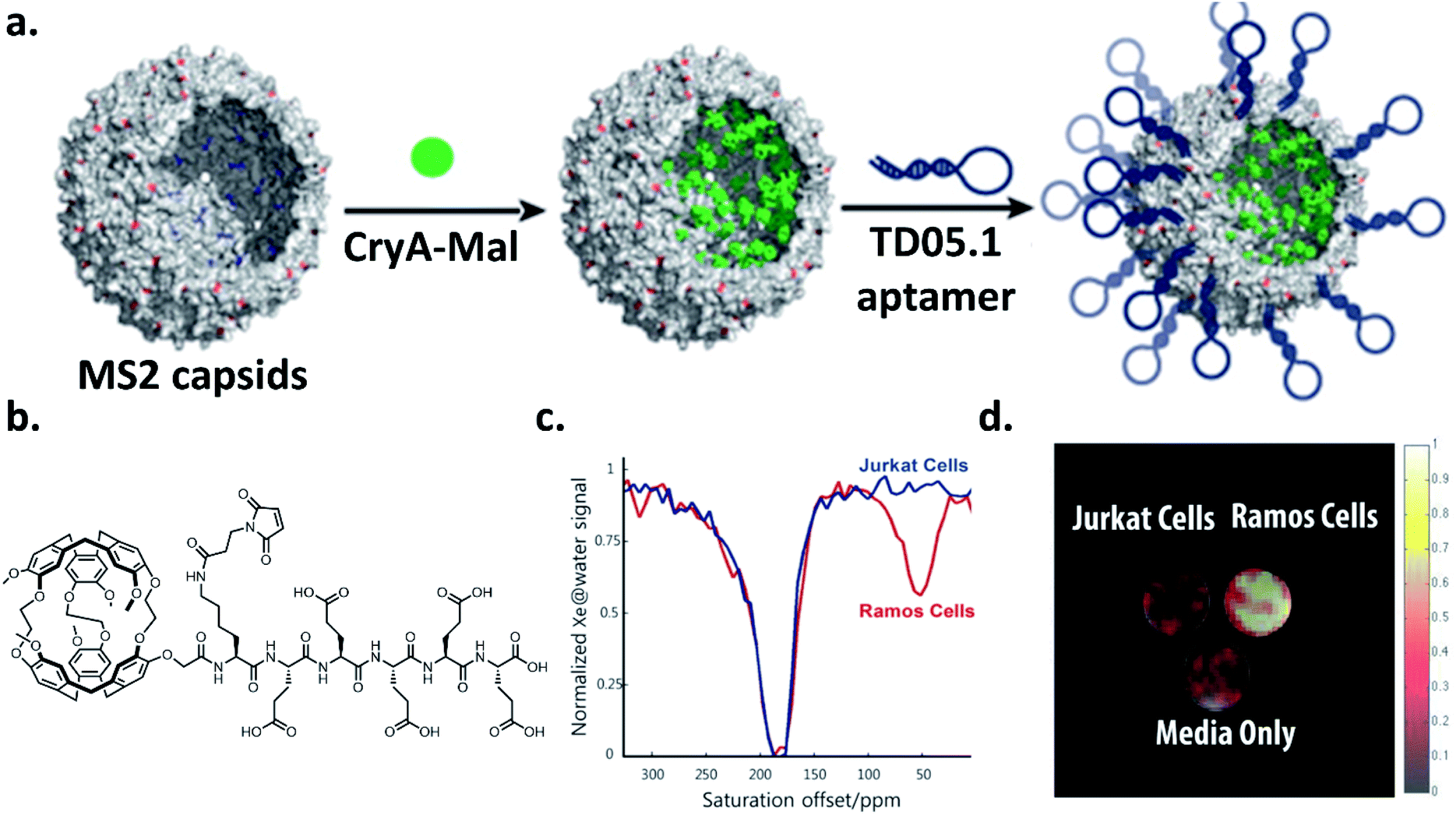 | ||
| Fig. 7 (a) Scheme of bacteriophage–cryptophane biosensor assembly. The capsid interior of bacteriophage MS2 was modified with cryptophane-A conjugated to maleimide and five glutamic acids to increase solubility (CryA-Mal, green circle) at ca. 110 N87C positions. The capsid exterior was modified with 54 units of the TD05.1 DNA aptamer for targeting mIgM markers on lymphoma cells. (b) Chemical structure of CryA-Mal. (c) 129Xe hyper-CEST NMR z-spectra of the biosensor (167 nM in capsids) incubated with 2 × 107 Ramos (mIgM+) or Jurkat (mIgM−) cells. (d) 129Xe hyper-CEST MRI superimposed on 1H MR image of tubes containing cell media only, and the two cell lines incubated with biosensor. Reproduced with permission from ref. 98. Copyright 2016 American Chemical Society. | ||
The prospect of using multiplexed cryptophane-based biosensors with distinct chemical shifts to detect different molecular targets is also a burgeoning area of research. Berthault et al. first demonstrated the possibility of multiplexed cryptophane imaging by performing 129Xe MRI with an NMR tube containing an aqueous solution of a carboxylated cryptophane layered on top of a solution of an organosoluble cryptophane in tetrachloroethane.99 To facilitate cryptophane functionality, Tyagi et al. showed that cryptophane-A can be functionalized with polyglycerol dendrons and still preserve its optimal Xe binding affinity and exchange rate.100 The multitude of hydroxyl groups at the outer surface of the cage allowed for high water solubility as well as for the potential of further functionalization with multiple targeting groups with high regioselectivity.101 Live cell multichannel 129Xe MRI was demonstrated by Klippel et al., who used cryptophane-A as well as perfluorooctyl bromide (PFOB) nanodroplets as Xe hosts.102 Because the 129Xe NMR chemical shift of cell-internalized cryptophane-A is approximately 40 ppm upfield of cell-internalized PFOB, it was possible to image mouse fibroblast cells via a frequency-selective saturation transfer approach, where observed contrast for the two Xe hosts was dependent upon the saturation frequency. Moreover, due to the 129Xe NMR chemical shift of cell-internalized cryptophane being nearly independent of cell type,80,91,93 these experiments illustrate the possibility of imaging varying cell types using this multichannel detection approach. Importantly, the use of cryptophanes in tandem with established contrast media such as PFOB nanodroplets is a valuable example of how the unique chemical environment, high detection sensitivity, and multifunctionality inherent to cryptophane contrast agents can drive their integration into modern biomedical applications.
5. Concluding remarks
