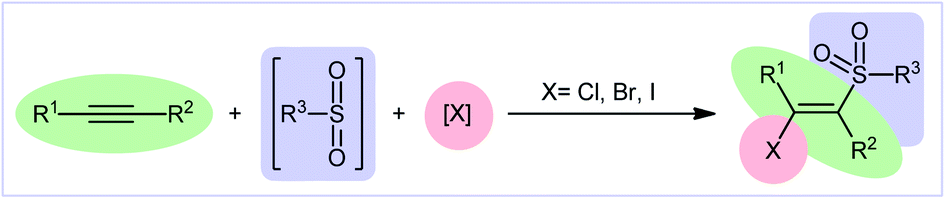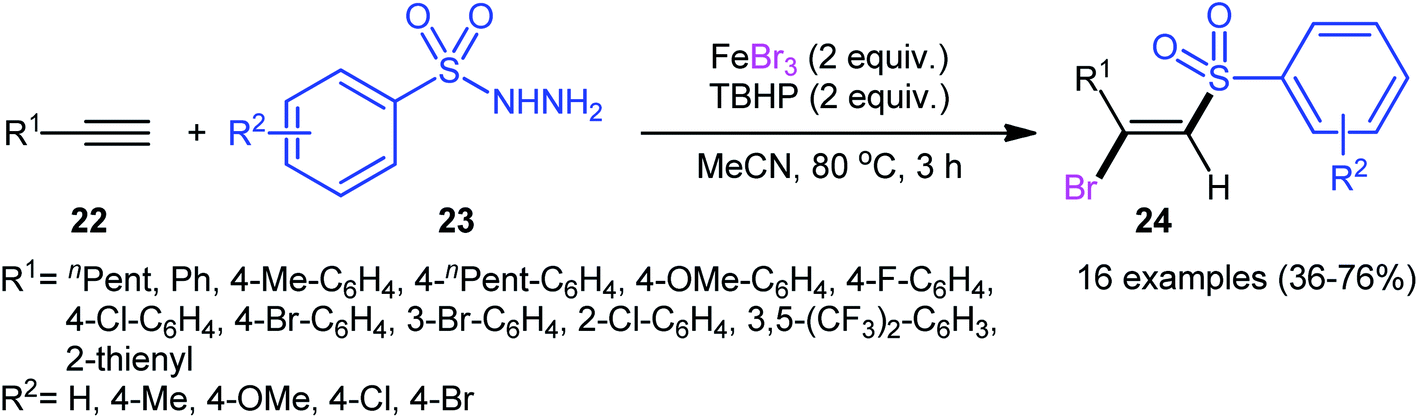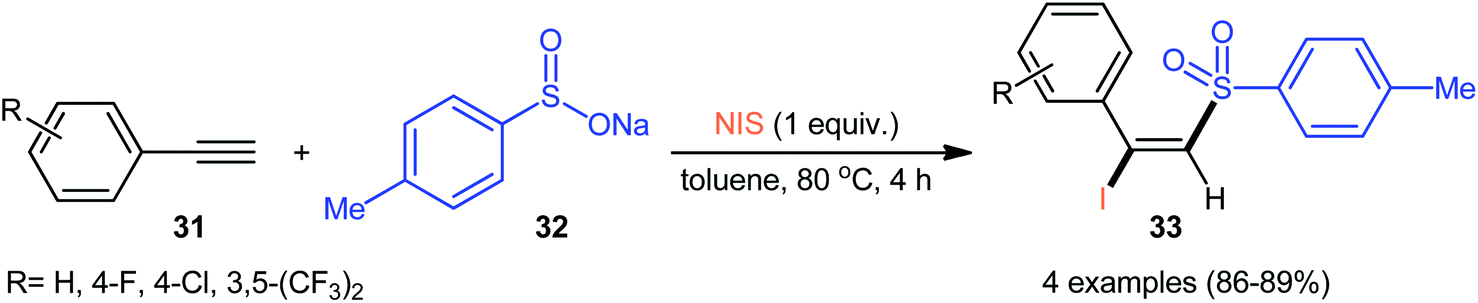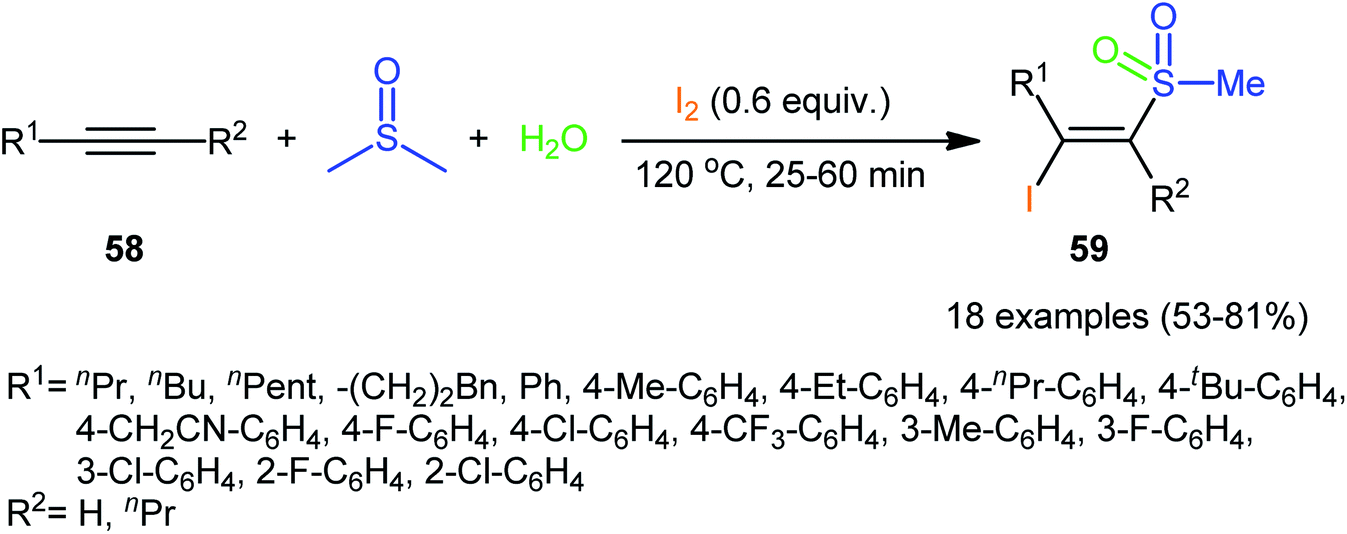DOI:
10.1039/D1RA03443J
(Review Article)
RSC Adv., 2021,
11, 33447-33460
Direct halosulfonylation of alkynes: an overview
Received
2nd May 2021
, Accepted 30th September 2021
First published on 13th October 2021
Abstract
The difunctionalization reactions of easily available and inexpensive alkynes have emerged as a reliable, powerful, and step-economical approach for the construction of highly substituted complex alkenes in a one-pot manner, without the need for isolation of intermediates. A wide variety of transformations based on this concept have been successfully achieved for the preparation of synthetically and biologically important β-halovinyl sulfone scaffolds. In this Review, we summarize the recent advances and developments in this field and present a comprehensive overview of halosulfonylation of alkyne substrates with emphasis on the mechanistic features of the reactions.
1. Introduction
Sulfones (RSO2R’) appeared as a useful and important class of organosulfur compounds due to the prevalence of this moiety in a great number of biologically active compounds1 as well as commercialized drugs, including apremilast, ceritinib, chlormezanone, dapsone, eletriptan, lifitegrast, tinidazole, and vismodegib.2 In addition, these compounds are very versatile synthetic intermediates and can function as suitable building blocks to synthesize various biologically active compounds or functional materials.3 In this family of compounds, β-halovinyl sulfones have attracted significant attention from the synthetic community because of their diverse reaction patterns and application in the assembly of various sulfonyl-substituted heterocycles4 and other value-added chemicals such as β-keto sulfones,5a,b β-amino sulfones,6 sulfonyl-substituted 1,3-enynes,7 acetylene sulfonyl compounds,8 1,3-bis(arylsulfonyl)allenes9 and many more. Therefore, it is always interesting to develop cost-effective facile synthesis strategies to prepare this special class of organosulfur compounds from easily available and inexpensive starting materials.
Recently, the direct halosulfonylation of easily accessible alkynes has emerged as an ideal strategy for the construction of the titled compounds which makes the synthetic route cleaner, greener, and faster than traditional methods that relay on the three-step sulfonation–zirconation–halogenation sequence.10 Although no reporting guideline exists for the direct fluoro-sulfonylation of alkynes and only a handful of methodologies have been reported for bromo-sulfonylation of alkynes, synthesis of β-chloro- and β-iodo-vinyl sulfones through the direct chloro- and iodo-sulfonylation of alkynes has been the subject of intense research efforts in recent years. In 2016, Luo and co-workers published an interesting review paper entitled “recent advances in the synthesis of vinyl sulfones” that covers most of the advances in the preparation of vinyl sulfone derivatives.11 However, synthesis of β-halovinyl sulfones through the direct halosulfonylation of simple and easily accessible alkynes was almost omitted, while this procedure has recently attracted a lot of attention because of its operational simplicity and high atom and step economy. In connection with our recent reviews on the synthesis of organosulfur compounds12 and new methodologies in modern organic synthesis,13–20 herein, we provide an overview of the direct halosulfonylation of C–C triple bonds (Fig. 1), with special emphasize on the mechanistic aspect of the reactions. For clarity, the reactions will be classified by the type of halogen atoms and sulfonylating agents.
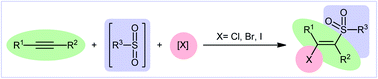 |
| | Fig. 1 Direct halosulfonylation of alkynes. | |
2. Chlorosulfonylation
In this section, the attention is paid to the synthesis of β-chlorovinyl sulfones from the corresponding alkynes by means of the concomitant formation of C–S and C–Cl bonds. The section is divided into two parts according to chlorosulfonylating reagents. The first includes the chlorosulfonylation of alkynes using bifunctional reagents, while the second contains the three-component reactions.
2.1. Two-component reactions
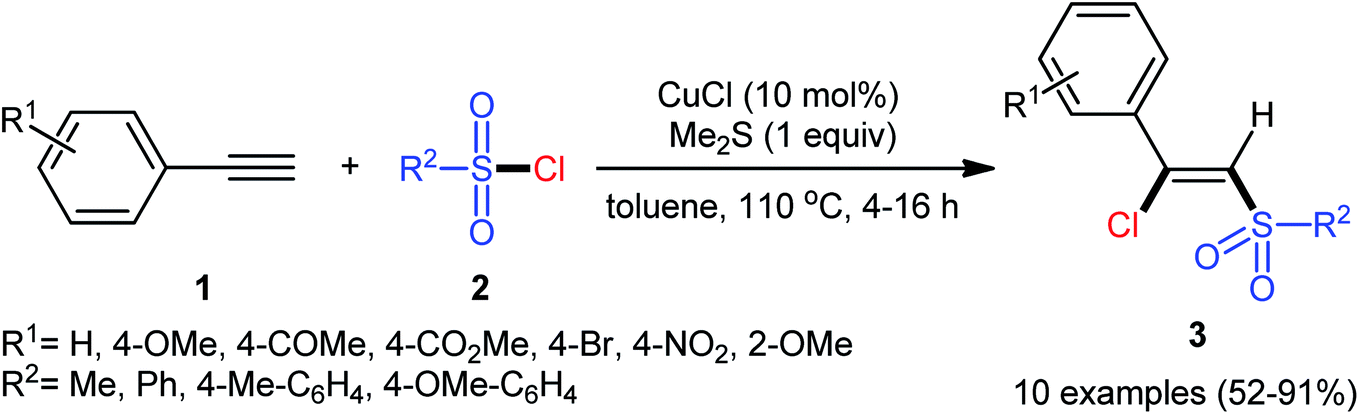
The reported examples of the chlorosulfonylation of alkynes are mainly relay on the use of sulfonyl chlorides as the both sulfonylating and chlorinating agents. As early as 1971, the first synthesis of β-chlorovinyl sulfones via the direct chlorosulfonylation of the corresponding alkynes with sulfonyl chlorides was realized by Amiel et al.21,22 However, since then, this synthetic strategy did not attract the attention of chemists for nearly 35 years. Since last three decades, several research groups investigated the scope and limitation of this transformation. In 2005, Liang and co-workers studied the possibility of stereoselective synthesis of β-chlorovinyl sulfones through the direct Cu-catalyzed chlorosulfonylation of phenylacetylene derivatives.23 By employing phenylacetylene and p-toluenesulfonyl chloride as the model substrates, the reaction variables such as catalysts, additives, and solvents were attentively screened. The results indicated that the merge of 10 mol% of CuCl with 1 equiv. of Me2S was the most appropriate catalytic system for this conversion and among the various aprotic solvents (e.g., THF, DMF, DCM, MeCN, toluene, dioxane); toluene was found to be the most suitable solvent. Under the optimized conditions, a diverse array of (Z)-β-chlorovinyl sulfones 3 were selectively obtained in good to excellent yields by reaction of various phenylacetylene derivatives 1 with sulfonyl chlorides 2 under an atmosphere of air at 110 °C (Scheme 1). An internal aryl–alkyl alkyne was also tested and gave the desired product but in moderate yield. The system was also amenable to the chlorosulfonylation of vinyl alkynes as exemplified by synthesis of (Z)-((2-chloro-2-(cyclohex-1-en-1-yl)vinyl)sulfonyl)benzene using 1-cyclohexeneacetylene as the reactant. However, diphenylacetylene, propargyl alkyl ether, and propiolate failed to participate in this transformation. The authors assumed that the Me2S is probably involved in stabilizing the intermediate A. Nevertheless, no comment was made by them regarding the plausible mechanistic pathway of this transformation.
