
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling ultrasmall gold nanoparticles with atomic precision
Nan
Xia
ab and
Zhikun
Wu
 *ab
*ab
aKey Laboratory of Materials Physics, Anhui Key Laboratory of Nanomaterials and Nanostructures, Institute of Solid State Physics, Chinese Academy of Sciences, Hefei 230031, P. R. China. E-mail: zkwu@issp.ac.cn
bInstitute of Physical Science and Information Technology, Anhui University, Hefei 230601, P. R. China
First published on 22nd December 2020
Abstract
Gold nanoparticles are probably the nanoparticles that have been best studied for the longest time due to their stability, physicochemical properties and applications. Controlling gold nanoparticles with atomic precision is of significance for subsequent research on their structures, properties and applications, which is a dream that has been pursued for many years since ruby gold was first obtained by Faraday in 1857. Fortunately, this dream has recently been partially realized for some ultrasmall gold nanoparticles (nanoclusters). However, rationally designing and synthesizing gold nanoparticles with atomic precision are still distant goals, and this challenge might rely primarily on rich atomically precise gold nanoparticle libraries and the in-depth understanding of metal nanoparticle chemistry. Herein, we review general synthesis strategies and some facile synthesis methods, with an emphasis on the controlling parameters determined from well-documented results, which might have important implications for future nanoparticle synthesis with atomic precision and facilitate related research and applications.
1. Introduction
Gold is a noble element with chemical inertness and enduring physical qualities, even after exposure to extreme conditions for tens or hundreds of years. Long after the discovery of gold in Bulgaria five thousand years ago, ancient Egypt and China started using colloidal gold for therapeutic and decorative purposes around the 5th or 4th century B.C.1–3 The scientific realization of such colloidal gold can be traced back to Michael Faraday's research in 1857 when he successfully prepared a ruby-red gold colloid through the reduction method.4 After a great deal of experimental and theoretical research was performed in the following 150 years, gold colloids (gold nanoparticles) were found to not only be beautiful but also to exhibit rich properties such as optics, sensing, catalysis, and biomedicine properties.1–3,5–7The properties of gold nanoparticles are often related to a set of physical parameters that may include the size, shape, composition, and structure.8,9 Over the past few decades, extensive efforts have been devoted to modulating gold nanoparticles to control their morphologies and compositions. Despite narrow dispersion in continuously improved synthesis, gold nanoparticles are still not atomically precise due to their polydisperse sizes and indefinable surface architecture. Controlling gold nanoparticles with atomic precision can be of great utility not only in tuning their properties accurately but also in obtaining an atomic-level understanding of their structure–property correlations (e.g. structure–catalytic activity relationships10). It has long been a major dream of nanochemists to prepare atomically precise gold nanoparticles, which was realized by the emergence of gold nanoclusters.
Gold nanoclusters (Au NCs), or ultrasmall gold nanoparticles, are aggregates of certain gold atoms protected by ligands with precise molecular formulae. Au NCs can be rigorously categorized as a branch of the broader class of gold nanoparticles and provide an ideal platform for the pursuit of atomically precise control of gold nanoparticles. In comparison with previously studied gold nanoparticles, Au NCs exhibit intriguing properties, such as discrete electronic energy levels, quantized charging, intrinsic chirality, strong luminescence, and excellent catalytic selectivity.11–13 The architecture of Au NCs usually consists of various numbers of metal atoms, types of metal atoms and ligands, surface bonding (staples), packing styles of metal atoms, etc. Thus, unique molecular-like structures and properties render Au NCs sensitive to one or a few atomic alterations and accessible for control by methods such as chemists modifying organic molecules through various synthetic methods.14–18 Nevertheless, rationally designing and synthesizing Au NCs are still challenging due to the limitation of synthesis methods and NC chemistry understanding.
This review aims to provide a comprehensive summary of the recent progress in precisely controlling Au NCs at the atomic level based on some existing synthesis methods, mainly including gold salt (complex) reduction, ligand induction, and antigalvanic reduction, among others. We will highlight the controlling parameters of how to influence the structures and properties of the resulting Au NCs. Finally, we conclude by presenting our perspective and suggestions for the further development of controlling Au NCs with atomic precision.
2. Synthesis strategy
Understanding the fundamentals of the underlying reaction process is extremely important to control Au NCs with atomic precision, which has attracted extensive attention in recent years. For example, Hainfeld et al. reported the preparation of size-controllable gold nanoparticles (2–6 nm) by tuning the polymeric Au(I) thiolate precursor size with pH control.19 Subsequently, Jin et al. achieved the controllable synthesis of NCs by tuning the kinetics of the formation of Au(I):SR intermediates, leading to the formation of Au25 NCs with high yield.20 They found that Au(I):SR intermediates formed at 0 °C, which, under very slow stirring conditions, were distributed in a narrow range of 100–400 nm, whereas those obtained at room temperature had a multimodal distribution with a broader range of 1–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nm. Soon afterwards, an important process – “size focusing’‘ – was revealed in the growth of Au25 by Jin and coworkers.21,22 This aging stage involves a slow conversion process of the initially formed mixture of differently sized Aun clusters into uniform Au25. Then, motivated by the formation of Au19 NCs using a weak reducing agent (borane tert-butylamine complex) instead of the strong reducing NaBH4 agent, Wu and Jin proposed a general strategy for the synthesis of Au NCs, involving kinetic control and thermodynamic selection processes (Scheme 1),23 which can be detailed as (i) the kinetic control of the initial size distribution of crude NCs by controlling the reduction speed, the ratio of Au:SR, and other parameters; and (ii) thermodynamic selection (or size focusing) during the aging process by controlling the aging time, temperature, and other parameters, leading to robust (thermodynamically favorable) cluster species. Xie et al. also demonstrated such a strategy in the synthesis of water-soluble Au25 in aqueous solution by using electrospray ionization mass spectrometry (ESI-MS) to monitor the reaction process. They adopted the NaOH-mediated NaBH4 reduction method to tune the formation kinetics of Au NCs and accelerate the thermodynamic selection of Au25.24 Another work by them is the usage of CO as a reducing agent that kinetically controls crude clusters, and Au25 NCs were also obtained after thermodynamically controlled size focusing.25 Although various synthetic methods have been developed in the last decade, the formation of atomically precise (narrow-sized) Au NCs can be mainly illustrated by kinetic control and thermodynamic selection (not limited to the gold salt (complex) reduction method (vide infra)).
000 nm. Soon afterwards, an important process – “size focusing’‘ – was revealed in the growth of Au25 by Jin and coworkers.21,22 This aging stage involves a slow conversion process of the initially formed mixture of differently sized Aun clusters into uniform Au25. Then, motivated by the formation of Au19 NCs using a weak reducing agent (borane tert-butylamine complex) instead of the strong reducing NaBH4 agent, Wu and Jin proposed a general strategy for the synthesis of Au NCs, involving kinetic control and thermodynamic selection processes (Scheme 1),23 which can be detailed as (i) the kinetic control of the initial size distribution of crude NCs by controlling the reduction speed, the ratio of Au:SR, and other parameters; and (ii) thermodynamic selection (or size focusing) during the aging process by controlling the aging time, temperature, and other parameters, leading to robust (thermodynamically favorable) cluster species. Xie et al. also demonstrated such a strategy in the synthesis of water-soluble Au25 in aqueous solution by using electrospray ionization mass spectrometry (ESI-MS) to monitor the reaction process. They adopted the NaOH-mediated NaBH4 reduction method to tune the formation kinetics of Au NCs and accelerate the thermodynamic selection of Au25.24 Another work by them is the usage of CO as a reducing agent that kinetically controls crude clusters, and Au25 NCs were also obtained after thermodynamically controlled size focusing.25 Although various synthetic methods have been developed in the last decade, the formation of atomically precise (narrow-sized) Au NCs can be mainly illustrated by kinetic control and thermodynamic selection (not limited to the gold salt (complex) reduction method (vide infra)).