The field of cryptophane biosensing has greatly evolved since the conception of the first xenon biosensor two decades ago. We have reviewed examples of the ultrasensitive detection and monitoring of proteins,35,41,42,45,46,55,56,61,73,78,79,92,97 ions,47–50,82,88 nucleic acids,51 biothiols,54,72 and even environmental conditions such as pH52,53,71 and temperature89 using direct and indirect detection methods. We have highlighted instances of innovative biosensor design, such as the use of a singular moiety for both fluorescence and 129Xe MRI readout,82 modulation of solubility after target binding,72 and loading up to thousands of individual cryptophane host molecules onto a single carrier.69,93–98 Finally, we have shown examples of selective and sensitive 129Xe MRI contrast attainable with these biosensors. This past decade of expanding the scope of 129Xe MRI using cryptophane-based biosensing has been extremely productive, and the future of this field remains bright.Considerable progress is still necessary for cryptophane–xenon biosensors and 129Xe MRI to achieve widespread use as imaging tools. Synthetic challenges including low overall yields from multistep syntheses and difficulties in performing large scale cryptophane syntheses due to the propensity of polymer formation at high concentrations are notable limiting factors.32 Also, translation of the end products into bioimaging applications can be challenging due to poor biosensor solubility and propensity for self-aggregation. We note that these limitations can be overcome with other small-molecule Xe hosts, such as members of the cucurbit[n]uril (CB[n]) family, which are commercially available and relatively inexpensive macrocycles with excellent Xe exchange kinetics103 and versatile host–guest chemistry.104 Indeed, detection of cucurbit[6]uril via 129Xe hyper-CEST MRI in the vasculature of a living rat was recently demonstrated by Albert and colleagues.105 However, the targeted biosensing capabilities of CB[n] are limited, as these compounds are difficult to functionalize and have high affinity for naturally occurring small molecules in living organisms, such as polyamines,103 necessitating the use of relatively high (mM) concentrations for in vivo imaging studies. Self-assembled metal–organic nanocapsules have recently emerged as promising candidates for 129Xe MRI applications due to their highly tunable characteristics,106,107 but further work is needed with respect to their functionalization in order to enable targeted imaging. Thus, at the current time, we believe that cryptophanes are the gold standard for ultrasensitive, targeted detection. The key experiment to validate their status as the definitive, biocompatible Xe host must be a demonstration of their detection via 129Xe MRI in a living organism, and we believe that the next several years of research should view this as a critical endeavor. For example, a study examining the biodistribution of unfunctionalized cryptophane-A in a mouse model, followed by a study with functionalized cryptophane targeting an overexpressed biomarker in transgenic mice would be essential in establishing cryptophane biocompatibility and targetability. In order to reach and progress past this milestone, efforts must be focused on optimizing syntheses of functionalized cryptophane. Recent progress in improving synthetic techniques to achieve shorter, scalable cryptophane syntheses with improved yields, milder reaction conditions, and easier purification procedures is promising,36,108 though efficient gram-scale syntheses are yet to be developed.