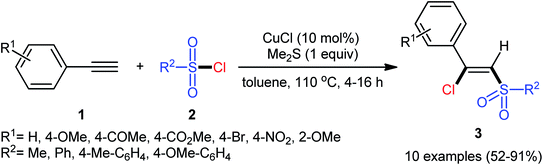 |
| | Scheme 1 Cu-catalyzed chlorosulfonylation of alkynes 1 with sulfonyl chlorides 2. | |
Seven years later, the iron-catalyzed version of this difunctionalization reaction was disclosed by Nakamura and colleagues,24 who revealed that the treatment of various aromatic and aliphatic terminal alkynes 4 with aromatic sulfonyl chlorides 5 in the presence of a combination of Fe(acac)2 (10 mol%) and (p-Tol)3P (10 mol%) in refluxing toluene afforded the corresponding β-chlorovinyl sulfones 6 in moderate to high yields and complete (E)-selectivities (Scheme 2a). According to authors proposed mechanistic pathways (Scheme 2b), this reaction proceeds through an electron transfer between iron(II) catalyst and sulfonyl chloride 5, following the radical addition of the resulting sulfonyl radical A to the alkyne 4 to give vinyl radical intermediate B, which could be trapped by in situ generated iron(III) chloride to yield (E)-β-chlorovinyl sulfone derivatives 6. According to the authors, the iron(III) chloride species acts as a bulky chlorine donor and is responsible for the excellent (E)-selectivity for the desired products. Subsequently, a similar reaction conditions were applied by Yang and Zhu along with their colleagues to chlorosulfonylation of a diverse array of alkynyl chlorides and the desired cis-1,2-dichlorovinylsulfones were obtained in good to excellent yields, ranging from 55% to 91%.25
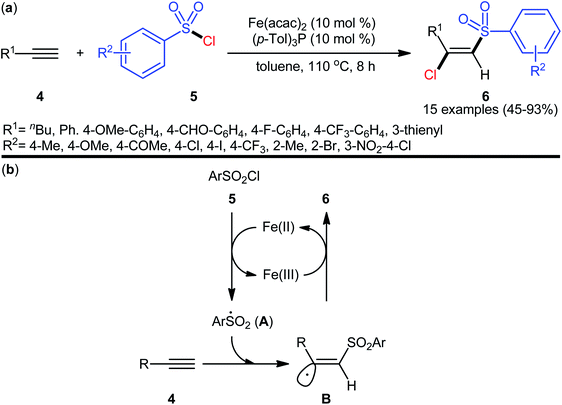 |
| | Scheme 2 (a) Fe-catalyzed chlorosulfonylation of terminal alkynes 4 with arylsulfonyl chlorides 5; (b) proposed mechanism for the formation of (E)-β-chlorovinyl sulfones 6. | |
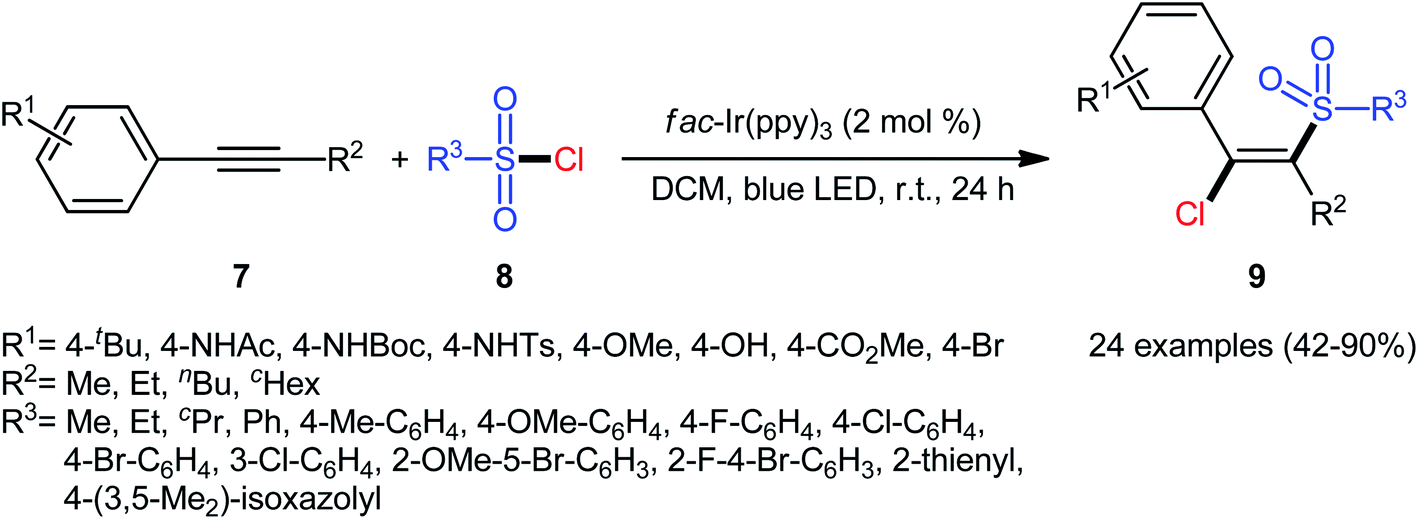
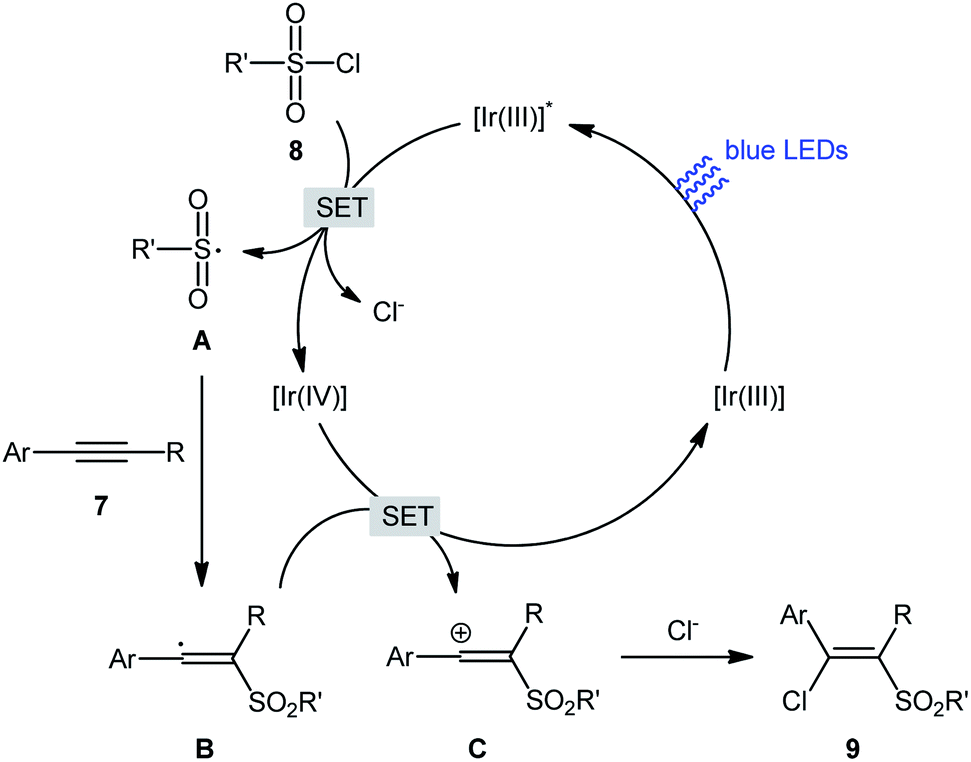
In 2018, Kim–Han's research group described a visible-light-mediated, fac-Ir(ppy)3-catalyzed regio- and stereo-selective chlorosulfonylation of internal aryl–alkyl alkynes 7 with various aliphatic, aromatic, and heteroaromatic sulfonyl chlorides 8.26 This synthetic transformation exhibited an efficient and attractive method for the high yielding synthesis of (E)-β-chlorovinyl sulfone derivatives 9 at room temperature under additive-free conditions (Scheme 3). Noteworthy, the reaction exhibited high degree of regioselectivity, in which the sulfonyl group is predominantly placed on the carbon atom adjacent to the alkyl group and Cl group added to the carbon atom near the aryl group. Interestingly, the reaction could be conducted on gram quantities without any difficulty. This procedure was also compatible with a variety of common functional groups including fluoro, chloro, bromo, hydroxy, methoxy, amide, sulfonamide, and ester functionalities, which are useful for further functionalization. According to the authors, this transformation involves a radical mechanism as shown in Scheme 4. In a related investigation, Reiser and co-workers reported the usefulness of well-established [Cu(dap)2]Cl photoredox catalyst for the direct chlorosulfonation of unsaturated hydrocarbons with sulfonyl chloride under green light irradiation.27 Thus, a small library of alkynes (terminal and internal) reacted with p-toluenesulfonyl chloride to give the respective β-chlorovinyl sulfone in moderate to almost quantitative yields. However, the regioselectivity of products was modest at best.
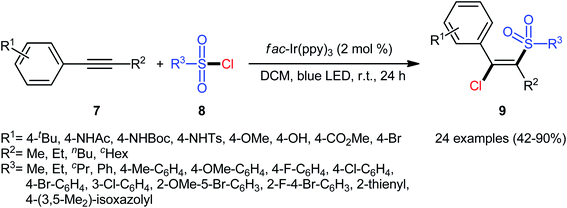 |
| | Scheme 3 Visible-light-mediated chlorosulfonylation of internal aryl–alkyl alkynes 7 with sulfonyl chlorides 8. | |
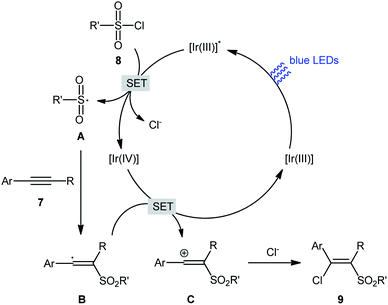 |
| | Scheme 4 Mechanistic explanation for the reaction in Scheme 3. | |
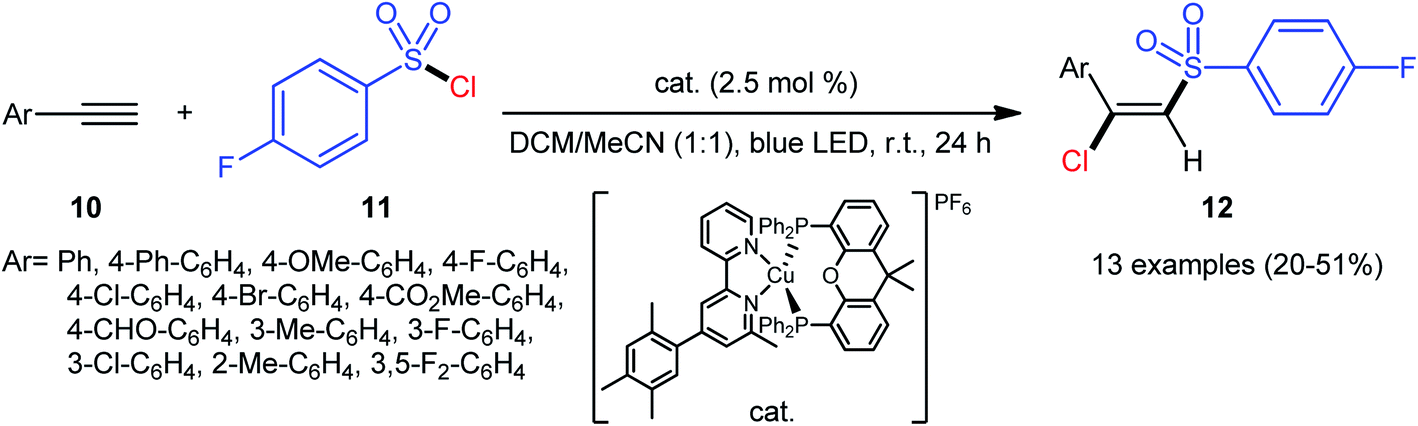
A similar strategy was then extended to a heteroleptic copper(I)-mediated chlorosulfonylation of terminal aryl alkynes 10 with para-fluorobenzylsulfonyl chloride 11 by the Hu group.28 As shown in Scheme 5, thirteen (E)-chlorosulfonylvinylbenzenes 12 were efficiently produced with excellent (E)-selectivity. However, internal aryl alkynes and aliphatic alkynes failed to undergo the reaction. It should be mentioned that the synthetic concept could also be extended to terminal alkene substrates. The authors stated that the outstanding (E)-selectivity in this difunctionalization reaction may originated from an outer-sphere mechanism where the radical termination step proceeds through the thermodynamically favored configuration.