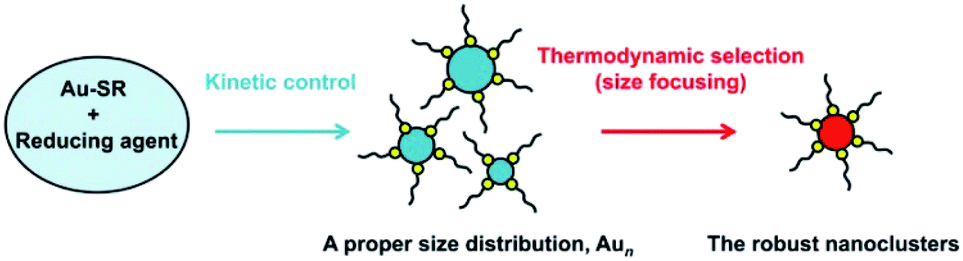 | ||
| Scheme 1 Illustration of the synthesis strategy “kinetic control and thermodynamic selection” for atomically monodisperse gold nanoclusters. | ||
3. Synthesis method and controlling factors
There are two basic routes used for synthesizing atomically precise gold nanoparticles in terms of the charge state of the starting gold (Scheme 2): one route is the reduction of gold salt (complex), and the other is the transformation of gold nanoparticles (including nanoclusters).18,26–28 For the first route, the synthesis method can be dubbed the “gold salt (complex) reduction method”; for the second route, there are two major synthesis methods, including the ligand induction method and anti-galvanic reduction method, in terms of inducing reagents. Below, we will specifically review the synthesis method and controlling parameters.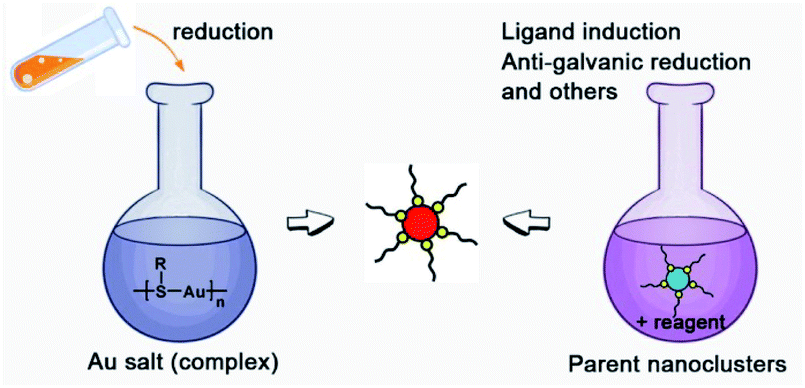 | ||
| Scheme 2 Schematic illustration of the synthesis route classification in terms of the charge state of the starting gold. | ||
3.1 Gold salt (complex) reduction method and controlling parameters
The gold salt (complex) reduction method is also known as a one-pot synthesis, and is one of the most versatile and best studied synthetic methods. This method is usually referred to as the direct reduction of a mixture of gold salt (complex) and other reactants, leading to final nanoparticles after essential purification but without any further disposal. The first gold colloid was synthesized by Faraday in 1857 from the aqueous reduction of gold chloride by phosphorus.4 An important advancement of this method was developed by Brust et al. (the Brust–Schiffrin method) in 1994.29 They exploited thiols for gold nanoparticle synthesis via a two-phase system in which gold chloride was transferred in toluene using a phase-transfer reagent. Following the footsteps of Brust et al., many groups, including Whetten, Murray, Tsukuda, Jin and other groups, conducted tremendous research on the synthesis of gold thiolate nanoparticles and nanoclusters. Gold salt (complex) reduction (often modified from the Brust–Schiffrin method) has been an efficient approach used to control the synthesis of Au NCs. The controlling parameters comprise the ligand, temperature, solvent, and others.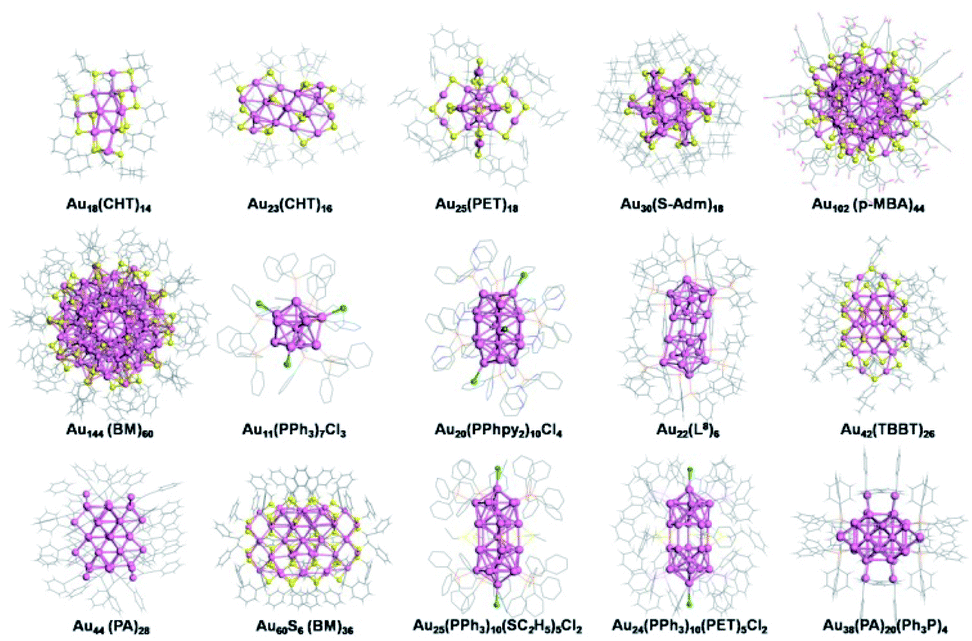 | ||
| Fig. 1 Representative crystal structures of Au NCs capped by different ligands using the gold salt (complex) reduction method. CHT-H: cyclohexanethiol; PET-H: phenylethanethiol; H-S-Adm: adamantanethiol; p-MBA-H: p-mercaptobenzoic acid; BM-H: benzyl mercaptan; PPh3: triphenylphosphine; PPhpy2: bis(2-pyridyl)-phenylphosphine; L8: 1,8-bis(diphenylphosphino) octane; TBBT-H: 4-tert-butylbenzenelthiol; PA: phenylacetylene; H-SC2H5: ethanethiol. | ||
Generally, smaller NCs are formed when using ligands with stronger steric repulsion, which can explain why the reported NCs protected by cyclohexanethiol, adamantanethiol and phosphines are often smaller than those protected by phenylethanethiol, 4-tert-butylbenzenelthiol and so on;30,31,34,36–38 Phosphine ligands (containing several benzene rings) often give rise to the formation of rod-shaped packing,41,44,45 and conjugated thiophenols or alkynes favor the formation of NCs with FCC structures,11 for example, most of the 4-tert-butylbenzenelthiolated gold nanoclusters obtained by reduction of gold salt (complex) are fcc-structured such as Au36(TBBT)24,47,48 Au44(TBBT)28,47,48 Au52(TBBT)32,49 Au56(TBBT)34,50 and Au92(TBBT)4451 (TBBT: 4-tert-butylbenzenelthiolate), while the phenylethanethiolated gold nanoclusters synthesized by reduction of gold salt (complex) generally adopt core–shell structures such as Au25(PET)18,32,33 Au38(PET)2437 and Au144(PET)6038 except for Au52(PET)3252 (PET: phenylethanethiolate).
The ligand type not only influences the atomic packing mode, but also affects structure distortion. For example, on the basis of density functional theory calculations of a series of [Au25(SR)18]− nanoclusters, Tlahuice-Flores et al. revealed that low-polarity R groups do not disturb the Au25S18 framework greatly, while in the case of p-thiolphenolate ligands (p-SPhX), a remarkable distortion of the Au25S18 framework destroys the inversion symmetry, the distortion increasing in the given order X = H, Cl, NO2 and CO2H. Similarly, the N-acetyl-cysteine ligand also notably distorts the framework.53
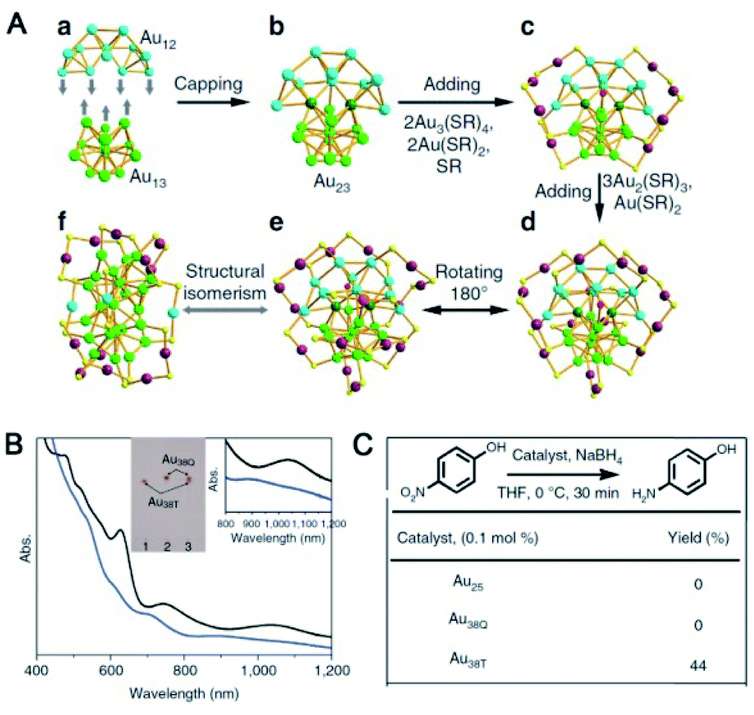 | ||
| Fig. 2 (A) Structures of Au38T (blue) and Au38Q (black). (B) UV-vis spectra of Au38T and Au38Q in toluene. (C) Catalytic activities of Au25, Au38T and Au38Q (reproduced from ref. 57 with permission from Springer Nature, Copyright (2015)). | ||
Note that, except for the kinetic influence, solvents also have a thermodynamic influence on the reaction, since solvents provide the environment and influence the stability of the reaction species.
 | ||
| Fig. 3 (A) The synthesis of differently sized Au NCs using the CO-reduction method by adjusting pH. (B) UV-vis absorption and fluorescence spectra of Au22(SG)18 (reproduced from ref. 58 and 59 with permission from the American Chemical Society, Copyright (2013, 2014)). | ||
Except for the type of reducing agent, the amount of the reducing agent could be considered for the control synthesis of gold nanoclusters.62,63 For example, Xie et al. investigated the synthesis process of water-soluble Au25 by using a stoichiometric amount of NaBH4. Due to the precise control of reducing electrons from NaBH4, a series of intermediate clusters (Au4–Au29) were confirmed with the help of the real-time mass spectrometry technique. Such stoichiometric62 or sub-stoichiometric63 controls on the amount of the reducing agent not only enrich the library of gold nanoclusters, but also largely contribute to the mechanistic understanding of the reaction.