A successful demonstration of in vivo detection of a cryptophane-based biosensor will surely give insight on many additional ways to optimize biosensor design, as we currently have no knowledge of how cryptophanes behave in multicellular organisms with respect to biodistribution. For example, the recently discovered cryptophane aggregation phenomenon62 suggests that cryptophane in vivo circulation time and stability may be modulated by the size of the nanoscale clusters. The facile nature of cryptophane functionalization should allow for a rapid response with respect to optimizing biosensor design to enhancing biocompatibility. Indeed, there are currently many innovative ways to tune cryptophane solution properties, including appending cryptophane with dendrons carrying numerous hydroxyl groups100 and functionalizing cryptophane with PEG moieties,109 and we expect additional techniques to emerge in the near future.
Continued research highlighting the use of 129Xe MRI and cryptophane-based biosensors for the in vivo detection of molecular markers of disease will facilitate the transition to the clinical realm. Based on numerous recent examples of the use of 129Xe MRI in assessing physiological processes in human lungs and kidneys,110–113 we believe that approval for hp 129Xe to be used in the clinical setting will progressively become increasingly widespread. At the time of this review, the use of hp 129Xe has not yet attained universal approval for use in general clinical settings, nor have cryptophane-based biosensors been studied in multicellular organisms. However, hp 129Xe is currently being used as an investigational medical product in humans at many institutions globally, and has received approval for routine clinical lung imaging in the United Kingdom.114 Xemed and Polarean, two prominent U.S.-based industry members in the field of hyperpolarized gas MRI, have filed requests with the United States Food and Drug Administration for the use of hp 129Xe in the clinical setting. Emerging new vendors, such as XeUS Technologies, are also contributing to the growth of the field by providing affordable instrumentation for clinical hp 129Xe production. In summary, it is clear that once clearance by regulatory agencies is granted, the infrastructure will be readily available for a rapid transition into the clinical realm.
Additionally, modern technological advancements, such as progress in generating larger quantities of isotopically enriched 129Xe and the development of 129Xe polarizers capable of achieving near-unity polarization levels115,116 will likely allow for significantly lower working concentrations of Xe and biosensor, thereby facilitating biocompatibility and lowering cost. The integration of cryptophane-based biosensors with established imaging tools such as PFOB nanodroplets102 and LUVs,93 and with common fluorescence imaging modalities, should also aid in the transition of cryptophanes into the clinical domain. Lastly, the continued development and optimization of Xe delivery systems, MRI scanners, and pulse sequences will undoubtedly expand the potential of 129Xe for in vivo imaging applications. With these advances on the horizon, we believe that the next decade of research into cryptophane-based biosensors with applications in 129Xe MRI promises to be even more rewarding than the last one.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH R35-GM-131907 to I. J. D.; S. D. Z. was supported by UPenn Chemistry Lynch Fellowship.References
- H. L. Clever, Krypton, Xenon and Radon - Gas Solubilities, Pergamon Press, New York, 1979 Search PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, 79th edn, CRC Press, Boca Raton, 2012 Search PubMed.
- D. Raftery, Annu. Rep. NMR Spectrosc., 2006, 57, 205–270 CrossRef CAS.
- R. M. Fairchild, A. I. Joseph, K. T. Holman, H. A. Fogarty, T. Brotin, J.-P. Dutasta, C. Boutin, G. Huber and P. Berthault, J. Am. Chem. Soc., 2010, 132, 15505–15507 CrossRef CAS.
- T. G. Walker and W. Happer, Rev. Mod. Phys., 1997, 69, 629 CrossRef CAS.
- B. M. Goodson, J. Magn. Reson., 2002, 155, 157–216 CrossRef CAS.
- D. A. Barskiy, A. M. Coffey, P. Nikolaou, D. M. Mikhaylov, B. M. Goodson, R. T. Branca, G. J. Lu, M. G. Shapiro, V. V. Telkki, V. V. Zhivonitko, I. V. Koptyug, O. G. Salnikov, K. V. Kovtunov, V. I. Bukhtiyarov, M. S. Rosen, M. J. Barlow, S. Safavi, I. P. Hall, L. Schröder and E. Y. Chekmenev, Chem.–Eur. J., 2017, 23, 725–751 CrossRef CAS.
- B. C. Anger, G. Schrank, A. Schoeck, K. A. Butler, M. S. Solum, R. J. Pugmire and B. Saam, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 78, 43406 CrossRef.