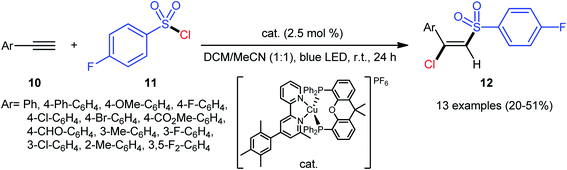 |
| | Scheme 5 Hu's synthesis of (E)-chlorosulfonylvinylbenzenes 12. | |
2.2. Three-component reactions
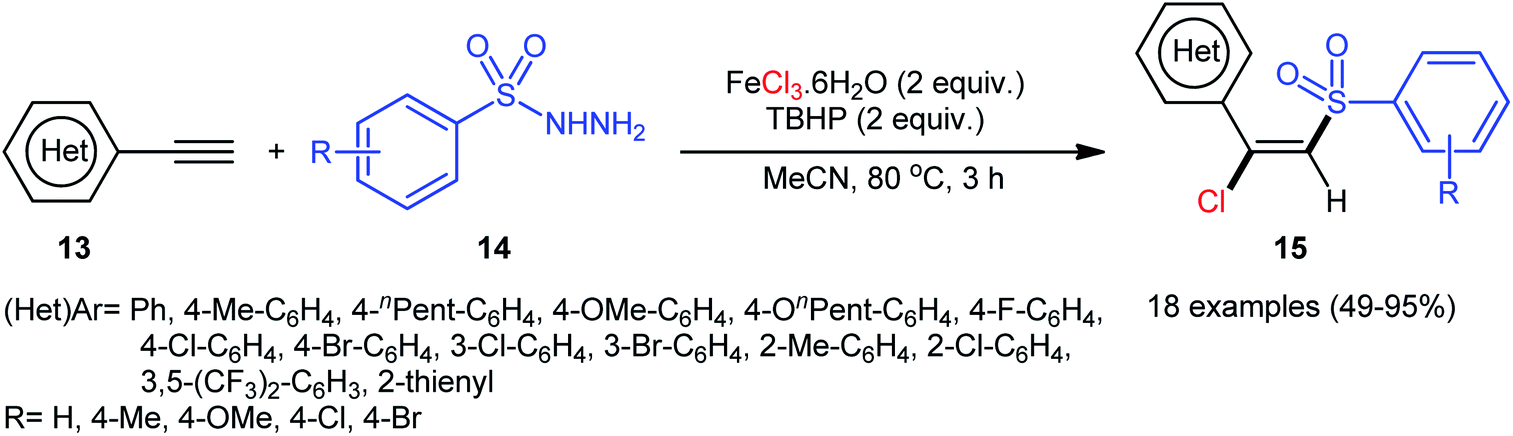
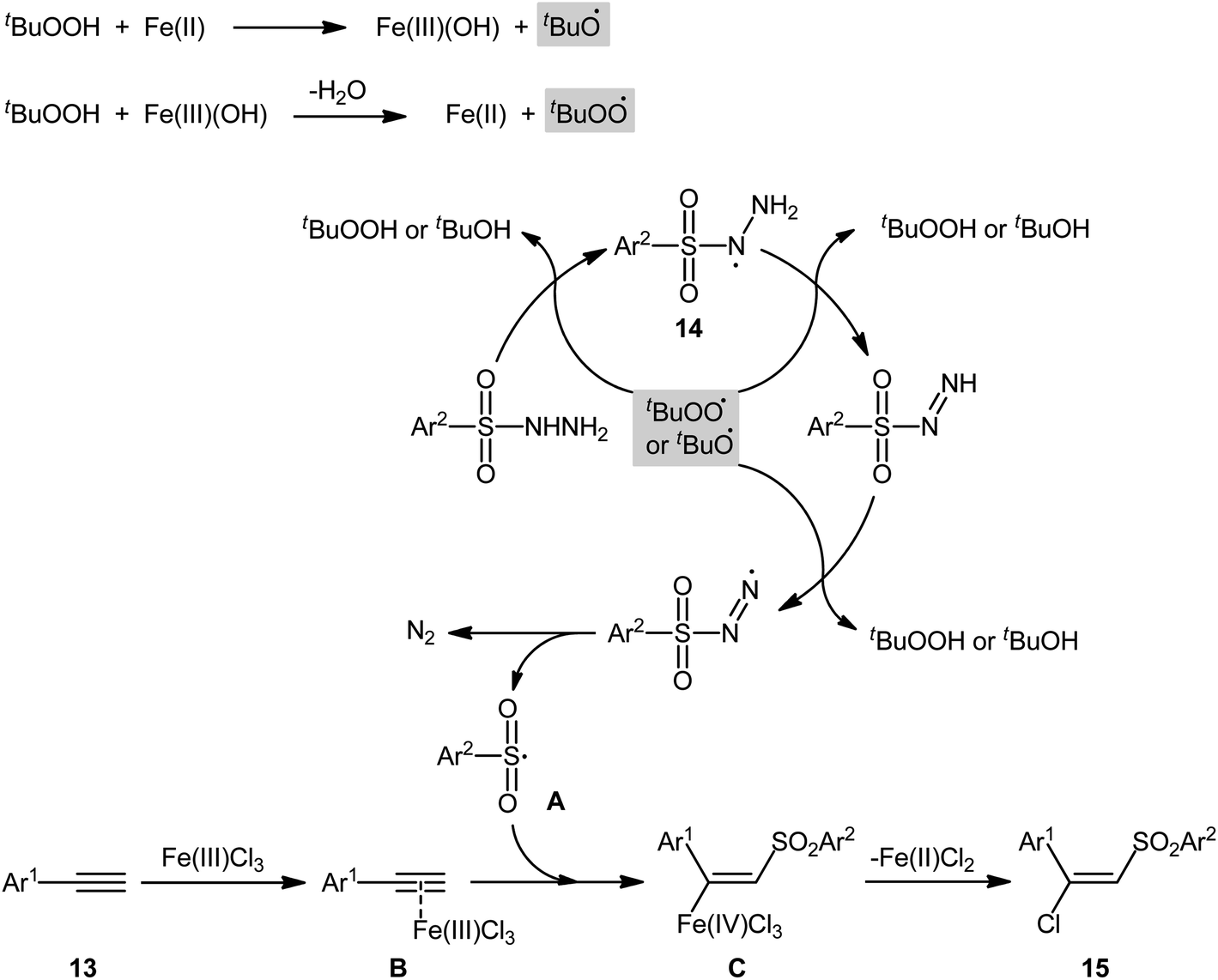
In 2013, Xu, Li, and colleagues disclosed the preparation of (E)-chlorosulfonylvinylarenes 15 with moderate to excellent yield (up to 95%) through iron-mediated regio- and stereo-selective chlorosulfonylation of terminal (hetero)aromatic alkynes 13 with arylsulfonyl hydrazides 14 (Scheme 6).29 The protocol uses iron(III) chloride hexahydrate as Cl source as well as mediator and tert-butyl hydroperoxide (tBuOOH; TBHP) as the oxidant. A series of important functional groups such as F, Cl, Br, CF3, and ether functionalities are found to be compatible under the reaction conditions. However, internal aryl alkynes were not effective in this system and applicability of alkylsulfonyl hydrazides did not investigated in this synthetic strategy. Hept-1-yne did not work well in the reaction and therefore no other aliphatic alkynes were examined in the protocol. Interestingly, by replacing FeCl3 with FeBr3, β-bromovinyl sulfones were afforded in high yields under the identical conditions. According to the control experiments and literature reports, the authors assumed that the reaction may starts with the generation of the tert-butoxyl and tert-butylperoxy radicals via decomposition of TBHP with the assistance of iron catalyst. Then, the resultant radicals abstract hydrogen atoms from sulfonyl hydrazide 14 to form sulfonyl radical A with the release of nitrogen. Subsequently, the selective addition of sulfonyl radical A to the Fe-coordinated alkyne B provides the Fe(IV) intermediate C, which after reductive elimination affords the (E)-β-halovinylsulfones 15 and regenerates the Fe(II) catalyst (Scheme 7).
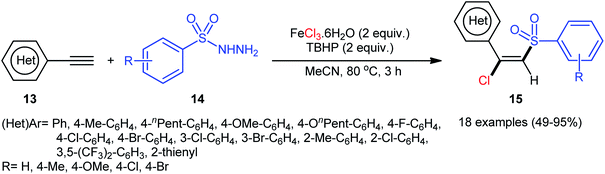 |
| | Scheme 6 Chlorosulfonylation of terminal (hetero)aromatic alkynes 13 with arylsulfonyl hydrazides 14 and FeCl3. | |
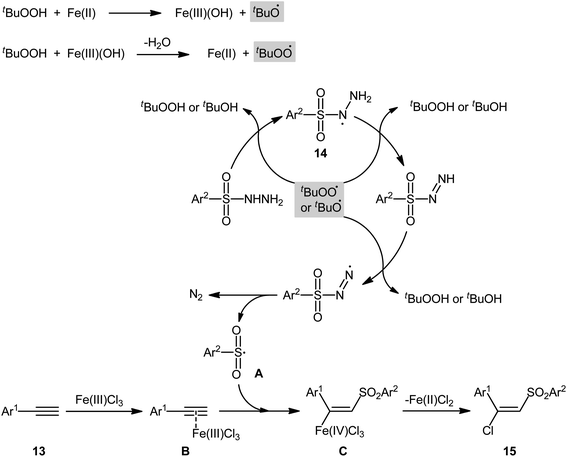 |
| | Scheme 7 Proposed Mechanism for the reaction in Scheme 6. | |
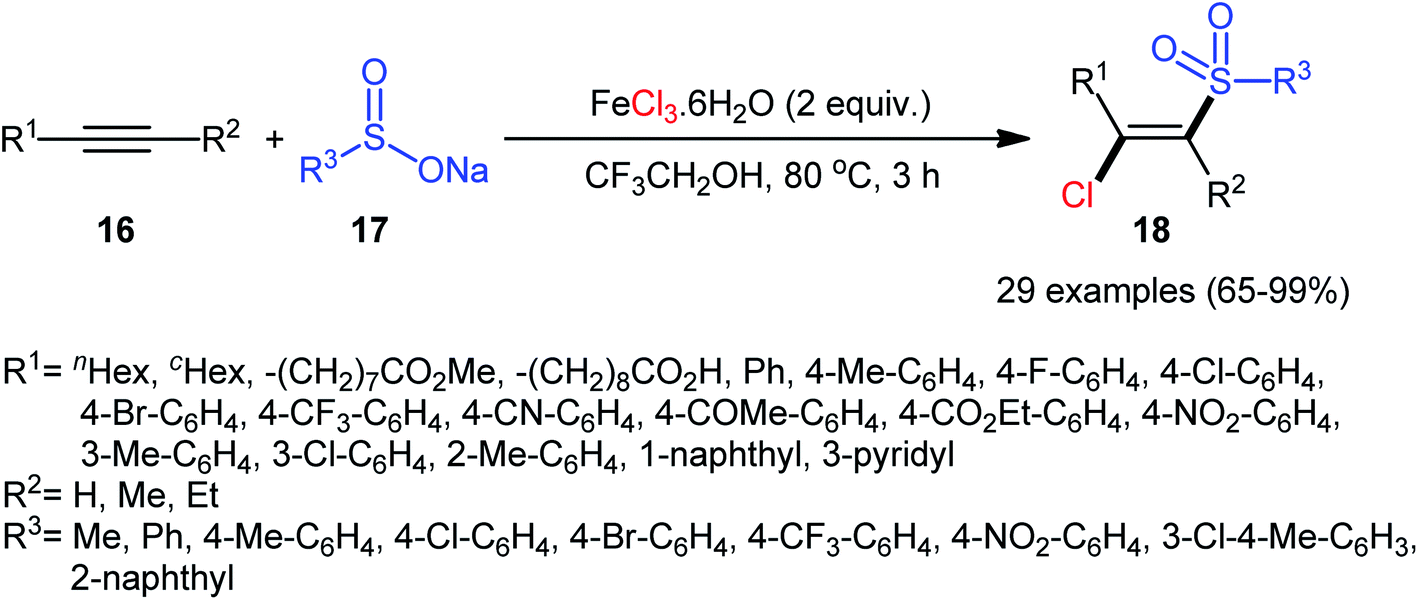
Four years later, Chen, Yin, and co-workers reported the related chlorosulfonylation of alkynes 16 with sodium sulfinates 17 and FeCl3·6H2O which did not require any oxidant or additive.30 The reactions were carried out in 2,2,2-trifluoroethanol at 80 °C under an inert atmosphere, tolerated both aromatic and aliphatic sodium sulfinates and various (hetero)aromatic and aliphatic terminal alkynes as well as internal aryl–alkyl alkynes, and generally delivered the expected (E)-β-chlorovinyl sulfones 18 in good to almost quantitative yields within 3 h (Scheme 8). However, just like Xu–Lis’ work, diaryl alkynes failed to enter into this reaction. Moreover, OMe-substituted aromatic alkynes did not respond the reaction under standard conditions.