Recently, the Wu group developed an acid-induction method via the addition of common acids (such as nitric acid, hydrochloric acid and acetic acid).52 Hydrogen ions that detached from the acid can accelerate the hydrolysis of NaBH4 and strengthen the reactivity of NaBH4. Meanwhile, they can interact with thiol groups, thus weakening the interaction between Au and thiolate and reducing the etching ability of thiolate. The influences of protons on the reaction kinetics and thermodynamics might be contrary to those of OH− reported by Xie et al.24 A novel Au52(PET)32 NC, whose kernel structure was revealed to be the isomer of the kernel structure of Au52(TBBT)32, was synthesized via this method. As shown in Fig. 4A–H, the gold atom fcc packing in Au52(PET)32 is a quadrangular prism-like core with defective top and bottom facets. The two defective facets consist of 6 gold atoms instead of 12 gold atoms for a complete facet. In Au52(TBBT)32, the 48-gold kernel comprises 4 × 4 × 6 Au atom layers along the X-, Y-, and Z-axes, respectively, and each layer has 8 gold atoms. The comparison of the UV/vis/NIR spectra indicated that such isomerization greatly influenced the electronic structure of the two Au52 NCs (Fig. 4I).
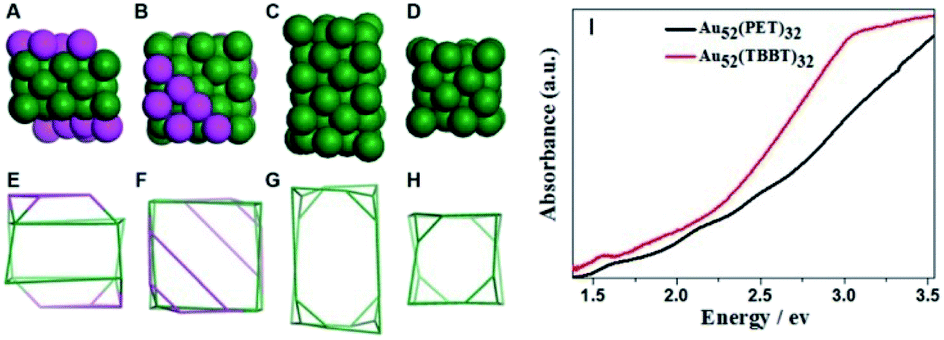 | ||
| Fig. 4 (A and E) The Au50 kernel of Au52(PET)32 with defective facets; (C and G) Au48 kernel of Au52(TBBT)32; (B, D, F, and H) aerial view of (A), (C), (E), and (G); (I) photon-energy plot of Au52(PET)32 and Au52(TBBT)32. | ||
When adding acetic acid in the synthesis of Au NCs protected by TBBT, Au42(TBBT)26 instead of Au36(TBBT)24 and Au44(TBBT)28 formed after aging for two hours.35 The Au34 kernel of Au42(TBBT)26 is composed of four cuboctahedra, which is identical to that of Au44(TBBT)28. Of note, the surface staples are totally different between these two clusters. In Au42(TBBT)26, the bridging thiolates and dimeric staples anchor the [100] facets, and each of the monomer staples anchor each [110] facet, with no staples anchoring the [111] facet. However, in Au44(TBBT)28, the [100], [111] and [110] facets are protected by bridging thiolates, a dimeric staple and monomeric staple, respectively. The clusters only exist with different staple styles and share the same kernel and ligands, which are termed homoligand–homokernel–heterostaple clusters. Moreover, the reduction of the two Au–SR units led to decreased stability of Au42(TBBT)26 compared with that of Au44(TBBT)28 and a decrease in photoluminescence.
Later, Au44(TBBT)26 and Au48(TBBT)28 were prepared via the acid-induction method.64 These two nanoclusters and Au38(PET)26 can be regarded as kernel homologues due to their similar kernel structures: one identical Au23 only differs in its number of capping gold atoms: 0, 6, and 8 (see Fig. 5). It is found by DFT calculations that the HOMOs-LUMOs are mainly distributed on the Au23 unit for every nanocluster, and the electrochemical properties of the three clusters are similar, both indicating that the Au23 unit acts as a “functional group”. In this work, the concepts of homologue, homology and functional groups were introduced into nanochemistry to help understand the structure–property relationship for the first time.
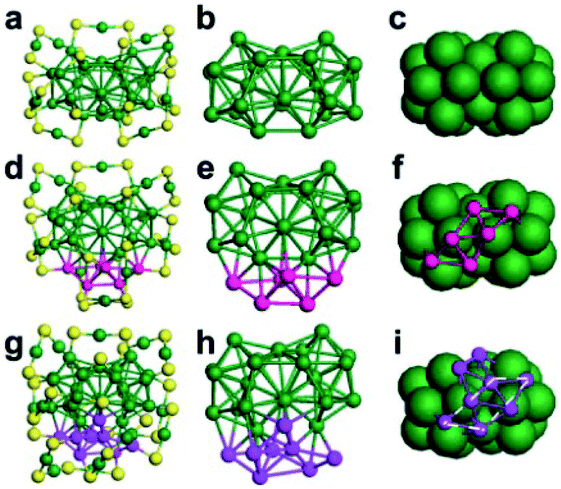 | ||
| Fig. 5 Comparison of the kernel structures of Au38(PET)24, Au44(TBBT)26 and Au48(TBBT)28 (reproduced from ref. 64 with permission from John Wiley and Sons, Copyright (2018)). | ||
Another assistant reagent is foreign metal ions. The co-reduction of Au and other metal ions would produce alloy NC intermediates and, finally, monometallic cluster products. For example, a non-fcc-structured Au42(TBBT)26 (Au42N) nanocluster was successfully synthesized in the presence of cadmium ions by Wu and coworkers.65 The addition of Cd may influence kinetics and thermodynamics in the production of Au42N nanoclusters by, for example, forming unstable Au/Cd intermediates, tuning the reducing ability of NaBH4, or influencing the etching rate of thiol. Au42N is the structural isomer of abovementioned Au42(TBBT)26 (Au42F) with an fcc packing kernel produced in the acid-induction method. As shown in Fig. 6, Au42N contains a non-fcc Au26 kernel that can be split into three parts consisting of 9, 9, and 8 Au atoms. The Au26 kernel is capped by several motifs, including four Au3(TBBT)4, four Au(TBBT)2 and two TBBT units. Wu et al. found that the photoluminescence emission of Au42F was approximately twice as extensive as that of Au42N, indicating that the fcc structure does not actually inhibit the emission compared with the non-fcc structure, which was further demonstrated by DFT calculations.