- A. Bifone, Y. Q. Song, R. Seydoux, R. E. Taylor, B. M. Goodson, T. Pietrass, T. F. Budinger, G. Navon and A. Pines, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12932–12936 CrossRef CAS.
- J. P. Mugler III and T. A. Altes, J. Magn. Reson. Imaging, 2013, 37, 313–331 CrossRef.
- G. Norquay, G. Leung, N. J. Stewart, G. M. Tozer, J. Wolber and J. M. Wild, Magn. Reson. Med., 2015, 74, 303–311 CrossRef CAS.
- M. S. Albert, G. D. Cates, B. Driehuys, W. Happer, B. Saam, C. S. Springer and A. Wishnia, Nature, 1994, 370, 199–201 CrossRef CAS.
- W. Ruan, J. Zhong, K. Wang, G. Wu, Y. Han, X. Sun, C. Ye and X. Zhou, J. Magn. Reson. Imaging, 2017, 45, 879–888 CrossRef.
- T. N. Matin, N. Rahman, A. H. Nickol, M. Chen, X. Xu, N. J. Stewart, T. Doel, V. Grau, J. M. Wild and F. V. Gleeson, Radiology, 2017, 282, 857–868 CrossRef.
- B. Driehuys, S. Martinez-Jimenez, Z. I. Cleveland, G. M. Metz, D. M. Beaver, J. C. Nouls, S. S. Kaushik, R. Firszt, C. Willis, K. T. Kelly, J. Wolber, M. Kraft and P. H. McAdams, Radiology, 2012, 262, 279–289 CrossRef.
- Y. Shukla, A. Wheatley, M. Kirby, S. Svenningsen, A. Farag, G. E. Santyr, N. A. M. Paterson, D. G. McCormack and G. Parraga, Acad. Radiol., 2012, 19, 941–951 CrossRef.
- J. P. Mugler III, B. Driehuys, J. R. Brookeman, G. D. Cates, S. S. Berr, R. G. Bryant, T. M. Daniel, E. E. De Lange, J. H. Downs, C. J. Erickson, W. Happer, D. P. Hinton, N. F. Kassel, T. Maier, C. D. Phillips, B. T. Saam, K. L. Sauer and M. E. Wagshul, Magn. Reson. Med., 1997, 37, 809–815 CrossRef.
- M. R. Rao, N. J. Stewart, P. D. Griffiths, G. Norquay and J. M. Wild, Radiology, 2018, 286, 659–665 CrossRef.
- M. R. Rao, N. J. Stewart, G. Norquay, P. D. Griffiths and J. M. Wild, Magn. Reson. Med., 2016, 75, 2227–2234 CrossRef CAS.
- J. P. Mugler III, B. Driehuys, J. R. Brookeman, G. D. Cates, S. S. Berr, R. G. Bryant, T. M. Daniel, E. E. De Lange, J. H. Downs, C. J. Erickson, W. Happer, D. P. Hinton, N. F. Kassel, T. Maier, C. D. Phillips, B. T. Saam, K. L. Sauer and M. E. Wagshul, Magn. Reson. Med., 1997, 37, 809–815 CrossRef.
- M. G. Shapiro, R. M. Ramirez, L. J. Sperling, G. Sun, J. Sun, A. Pines, D. V. Schaffer and V. S. Bajaj, Nat. Chem., 2014, 6, 629–634 CrossRef CAS.
- Y. Wang, B. W. Roose, E. J. Palovcak, V. Carnevale and I. J. Dmochowski, Angew. Chem., Int. Ed., 2016, 55, 8984–8987 CrossRef CAS.
- B. W. Roose, S. D. Zemerov and I. J. Dmochowski, Chem. Sci., 2017, 8, 7631–7636 RSC.
- B. W. Roose, S. D. Zemerov, Y. Wang, M. Kasimova, V. Carnevale and I. J. Dmochowski, ChemPhysChem, 2019, 20, 260–267 CrossRef CAS.
- S. D. Zemerov, B. W. Roose, K. L. Farenhem, Z. Zhao, M. A. Stringer, A. R. Goldman, D. W. Speicher and I. J. Dmochowski, Anal. Chem., 2020, 92, 12817–12824 CrossRef CAS.
- Z. Zhao, B. W. Roose, S. D. Zemerov, M. A. Stringer and I. J. Dmochowski, Chem. Commun., 2020, 56, 11122–11125 RSC.
- O. Taratula and I. J. Dmochowski, Curr. Opin. Chem. Biol., 2010, 14, 97–104 CrossRef CAS.
- Y. Wang and I. J. Dmochowski, Acc. Chem. Res., 2016, 49, 2179–2187 CrossRef CAS.