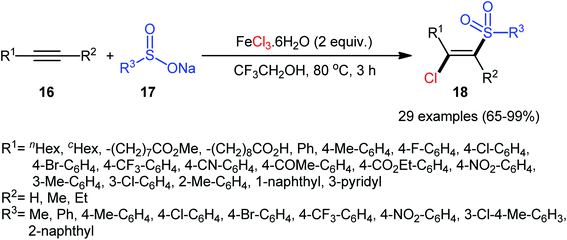 |
| | Scheme 8 Synthesis of (E)-β-chlorovinyl sulfones 18 via three-component reaction between alkynes 16, sodium sulfinates 17 and FeCl3·6H2O. | |
3. Bromosulfonylation
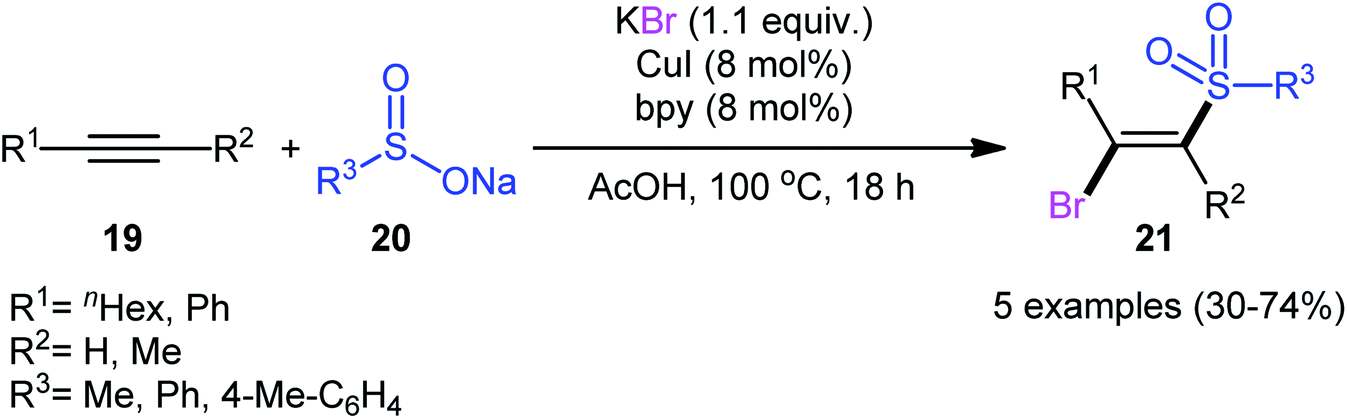
After pioneering works by Amiel's31 and Vasin's32 research teams on bromosulfonylation of phenylacetylene with a small series of sulfonyl bromides, the first general protocol for the direct bromosulfonylation of C–C triple bonds was published in 2011 by Taniguchi et al.33 who showed that three-component reaction between alkynes 19, sodium sulfinates 20, and KBr in the presence of catalytic amounts of CuI and pby in AcOH under open air resulted in the formation of (E)-β-bromovinyl sulfones 21 in good yields (Scheme 9). The synthetic broad scope of this methodology was established by using both aromatic and aliphatic sodium sulfinates and a library of various terminal and aryl–alkyl internal alkynes. However, alkyl–alkyl internal alkynes did not work well in this process and aryl–aryl internal alkynes were quite inert under standard conditions. A plausible mechanism for this Cu-catalyzed difunctionalization reaction was suggested by the authors, as shown in Scheme 10. It involved single-electron oxidation of sodium sulfinate 20 by in situ generated Cu(II) species to generate the sulfonyl radical A, which was then experienced the second oxidation to furnish sulfonyl cation B. Finally, this intermediate underwent stereoselective anti-addition to alkyne 19 to form the target product 21.
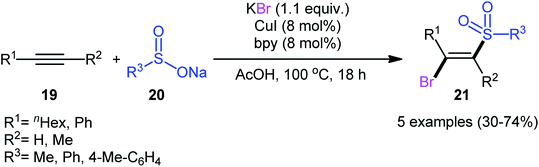 |
| | Scheme 9 Cu-catalyzed bromosulfonylation of alkynes 19 with sodium sulfinates 20 and KBr. | |
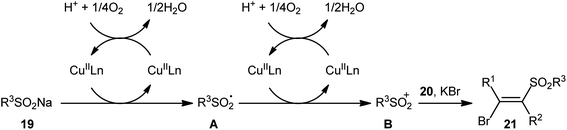 |
| | Scheme 10 Plausible reaction pathway for the formation of (E)-β-bromovinyl sulfones 21. | |
In 2013, in the same paper describing the direct chlorosulfonylation of alkynes with arylsulfonyl hydrazides as the sulfonyl precursor and FeCl3 as the chloride source in the presence of TBHP, Li and Xu reported the successful FeBr3-mediated preparation of (E)-β-bromovinyl sulfones 24 from alkynes 22 and sulfonyl hydrazides 23 (Scheme 11).29 Generally, bromosulfonylation exhibited relatively poorer reactivity compared to chlorosulfonylation. The authors speculated that the difference in the reactivity between chlorosulfonylation and bromosulfonylation might result from the easier abstraction of Br than Cl by sulfonyl radicals. Of note, the formation of both sulfonyl chloride and bromide by-products was detected in the halosulfonylation.
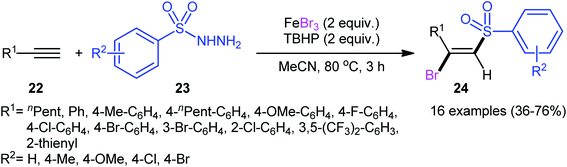 |
| | Scheme 11 Regio- and stereo-selective synthesis of (E)-β-bromovinyl sulfones 24 reported by Li and Xu. | |
Following these works, Wu and Jiang along with their co-workers reported an effective metal-free bromosulfonylation reaction of terminal alkynes 25 with sodium arylsulfinates 26.27 This difunctionalization reaction, accomplished with 1 equiv. of N-bromosuccinimide (NBS) as both a trigger and a bromine source, exhibited high reactivity and broad substrate scope with respect to both alkynes and sodium sulfinates. Twenty-one β-bromovinyl sulfones 27 were synthesized in good yields (76–88%) and excellent regio- and stereoselectives under the reaction conditions (Scheme 12). This procedure was further successfully applied to the synthesis of (E)-β-iodovinyl sulfones by using NIS instead of NBS. The authors also investigated the synthetic utility of the prepared compounds as electrophilic coupling partners in Suzuki and Sonogashira cross-coupling reactions.
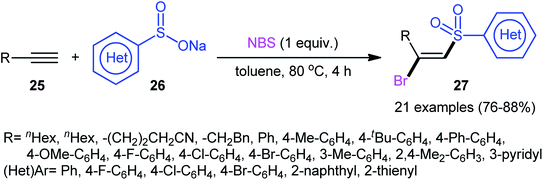 |
| | Scheme 12 Catalyst-free direct bromosulfonylation of terminal alkynes 25 with sodium arylsulfinates 26 employing NBS as the bromine source. | |
4. Iodosulfonylation
In this section we would like to focus on the direct introduction of iodine and sulfonyl groups into alkynes. Note that the section is divided into two major sub-sections according to iodosulfonylating reagents. The first includes the iodosulfonylation of alkyne substrates using bifunctional reagents, while the second contains the three-component reactions.
4.1. Two-component reactions
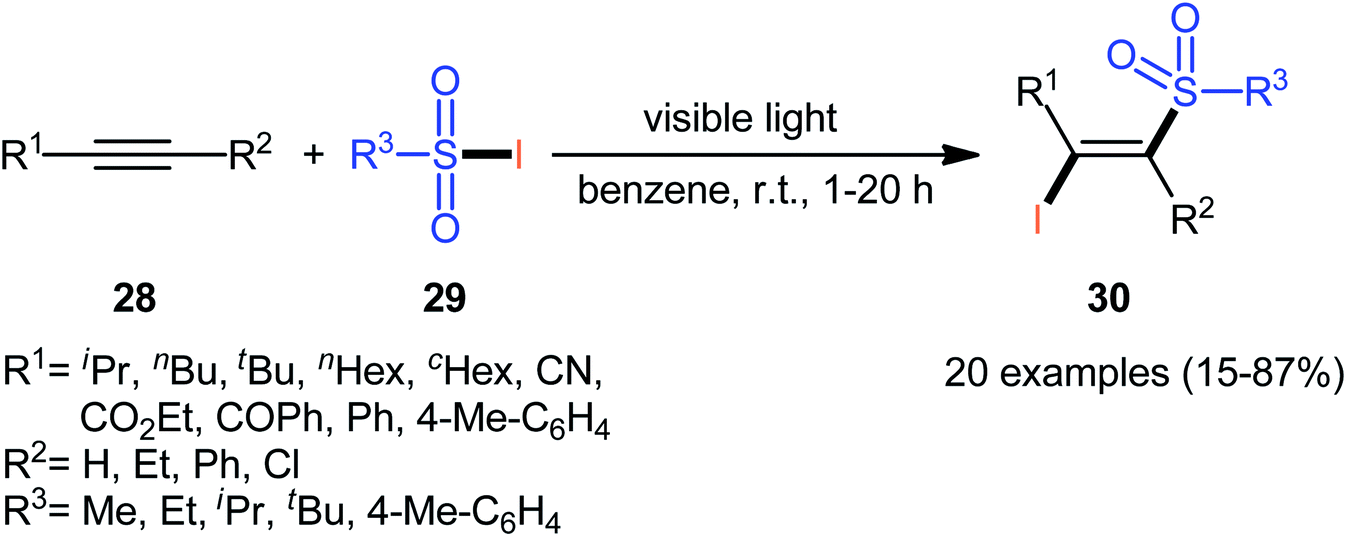
In 1971, Truce and Wolf reported the first example of the direct iodosulfonylation of alkynes 28 employing sulfonyl iodides 29 as both the iodine and sulfonyl sources.35 The reaction was run under irradiation of visible light in the absence of any catalyst or oxidant, tolerated various terminal and internal alkynes, and generally provided (E)-β-iodovinyl sulfone products 30 in relatively poor to high yields as exclusively Markovnikov regioisomers (Scheme 13). Analysis by X-ray diffraction clearly indicated that the stereochemistry of the products was E. Concerning the substrate scope, the reaction was highly dependent on the steric- and electronic-factors of both substrates. Generally, sterically less hindered sulfonyl iodides gave higher yields as compared to bulkier sulfonyl iodides. The relative reaction rates of alkynes in this transformation followed the order: terminal aromatic alkynes ≥ terminal aliphatic alkynes ≈ internal alkyl–alkyl alkynes ≫ internal aryl–aryl alkynes > propiolates > propiolonitriles. It is noteworthy that the authors applied the prepared iodovinyl sulfones as starting materials in high yielding synthesis of acetylenic sulfones via simple dehydrohalogenation by treatment with K2CO3 in methanol–water.
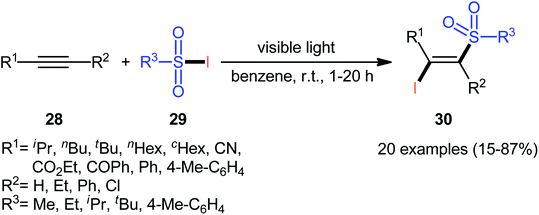 |
| | Scheme 13 Truce's synthesis of (E)-β-iodovinyl sulfones 30. | |
Sixteen years later, Corréa and co-workers reported further examples of β-iodovinyl sulfones synthesis under Truce's standard reaction condition.36 Although, the story of direct iodosulfonylation of C–C triple bonds was began by using sulfonyl iodides as bifunctional iodosulfonylating reagents, the instability of these compounds greatly limited the application of this method to construct β-iodovinyl sulfones.37
4.2. Three-component reactions
4.2.1. Sodium sulfinates as sulfonylation agents. As early as in the beginning of the 2000s, Nair and co-workers observed the formation of (E)-β-iodovinyl sulfones by cerium(IV) ammonium nitrate(CAN)-mediated direct iodosulfonylation of phenylacetylene derivatives with sodium arylsulfinates and sodium iodide.38,39 Later, Wu–Jiang's research group reported a catalyst-free iodosulfonylation reaction via three-component reaction of a small library of terminal aromatic alkynes 31, sodium p-toluenesulfinate 32, and N-iodosuccinimide (NIS).34 The reaction proceeds with excellent regio- and stereoselectivity to afford (E)-β-iodovinyl sulfones 33 in high yields, ranging from 86% to 89% (Scheme 14).