 | ||
| Fig. 6 The kernel structure of Au42N (reproduced from ref. 65 with permission from John Wiley and Sons, Copyright (2019)). | ||
3.2 Ligand induction method and controlling parameters
In early work, the ligand induction method typically involved the partial exchange of ligands for the functionalization and tailoring of the surface properties of nanoparticles, but changes in the nanoparticle size are often neglected due to the lack of structural determination.68,69 Since the accessible structure of Au NCs was developed, great advances in ligand induction methods have been made mainly by Jin, Tsukuda, etc.70,71 They introduced thermal conditions and the addition of a large excess of thiol into this method to control the size and structure of Au NCs. As one of the most important methods for synthesizing NCs, ligand induction often includes several step ligand exchange, structural transformation and size-focusing on the basis of the stability of differently sized NCs. Here, the precursor nanoclusters are not limited to monodispersed NCs and can be extended to mixed-sized NCs. | ||
| Fig. 7 Reaction pathway for the conversion of Au38(PET)24 to Au36(TBBT)24 (reproduced from ref. 72 with permission from the American Chemical Society, Copyright (2013)). | ||
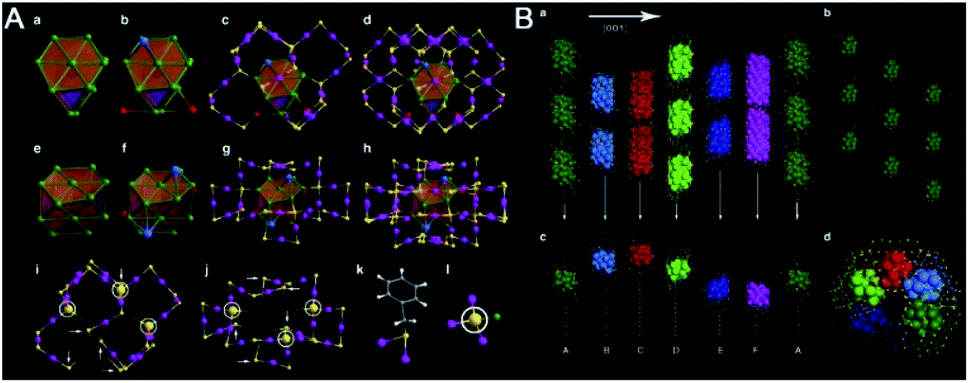 | ||
| Fig. 8 (A) Anatomy of the atomic structure and (B) crystallographic arrangement of the Au60S6 nanocluster (reproduced from ref. 43 with permission from Springer Nature, Copyright (2015)). | ||
Another example is the ligand exchange of Au25(PET)18. Au28(TBBT)20 nanoclusters can be synthesized by reacting Au25 with excess TBBT thiol.73 As shown in Fig. 9A, Au28(TBBT)20 is chiral and can be viewed as an fcc Au20 kernel capped by four Au2(SR)3 dimeric staples and eight SR bridging thiolates. The Au20 kernel is composed of two fused cuboctahedra sharing six common atoms. The Au24(SCH2Ph)20 nanocluster was obtained after mixing Au25 with benzyl mercaptan.74 Au24 consists of a bitetrahedral Au8 kernel with an fcc-based antiprismatic shape and four Au4(SR)5 tetrameric staples (Fig. 9B). More significantly, the strong fluorescence of Au24 NCs was rationally attributed to their particular structures compared with some other common nanoclusters. The interlocked Au4(SR)5 staples reduce emission loss by vibration, and the fully thiolate-bound kernel style strengthens electron transfer from the thiolates to the kernel through the Au–S bonds, thus leading to enhanced fluorescence.
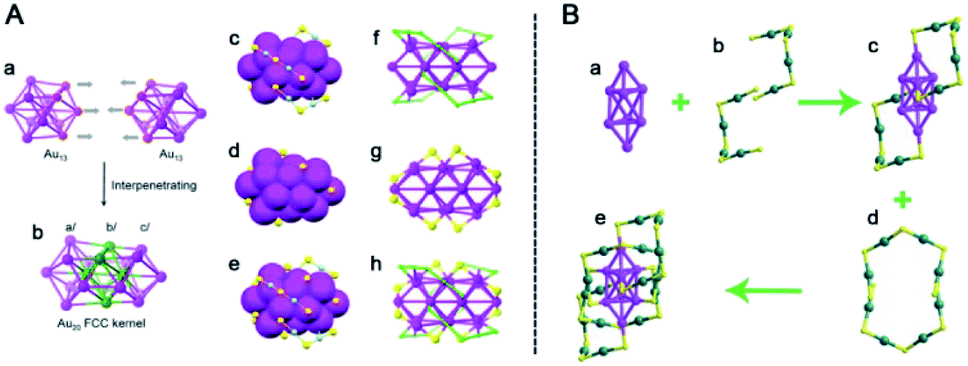 | ||
| Fig. 9 (A) The Au20 kernel structure (a and b) and the thiolate-binding modes (c–h) in Au28; (B) analysis of the Au24(SCH2Ph)20 nanocluster structure (reproduced from ref. 73 with permission from the American Chemical Society, Copyright (2013) and ref. 74 with permission from John Wiley and Sons, Copyright (2016)). | ||
Dass reported the synthesis of Au99(SPh)42 nanoclusters by etching Au144(PET)60 with benzenethiol.75 They found the instability of Au144 upon reaction with aromatic ligands, indicating that Au NCs protected by aromatic ligands favored different cores from those protected by aliphatic ligands. However, Jin and coworkers found that Au144(PET)60 converted to Au133(TBBT)52 after treatment with excess 4-tert-butylbenzenethiol at 80 °C for 4 days.76 Au133 has a four-shell structure. The central atom and the first shell form a 13-atom icosahedron that is wrapped by a 42-atom icosahedral shell, generating 55-atom Mackay icosahedra (MI). The third 52-atom shell is a transition layer between the Au55 MI and the outermost protecting layer. The resultant Au107 kernel is quasi-spherical and further protected by 26 monomeric staples. Notably, the surface gold-thiolate staples exhibit a self-organized helical “stripe” pattern. There are four such stripes that are bound with a curved surface clockwise or anticlockwise in the two chiral isomers.
An interesting study on the ligand effect was reported by Jin and coworkers in 2015.77 They explored the control of the magic sizes of Au NCs by the subtle structural change of the surface protecting ligands. After etching the polydispersed Aun clusters with isomeric methylbenzenethiols (MBT), Au130(p-MBT)50 (Au130), Au104(m-MBT)41 (Au104) and Au40(o-MBT)24 (Au40) NCs are obtained using para-, meta- and ortho-methylbenzenethiol, respectively (Fig. 10). This work revealed that the magic size and stability of Au NCs were sensitive to the surface protecting thiols. Later, the structural determination of Au130 and Au40 was also achieved.78,79 Au130 is constructed in a four-shell manner with a barrel shape, while Au40 exhibits a Kekulé-like ring shape composed of a series of four-atom tetrahedral units.