- L. Garel, J.-P. Dutasta and A. Collet, Angew. Chem., Int. Ed., 1993, 32, 1169–1171 CrossRef.
- G. Huber, T. Brotin and L. Dubois, J. Am. Chem. Soc., 2006, 128, 6239–6246 CrossRef CAS.
- J. Gabard and A. Collet, J. Chem. Soc., Chem. Commun., 1981, 1137 RSC.
- T. Brotin and J.-P. Dutasta, Chem. Rev., 2009, 109, 88–130 CrossRef CAS.
- T. Brotin and J.-P. Dutasta, Compr. Supramol. Chem. II, 2017, 1, 317–335 CAS.
- K. Bartik, M. Luhmer, J.-P. Dutasta, A. Collet and J. Reisse, J. Am. Chem. Soc., 1998, 120, 784–791 CrossRef CAS.
- M. M. Spence, S. M. Rubin, I. E. Dimitrov, E. J. Ruiz, D. E. Wemmer, A. Pines, S. Q. Yao, F. Tian and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10654–10657 CrossRef CAS.
- O. Taratula, P. A. Hill, Y. Bai, N. S. Khan and I. J. Dmochowski, Org. Lett., 2011, 13, 1414–1417 CrossRef CAS.
- P. A. Hill, Q. Wei, R. G. Eckenhoff and I. J. Dmochowski, J. Am. Chem. Soc., 2007, 129, 9262–9263 CrossRef CAS.
- P. A. Hill, Q. Wei, T. Troxler and I. J. Dmochowski, J. Am. Chem. Soc., 2009, 131, 3069–3077 CrossRef CAS.
- Y. Bai, P. A. Hill and I. J. Dmochowski, Anal. Chem., 2012, 84, 9935–9941 CrossRef CAS.
- D. R. Jacobson, N. S. Khan, R. Colle, R. Fitzgerald, L. Laureano-Perez, Y. Bai and I. J. Dmochowski, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10969–10973 CrossRef CAS.
- G. K. Seward, Y. Bai, N. S. Khan and I. J. Dmochowski, Chem. Sci., 2011, 2, 1103–1110 RSC.
- J. M. Chambers, P. A. Hill, J. A. Aaron, Z. Han, D. W. Christianson, N. N. Kuzma and I. J. Dmochowski, J. Am. Chem. Soc., 2009, 131, 563–569 CrossRef CAS.
- J. A. Aaron, J. M. Chambers, K. M. Jude, L. Di Costanzo, I. J. Dmochowski and D. W. Christianson, J. Am. Chem. Soc., 2008, 130, 6942–6943 CrossRef CAS.
- O. Taratula, Y. Bai, E. L. D'Antonio and I. J. Dmochowski, Supramol. Chem., 2015, 27, 65–71 CrossRef CAS.
- Q. Wei, G. K. Seward, P. A. Hill, B. Patton, I. E. Dimitrov, N. N. Kuzma and I. J. Dmochowski, J. Am. Chem. Soc., 2006, 128, 13274–13283 CrossRef CAS.
- A. Schlundt, W. Kilian, M. Beyermann, J. Sticht, S. Günther, S. Höpner, K. Falk, O. Roetzschke, L. Mitschang and C. Freund, Angew. Chem., Int. Ed., 2009, 48, 4142–4145 CrossRef CAS.
- N. Kotera, N. Tassali, E. Léonce, C. Boutin, P. Berthault, T. Brotin, J.-P. Dutasta, L. Delacour, T. Traoré, D. A. Buisson, F. Taran, S. Coudert and B. Rousseau, Angew. Chem., Int. Ed., 2012, 51, 4100–4103 CrossRef CAS.
- N. Tassali, N. Kotera, C. Boutin, E. Léonce, Y. Boulard, B. Rousseau, E. Dubost, F. Taran, T. Brotin, J.-P. Dutasta and P. Berthault, Anal. Chem., 2014, 86, 1783–1788 CrossRef CAS.
- J. Zhang, W. Jiang, Q. Luo, X. Zhang, Q. Guo, M. Liu and X. Zhou, Talanta, 2014, 122, 101–105 CrossRef CAS.
- K. Jeong, C. C. Slack, C. C. Vassiliou, P. Dao, M. D. Gomes, D. J. Kennedy, A. E. Truxal, L. J. Sperling, M. B. Francis, D. E. Wemmer and A. Pines, ChemPhysChem, 2015, 16, 3573–3577 CrossRef CAS.
- V. Roy, T. Brotin, J.-P. Dutasta, M. H. Charles, T. Delair, F. Mallet, G. Huber, H. Desvaux, Y. Boulard and P. Berthault, ChemPhysChem, 2007, 8, 2082–2085 CrossRef CAS.