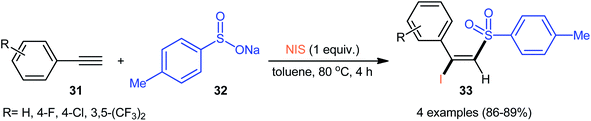 |
| | Scheme 14 Catalyst-free iodosulfonylation of terminal aromatic alkynes 31 with sodium p-toluenesulfinate 32 and NIS. | |
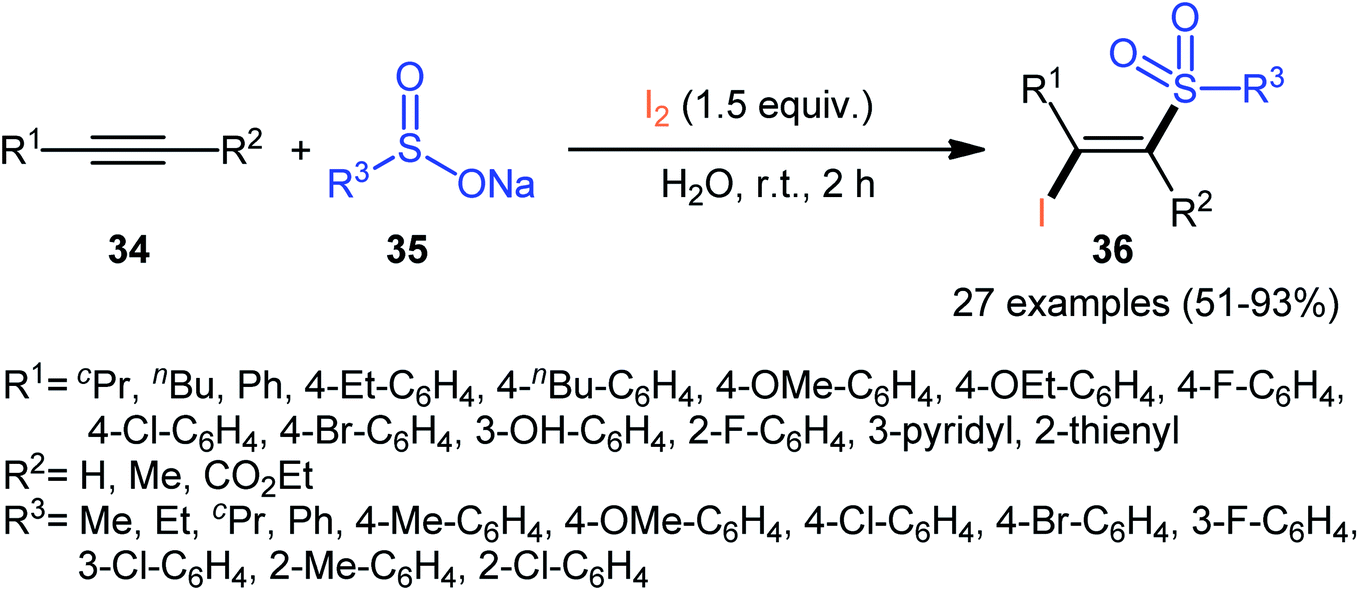
In 2017, with the objective of designing a milder and greener procedure to β-iodovinyl sulfones through iodosulfonylation of the respective alkynes, Sun and Liu along with their colleagues were able to demonstrated that a diverse range of (E)-β-iodovinyl sulfones 36 could be obtained in moderate to excellent yields from the reaction of alkynes 34 (terminal and internal) with sodium sulfinates 35 (aromatic and aliphatic) and molecular iodine in the most environmentally friendly solvent, water, at room temperature under catalyst- and additive-free conditions (Scheme 15).40 In addition, a scale-up experiment (20 mmol scale) and a few synthetic applications based on the known cross-coupling reactions highlighted the usefulness of this methodology. A few control experiments were carried out to gain mechanistic insights for this appealing reaction. With the use of the radical scavenger butylated hydroxytoluene (BHT), no prohibition of the reaction was occurred. However, the reaction was completely inhibited in the presence of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), which indicated that the reaction presumably underwent a radical pathway (Scheme 16). Initially, a sulfonyl iodide intermediate A was formed via the reaction of sulfinate sodium salt 35 with iodine. In the next step, this intermediate A underwent homolytic cleavage and a sulfonyl radical B along with an iodine radical was generated. Thereafter, the newly formed radical B was added chemoselectively to the alkyne 34 and provided the vinyl sulfone radicals C and D. Finally, electrophilic addition of I˙ the vinyl sulfone radical D gave the desired product 36. Subsequently, Bi and co-workers slightly improved the efficiency of this reaction in the term of product yields by performing the process in the presence of 2 equiv. of di-tert-butyl peroxide (DTBP) as an oxidant in the binary solvent EtOH/H2O with ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 60 °C.41
:1 at 60 °C.41
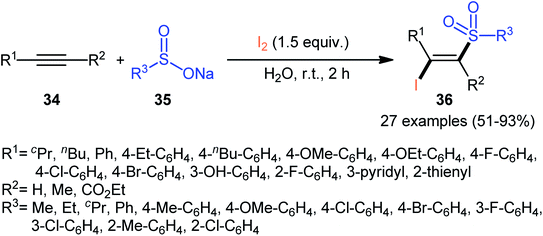 |
| | Scheme 15 Catalyst-free synthesis of (E)-β-iodovinyl sulfones 36 via the reaction of alkynes 34 with sodium sulfinates 35 and I2. | |
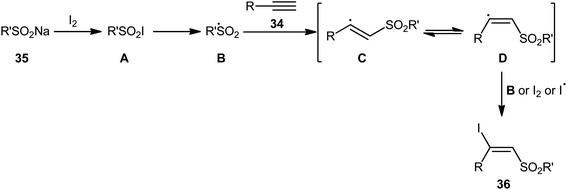 |
| | Scheme 16 A proposed pathway for the formation of (E)-β-iodovinyl sulfones 36. | |
Based on these developments, Reddy and co-workers developed a metal-free iodosulfonylation reaction of acetylenic ketones 37 using sodium sulfinates 38 and molecular iodine as the sulfonyl and iodine sources, respectively.42 In the presence of TBHP as oxidant, a wide range of internal acetylenic ketones successfully underwent the reaction to generate polyfunctional tetrasubstituted olefins 39 in moderate to excellent yields with complete regio- and stereoselectively (Scheme 17). Regarding the regioselectivity of this transformation, in all examples the sulfonyl group selectively attacked to the carbon atom adjacent to the carbonyl group and iodine placed on the carbon atom near the aryl/alkyl substituents. A wide range of functionalities both in the acetylenic ketones and sodium sulfinates were well tolerated with these reaction conditions. However, nitro-substituted sodium arylsulfinates did not respond this reaction. Recently, the Zhang–Xie group successfully extended this chemistry to propargyl alcohols using a 1:2 mixture of ClCH2CH2Cl:H2O as the reaction medium.43 Noteworthy, the reaction proceeded effectively without addition of any catalyst or oxidant, ultimately minimizing the production of waste streams.
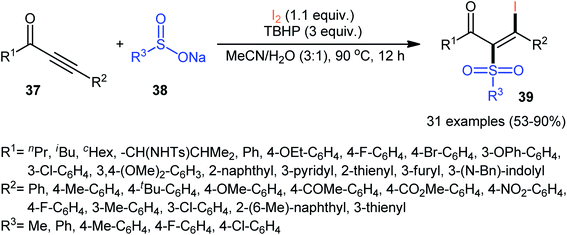 |
| | Scheme 17 Metal-free direct iodosulfonylation of acetylenic ketones 37 with sodium sulfinates 38 and I2 developed by Reddy. | |
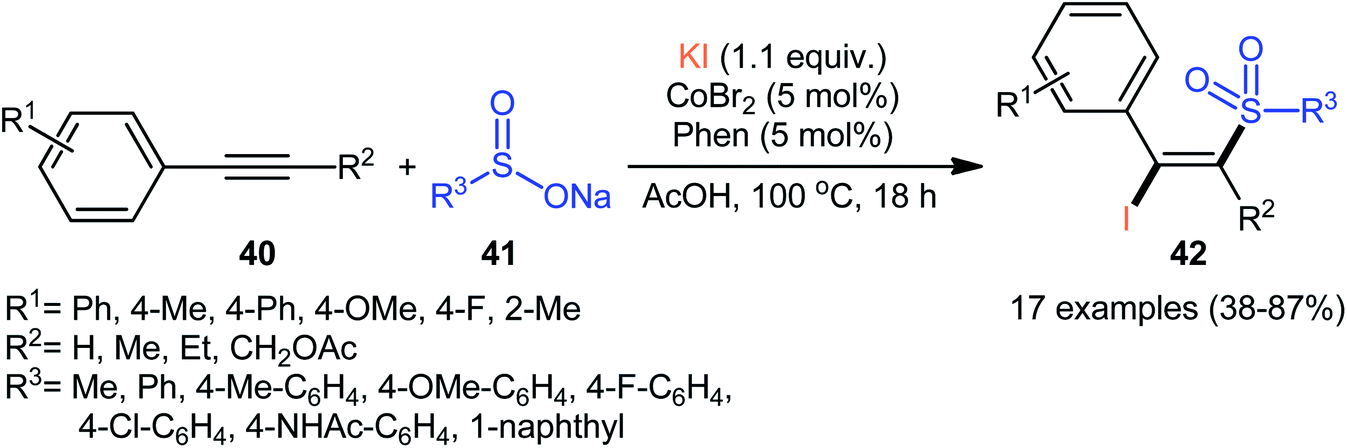
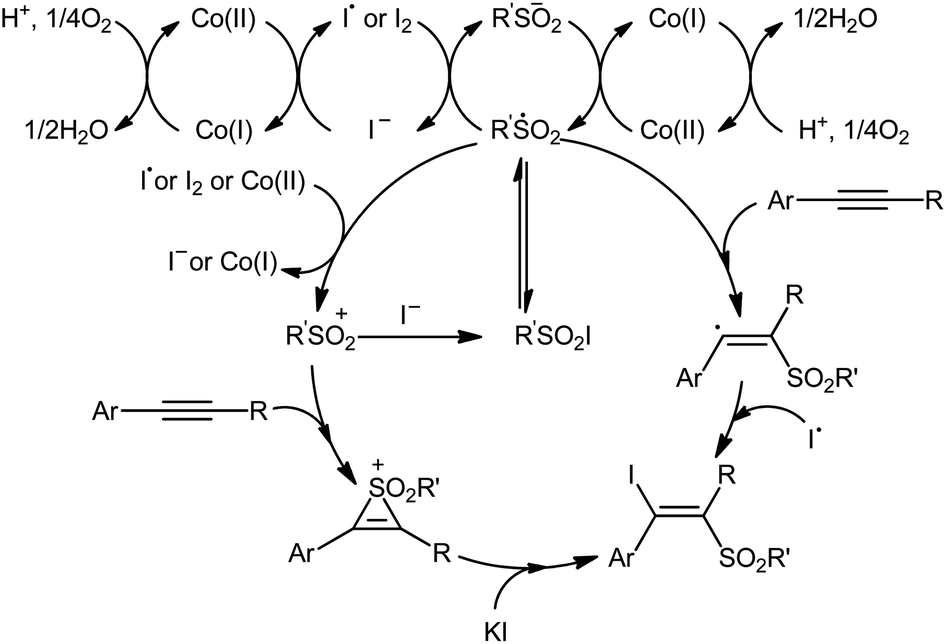
In 2018, Taniguchi reported that cobalt salts could act as efficient catalysts for iodosulfonylation reactions of alkynes with sodium sulfinates in which potassium iodide (KI) was used as iodination reagent.44 Among the various cobalt catalysts like CoCl2, CoBr2, Co(OAc)2; low-cost CoBr2 was the most efficient for the transformation. In the presence of 5 mol% of CoBr2 and 5 mol% of Phen·H2O (Phen = phenanthroline) in AcOH under air, a library of aromatic terminal and aryl–alkyl internal alkynes 40 smoothly underwent iodosulfonylation with sodium aryl/alkyl-sulfinates 41 and KI to give structurally diverse (E)-β-iodovinyl sulfones 42 in modestly to high yields (Scheme 18). It should be mentioned that iodosulfonylation of a vinylacetylene was also achieved with use of similar reaction conditions. However, this synthetic strategy was found not to be applicable to terminal alkyl and internal aryl–aryl alkynes. Interestingly, when the reaction was performed in the absence of sodium sulfinates, the corresponding diiodoalkenes were obtained in high yields. The mechanism proposed for the formation of β-iodovinyl sulfones is depicted in Scheme 19.