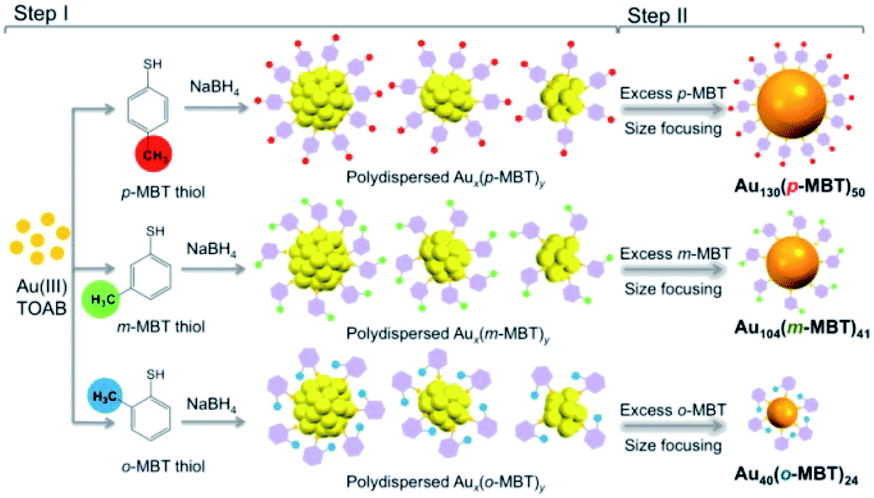 | ||
| Fig. 10 The synthesis of differently magic-sized nanoclusters capped by three isomeric thiol ligands: p-MBT, m-MBT, and o-MBT (reproduced from ref. 77 with permission from the American Chemical Society, Copyright (2015)). | ||
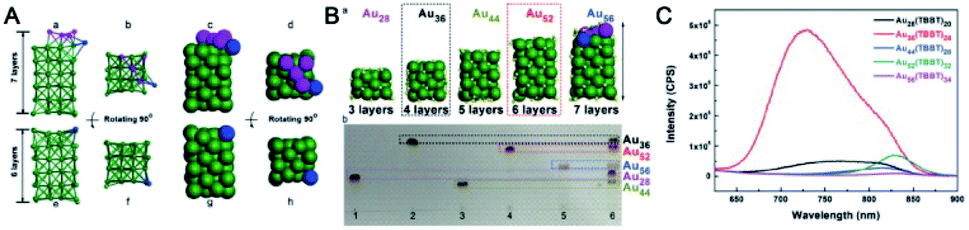 | ||
| Fig. 11 (A) The comparison of the kernels of Au56(TBBT)34 and Au52(TBBT)32; (B) structure evolution (a) and thin-layer chromatography (b); (C) the photoluminescence spectra of Au28(TBBT)20, Au36(TBBT)24, Au44(TBBT)28, Au52(TBBT)32, and Au56(TBBT)34 (reproduced from ref. 50 with permission from John Wiley and Sons, Copyright (2020)). | ||
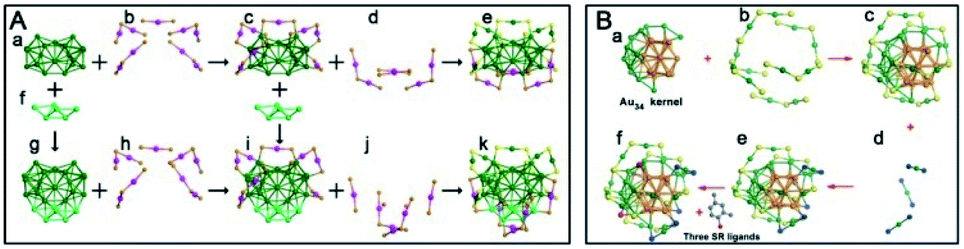 | ||
| Fig. 12 (A) Anatomy of the structures of Au38Q and Au44(2,4-DMBT)26 nanoclusters; (B) anatomy of the structure of the Au49(2,4-DMBT)27 nanocluster (reproduced from ref. 81 with permission from John Wiley and Sons, Copyright (2017)). | ||
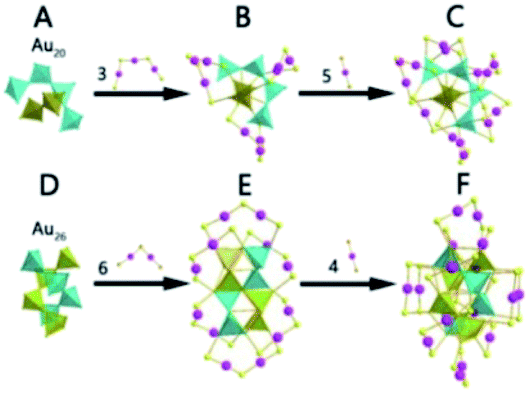 | ||
| Fig. 13 The kernel, total structure and growth mode of Au28(CHT)20, Au34(CHT)22 and Au42(CHT)26. | ||
3.3 Anti-galvanic reduction (AGR) method and controlling parameters
Anti-galvanic reduction (AGR), which was first proposed in 2012 by Wu,88 involves the reduction of metal ions by less reactive (more noble) metals, which is the opposite of the famous GR discovered by Italian scientist Luigi Galvani. The driving force of AGR is that the oxidation potential of ultrasmall metal NPs greatly decreases to a potential that is even lower than the reduction potential of some less noble metal ions. AGR has been used for metal ion sensing, antioxidation, synthesis of atomically precise alloy NCs or monometal NCs, etc. For more information, readers are recommended to read a recent review article, ref. 89. In this section, we mainly focus on the controlling parameters in the AGR synthesis method.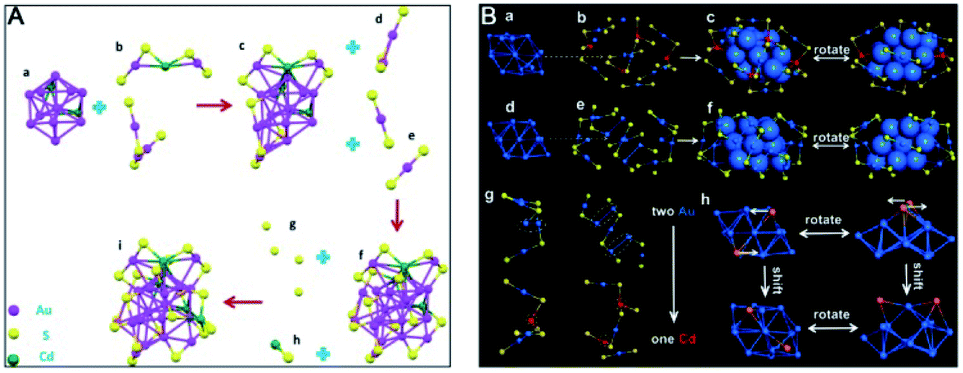 | ||
| Fig. 14 Anatomy of the structure of (A) Au20Cd4(SH)(SR)19 and (B) Au26Cd4(CHT)22 nanoclusters (reproduced from ref. 92 with permission from John Wiley and Sons, Copyright (2018) and ref. 93 with permission from the American Chemical Society, Copyright (2019)). | ||
Recently, Zhu et al. carefully investigated the Ag doping position of TBBT-protected Au28 and Au36 NCs by reacting with the Ag(I)SR complex.94 Compared with the general doping of Ag atoms in the isotropic nanocluster (e.g., Au25(SR)18), only specific Au atoms of Au28 and Au36 (e.g., Au atoms at the vertex sites in the metal core or in the staples) can be replaced by Ag atoms, which was attributed to the electrophilic effect revealed by DFT calculations.
:Au atomic ratio reaches 2:1.
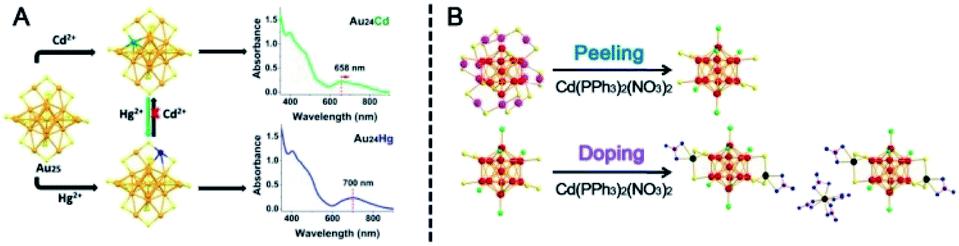 | ||
| Fig. 15 (A) The doping position and UV-vis spectra of Au24Cd and Au24Hg. (B) Structural evolution from Au25 to Au26Cd5 (reproduced from ref. 97 with permission from the American Chemical Society, Copyright (2019) ref. 98 with permission from John Wiley and Sons, Copyright (2017)). | ||
When a special ion precursor–Au complex is introduced to react with Au NCs (such reactions were dubbed quasi- or pseudo-AGR), the parent NCs will convert into others by adding or losing Au atoms. Wu et al. found that fluorescent Au24(PET)20 was easily synthesized by reacting Au25 with the Au–PET complex and revealed that the electrochemical gap was enlarged with decreasing size.99 Very recently, the same group reported the simultaneous synthesis of two Au28(CHT)20 isomers by mixing Au23(CHT)16 with the Au–CHT complex.100 The two isomers, consisting of the same Au20(CHT)8 kernel and different outer staples, can reversibly transform into each other through dissolution and crystallization processes with oscillating fluorescence. Wu et al. also revealed a module replacement (MR)-like process during the pseudo-AGR of Au48(CHT)26.101 The MR-like process involves the replacement of the local structural unit (including three or more Au atoms) by a new one. As-obtained Au37(CHT)23 and Au48(CHT)26 share the same Au31(CHT)12 section as the other section (Au6(CHT)11vs. Au17(CHT)14). Au37(CHT)23 exhibited stronger photoluminescence but weaker photothermy than Au48(CHT)26, indicating that photoluminescence and photothermy are balanced and can be at least partly converted into each other.
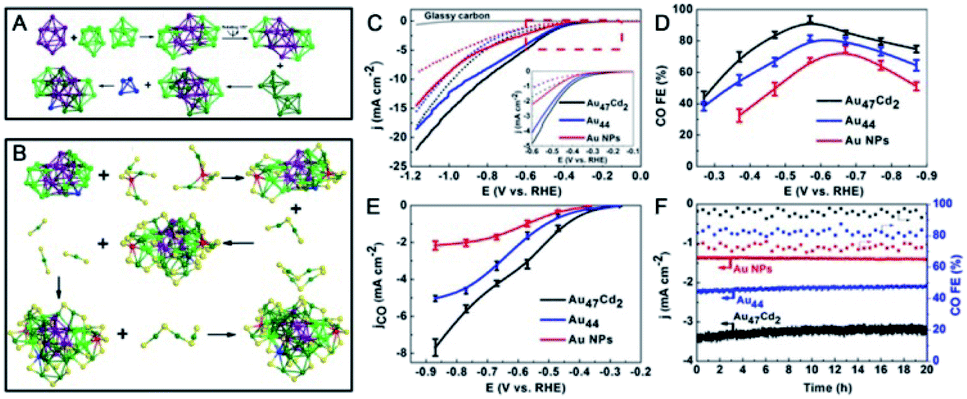 | ||
| Fig. 16 (A) Kernel structure and (B) anatomy of the structure of Au47Cd2(TBBT)31. (C) LSV curves of Au47Cd2(TBBT)31, Au44(TBBT)28 and the ca. 1.5 nm Au NPs in an Ar-saturated (dotted line) and a CO2-saturated (full line) 0.5 M KHCO3 solution. (D) CO faradaic efficiency of the catalysts examined with different applied potentials. (E) The corresponding CO partial current density. (F) The stability test (reproduced from ref. 102 with permission from John Wiley and Sons, Copyright (2020)). | ||
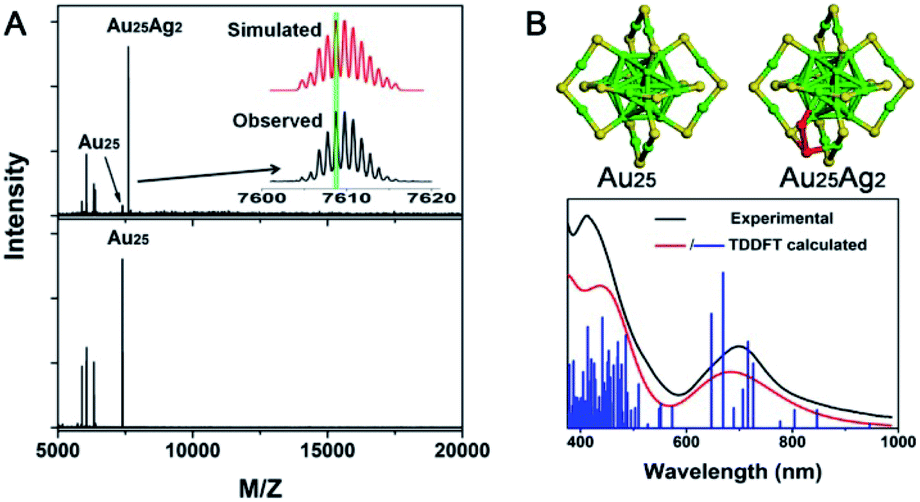 | ||
| Fig. 17 (A) MALDI-MS spectra of Au25Ag2 and Au25. (B) Structure of Au25 and Au25Ag2 and the comparison of the experimental and calculated spectra of Au25Ag2 (reproduced from ref. 103 with permission from the American Chemical Society, Copyright (2015)). | ||
Since trimetallic nanoclusters can be achieved by successive AGR reactions, the sequence of the usage of metal complexes also affects the product type. For example, Au25(PET)18 first reacted with Hg2+ ions and then with the Ag(I)–PET complex, leading to the formation of Au16.8Ag7.2Hg1(PET)18 nanoclusters.104 When the sequence of Hg2+ ions and the Ag(I)-PET complex was exchanged, Au24−xAgxHg1(PET)18 (x = 2–6) was obtained with a relatively low Ag content.105
3.4 Other methods and controlling parameters
Except for the abovementioned methods, there are also some other efficient approaches for synthesizing and controlling Au NCs, including oxidation/reduction-driven transformation, intercluster reactions, acid etching, and solvent induction, to name a few.In the oxidation/reduction-driven method, the charge or even the structure of the parent NCs was often altered, and some examples are shown below. Early in 2008, Jin et al. reported the transformation from the [Au25(PET)18]− (Au25−) anionic cluster into a charge neutral cluster [Au25(PET)18]0 (Au250) via air oxidation or H2O2 oxidation.106 Despite the similar structures, the staples of Au250 have some apparent distortions; for instance, every two opposite Au2(SR)3 staples in Au250 are completely coplanar with the symmetry plane of the icosahedron kernel. Negishi et al. studied the oxidation of hexanethiol-capped Au25 with the strong oxidizing agent Ce(SO4)2 and obtained positive Au25.107 In 2017, Jin and coworkers reported the structural transformation from another anionic Au NC (Au23(CHT)16) into Au28(CHT)20 by H2O2 oxidation.108 Unlike the case of Au25− transformation to Au250, the metal core and surface staples of Au28 are completely different from those of Au23, indicating that oxidation-induced transformation is a potential method to access new structures of Au NCs. Wu et al. also found the conversion from Au23(CHT)16 to Au28(CHT)20 using Cd(NO3)2 as the oxidant.92 Reduction-induced transformation has also been reported. For example, Jin and Zhu reported the transformation from [Au25(SePh)18]− to [Au23(SePh)16]− in the presence of reductive NaBH4.109 The direct loss of two Au–SR units in the parent Au25 was revealed by experiments and theoretical calculations.
Pradeep's group reported the first example of intercluster reactions between Au25(SR)18 and Ag44(SR)30 (SR: alkyl/aryl thiolate) (Fig. 18A).110,111 The mass data demonstrated the exchange of the metal atoms as well as the metal–thiolate fragments. Of note, the number of Ag dopants added to Au25 could reach up to 20. The thermodynamic feasibility of the reaction has also been revealed using DFT calculations. Almost at the same time, inspired by AGR,88,89 Wu et al. reported the reaction between hydrophilic metal nanoparticles (including nanoclusters) (Fig. 18B).112 Captopril-protecting Au25(Capt)18 (Capt: Captopril) was chosen to react with Ag30(Capt)18, and a novel alloy NC (Au20Ag5(Capt)18) was demonstrated by mass spectrometry and X-ray photoelectron spectroscopy. Au20Ag5(Capt)18 showed enhanced photoluminescence with a quantum yield of ca. 7.6% relative to rhodamine 6G, which is 130 times and 38 times higher than that of Au25(Capt)18 or Ag30(Capt)18, respectively. Wu et al. further investigated the reaction between Au25(Capt)18 and silver (copper) nanoparticles and observed the mass peaks of Ag- or Cu-doped Au25, indicating the universality of the interparticle reaction of water-soluble nanoparticles. In another example, Tsukuda et al. reported that the reaction between an equal amount of [PtAu24]0 and [PtAu24]2– could produce two equivalents of [PtAu24]−, indicating possible electron transfer between differently valenced nanoclusters.113
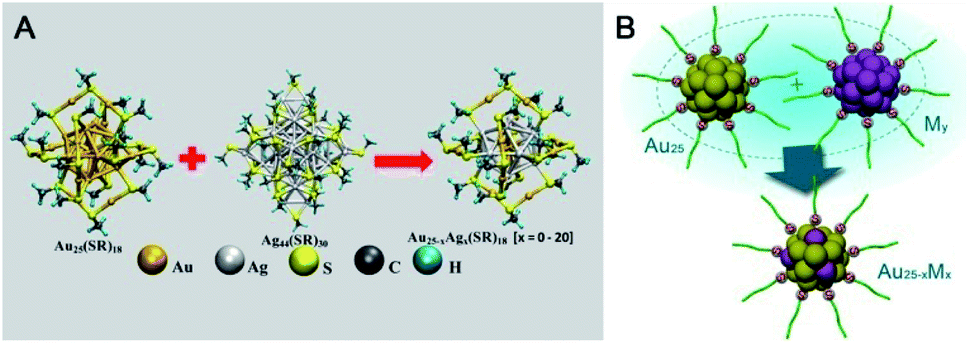 | ||
| Fig. 18 (A) Intercluster reactions between Au25(SR)18 and Ag44(SR)30 (SR: alkyl/aryl thiolate) (reproduced from ref. 110 with permission from the American Chemical Society, Copyright (2016)). (B) The reaction between Au25 and Ag (Cu) nanoclusters or nanoparticles. | ||
Bulk gold is inert in common acids; however, nanoscale gold is not the same as bulk gold. Utilizing the activity of gold toward acid, Wu and coworkers introduced the acid-etching synthesis method and revealed the size-dependent reactivity of PET- and TBBT-capped gold nanoparticles with acetic acid:114 large nanoparticles are still stable in acetic acid, while small nanoparticles (e.g., Au25 and Au38) show distinct reactivity with acetic acid. A new Au38(PET)26 nanocluster was obtained after etching Au25 in acetic acid overnight. Au38(PET)26 exhibited great disparities from the reported Au38(PET)24 in terms of both optical and electrochemical properties. The etching mechanism was also studied, and Wu et al. observed some intermediate species, such as Aun(PET)n+1Cl2(HAc) (n = 1–3, HAc: acetic acid), from mass spectra, indicating the possible attack of the staples by acetic acid.
More attention has been given to the structural transformation of Au NCs in solvents. Wei and coworkers reported an icosahedral-to-cuboctahedral structural transformation of Au13 driven by hexane.115 The transformation process consists of the hexane-induced selective removal of the thiolate ligands from the Au13 surface, and the subsequent structure conversion process was monitored by X-ray absorption fine structure (XAFS) analyses. Xie et al. also reported the size-conversion reaction from [Au23(SR)16]− to [Au25(SR)18]− (SR: para-mercaptobenzoic acid) by changing the solvent polarity (e.g., from water/ethanol to pure water).116 A surface-motif-exchange-induced mechanism was revealed by tandem mass analyses. The Wu group recently found that one of the Au28(CHT)20 isomers (Au28i) could be transformed into another (Au28ii) in diverse solvents.100 The dielectric-constant effect and deuteration effect were observed for the first time in the solvent-induced conversion process of Au NCs, which was also confirmed by the adsorption energies and free energies obtained from DFT calculations.
4. Summary and future outlook
The past two decades have witnessed the rapid development of the synthesis of Au NCs, undergoing the stages of the acquisition of Au NCs, the pursuit of monodispersity, structural determination, and controlling the composition and structure with atomic precision. In this review, we have introduced important research on controlling the synthesis of Au NCs by categorizing the present synthetic methods into three main routes, (i) gold salt (complex) reduction, (ii) ligand induction, and (iii) antigalvanic reduction, noting the influences of various controlling parameters in the synthesis process. Although many types of Au NCs with diverse structures and compositions have been reported and explored, there are still several challenges for controlling Au NCs. At the end of this review, we attempt to propose several potential studies to address these issues:(1) One to a few metal atom (ligand) manipulation. With the growing synthesis skill, we might expect the directed and precise removal, replacement or addition of one to a few metal atoms in NCs, with the remnant structures essentially being unchanged. Similarly, we also expect the manipulation of one to a few ligands (including reduction, addition and replacement) on the NC surface, with metal atom packing remaining untouched. Such a manipulation is of great significance not only for fundamental structure (composition)–property studies but also for improving the properties targeted for potential applications.
(2) Property-targeted control. Recently, Au NCs have attracted increasing attention due to their potential application in catalysis, sensing, and biology. Thus, the research community in this field is suggested to rationally control Au NCs with enhanced properties (e.g., catalytic activity and fluorescence) based on the existing structure–property correlation.
(3) Development of doping methods. The precisely controlled synthesis of multi-metal (ligand) Au NCs is crucial for obtaining unique properties and practical applications due to the lower cost or newly imparted properties generated by foreign metal atoms (ligands) or synergistic effects. More metal elements, especially relatively active metals,117 are expected to be doped into Au NCs. Controllable ligand doping has been rarely reported.
In brief, the future controlled synthesis of Au NCs will be developed not only with enriched sizes, compositions, and structures but also with attractive and selectable properties for applications.
Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21829501, 21701179, 21771186, 21925303, 21222301, 21603234, 21171170, and 21528303), Natural Science Foundation of Anhui Province (1708085QB36), CASHIPS Director's Fund (BJPY2019A02), Key Program of the 13th five year plan, CASHIPS (2020HSC-CIP005), and Innovative Program of Development Foundation of Hefei Center for Physical Science and Technology (2020HSC-CIP005) and CAS/SAFEA International Partnership Program for Creative Research Teams.References
- M. Haruta, Chem. Rec., 2003, 3, 75–87 CrossRef CAS
.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145 CrossRef
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS
- M. B. Cortie and A. M. McDonagh, Chem. Rev., 2011, 111, 3713–3735 CrossRef CAS
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS
- T. Higaki, Q. Li, M. Zhou, S. Zhao, Y. Li, S. Li and R. Jin, Acc. Chem. Res., 2018, 51, 2764–2773 CrossRef CAS
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS
- S. Wang, Q. Li, X. Kang and M. Zhu, Acc. Chem. Res., 2018, 51, 2784–2792 CrossRef CAS
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS
- Q. Yao, X. Yuan, T. Chen, D. T. Leong and J. Xie, Adv. Mater., 2018, 30, 1870358 CrossRef
- R. P. Briñas, M. Hu, L. Qian, E. S. Lymar and J. F. Hainfeld, J. Am. Chem. Soc., 2008, 130, 975–982 CrossRef
- M. Zhu, E. Lanni, N. Garg, M. E. Bier and R. Jin, J. Am. Chem. Soc., 2008, 130, 1138–1139 CrossRef CAS
- Z. Wu, J. Suhan and R. Jin, J. Mater. Chem., 2009, 19, 622–626 RSC
- H. Qian, Y. Zhu and R. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS
- Z. Wu, M. A. MacDonald, J. Chen, P. Zhang and R. Jin, J. Am. Chem. Soc., 2011, 133, 9670–9673 CrossRef CAS
- X. Yuan, B. Zhang, Z. Luo, Q. Yao, D. T. Leong, N. Yan and J. Xie, Angew. Chem., Int. Ed., 2014, 53, 4623–4627 CrossRef CAS
- Z. Luo, V. Nachammai, B. Zhang, N. Yan, D. T. Leong, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 10577–10580 CrossRef CAS
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1–29 CrossRef CAS
- X. Yuan, X. Dou, K. Zheng and J. Xie, Part. Part. Syst. Charact., 2015, 32, 613–629 CrossRef
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC
- A. Das, C. Liu, H. Y. Byun, K. Nobusada, S. Zhao, N. Rosi and R. Jin, Angew. Chem., Int. Ed., 2015, 54, 3140–3144 CrossRef CAS
- A. Das, T. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 18264–18267 CrossRef CAS
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS
- T. Higaki, C. Liu, C. Zeng, R. Jin, Y. Chen, N. L. Rosi and R. Jin, Angew. Chem., Int. Ed., 2016, 55, 6694–6697 CrossRef CAS
- S. Zhuang, L. Liao, Y. Zhao, J. Yuan, C. Yao, X. Liu, J. Li, H. Deng, J. Yang and Z. Wu, Chem. Sci., 2018, 9, 2437–2442 RSC
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS
- H. Qian and R. Jin, Nano Lett., 2009, 9, 4083–4087 CrossRef CAS
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS
- X.-K. Wan, Z.-W. Lin and Q.-M. Wang, J. Am. Chem. Soc., 2012, 134, 14750–14752 CrossRef CAS
- J. Chen, Q.-F. Zhang, T. A. Bonaccorso, P. G. Williard and L.-S. Wang, J. Am. Chem. Soc., 2014, 136, 92–95 CrossRef CAS
- X.-K. Wan, Z.-J. Guan and Q.-M. Wang, Angew. Chem., Int. Ed., 2017, 56, 11494–11497 CrossRef CAS
- Z. Gan, J. Chen, J. Wang, C. Wang, M. B. Li, C. Yao, S. Zhuang, A. Xu, L. Li and Z. Wu, Nat. Commun., 2017, 8, 14739 CrossRef CAS
- Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef CAS
- A. Das, T. Li, K. Nobusada, Q. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2012, 134, 20286–20289 CrossRef CAS
- X.-K. Wan, J.-Q. Wang, Z.-A. Nan and Q.-M. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef CAS
- L. Liao, C. Yao, C. Wang, S. Tian, J. Chen, M.-B. Li, N. Xia, N. Yan and Z. Wu, Anal. Chem., 2016, 88, 11297–11301 CrossRef CAS
- C. Zeng, Y. Chen, C. Liu, K. Nobusada, N. L. Rosi and R. Jin, Sci. Adv., 2015, 1, e1500425 CrossRef
- L. Liao, C. Wang, S. Zhuang, N. Yan, Y. Zhao, Y. Yang, J. Li, H. Deng and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 731–734 CrossRef CAS
- L. Liao, J. Chen, C. Wang, S. Zhuang, N. Yan, C. Yao, N. Xia, L. Li, X. Bao and Z. Wu, Chem. Commun., 2016, 52, 12036–12039 RSC
- S. Zhuang, L. Liao, M.-B. Li, C. Yao, Y. Zhao, H. Dong, J. Li, H. Deng, L. Li and Z. Wu, Nanoscale, 2017, 9, 14809–14813 RSC
- A. Tlahuice-Flores, R. L. Whetten and M. Jose-Yacaman, J. Phys. Chem. C, 2013, 117, 20867–20875 CrossRef CAS
- T. Chen, Q. Yao, X. Yuan, R. R. Nasaruddin and J. Xie, J. Phys. Chem. C, 2017, 121, 10743–10751 CrossRef CAS
- S. K. Katla, J. Zhang, E. Castro, R. A. Bernal and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 75–82 CrossRef CAS
- H. Qian and R. Jin, Chem. Mater., 2011, 23, 2209–2217 CrossRef CAS
- S. Tian, Y.-Z. Li, M.-B. Li, J. Yuan, J. Yang, Z. Wu and R. Jin, Nat. Commun., 2015, 6, 8667 CrossRef CAS
- Y. Yu, X. Chen, Q. Yao, Y. Yu, N. Yan and J. Xie, Chem. Mater., 2013, 25, 946–952 CrossRef CAS
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS
- A. Ghosh, T. Udayabhaskararao and T. Pradeep, J. Phys. Chem. Lett., 2012, 3, 1997–2002 CrossRef CAS
- F. Bertorelle, I. Russier-Antoine, C. Comby-Zerbino, F. Chirot, P. Dugourd, P.-F. Brevet and R. Antoine, ACS Omega, 2018, 3, 15635–15642 CrossRef CAS
- T. Chen, V. Fung, Q. Yao, Z. Luo, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2018, 140, 11370–11377 CrossRef CAS
- T. Chen, Q. Yao, Y. Cao and J. Xie, Cell Rep. Phys. Sci., 2020, 1, 100206 CrossRef
- S. Zhuang, L. Liao, J. Yuan, C. Wang, Y. Zhao, N. Xia, Z. Gan, W. Gu, J. Li, H. Deng, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2018, 57, 15450–15454 CrossRef CAS
- S. Zhuang, L. Liao, J. Yuan, N. Xia, Y. Zhao, C. Wang, Z. Gan, N. Yan, L. He, J. Li, H. Deng, Z. Guan, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 4510–4514 CrossRef CAS
- X. Meng, Z. Liu, M. Zhu and R. Jin, Nanoscale Res. Lett., 2012, 7, 277 CrossRef
- X. Zhu, S. Jin, S. Wang, X. Meng, C. Zhu, M. Zhu and R. Jin, Chem.–Asian J., 2013, 8, 2739–2745 CrossRef CAS
- J. Yang, J. Y. Lee and J. Y. Ying, Chem. Soc. Rev., 2011, 40, 1672–1696 RSC
- G. H. Woehrle, L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 2005, 127, 2172–2183 CrossRef CAS
- Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef CAS
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS
- C. Zeng, C. Liu, Y. Pei and R. Jin, ACS Nano, 2013, 7, 6138–6145 CrossRef CAS
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS
- Z. Gan, Y. Lin, L. Luo, G. Han, W. Liu, Z. Liu, C. Yao, L. Weng, L. Liao, J. Chen, X. Liu, Y. Luo, C. Wang, S. Wei and Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 11567–11571 CrossRef CAS
- P. R. Nimmala and A. Dass, J. Am. Chem. Soc., 2014, 136, 17016–17023 CrossRef CAS
- C. Zeng, Y. Chen, K. Kirschbaum, K. Appavoo, M. Y. Sfeir and R. Jin, Sci. Adv., 2015, 1, e1500045 CrossRef
- Y. Chen, C. Zeng, D. R. Kauffman and R. Jin, Nano Lett., 2015, 15, 3603–3609 CrossRef CAS
- Y. Chen, C. Zeng, C. Liu, K. Kirschbaum, C. Gayathri, R. R. Gil, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2015, 137, 10076–10079 CrossRef CAS
- C. Zeng, Y. Chen, C. Liu, K. Nobusada, N. L. Rosi and R. Jin, Sci. Adv., 2015, 1, e1500425 CrossRef
- L. Liao, S. Zhuang, C. Yao, N. Yan, J. Chen, C. Wang, N. Xia, X. Liu, M.-B. Li, L. Li, X. Bao and Z. Wu, J. Am. Chem. Soc., 2016, 138, 10425–10428 CrossRef CAS
- L. Liao, S. Zhuang, P. Wang, Y. Xu, N. Yan, H. Dong, C. Wang, Y. Zhao, N. Xia, J. Li, H. Deng, Y. Pei, S.-K. Tian and Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 12644–12648 CrossRef CAS
- H. Dong, L. Liao, S. Zhuang, C. Yao, J. Chen, S. Tian, M. Zhu, X. Liu, L. Li and Z. Wu, Nanoscale, 2017, 9, 3742–3746 RSC
- P. Liu, W. Han, M. Zheng and W. W. Xu, Nanoscale, 2020, 12, 20677–20683 RSC
- A. Tlahuice-Flores, J. Phys. Chem. C, 2019, 123(17), 10831–10841 CrossRef CAS
- A. Tlahuice-Flores, M. Jose-Yacamán and R. L. Whetten, Phys. Chem. Chem. Phys., 2013, 15, 19557–19560 RSC
- A. Tlahuice and I. L. Garzón, Phys. Chem. Chem. Phys., 2012, 14, 3737–3740 RSC
- W. W. Xu, X. C. Zeng and Y. Gao, Acc. Chem. Res., 2018, 51, 2739–2747 CrossRef CAS
- Z. Wu, Angew. Chem., Int. Ed., 2012, 51, 2934–2938 CrossRef CAS
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS
- Z. Wu, M. Wang, J. Yang, X. Zheng, W. Cai, G. Meng, H. Qian, H. Wang and R. Jin, Small, 2012, 8, 2028–2035 CrossRef CAS
- S. Tian, C. Yao, L. Liao, N. Xia and Z. Wu, Chem. Commun., 2015, 51, 11773–11776 RSC
- Z. Min, W. Pu, Y. Nan, C. Xiaoqi, H. Lizhong, Z. Yan, X. Nan, Y. Chuanhao, L. Jin, D. Haiteng, Z. Yan, P. Yong and W. Zhikun, Angew. Chem., Int. Ed., 2018, 57, 4500–4504 CrossRef
- W. Zhang, S. Zhuang, L. Liao, H. Dong, N. Xia, J. Li, H. Deng and Z. Wu, Inorg. Chem., 2019, 58, 5388–5392 CrossRef CAS
- S. Wang, L. Xiong, G. Sun, L. Tang, J. Zhang, Y. Pei and M. Zhu, Nanoscale Adv., 2020, 2, 664–668 RSC
- M.-B. Li, S.-K. Tian, Z. Wu and R. Jin, Chem. Commun., 2015, 51, 4433–4436 RSC
- S. Wang, Y. Song, S. Jin, X. Liu, J. Zhang, Y. Pei, X. Meng, M. Chen, P. Li and M. Zhu, J. Am. Chem. Soc., 2015, 137, 4018–4021 CrossRef CAS
- C. Yao, Y.-J. Lin, J. Yuan, L. Liao, M. Zhu, L.-H. Weng, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 15350–15353 CrossRef CAS
- M.-B. Li, S.-K. Tian and Z. Wu, Chin. J. Chem., 2017, 35, 567–571 CrossRef CAS
- C. Yao, S. Tian, L. Liao, X. Liu, N. Xia, N. Yan, Z. Gan and Z. Wu, Nanoscale, 2015, 7, 16200–16203 RSC
- N. Xia, J. Yuan, L. Liao, W. Zhang, J. Li, H. Deng, J. Yang and Z. Wu, J. Am. Chem. Soc., 2020, 142, 12140–12145 CrossRef CAS
- F. Jin, H. Dong, Y. Zhao, S. Zhuang, L. Liao, N. Yan, W. Gu, J. Zha, J. Yuan, J. Li, H. Deng, Z. Gan, J. Yang and Z. Wu, Acta Chim. Sin., 2020, 78, 407–411 CrossRef
- S. Zhuang, D. Chen, L. Liao, Y. Zhao, N. Xia, W. Zhang, C. Wang, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 3073–3077 CrossRef CAS
- C. Yao, J. Chen, M.-B. Li, L. Liu, J. Yang and Z. Wu, Nano Lett., 2015, 15, 1281–1287 CrossRef CAS
- N. Yan, L. Liao, J. Yuan, Y.-J. Lin, L.-H. Weng, J. Yang and Z. Wu, Chem. Mater., 2016, 28, 8240–8247 CrossRef CAS
- S. Yang, S. Wang, S. Jin, S. Chen, H. Sheng and M. Zhu, Nanoscale, 2015, 7, 10005–10007 RSC
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CrossRef CAS
- Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten and T. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323 CrossRef CAS
- T. Higaki, C. Liu, Y. Chen, S. Zhao, C. Zeng, R. Jin, S. Wang, N. L. Rosi and R. Jin, J. Phys. Chem. Lett., 2017, 8, 866–870 CrossRef CAS
- Y. Song, H. Abroshan, J. Chai, X. Kang, H. J. Kim, M. Zhu and R. Jin, Chem. Mater., 2017, 29, 3055–3061 CrossRef CAS
- K. R. Krishnadas, A. Ghosh, A. Baksi, I. Chakraborty, G. Natarajan and T. Pradeep, J. Am. Chem. Soc., 2016, 138, 140–148 CrossRef CAS
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan, A. Som and T. Pradeep, Acc. Chem. Res., 2017, 50, 1988–1996 CrossRef CAS
- N. Xia and Z. Wu, J. Mater. Chem. C, 2016, 4, 4125–4128 RSC
- M. Suyama, S. Takano, T. Nakamura and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 14048–14051 CrossRef CAS
- N. Xia, Z. Gan, L. Liao, S. Zhuang and Z. Wu, Chem. Commun., 2017, 53, 11646–11649 RSC
- Y. Li, H. Cheng, T. Yao, Z. Sun, W. Yan, Y. Jiang, Y. Xie, Y. Sun, Y. Huang, S. Liu, J. Zhang, Y. Xie, T. Hu, L. Yang, Z. Wu and S. Wei, J. Am. Chem. Soc., 2012, 134, 17997–18003 CrossRef CAS
- Q. Yao, V. Fung, C. Sun, S. Huang, T. Chen, D.-e. Jiang, J. Y. Lee and J. Xie, Nat. Commun., 2018, 9, 1979 CrossRef
- A. Tlahuice-Flores, J. Nanopart. Res., 2013, 15, 1771 CrossRef
| This journal is © The Royal Society of Chemistry 2021 |