- P. Berthault, H. Desvaux, T. Wendlinger, M. Gyejacquot, A. Stopin, T. Brotin, J.-P. Dutasta and Y. Boulard, Chem.–Eur. J., 2010, 16, 12941–12946 CrossRef CAS.
- E. Léonce, J. P. Dognon, D. Pitrat, J. C. Mulatier, T. Brotin and P. Berthault, Chem.–Eur. J., 2018, 24, 6534–6537 CrossRef.
- S. Yang, W. Jiang, L. Ren, Y. Yuan, B. Zhang, Q. Luo, Q. Guo, L.-S. Bouchard, M. Liu and X. Zhou, Anal. Chem., 2016, 88, 5835–5840 CrossRef CAS.
- N. Kotera, E. Dubost, G. Milanole, E. Doris, E. Gravel, N. Arhel, T. Brotin, J.-P. Dutasta, J. Cochrane, E. Mari, C. Boutin, E. Leonce, P. Berthault and B. Rousseau, Chem. Commun., 2015, 51, 11482–11484 RSC.
- E. Mari, Y. Bousmah, C. Boutin, E. Léonce, G. Milanole, T. Brotin, P. Berthault and M. Erard, ChemBioChem, 2019, 20, 1450–1457 CrossRef CAS.
- M. M. Spence, E. J. Ruiz, S. M. Rubin, T. J. Lowery, N. Winssinger, P. G. Schultz, D. E. Wemmer and A. Pines, J. Am. Chem. Soc., 2004, 126, 15287–15294 CrossRef CAS.
- T. Brotin, R. Barbe, M. Darzac and J.-P. Dutasta, Chem.–Eur. J., 2003, 9, 5784–5792 CrossRef CAS.
- L. Gao, W. Liu, O.-S. Lee, I. J. Dmochowski and J. G. Saven, Chem. Sci., 2015, 6, 7238–7248 RSC.
- O. Taratula, P. A. Hill, N. S. Khan, P. J. Carroll and I. J. Dmochowski, Nat. Commun., 2010, 1, 148 CrossRef.
- B. A. Riggle, M. L. Greenberg, Y. Wang, R. F. Wissner, S. D. Zemerov, J. Petersson and I. J. Dmochowski, Org. Biomol. Chem., 2017, 15, 8883–8887 RSC.
- S. D. Zemerov, B. W. Roose, M. L. Greenberg, Y. Wang and I. J. Dmochowski, Anal. Chem., 2018, 90, 7730–7738 CrossRef CAS.
- T. J. Lowery, S. Garcia, L. Chavez, E. J. Ruiz, T. Wu, T. Brotin, J.-P. Dutasta, D. S. King, P. G. Schultz, A. Pines and D. E. Wemmer, ChemBioChem, 2006, 7, 65–73 CrossRef CAS.
- D. N. Sears and C. J. Jameson, J. Chem. Phys., 2003, 119, 12231–12244 CrossRef CAS.
- S. D. Zemerov, Y. Lin and I. J. Dmochowski, Anal. Chem., 2021, 93, 1507–1514 CrossRef CAS.
- K. K. Palaniappan, M. B. Francis, A. Pines and D. E. Wemmer, Isr. J. Chem., 2014, 54, 104–112 CrossRef CAS.
- G. K. Seward, Q. Wei and I. J. Dmochowski, Bioconjugate Chem., 2008, 19, 2129–2135 CrossRef CAS.
- L. Schröder, T. J. Lowery, C. Hilty, D. E. Wemmer and A. Pines, Science, 2006, 314, 446–449 CrossRef.
- T. Meldrum, K. L. Seim, V. S. Bajaj, K. K. Palaniappan, W. Wu, M. B. Francis, D. E. Wemmer and A. Pines, J. Am. Chem. Soc., 2010, 132, 5936–5937 CrossRef CAS.
- Y. Bai, Y. Wang, M. Goulian, A. Driks and I. J. Dmochowski, Chem. Sci., 2014, 5, 3197–3203 RSC.
- B. A. Riggle, Y. Wang and I. J. Dmochowski, J. Am. Chem. Soc., 2015, 137, 5542–5548 CrossRef CAS.
- Q. Zeng, Q. Guo, Y. Yuan, Y. Yang, B. Zhang, L. Ren, X. Zhang, Q. Luo, M. Liu, L.-S. Bouchard and X. Zhou, Anal. Chem., 2017, 89, 2288–2295 CrossRef CAS.