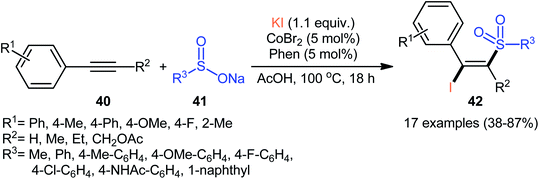 |
| | Scheme 18 Co-catalyzed iodosulfonylation of alkynes 40 with sodium sulfinates 41 and KI. | |
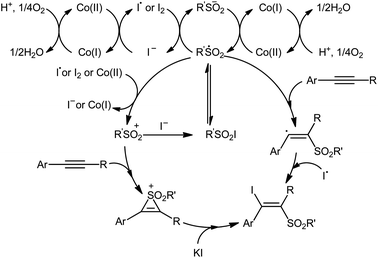 |
| | Scheme 19 The proposed mechanism for the reaction in Scheme 18. | |
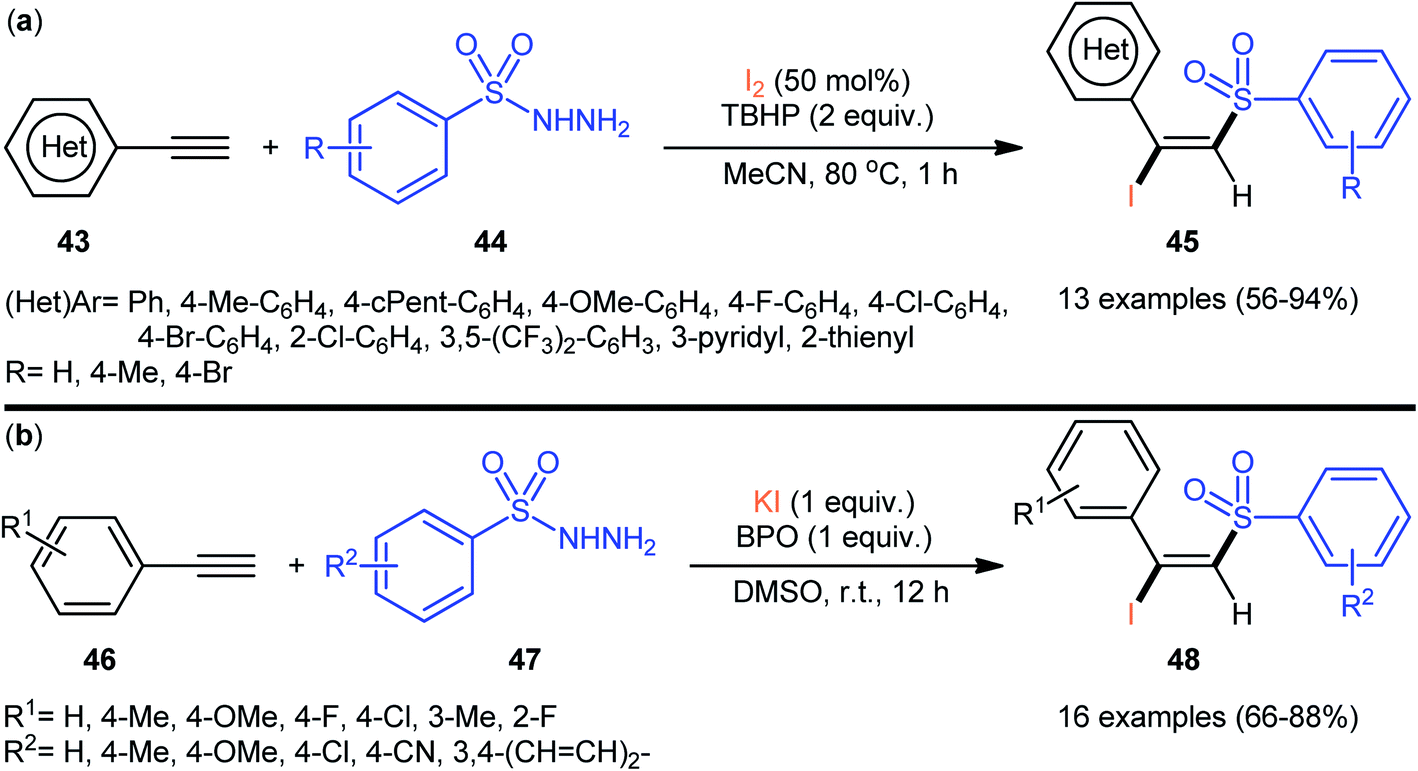
4.2.2. Sulfonyl hydrazides as sulfonylation agents. In 2013, Li–Xu's group reported the first iodosulfonylation of alkynes with sulfonyl hydrazides and molecular iodine.45 In this work, a regio- and stereoselective iodosulfonylation reaction occurs in the presence of TBHP as the oxidant. A wide range of (hetero)aromatic terminal alkynes 43 could be reacted with arylsulfonyl hydrazides 44 under the optimized conditions to give the (E)-β-iodovinyl sulfone products 45 in moderate to almost quantitative yields (Scheme 20a). Aliphatic terminal alkynes, such as hept-1-yne, could also be reacted under the identical conditions. However, the desired products were obtained in poor yields. The authors proposed a mechanism analogous to that of their chlorosulfonylation strategy. Drawing inspiration from this work, Yang and co-workers reported the synthesis of a library of (E)-β-iodovinyl sulfone products 48 shown in Scheme 20b.46 These compounds were formed by reaction of the corresponding alkynes 46 with arylsulfonyl hydrazides 47 using KI as iodine source and benzoic peroxyanhydride (BPO) as an oxidant. Subsequently, Tong et al. utilized TBAI as iodine source, and developed a K2S2O8-mediated reaction of alkynes with sulfonyl hydrazides for the stereoselective synthesis of (E)-β-iodovinyl sulfones.47
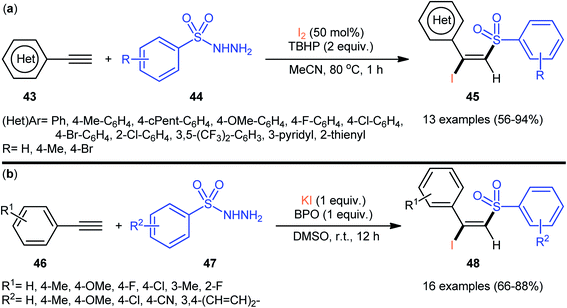 |
| | Scheme 20 (a) TBHP-mediated iodosulfonylation of terminal alkynes 43 with arylsulfonyl hydrazides 44 and I2; (b) BPO-mediated three-component reaction between alkynes 46, arylsulfonyl hydrazides 47, and KI. | |
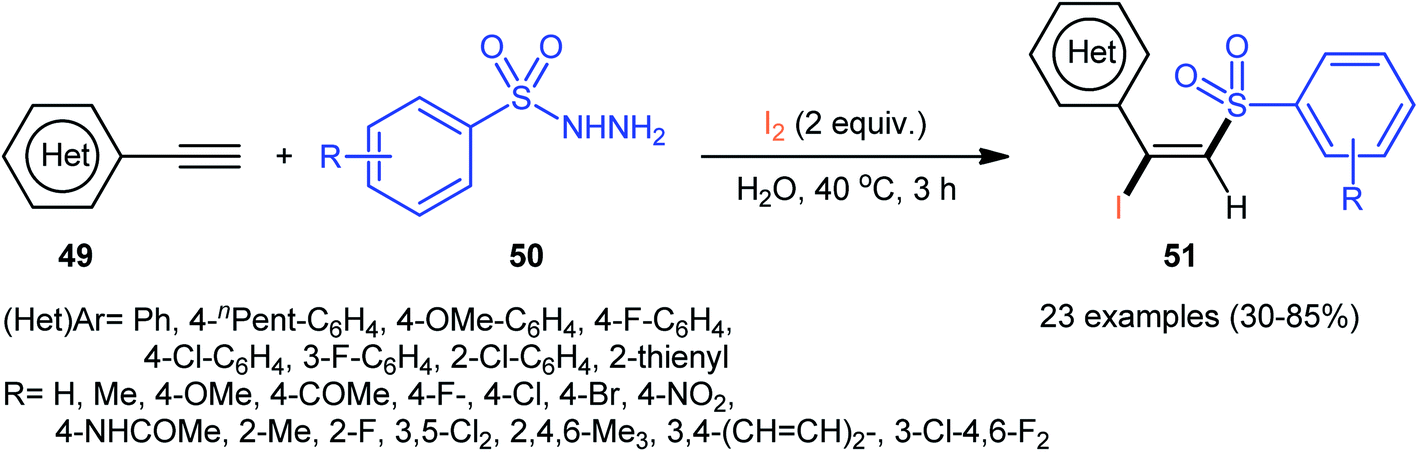
Following these works, an elegant catalyst- and additive-free reaction of preparing (E)-β-iodovinyl sulfone scaffolds 51 from terminal aromatic alkynes 49, arylsulfonyl hydrazides 50, and molecular iodine using environmentally benign water as the solvent under room temperature was demonstrated by Wang and Yan (Scheme 21).37 The results indicated that the phenylacetylenes with electron-donating substituents gave higher yields compared to the electron-withdrawing group containing phenylacetylenes. Unfortunately, the alkynes bearing nitro-substitution were incompatible in this transformation. Intriguingly, the electronic character of the substituents on the arylsulfonyl hydrazides had a little impact on the rate of this reaction. However, nitro-substituted arylsulfonyl hydrazides did not work well in the methodology. The mechanism investigation of the reaction using the radical scavengers indicated that a radical pathway could be involved. Shortly afterwards, Sun and co-workers extended the substrate scope of this iodosulfonylation reaction to internal alkynes by preforming the process in the presence of over-stoichiometric amounts of H2O2 using KI as iodine source.48 Interestingly, when the same reaction was performed in 2,2,2-trifluoroethanol (TFE), aggregation induced-emission (AIE)-active benzo[b]thiophene-1,1-dioxides were obtained instead of the desired iodovinyl sulfones through intra-molecular cyclization of the vinyl radical intermediate (generated from the addition of sulfonyl radical to alkyne). Along this line, Wei and Wang along with their colleagues demonstrated the synthesis of multisubstituted α,β-enones through the direct selective iodosulfonylation of alkylynones with arylsulfonyl hydrazides and iodine pentoxide (I2O5).49
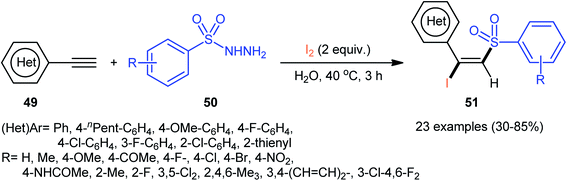 |
| | Scheme 21 Catalyst-free iodosulfonylation of alkynes 49 with arylsulfonyl hydrazides 50 and I2 in water. | |
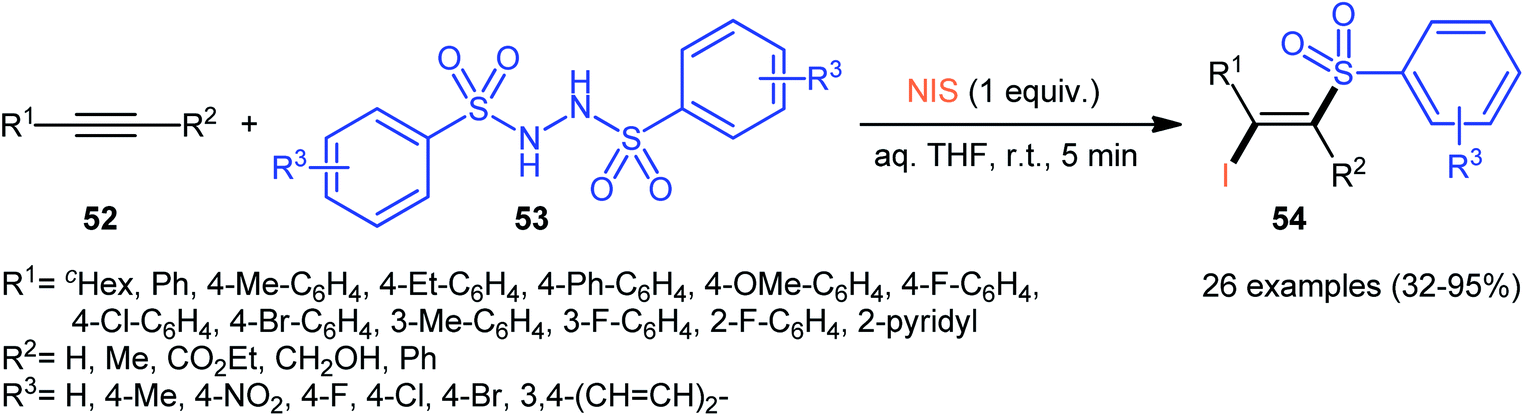
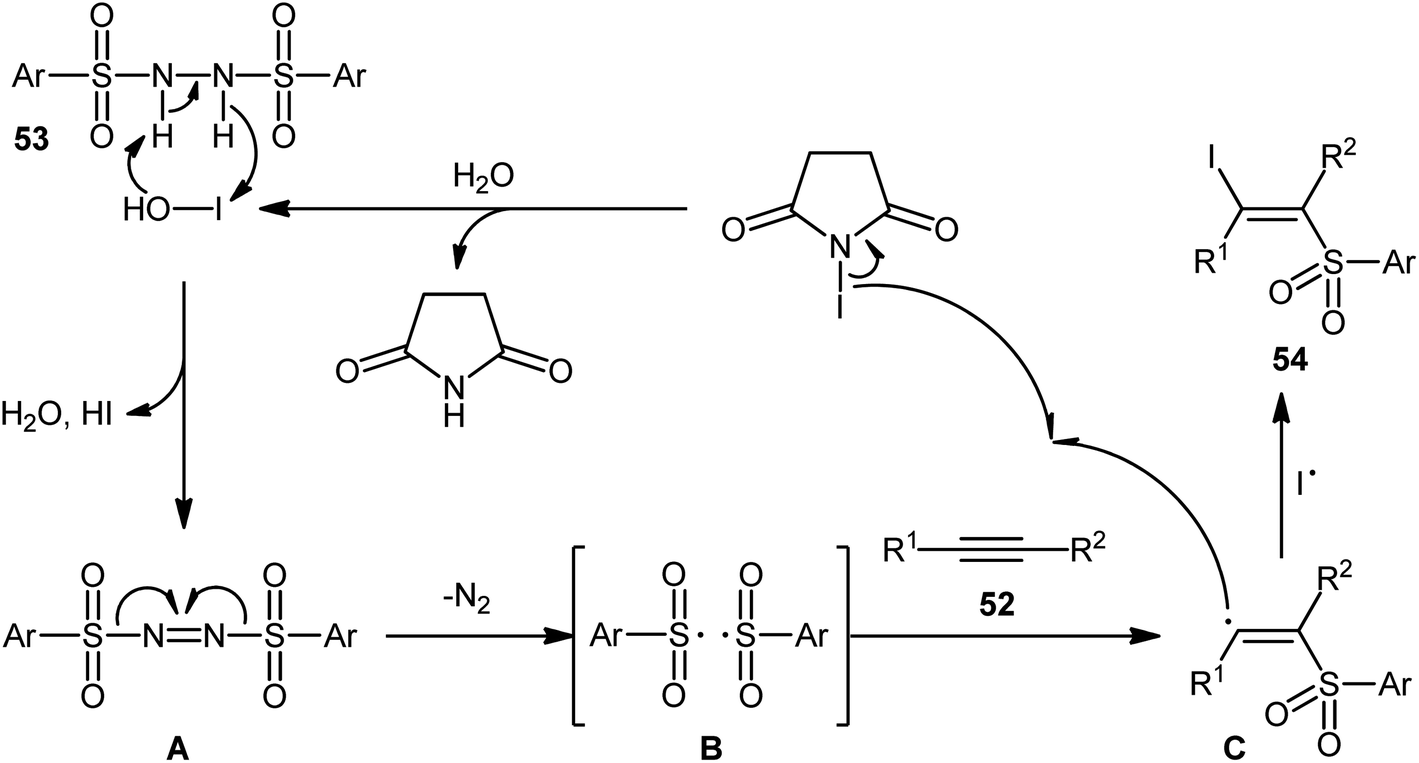
Very recently, the group of Wang and Hu informed for the first time the usefulness of N,N′-disulfonylhydrazines as novel sulfonylating reagents for halosulfonylation reactions.50 Thus, in the absence of any catalyst or additive in aqueous THF at room temperature, the reaction of various terminal and internal alkynes 52 with N,N′-disulfonylhydrazines 53 and NIS furnished the corresponding (E)-β-iodovinyl sulfones 54 in moderate to excellent yields within only 5 min (Scheme 22). Indeed, NIS played a dual role in this transformation; the iodine source and the oxidant. Mechanistically, the reaction starts with the formation of HOI oxidant through the hydrolysis of NIS by H2O, which subsequently oxidizes N,N′-disulfonyl-hydrazine 53 into the corresponding azo-intermediate A. Next, this intermediate undergoes a spontaneous decomposition to give two sulfonyl radicals B. Thereafter, the newly formed radical selectively attacks to the less hindered end of alkyne 52 to yield a vinyl radical C that, after getting an iodine radical from NIS affords the expected products 54 (Scheme 23).
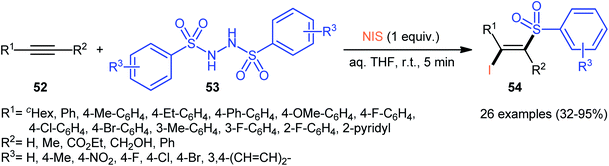 |
| | Scheme 22 Synthesis of (E)-β-iodovinyl sulfones 54 via the reaction of alkynes 52 with N,N′-disulfonylhydrazines 53 and NIS. | |
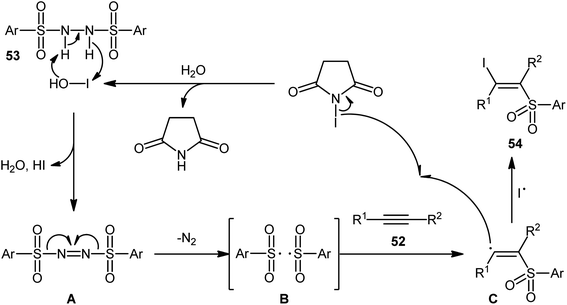 |
| | Scheme 23 Mechanism that accounts for the generation of (E)-β-iodovinyl sulfones 54. | |
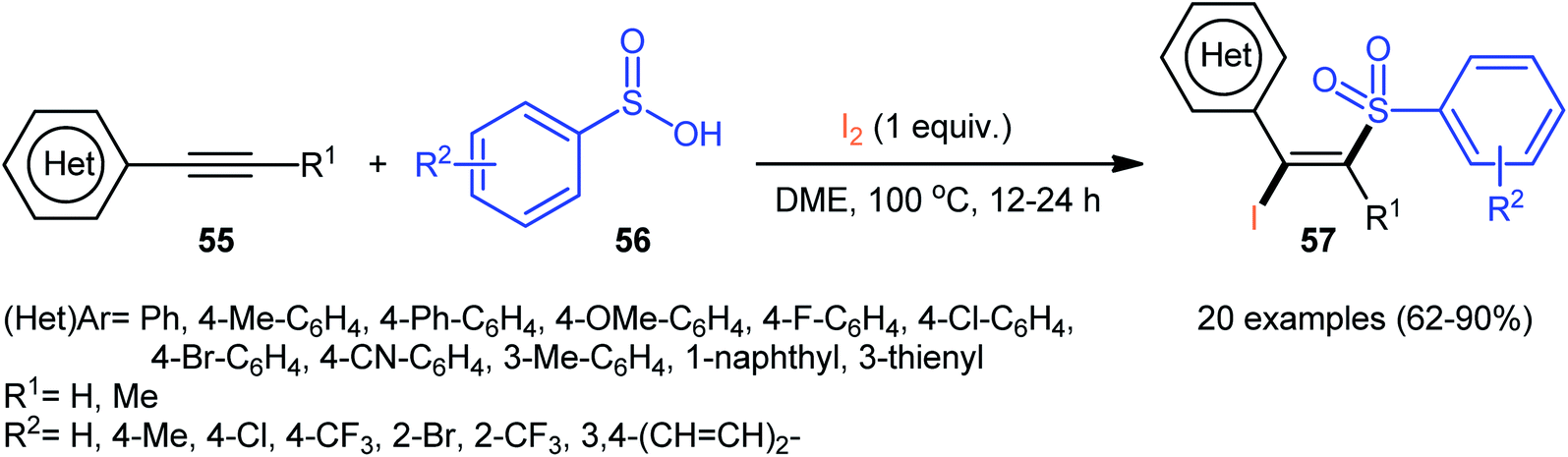
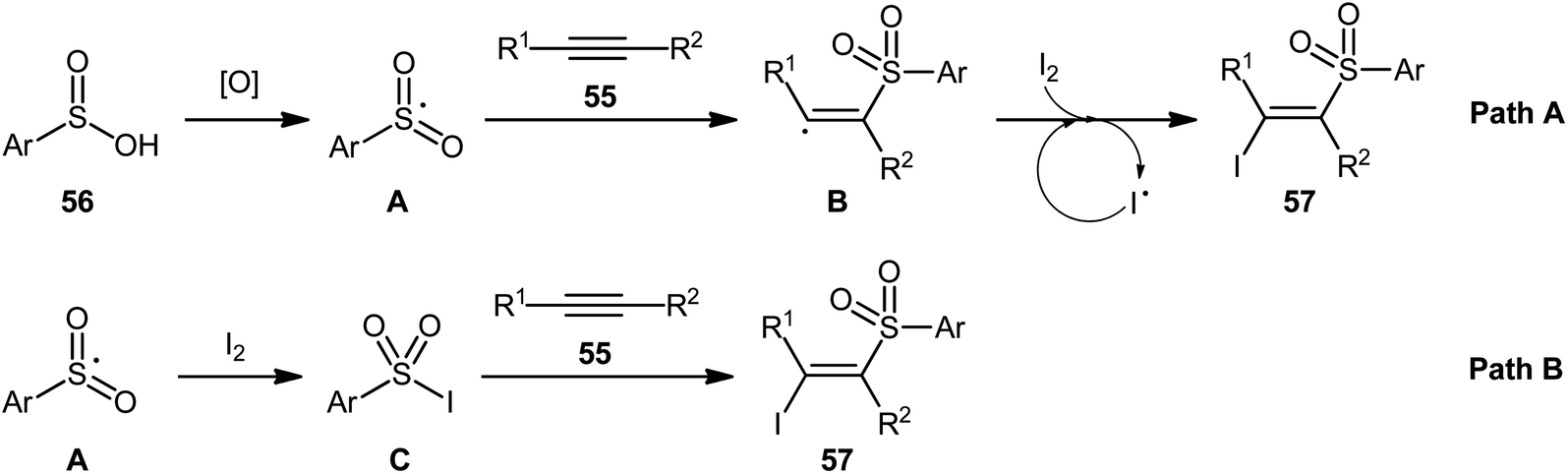
4.2.3. Miscellaneous reactions. In 2015, the Wang group realized that sulfinic acids could serve as good sulfonyl sources in halosulfonylation reactions of alkynes.51 They showed that three-component reaction between aromatic alkynes 55, sulfinic acids 56, and molecular iodine in the absence of any catalyst or additive formed a series corresponding (E)-β-iodovinyl sulfones 57 in good to excellent yields with outstanding stereo- and regio-selectivities (Scheme 24). Of note, propiolates could also be participated in this reaction, albeit with lower efficiencies. However, when 1-hexyne (an aliphatic alkyne) was employed under the identical reaction conditions, the desired product was isolated obtained in only 12% yield. Interestingly, when the reactions were performed in the presence of stoichiometric amounts of K2CO3, the corresponding acetylenic sulfones were obtained in high yields. Based on a series of control experiments, a plausible mechanism was suggested for the formation of (E)-β-iodovinyl sulfones, as depicted in Scheme 25. Initially, the sulfinic acid 56 was oxidized by air oxygen to form the sulfonyl radical A, followed by addition to alkyne 55 to generate the alkenyl radical B. Finally, the interaction of intermediate B with molecular iodine delivered the desired β-iodovinyl sulfone 57 (Scheme 25, path a). In another possibility, the direct iodosulfonylation of alkyne 55 with in situ generated sulfonyl iodides C (from sulfinic acid 56 and molecular iodine) led to the formation of the target products 57 (Scheme 25, path b).
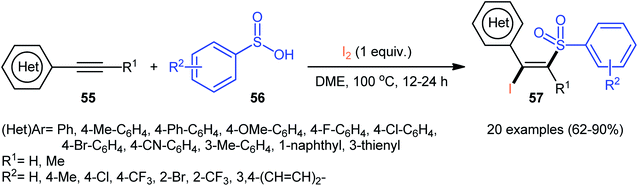 |
| | Scheme 24 Wang's synthesis of (E)-β-iodovinyl sulfones 57. | |
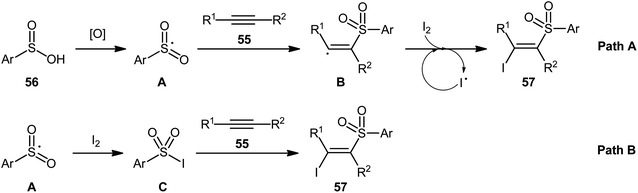 |
| | Scheme 25 Possible reaction pathway for the formation (E)-β-iodovinyl sulfones 57. | |
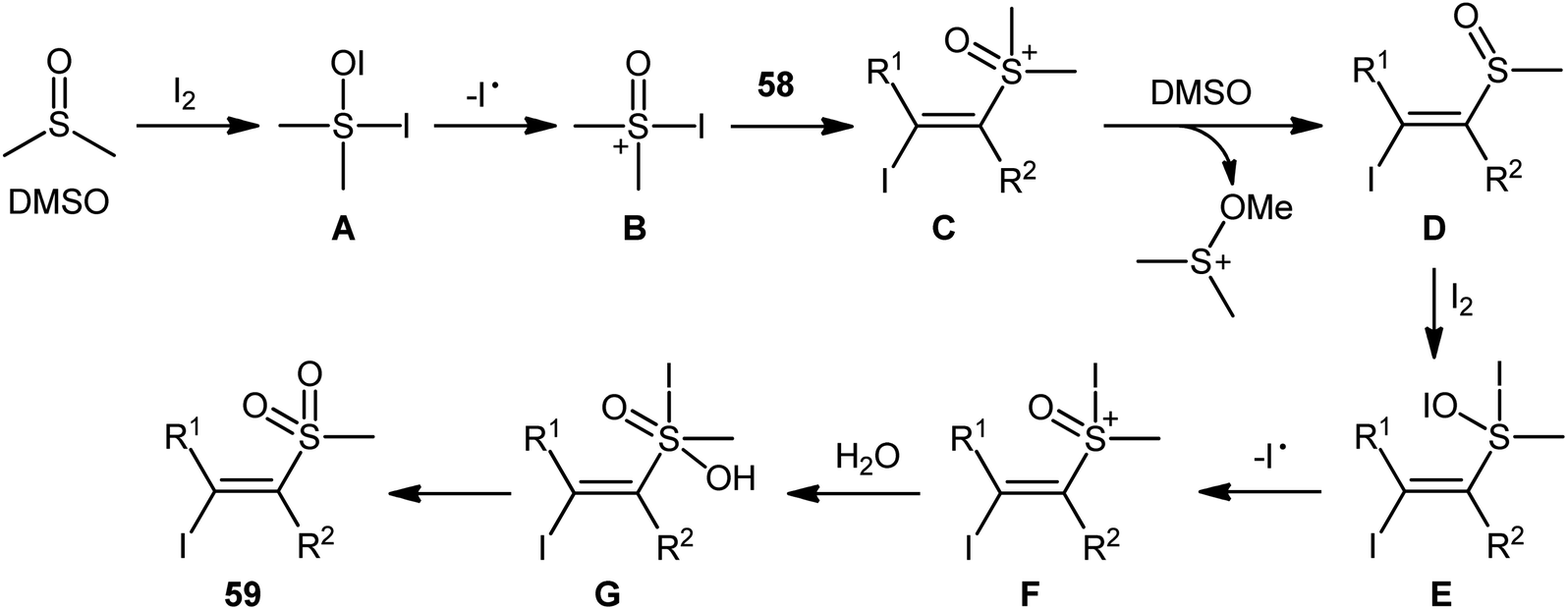
Recently, in a significant contribution in this field, Pan and Liu developed an efficient four-component synthesis of (E)-α-iodo-β-methylsulfonylalkenes 59 from alkynes 58, dimethyl sulfoxide (DMSO), molecular iodine and water without any added catalyst and solvent (Scheme 26).52 This eco-friendly synthetic strategy is distinguished by its generality, accommodating aliphatic and aromatic terminal alkynes, and even internal alkyl–alkyl alkynes in the presence of various sensitive functional groups (e.g., F, Cl, CF3, CN). A possible mechanism for this transformation was proposed by the authors, as shown in Scheme 27.
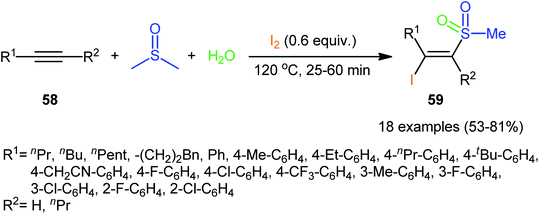 |
| | Scheme 26 Catalyst-free four-component synthesis of (E)-α-iodo-β-methylsulfonylalkenes 59 from alkynes 58, DMSO, I2, and H2O. | |
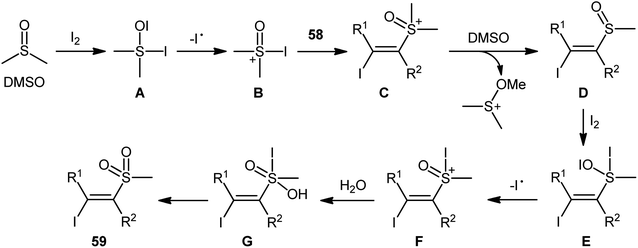 |
| | Scheme 27 Possible pathway of (E)-α-iodo-β-methylsulfonylalkenes 59 formation. | |
Finally, it should be noted that transition metals play main role in direct halosulfonylation of alkynes.53–61
5. Conclusion
Difunctionalization of alkyne feedstocks is a specific class of important synthetic transformations that allow for the buildup of molecular complexity within a single click.62 In this context, the direct halosulfonylation of alkynes which provide a powerful strategy for the preparation of β-halovinyl sulfone derivatives has devoted a great deal of attention. As illustrated, most of the halosulfonylation reaction of alkynes were performed under catalyst- and additive-free conditions. Despite the remarkable progress that has been achieved in this interesting research arena, some challenging problems remain unsolved yet. For example: (i) generally, the regioselectivity of reactions is dominated by the Markovnikov-type products. Thus, many more studies are further needed to development of efficient procedures that allow halosulfonylation reactions in an anti-Markovnikov-type manner; (ii) the number of reported examples in bromosulfonylation reactions are very narrow and, of course, there is a need to study the scope and limitations of these reactions; and (iii) fluorosulfonylation of alkynes should be explored.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was sponsored by Hanshan Normal University.
References
-
(a) D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793–814 CrossRef CAS PubMed
 ;
(b) M. A. Alam, K. Shimada, A. Jahan, M. W. Khan, M. M. Bhuiyan, M. S. Alam and M. M. Matin, Nat. Prod. Chem. Res., 2018, 6, 4–11 Search PubMed ;
(c) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed .
;
(b) M. A. Alam, K. Shimada, A. Jahan, M. W. Khan, M. M. Bhuiyan, M. S. Alam and M. M. Matin, Nat. Prod. Chem. Res., 2018, 6, 4–11 Search PubMed ;
(c) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed . - K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2019, 376, 5 CrossRef .
-
(a) N. W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 48, 1939–1973 CrossRef CAS ;
(b) A. N. R. Alba, X. Companyó and R. Rios, Chem. Soc. Rev., 2010, 39, 2018–2033 RSC ;
(c) E. N. Prilezhaeva, Russ. Chem. Rev., 2000, 69, 367–408 CrossRef CAS .
-
(a) K. M. Short and C. B. Ziegler Jr, Tetrahedron Lett., 1995, 36, 355–356 CrossRef CAS ;
(b) R. J. Reddy, J. J. Kumar, A. H. Kumari and G. R. Krishna, Adv. Synth. Catal., 2020, 362, 1317–1322 CrossRef CAS .
-
(a) R. J. Reddy, J. J. Kumar and A. H. Kumari, Eur. J. Org. Chem., 2019, 3771–3775 CrossRef CAS ;
(b) S. H. Suzol, A. H. Howlader, Z. Wen, Y. Ren, E. E. Laverde, C. Garcia, Y. Liu and S. F. Wnuk, ACS Omega, 2018, 3, 4276–4288 CrossRef CAS .
- Y. Liang, S. H. Suzol, Z. Wen, A. G. Artiles, L. Mathivathanan, R. G. Raptis and S. F. Wnuk, Org. Lett., 2016, 18, 1418–1421 CrossRef CAS .
- X. X. Gu, M. H. Xie, X. Y. Zhao, Y. Sun, W. Zhang, F. D. Xie and S. W. Wang, Chin. J. Chem., 2008, 26, 1625–1629 CrossRef CAS .
- N. Iwata, T. Morioka, T. Kobayashi, T. Asada, H. Kinoshita and K. Inomata, Bull. Chem. Soc. Jpn., 1992, 65, 1379–1388 CrossRef CAS .
- J. R. Bull, N. S. Desmond-Smith, S. J. Heggie, R. Hunter and F. C. Tien, Synlett, 1998, 900–902 CrossRef CAS .
-
(a) X. Huang, D. Duan and W. Zheng, J. Org. Chem., 2003, 68, 1958–1963 CrossRef CAS PubMed ;
(b) R. R. Wolff, V. Basava, R. M. Giuliano, W. J. Boyko and J. H. Schauble, Can. J. Chem., 2006, 84, 667–675 CrossRef CAS .
- Y. Fang, Z. Luo and X. Xu, RSC Adv., 2016, 6, 59661–59676 RSC .
- Selected reviews:
(a) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 1–32 CrossRef ;
(b) X. G. Liang, J. S. Hua, S. Xiangdong and F. Behmagham, J. Fluorine Chem., 2020, 235, 109524 CrossRef ;
(c) A. Bakhtiary, M. R. P. Heravi, A. Hassanpour, I. Amini and E. Vessally, RSC Adv., 2021, 11, 470–483 RSC ;
(d) M. R. J. Sarvestani, N. Mert, P. Charehjou and E. Vessally, J. Chem. Lett., 2020, 1, 93–102 Search PubMed ;
(e) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed ;
(f) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed .
- Z. Liu, A. Ebadi, M. Toughani, N. Mert and E. Vessally, RSC Adv., 2020, 10, 37299–37313 RSC .
- A. Hassanpour, M. R. P. Heravi, A. Ebadi, A. Hosseinian and E. Vessally, J. Fluorine Chem., 2021, 245, 109762 CrossRef CAS .
- W. Xu, D. Guo, A. G. Ebadi, M. Toughani and E. Vessally, J. CO2 Util., 2021, 45, 101403 CrossRef CAS .
- X. Wang, W. Ping, A. G. Ebadi, S. Majedi, Z. Hossaini and M. Toughani, J. CO2 Util., 2021, 50, 101592 CrossRef CAS .
- X. Ma, Z. Kexin, W. Yonggang, A. G. Ebadi and M. Toughani, Iran. J. Chem. Chem. Eng., 2021, 40, 1364–1374 Search PubMed .
- S. Ahmadi, A. Hosseinian, P. D. Kheirollahi Nezhad, A. Monfared and E. Vessally, Iran. J. Chem. Chem. Eng., 2019, 38, 1–19 CAS .
- E. Vessally, S. Mohammadi, M. Abdoli, A. Hosseinian and P. Ojaghloo, Iran. J. Chem. Chem. Eng., 2020, 39, 11–19 CAS .
- Y. Liu, A. G. Ebadi, L. Youseftabar-Miri, A. Hassanpour and E. Vessally, RSC Adv., 2019, 9, 25199–25215 RSC .
- Y. Amiel, J. Org. Chem., 1971, 36, 3697–3702 CrossRef CAS .
- Y. Amiel, Tetrahedron Lett., 1971, 12, 661–663 CrossRef .
- X. Liu, X. Duan, Z. Pan, Y. Han and Y. Liang, Synlett, 2005, 1752–1754 CAS .
- X. Zeng, L. Ilies and E. Nakamura, Org. Lett., 2012, 14, 954–956 CrossRef CAS PubMed .
- L. Wang, H. Zhu, J. Che, Y. Yang and G. Zhu, Tetrahedron Lett., 2014, 55, 1011–1013 CrossRef CAS .
- P. Chakrasali, K. Kim, Y. S. Jung, H. Kim and S. B. Han, Org. Lett., 2018, 20, 7509–7513 CrossRef CAS .
- A. Hossain, S. Engl, E. Lutsker and O. Reiser, ACS Catal., 2018, 9, 1103–1109 CrossRef .
- M. Alkan-Zambada and X. Hu, J. Org. Chem., 2019, 84, 4525–4533 CrossRef CAS PubMed .
- X. Li, X. Shi, M. Fang and X. Xu, J. Org. Chem., 2013, 78, 9499–9504 CrossRef CAS .
- K. Zeng, L. Chen, Y. Chen, Y. Liu, Y. Zhou, C. T. Au and S. F. Yin, Adv. Synth. Catal., 2017, 359, 841–847 CrossRef CAS .
- Y. Amiel, J. Org. Chem., 1974, 39, 3867–3870 CrossRef CAS .
- V. Vasin, I. Bolusheva and V. Razin, Sulfur Lett., 2003, 26, 101–107 CrossRef CAS .
- N. Taniguchi, Synlett, 2011, 1308–1312 CrossRef CAS .
- Y. Gao, W. Wu, Y. Huang, K. Huang and H. Jiang, Org. Chem. Front., 2014, 1, 361–364 RSC .
- W. E. Truce and G. C. Wolf, J. Org. Chem., 1971, 36, 1727–1732 CrossRef CAS .
- C. M. da Silva Corrêa and M. D. C. Fleming, J. Chem. Soc., Perkin Trans. 2, 1987, 103–107 RSC .
- Y. Hou, L. Zhu, H. Hu, S. Chen, Z. Li, Y. Liu and P. Gong, New J. Chem., 2018, 42, 8752–8755 RSC .
- V. Nair, A. Augustine, T. G. George and L. G. Nair, Tetrahedron Lett., 2001, 42, 6763–6765 CrossRef CAS .
- V. Nair, A. Augustine and T. D. Suja, Synthesis, 2002, 2259–2265 CrossRef CAS .
- Y. Sun, A. Abdukader, D. Lu, H. Zhang and C. Liu, Green Chem., 2017, 19, 1255–1258 RSC .
- W. Bi, C. Ren, L. Jia, X. Xia, X. Chen, X. Chen and Y. Zhao, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 391–396 CrossRef CAS .
- R. Kumar, V. Dwivedi and M. S. Reddy, Adv. Synth. Catal., 2017, 359, 2847–2856 CrossRef CAS .
- J. Zhang, Z. Liang, J. Wang, Z. Guo, C. Liu and M. Xie, ACS Omega, 2018, 3, 18002–18015 CrossRef CAS PubMed .
- N. Taniguchi, Tetrahedron, 2018, 74, 1454–1460 CrossRef CAS .
- X. Li, X. Xu and X. Shi, Tetrahedron Lett., 2013, 54, 3071–3074 CrossRef CAS .
- L. Yang, D. Hu, L. Wei and Y. Liu, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 1301–1304 CrossRef CAS .
- C. Tong, B. Gan, Y. Yan and Y. Y. Xie, Synth. Commun., 2017, 47, 1927–1933 CrossRef CAS .
- Y. Ma, K. Wang, D. Zhang and P. Sun, Adv. Synth. Catal., 2019, 361, 597–602 CrossRef CAS .
- H. H. Cui, C. L. He, D. S. Yang, H. L. Yue, W. Wei and H. Wang, Synlett, 2018, 29, 830–834 CrossRef CAS .
- D. Luo, L. Min, W. Zheng, L. Shan, X. Wang and Y. Hu, Chin. Chem. Lett., 2020, 31, 1877–1880 CrossRef CAS .
- W. Wei, J. Wen, D. Yang, H. Jing, J. You and H. Wang, RSC Adv., 2015, 5, 4416–4419 RSC .
- P. Zhou, Y. Pan, H. Tan and W. Liu, J. Org. Chem., 2019, 84, 15662–15668 CrossRef CAS .
- Q. Hu, W. Zhang, Q. Yin, Y. Wang and H. Wang, Spectrochim. Acta, Part A, 2021, 244, 118864 CrossRef CAS PubMed .
- H. Zhang, W. Guan, L. Zhang, X. Guan and S. Wang, ACS Omega, 2020, 5, 18007–18012 CrossRef CAS PubMed .
- L. Zhang, M. Zhang, S. You, D. Ma, J. Zhao and Z. Chen, Sci. Total Environ., 2021, 780, 146505 CrossRef CAS PubMed .
- M. Zou, Y. Qi, R. Qu, G. Al-Basher, X. Pan, Z. Wang and F. Zhu, Sci. Total Environ., 2021, 771, 144743 CrossRef CAS PubMed .
- W. Cao, N. Wu, R. Qu, R. C. Sun, Z. Huo, J. S. Ajarem and F. Zhu, Environ. Sci. Pollut. Res., 2021, 28, 31301–31311 CrossRef CAS PubMed .
- Z. Chen, H. Zhang, X. He, G. Fan, X. Li, Z. He, Z. He, G. Wang and L. Zhang, BioResources, 2021, 16, 2644–2654 CAS .
- Z. Farasat, R. Panahi and B. Mokhtarani, Water Conservation and Management, 2017, 1, 7–9 CrossRef .
- H. Zhang, M. Sun, L. Song, J. Guo and L. Zhang, Biochem. Eng. J., 2019, 147, 146–152 CrossRef CAS .
- W. Zhang, Y. Hu, J. Liu, H. Wang, J. Wei, P. Sun, L. Wu and H. Zheng, Saudi J. Biol. Sci., 2020, 27, 1667–1673 CrossRef CAS PubMed .
-
(a) H. Yao, W. Hu and W. Zhang, Molecules, 2021, 26, 105 CrossRef CAS PubMed ;
(b) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Chin. J. Chem., 2019, 37, 292–301 CAS .
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b
b