- G. Milanole, B. Gao, A. Paoletti, G. Pieters, C. Dugave, E. Deutsch, S. Rivera, F. Law, J. L. Perfettini, E. Mari, E. Léonce, C. Boutin, P. Berthault, H. Volland, F. Fenaille, T. Brotin and B. Rousseau, Bioorg. Med. Chem., 2017, 25, 6653–6660 CrossRef CAS.
- A. M. Oros and N. J. Shah, Phys. Med. Biol., 2004, 49, R105–R153 CrossRef CAS.
- M. S. Albert and D. Balamore, Nucl. Instrum. Methods Phys. Res., Sect. A, 1998, 402, 441–453 CrossRef CAS.
- P. Berthault, G. Huber and H. Desvaux, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 55, 35–60 CrossRef CAS.
- C. Boutin, A. Stopin, F. Lenda, T. Brotin, J.-P. Dutasta, N. Jamin, A. Sanson, Y. Boulard, F. Leteurtre, G. Huber, A. Bogaert-Buchmann, N. Tassali, H. Desvaux, M. Carrière and P. Berthault, Bioorg. Med. Chem., 2011, 19, 4135–4143 CrossRef CAS.
- H. M. Rose, C. Witte, F. Rossella, S. Klippel, C. Freund and L. Schröder, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11697–11702 CrossRef CAS.
- C. Witte, V. Martos, H. M. Rose, S. Reinke, S. Klippel, L. Schröder and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2015, 54, 2806–2810 CrossRef CAS.
- S. Klippel, J. Döpfert, J. Jayapaul, M. Kunth, F. Rossella, M. Schnurr, C. Witte, C. Freund and L. Schröder, Angew. Chem., Int. Ed., 2014, 53, 493–496 CrossRef CAS.
- N. S. Khan, B. A. Riggle, G. K. Seward, Y. Bai and I. J. Dmochowski, Bioconjugate Chem., 2015, 26, 101–109 CrossRef CAS.
- S. Yang, Y. Yuan, W. Jiang, L. Ren, H. Deng, L. S. Bouchard, X. Zhou and M. Liu, Chem.–Eur. J., 2017, 23, 7648–7652 CrossRef CAS.
- M. Schnurr, C. Witte and L. Schröder, Phys. Chem. Chem. Phys., 2013, 15, 14178–14181 RSC.
- R. D. Booker and A. K. Sum, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 1347–1356 CrossRef CAS.
- G. L. Britton, H. Kim, P. H. Kee, J. Aronowski, C. K. Holland, D. D. McPherson and S. L. Huang, Circulation, 2010, 122, 1578–1587 CrossRef CAS.
- M. P. Dandekar, T. Peng, D. D. McPherson, J. Quevedo, J. C. Soares and S. L. Huang, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2018, 86, 140–149 CrossRef CAS.
- Y. F. Miao, T. Peng, M. R. Moody, M. E. Klegerman, J. Aronowski, J. Grotta, D. D. McPherson, H. Kim and S. L. Huang, Sci. Rep., 2018, 8, 1–12 CrossRef.
- Q. Guo, Q. Zeng, W. Jiang, X. Zhang, Q. Luo, X. Zhang, L.-S. Bouchard, M. Liu and X. Zhou, Chem.–Eur. J., 2016, 22, 3967–3970 CrossRef CAS.
- F. Schilling, L. Schröder, K. K. Palaniappan, S. Zapf, D. E. Wemmer and A. Pines, ChemPhysChem, 2010, 11, 3529–3533 CrossRef CAS.
- M. Kunth, J. Döpfert, C. Witte, F. Rossella and L. Schröder, Angew. Chem., Int. Ed., 2012, 51, 8217–8220 CrossRef CAS.
- F. Rossella, H. M. Rose, C. Witte, J. Jayapaul and L. Schröder, ChemPlusChem, 2014, 79, 1463–1471 CrossRef CAS.
- A. Piontek, C. Witte, H. M. Rose, M. Eichner, J. Protze, G. Krause, J. Piontek and L. Schröder, Ann. N. Y. Acad. Sci., 2017, 1397, 195–208 CrossRef CAS.
- M. Schnurr, K. Sydow, H. M. Rose, M. Dathe and L. Schröder, Adv. Healthcare Mater., 2015, 4, 40–45 CrossRef CAS.
- M. Schnurr, I. Volk, H. Nikolenko, L. Winkler, M. Dathe and L. Schröder, Adv. Biosyst., 2020, 4, 1900251 CrossRef.
- J. L. Mynar, T. J. Lowery, D. E. Wemmer, A. Pines and J. M. J. Fréchet, J. Am. Chem. Soc., 2006, 128, 6334–6335 CrossRef CAS.
- T. K. Stevens, K. K. Palaniappan, R. M. Ramirez, M. B. Francis, D. E. Wemmer and A. Pines, Magn. Reson. Med., 2013, 69, 1245–1252 CrossRef CAS.
- K. K. Palaniappan, R. M. Ramirez, V. S. Bajaj, D. E. Wemmer, A. Pines and M. B. Francis, Angew. Chem., Int. Ed., 2013, 52, 4849–4853 CrossRef CAS.
- K. Jeong, C. Netirojjanakul, H. K. Munch, J. Sun, J. A. Finbloom, D. E. Wemmer, A. Pines and M. B. Francis, Bioconjugate Chem., 2016, 27, 1796–1801 CrossRef CAS.
- P. Berthault, A. Bogaert-Buchmann, H. Desvaux, G. Huber and Y. Boulard, J. Am. Chem. Soc., 2008, 130, 16456–16457 CrossRef CAS.
- R. Tyagi, C. Witte, R. Haag and L. Schröder, Org. Lett., 2014, 16, 4436–4439 CrossRef CAS.
- S. Malhotra, M. Calderón, A. K. Prasad, V. S. Parmar and R. Haag, Org. Biomol. Chem., 2010, 8, 2228–2237 RSC.
- S. Klippel, C. Freund and L. Schröder, Nano Lett., 2014, 14, 5721–5726 CrossRef CAS.
- Y. Wang and I. J. Dmochowski, Chem. Commun., 2015, 51, 8982–8985 RSC.
- Y. Wang, B. W. Roose, J. P. Philbin, J. L. Doman and I. J. Dmochowski, Angew. Chem., Int. Ed., 2016, 55, 1733–1736 CrossRef CAS.
- F. T. Hane, T. Li, P. Smylie, R. M. Pellizzari, J. A. Plata, B. DeBoef and M. S. Albert, Sci. Rep., 2017, 7, 41027 CrossRef CAS.
- K. Du, S. D. Zemerov, S.-H. Parra, J. M. Kikkawa and I. J. Dmochowski, Inorg. Chem., 2020, 59, 13831–13844 CrossRef CAS.
- K. Du, S. D. Zemerov, P. J. Carroll and I. J. Dmochowski, Inorg. Chem., 2020, 59, 12758–12767 CrossRef CAS.
- T. Traoré, L. Delacour, S. Garcia-Argote, P. Berthault, J.-C. Cintrat and B. Rousseau, Org. Lett., 2010, 12, 960–962 CrossRef.
- L. Delacour, N. Kotera, T. Traoré, S. Garcia-Argote, C. Puente, F. Leteurtre, E. Gravel, N. Tassali, C. Boutin, E. Léonce, Y. Boulard, P. Berthault and B. Rousseau, Chem.–Eur. J., 2013, 19, 6089–6093 CrossRef CAS.
- M. Chen, O. Doganay, T. Matin, A. McIntyre, N. Rahman, D. Bulte and F. Gleeson, Eur. Radiol., 2020, 30, 1145–1155 CrossRef.
- J. Chacon-Caldera, A. Maunder, M. R. Rao, G. Norquay, O. I. Rodgers, M. Clemence, C. Puddu, L. R. Schad and J. M. Wild, Magn. Reson. Med., 2020, 83, 262–270 CrossRef CAS.
- E. J. Song, C. R. Kelsey, B. Driehuys and L. Rankine, J. Med. Imaging Radiat. Oncol., 2018, 62, 91–93 CrossRef.
- L. Ebner, M. He, R. S. Virgincar, T. Heacock, S. S. Kaushik, M. S. Freemann, H. P. McAdams, M. Kraft and B. Driehuys, Invest. Radiol., 2017, 52, 120–127 CrossRef.
- J. M. Wild, G. Collier, H. Marshall, L. Smith, G. Norquay, A. J. Swift, F. C. Horn, F. Chan, N. J. Stewart, L. C. Hutchison, M. R. Rao, I. Sabbroe, R. Niven, A. Horsley, S. Siddiqui, K. Ugonna and R. Lawson, Thorax, 2015, 70, A220–A221 CrossRef.
- J. R. Birchall, P. Nikolaou, A. M. Coffey, B. E. Kidd, M. Murphy, M. Molway, L. B. Bales, B. M. Goodson, R. K. Irwin, M. J. Barlow and E. Y. Chekmenev, Anal. Chem., 2020, 92, 4309–4316 CrossRef CAS.
- J. R. Birchall, R. K. Irwin, P. Nikolaou, A. M. Coffey, B. E. Kidd, M. Murphy, M. Molway, L. B. Bales, K. Ranta, M. J. Barlow, B. M. Goodson, M. S. Rosen and E. Y. Chekmenev, J. Magn. Reson., 2020, 319, 106813 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |