
Chitosan-based smart hybrid materials: a physico-chemical perspective†
Giuseppe
Cavallaro
 a,
Samantha
Micciulla
*b,
Leonardo
Chiappisi
*b and
Giuseppe
Lazzara
*a
a,
Samantha
Micciulla
*b,
Leonardo
Chiappisi
*b and
Giuseppe
Lazzara
*a
aDipartimento di Fisica e Chimica, Università degli Studi di Palermo, Viale delle Scienze pad 17, 90128 Palermo, Italy. E-mail: giuseppe.lazzara@unipa.it
bInstitut Max von Laue – Paul Langevin, 71 avenue des Martyrs, 38042 Grenoble, France. E-mail: micciulla@ill.eu; chiappisil@ill.eu
First published on 19th November 2020
Abstract
Chitosan is one of the most studied cationic polysaccharides. Due to its unique characteristics of being water soluble, biocompatible, biodegradable, and non-toxic, this macromolecule is highly attractive for a broad range of applications. In addition, its complex behavior and the number of ways it interacts with different components in a system result in an astonishing variety of chitosan-based materials. Herein, we present recent advances in the field of chitosan-based materials from a physico-chemical perspective, with focus on aqueous mixtures with oppositely charged colloids, chitosan-based thin films, and nanocomposite systems. In this review, we focus our attention on the physico-chemical properties of chitosan-based materials, including solubility, mechanical resistance, barrier properties, and thermal behaviour, and provide a link to the chemical peculiarities of chitosan, such as its intrinsic low solubility, high rigidity, large charge separation, and strong tendency to form intra- and inter-molecular hydrogen bonds.
 Giuseppe Cavallaro | Giuseppe Cavallaro is an assistant professor at the Department of Physics and Chemistry, University of Palermo, Italy. He was a Research Associate at the Institute of Micromanifacturing, Lousiana Tech University, (USA) and Institut fur Chemie, Technische Universitat Berlin (Germany). His research activities focus on nanoclays and polymer/nanoparticle interactions. He is the author of more than 75 publications in peer-reviewed international journals. |
 Samantha Micciulla | Samantha Micciulla is a research and instrument scientist at the Institut Laue-Langevin in Grenoble, France. In 2016, she received her PhD degree in Applied Physical Chemistry at the Technische Universität Berlin, Germany, and then she was a Post-Doc fellow at the Max-Planck Institut for Colloid and Interface Science in Potsdam, Germany. With a background in polymer science and physical chemistry of soft matter at interfaces, she has been in charge of projects on the design of multi-functional responsive coatings based on polymers, including hybrid organic–inorganic composites, and of technical strategies for the investigation of separation-dependent structures of interacting soft interfaces. |
 Leonardo Chiappisi | Leonardo Chiappisi received his PhD in 2015 in Physical Chemistry from the Technische Universität Berlin. In his PhD thesis, he investigated chitosan-based responsive assemblies with oppositely charged surfactants. Since 2015 he has been working at the Institut Laue-Langevin, the European flagship for neutron science. His research activity is focussed on the investigation of complex colloidal systems, both in bulk and at interfaces. |
 Giuseppe Lazzara | Giuseppe Lazzara has been an associate professor at the Department of Physics and Chemistry, University of Palermo, Italy, since 2015. He received his PhD degree in Chemistry in 2007. He was a Postdoc fellow at the Chemistry Department, Lund University (Sweden). He is involved in several projects on halloysite clay nanotubes for drug delivery and materials for the conservation of cultural heritage. Lazzara has more than 150 publications in peer-reviewed international journals, an edited book and 2 patents in the field of nanomaterials for cultural heritage. |
Introduction
Chitosan is commonly obtained from the deacetylation of chitin, the second most abundant natural polymer on earth after cellulose.1 The primary sources of chitin are crustaceans such as crabs, shrimps and lobsters, which are highly abundant waste products from the food, beverage and canning industries.2,3 The backbone of chitosan is very similar to that of cellulose, with the hydroxyl group at the C2 position replaced by either an amino or an acetylamino group. Thus, chitosan is a copolymer consisting of N-acetyl-2-amino-2-deoxy-D-glucopyranose and 2-amino-2-deoxy-D-glucopyranose, where the two types of repeating units are linked by (1 → 4)-β-glycosidic bonds. The chemical structure of chitosan is shown in Fig. 1a. It is available within a large range of molecular weights and degrees of deacetylation. These two parameters largely alter the physico-chemical properties of the biopolymer and therefore a variety of specific applications can be considered based on viscosity, biological activity, biodegradability, wettability, colloidal stability and pH responsive features. Chitosan is readily soluble in dilute acidic solutions below pH 6.0 due to the protonation of the amine groups (a pKa value of 6.3). Furthermore, it has gel, fiber and film forming properties. From the biological point of view it shows antimicrobial activity and good compatibility with living tissue.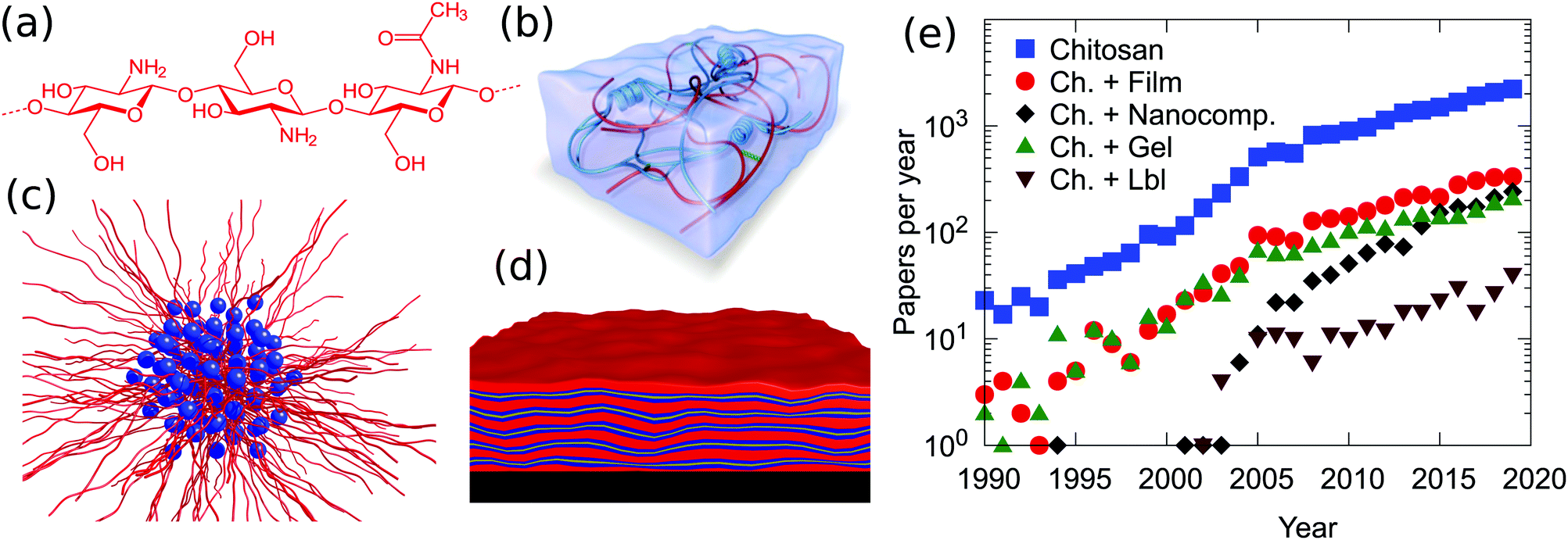 | ||
| Fig. 1 (a) Chemical structure of chitosan and schematic representations of (b) a hybrid hydrogel from ref. 12, (c) chitosan/surfactant complexes, (d) chitosan-based layered coatings and (e) the number of publications per year on chitosan based materials. Data are from Scopus and they were obtained on 1 June 2020 using as searching string TITLE-ABS-KEY (“Chitosan” and “material”) or (“Chitosan” and “material” and “film”), (“Chitosan” and “material” and “gel”), (“Chitosan” and “material” and “LbL”) or (“Chitosan” and “material” and “nanocomposite”). | ||
Chitosan exhibits a combination of physico-chemical features which make this polymer a fundamental component in materials science. With the exception of cationically modified cellulose, it is one of the very few cationic biopolymers available. The saccharidic backbone provides this macromolecule with three fundamental peculiarities, not found in other polymers: (i) a high intrinsic rigidity, with reports of persistence lengths which vary between 5 and 30 nm;4,5 (ii) a relatively large spacing between the charges, with a maximum of 1 change per 5 Å in the case of a fully deacetylated polymer. The actual value is in reality much closer to the Bjerrum length in water of 7.1 Å, i.e., the distance at which the magnitude between electrostatic interactions approaches the thermal energy. The consequence of the large separation, in combination with the high intrinsic rigidity, is the fact that the polymer conformation in solution is less affected by the type and concentration of counterions or by the binding to oppositely charged colloids compared to high charge density, flexible polymers. (iii) A high tendency of forming intra- and inter-molecular hydrogen bonds. Such hydrogen bonds increase, on one hand, the rigidity of the polymer backbone and are, on the other hand, at the origin of the very low tendency of mixing between polysaccharides and other polymers.
Chitosan-based materials have been reviewed in different fields, from molecular separation to food packaging films, from artificial skin to bone substitutes and water treatment.3,6,7 Most of the reviews are focused on a given application of the chitosan-based materials6–10 or in some other cases they explore a wide range of biopolymers, including chitosan, for some specific applications.11 How these and further peculiarities affect the properties of soft, chitosan hybrid compounds in aqueous environments, in thin films, and in nanocomposite systems, schematically represented in Fig. 1b–d, is discussed hereafter.
It is also useful to recall that chitin, the precursor of chitosan and the main structural component of the exoskeleton of crustaceans, is, by design, a very poorly soluble polymer. This intrinsic property is retained in chitosan, the solubility of which in acidic aqueous environment is simply given by the translational entropy of the counterions. In other terms, as soon as the soluble counterions, often acetate ones, are exchanged with much less soluble macroions such as polymers, micelles, or clay particles, the formation of an insoluble complex is observed. We find this general tendency throughout the physico-chemistry of chitosan-based systems, and, in the course of this review, we will highlight how this phenomenon is exploited and which strategies were developed to increase the solubility.
As illustrated in Fig. 1e, chitosan is highly employed in materials science. Film and gel formulations are the most traditional investigated materials, since 2005 nanocomposites have been growing. Until now, less explored but promising, is the possibility to prepare a layer-by-layer system using chitosan as the cationic biopolymer. The observed trends suggest that although the applications of this polymer have been established, some properties, such as mechanical strength, thermal stability, low water content and gas barrier properties, were not good enough to meet this wide range of applications. The preparation of hybrid materials based on chitosan, with both organic and/or inorganic fillers, overcomes some intrinsic limitations and opportunely tunes the physico/chemical properties of the material.
Soft materials in aqueous media
The above-mentioned properties of chitosan make this polymer a unique building block in the field of colloidal chemistry. Accordingly, substantial efforts have been made to characterize chitosan hydrogels or complex mixtures of chitosan with surfactants, polymers, emulsions, etc.Chitosan/surfactant systems
Mixtures of chitosan and surfactants have been a matter of intensive studies.13–15 Given its cationic nature, particular attention has been paid to mixtures of chitosan with oppositely charged anionic surfactants. It can be safely stated that chitosan forms insoluble complexes with strongly ionic surfactants over a wide range of concentrations and mixing ratios,13,16–18 with the formation of insoluble complexes even at very low surfactant concentrations (mM) and large polymer excess.15 A clear explanation of this extremely pronounced low solubility has not been found yet, and the experimental results point towards a kinetically trapped state and highly cooperative binding. The high tendency to form water-insoluble complexes has been exploited for the preparation of beads,13,19 the size of which can be controlled by the preparation method and varies between a few hundred nanometers and a few centimeters. The thickness of the bead wall shows an initial growth with the square root of time, indicating a diffusion controlled process.13 Such beads are highly promising for pollutant recovery applications.19,20 Few studies have been focused on the interaction of chitosan with fatty acids.21–24 Due to the fact that chitosan is soluble in a slightly acidic medium, while long and medium chain fatty acids are solubilized in alkaline conditions, soluble mixtures are found only when chitosan is mixed with short chain carboxylic acids, such as formic, acetic, butyric and valeric acid.21,25 Due to the short alkyl chain, the interactions in these systems are purely electrostatic.21 As soon as the alkyl chain is long enough, lateral, hydrophobic interactions favor the spontaneous formation of micelles, and a much more complex behavior in mixtures with chitosan is observed. This is the case, for instance, of mixtures with undecylenic acid, which exhibit the formation of supramolecular aggregates with a typical size of a few hundred nanometers.26 Mixtures of chitosan with long chain acids, such as oleic, linoleic, palmitic, and stearic acid, have also been studied,22,27 despite their low solubility, as mentioned before. A schematic representation of the structures formed in carboxylic acid/chitosan mixed systems is provided in Fig. 2. However, water soluble complexes can be obtained when chitosan oligosaccharides, which are soluble also in alkaline conditions, are used. An example is mixtures of chitosan oligosaccharide with oleic acid vesicles.28,29 The coating by chitosan decreased the fluidity of the membrane and increased the stability of the liposomes towards shear and flow stresses.28 | ||
| Fig. 2 Schematic representation of structures and the interaction mechanism found in chitosan/carboxylic acids as a function of the acidity of the solution and of the length of the alkyl chain. Insets present the structures observed in the mixtures and provide an insight into the ionic hydrogen bond formed between the carboxylic acid termination and the amine moiety of chitosan. Adapted from ref. 15, copyright 2020, with permission from Elsevier. | ||
The strong limitation found for the incompatible difference in solubilities of chitosan and fatty acids is overcome when the fatty acids are chemically modified to include an oligoethylene oxide block between the aliphatic chain and the carboxylic acid termination. So-called alkyl ether carboxylates were shown to co-assemble with chitosan in a broad variety of structures which are highly responsive to external stimuli.30–34 In particular, depending on the solution acidity and the molecular architecture of the surfactant, multilayer vesicles or compact aggregates embedding small surfactant micelles are obtained.14,31,34 To the best of our knowledge, these surfactant/chitosan systems show the largest structural variety with an exquisite response to pH variations. The large structural variety derives from the high control over the surfactant packing parameter, depending on pH and the ratio between the alkyl chain and the size of the oligo ethylene oxide block. The strong response to even very little changes in solution acidity derives from the very specific ionic hydrogen bond between the carboxylic surfactant termination and the amine moiety of the macromolecule with an estimated strength of 10 kBT.32,35 In contrast to generic electrostatic interactions, the ionic hydrogen bridges are extremely localized and their strength is strongly dependent on pH. The peculiarity of this bond is probably also at the origin of the very unique observation of chitosan–fatty acid mixtures becoming less soluble upon the addition of a nonionic surfactant.32 Finally, the strong structural response of these systems towards mild variations in pH can be exploited for the formulation of environmentally friendly delivery and recovery systems.34
Chitosan-based polyelectrolyte complexes
The formation of supramolecular complexes in mixtures with anionic polyelectrolytes has been extensively studied, and we address the readers to some extensive reviews.36,37 As indicated in the introduction, chitosan is an intrinsically insoluble polymer, and its solubility is provided by the counterion cloud. In mixtures with oppositely charged polyelectrolytes, these many, soluble counterions are exchanged with an ionic macroion. Accordingly, the most common finding is that an insoluble coacervate is formed. This condition has motivated the establishment of various strategies to prepare more water-soluble systems, such as dispersed colloidal complexes or hydrogels. The formation of complexes can also be exploited for the formation of thin films, which are discussed in the next section of this review. A schematic description of the different typologies of chitosan-based polyelectrolyte complexes is given in Fig. 3. | ||
| Fig. 3 Schematic representation of from left to right: complex coacervates, soluble complexes, solid supported thin films, and gellified systems obtained from mixtures of chitosan (in) and oppositely charged polyanions (in blue). | ||
Mixtures of chitosan with virtually every other available polyanion were investigated. However, particular attention has been paid to complexes with polynucleotides, such as DNA38–42 or RNA,43–46 with anionic polysaccharides, such as alginate,9,47–50 hyaluronic acid,51,52 dextran sulfate,53,54 or synthetic polyelectrolytes, such as poly acrylic acid.55–58
The main objective of studying chitosan–polynucleotide mixtures is the understanding and improvement of gene delivery systems.59,60 In this sense, it is essential to determine the factors affecting the affinity between chitosan and the polynucleotide, in order to be able to balance the stability of the complexes and the delivery efficiency. A complex with a too high binding constant cannot release the gene to the target cell. In contrast, a complex with a too low affinity is not able to transport the gene sequence to the target cell. In this sense, isothermal titration calorimetry provides valuable insights into the binding affinity of chitosan and DNA or RNA.40,42,46 The binding is mainly due to the electrostatic interaction between the charged amine group of the polysaccharide and the phosphate unit of the nucleic base41,42 and is therefore affected by the degree of acetylation and the degree of ionization of chitosan.
Given the biocompatible properties of anionic polysaccharides, such as alginate and hyaluronate, their complexes with chitosan have been extensively investigated, mainly as scaffolds for tissue engineering,37,47,48 for drug delivery,47,49 or for pollutant recovery,9,50 to mention some of the most relevant applications. The properties of chitosan/alginate scaffolds can be further improved when a third component is added to the mixture, such as inorganic nanoparticles,61–63 or using chemical cross-linking agents.64,65 Similarly, chitosan/hyaluronic acid complexes are highly interesting in tissue engineering applications due to the combined flexible nature and antibacterial properties of such complexes. In particular, hydrogels can be formed in situ upon injection to the desired tissue due to the slow kinetics of gel formation.66 When chitosan is mixed with dextran sulfate, a strongly charged polyion, the formation of capsules and beads is observed even in large excess of one of the components,53,54 similarly as for mixtures of chitosan with sulfated surfactants described earlier.
Poly(acrylic acid) (PAA) is one of the most relevant synthetic weakly anionic polymers and complexes with chitosan were probed, mainly with the aim of designing pH-responsive delivery systems. The preparation of chitosan/PAA complexes follows two main routes: non-cross linked particles are prepared by mixing chitosan and PAA in different stoichiometries and experimental conditions;55,56 cross-linked chitosan PAA particles are obtained when acrylic acid is polymerized in the presence of chitosan.57
Chitosan-based hydrogels
The challenge of preparing chitosan-based hydrogels lies in the fact that a significant amount of water needs to be retained in the system. Chitosan hydrogels are generally prepared by physical or chemical cross-linking of the polymer chains, keeping enough charges and/or hydrophilic moieties to guarantee sufficient hydration in the network. There are numerous protocols for the preparation of simple and hybrid chitosan hydrogels, which attract huge interest, in particular in the fields of biomedical applications12,67,68 and wastewater treatment.69Physically cross-linked simple hydrogels
Chitosan-based hydrogels are formed through physical or chemical cross-linking between the polymer chains. The simplest procedure consists in increasing the pH of the chitosan solution, thus strongly inducing the solubility of the polymer.70,71 In practice, a concentrated solution of chitosan is brought into contact with an alkaline environment. The swelling of this precipitate/hydrogel is determined by the osmotic pressure of the counterions of the residual charges on the chitosan backbone. In particular, Enache et al. showed that the advancement of the gelation front can be adequately described with Fick's second law.71 Moreover, different studies have reported that the chitosan hydrogel structure becomes more heterogeneous the larger the distance from the hydrogel surface.70,71A different approach consists in using multivalent, negative ions to physically cross-link chitosan via electrostatic interactions. While tripolyphosphate is the most common anionic cross-linking agent,72–75 examples of ionotropic gelation of chitosan by molybdate,76 polyoxometalates,77 sulfate,78 citrate,78 or phytate12 have been also reported. A clear advantage of using tripolyphosphate as a cross-linking agent is the high mechanical stability of the obtained particles. For instance, it was shown that the mechanical strength of chitosan/tripolyphosphate gel beads is approximately ten times higher than that of the analogous beads prepared by cross-linking the polymer with sulfate and citrate.78 Noteworthily, the chitosan/β-glycerophosphate system shows thermally induced gelation when the system is heated at 37 °C, thus being ideally suited for the preparation of injectable chitosan hydrogels.79,80 A further interesting example is the formation of a hybrid chitosan–gelatin hydrogel, the mechanical properties of which are strongly enhanced upon the addition of phytate, a multivalent negatively charged ion, to the hydrogel12 (as depicted in Fig. 4). This system provides an excellent example of how the physico-chemical properties of chitosan are linked to the hydrogel features. In fact, when chitosan is neutralized with sodium phytate, a rather dense precipitate is formed due to the high charge density and stiffness of the polysaccharide. In contrast, a well hydrated, elastic hydrogel is formed when chitosan is co-crosslinked with a flexible, hydrophilic, and loosely charged polymer such as gelatin. Finally, an important property found in chitosan hydrogels formed through ionic cross-linking with anionic polysaccharides is the self-healing capacity of these gels, which originates from the dynamic nature of the ionic cross-linking point.61,81
 | ||
| Fig. 4 Schematic representation of chitosan (A) and gelatin (B) solutions which form a composite hydrogel upon mixing and cooling (E). Optical photos of the dense precipitate formed by chitosan (C) and of the loose precipitate formed by gelatin (D) upon the addition of sodium phytate. The different degree of hydration of the precipitate is a direct consequence of the different charge density and stiffness of chitosan and gelatin. (F) Schematic representation of the chitosan, gelatin, phytate conjoined-network hydrogel. The inset illustrates the structure of the network consisting of physical bridging between the polymer chains cross-linked by multivalent counterions. The cross-bridging of the two networks allows a unique combination of high compressive modulus and toughness to be obtained. Reprinted with permission from ref. 12. | ||
Chemically cross-linked simple hydrogels
Covalent cross-linking of chitosan is also performed in a routine fashion. In many cases, small molecules such as dialdehydes82,83 or Genipin84,85 are used. To improve the elasticity of the gel, polymeric cross-linking agents such as diepoxy polyethylene glycol or dicarboxylic acid polyethylene glycol are also employed.86,87Hybrid hydrogels
To add new functionalities and to adapt the mechanical properties of the hydrogels to the desired needs, the formation of hybrid chitosan-based hydrogels has been extensively probed. The strong adhesive, anti-inflammatory, hemostatic, and bactericidal properties of chitosan make this polysaccharide an excellent candidate for a broad range of biomedical applications of hydrogels. We address the reader to some recent reviews on the topic.6,67,68We have mentioned above the formation of hydrogels based on chitosan and hyaluronic acid for biomedical purposes.52,66,88 To provide mechanical stability to these hydrogels, a covalent cross-linking between the two polysaccharides can be obtained via a Schiff base reaction. Chemical pre-functionalisation of chitosan with a N-succinyl group and of hyaluronic acid with an aldehyde one allows the reaction to occur in situ without the need for additional chemicals.88 Similar cross-linking procedures are applied to other polysaccharide based hybrid hydrogels.89,90 A plethora of different chemical modifications of chitosan to enable the chemical cross-linking within chitosan/polysaccharide networks is described in the literature91,92 and the right choice must be dictated by the field of application, the nature of the components, and the desired physical properties of the resulting hydrogel. Of note are externally-triggered cross-linking reactions, e.g., by photoirradiation.93,94
In summary, extensive work has been performed on the characterization of chitosan-based hydrogels and the understanding of the correlation between molecular properties (the degree of substitution, charge density, complex solubility, and the final gel characteristics). In particular, their use in the field of tissue engineering seems to be highly promising, given the 3D network structure of the gel and the tunable mechanical properties, associated with the strongly adhesive, anti-inflammatory, and anti-bacterial properties of chitosan.
Self-assembly of chitosan-based thin films
Besides hydrogels, polysaccharide assemblies in the form of thin films are very well suited for the design of functional coatings dedicated to biomedical and biotechnological purposes.95–99 Among them, chitosan-based thin films have been widely applied for drug delivery systems,100–102 antibacterial103–106 and antifungal surfaces,102,107,108 food protection and paper packaging,109,110 as well as wound healing.111,112 The broad range of applications is possible, thanks to the excellent biocompatibility, biodegradability, low toxicity and high availability of chitosan, as well as to the tunable film properties (structure, elasticity, porosity) by adjusting both molecular composition and assembly conditions.13One of the most commonly employed strategies for the formation of chitosan-based thin films is to exploit the ionic character of the polysaccharide to form polyelectrolyte multilayers (PEMs).102,113–115 The simplest preparation approach consists in the alternate adsorption of oppositely charged species onto a charged substrate, known as layer-by-layer (LbL) deposition116 (see Fig. 5A). The main driving force for the complexation of oppositely charged macromolecules is the entropic gain associated to the release of counterions,117 a universal prerequisite for both synthetic and natural colloidal species.
 | ||
| Fig. 5 Schematic illustration of different methods for the LbL assembly process: (A) dipping method, (B) consecutive spin-coating, and (C) spraying of oppositely charged polyelectrolytes. From ref. 113. Reprinted with permission from AAAS. | ||
The ease of preparation and extreme versatility of the method come with a remarkably fine control of the film properties, such as thickness, roughness, internal layer structure, elasticity, and porosity, by selecting materials, solvent quality, pH, and ionic strength.118,119
Among the most popular combinations of chitosan with natural polyelectrolytes, we find alginate120–123 and hyaluronic acid.102,106,124 Alginate has the ability to form gels by ion bridges in the presence of multivalent cations, e.g., calcium, due to the presence of carboxyl groups along the polymer backbone. Due to the porous structure and high water-absorption capacity of alginate-based assemblies, it is a promising and largely employed material for the design of wound dressings.125 Concerning hyaluronic acid, it is almost ubiquitous in the human body and serves as an essential component mediating cellular signaling, wound repair, morphogenesis and matrix organization.126 Furthermore, it is popular for its high flexibility, which becomes more relevant with increasing molecular weight.127 Multilayers prepared from its combination with chitosan, which is known to participate in the up-regulation of genes related to calcium binding and mineralization promoting bone formation,128 have been proposed to be used in potential “generic” surface treatment, since a simple variation of scaffold morphology, protein attachment and additive incorporation renders such films suitable for most tissue engineering applications.112 Interestingly, the properties of the individual polyelectrolytes are partially retained in their surface properties, as evidenced by the surface hydrophilicity measured by water contact angle106 (Fig. 6g), and it represents a convenient way to monitor the subsequent layer adsorption. A water contact angle between 80 and 100° is most commonly reported for chitosan-terminated PEMs, while the value decreases to 40–50° for alginate or hyaluronic acid termination.
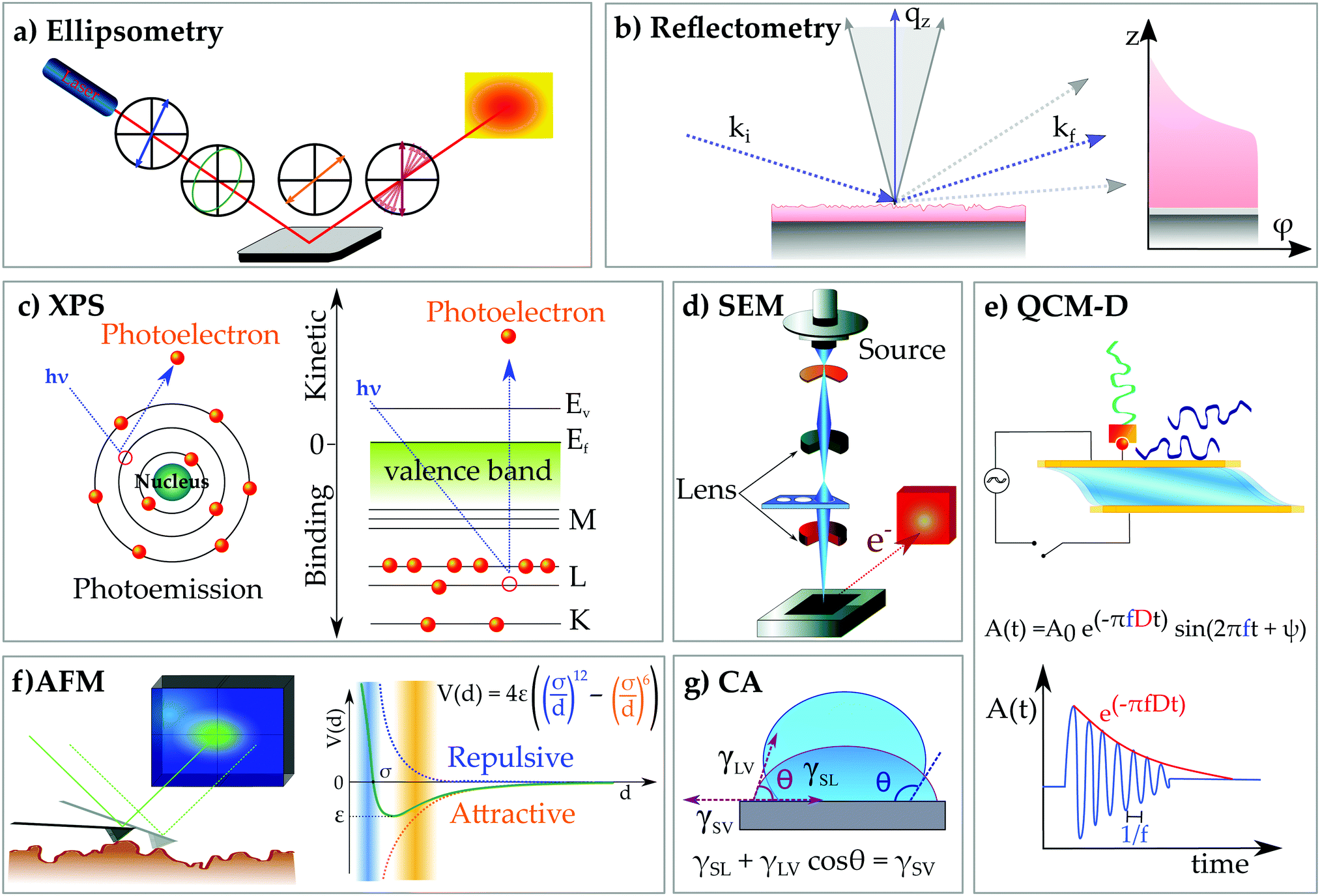 | ||
Fig. 6 Typical techniques for the characterization of thin films. (a) Ellipsometry is based on the detection of the change of polarization of light upon reflection from a substrate, which is described by the ellipsometric angles Δ and ψ. These parameters are related to the amplitude and the phase of the electromagnetic wave according to the fundamental equation: tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ψ·expiΔ = rp/rs with rp and rs being the reflection coefficients of the perpendicular and parallel components of light, respectively. A layer model which describes the sample composition and its optical properties allow us to obtain the thickness d and refractive index n of the thin film from the experimental measurements. (b) Neutron or X-ray reflectometry provides information on the volume fraction distribution of molecules at interfaces. By measuring the reflected intensity of a probe (photons or neutrons) interacting with a solid surface at outgoing angles equal to the incoming one, a reflectivity profile in the reciprocal space, qz, can be reconstructed. Characteristic structural parameters of a film like thickness (d), roughness σ and refractive index can be obtained by fitting the data to a proper model. The scattering properties of the components with respect to the incoming probe are described by the scattering length density ρ, which is correlated to the optical properties of the system by ψ·expiΔ = rp/rs with rp and rs being the reflection coefficients of the perpendicular and parallel components of light, respectively. A layer model which describes the sample composition and its optical properties allow us to obtain the thickness d and refractive index n of the thin film from the experimental measurements. (b) Neutron or X-ray reflectometry provides information on the volume fraction distribution of molecules at interfaces. By measuring the reflected intensity of a probe (photons or neutrons) interacting with a solid surface at outgoing angles equal to the incoming one, a reflectivity profile in the reciprocal space, qz, can be reconstructed. Characteristic structural parameters of a film like thickness (d), roughness σ and refractive index can be obtained by fitting the data to a proper model. The scattering properties of the components with respect to the incoming probe are described by the scattering length density ρ, which is correlated to the optical properties of the system by  with λ being the probe wavelength. (c) X-ray photoelectron spectroscopy provides information on the chemical composition of a specimen adsorbed onto a surface. It is based on the photoemission of electrons from an excited sample, the kinetic energy distribution of which is measured to derive the chemical composition and electronic state of the sample surface. By total energy transfer from the photons to the core-level electrons of atomic or molecular orbitals, these electrons are emitted from the sample surface and separated according to their energy, and so quantified. (d) Scanning electron microscopy uses a beam of electrons generated by an electron gun and guided through within vacuum in an electromagnetic field towards the specimen surface to study the morphological properties of a surface. When the beam strikes the specimen surface, its high energy electrons interact with the valence electrons of the atoms in the sample, and these are ejected (secondary electrons). These electrons are then translated in changes of the brightness of the corresponding point onto the screen, leading to the contrast of light and dark areas. (e) Quartz-crystal microbalance with dissipation monitoring (QCM-D) is a highly sensitive balance, which is able to detect nanograms of mass coupled to a substrate by detecting changes of the oscillation frequency of a quartz crystal. QCM-D uses the inverse piezoelectric effect, meaning that a driving sinusoidal potential is applied to a quartz crystal to excite the crystal oscillation forward and backward to its resonance frequency and higher overtones. When the potential is cut off, the damped amplitude of the freely decaying oscillation is recorded, and the frequency shift, Δf, and the dissipation energy, D, quantify the coupled mass and energy dissipated by the specimen. (f) Atomic force microscopy (AFM) measures the interaction between a specimen and a solid probe scanning over the surface in either static or dynamic (oscillating) mode. From its deflection of the probe, morphological features from the atomic to the macroscopic (millimeters) scale can be explored. The mapping of the surface topology is a reconstruction of the interaction forces, with near-field forces approximated to Lennard-Jones potentials. (g) Contact angle measurements are performed by depositing a droplet of a liquid, in most cases water, onto a bare or modified surface to determine its wetting behavior and therefore the surface hydrophilicity. The contact angle is defined as the geometrical angle between the substrate and the droplet, and it is correlated to the surface tension of all the involved interfaces by the Young equation. with λ being the probe wavelength. (c) X-ray photoelectron spectroscopy provides information on the chemical composition of a specimen adsorbed onto a surface. It is based on the photoemission of electrons from an excited sample, the kinetic energy distribution of which is measured to derive the chemical composition and electronic state of the sample surface. By total energy transfer from the photons to the core-level electrons of atomic or molecular orbitals, these electrons are emitted from the sample surface and separated according to their energy, and so quantified. (d) Scanning electron microscopy uses a beam of electrons generated by an electron gun and guided through within vacuum in an electromagnetic field towards the specimen surface to study the morphological properties of a surface. When the beam strikes the specimen surface, its high energy electrons interact with the valence electrons of the atoms in the sample, and these are ejected (secondary electrons). These electrons are then translated in changes of the brightness of the corresponding point onto the screen, leading to the contrast of light and dark areas. (e) Quartz-crystal microbalance with dissipation monitoring (QCM-D) is a highly sensitive balance, which is able to detect nanograms of mass coupled to a substrate by detecting changes of the oscillation frequency of a quartz crystal. QCM-D uses the inverse piezoelectric effect, meaning that a driving sinusoidal potential is applied to a quartz crystal to excite the crystal oscillation forward and backward to its resonance frequency and higher overtones. When the potential is cut off, the damped amplitude of the freely decaying oscillation is recorded, and the frequency shift, Δf, and the dissipation energy, D, quantify the coupled mass and energy dissipated by the specimen. (f) Atomic force microscopy (AFM) measures the interaction between a specimen and a solid probe scanning over the surface in either static or dynamic (oscillating) mode. From its deflection of the probe, morphological features from the atomic to the macroscopic (millimeters) scale can be explored. The mapping of the surface topology is a reconstruction of the interaction forces, with near-field forces approximated to Lennard-Jones potentials. (g) Contact angle measurements are performed by depositing a droplet of a liquid, in most cases water, onto a bare or modified surface to determine its wetting behavior and therefore the surface hydrophilicity. The contact angle is defined as the geometrical angle between the substrate and the droplet, and it is correlated to the surface tension of all the involved interfaces by the Young equation. | ||
Similar to synthetic weak polyelectrolytes,129,130 the properties of both chitosan/alginate and chitosan/hyaluronic acid pairs are pH dependent, which allows the properties of the resulting films to be tuned. In particular, thicker and rougher layers can be formed at a pH between 3 and 5,121 where the charge mismatch between low charge density chitosan and high charge density alginate (or hyaluronic acid) chains leads to the adsorption in a more coiled conformation. Such charge mismatch induces also larger mass adsorption to achieve charge neutralization.131 The increment of surface roughness when chitosan/hyaluronan multilayers are used to coat solid substrates is very advantageous for cell adhesion, proliferation and differentiation, making them excellent bone scaffolds.132 Regardless of the pH, the increase of ionic strength always results in the formation of thicker layers due to a stronger extrinsic charge compensation123,133,134 and, analogous to synthetic polymers, ionic strengths below 10−4 mol L−1 prevent layer growth.135
The ensemble of characteristics of a deposition protocol, namely polymer molecular weight and architecture, ionic strength and type of ions, pH, and temperature, always defines the growth mechanism and the final properties of the multilayer, with the growth mechanism being an indication of the interaction strength within complexes. The low charge density, a consequence of the large intrinsic persistence length characteristic of chitosan, is the reason for the non-linear growth regime observed during the LbL deposition of several chitosan-based PEMs.105,131 When the chitosan chains are loosely bound, they diffuse within the film, which enhances the mass uptake per adsorption cycle. The growth mechanism is therefore governed by the ability of chitosan to diffuse through the pre-adsorbed layers (in/out model),136 with high MW chains diffusing less than their low MW equivalents.131 A convenient way to highlight this two-step layer growth (adsorption and diffusion) is by monitoring the polyelectrolyte adsorption in situ by quartz crystal microbalance with dissipation monitoring (QCM-D) (Fig. 6e). The working principle of the technique exploits the piezoelectric properties of a quartz crystal under the application of an oscillating shear stress. The freely decaying damped sinusoidal oscillation of the crystal measured between subsequently applied shear deformations is registered and analysed with respect to the oscillation of an unloaded crystal. As a result of the mass coupled to the surface, the oscillation frequency f decreases, while the speed of the amplitude decay can be interpreted as a measure of the energy dissipated by the system coupled to the surface, i.e. the viscoelasticity of the film. The oscillation of the sensed mass has been a valid parameter to identify a typical behavior of highly diffusive polymers, which give very large mass uptake upon each adsorption step. This is the case of chitosan in combination with both alginate and hyaluronan, the layer growth of which can show a characteristic odd–even effect.112,137 In addition to the diffusion through the film, such a behavior can also be explained by the pronounced hydration of chitosan layers, responsible for the higher mass uptake (hydration water) and pronounced layer swelling,114,121 therefore being well identifiable from the frequency shift/dissipation change upon layer formation and in combination with measurements of the “dry” mass by ellipsometry (Fig. 6a). Differently from chitosan, high charge density polymers like poly-L-lysine lead to a denser sequence of interaction sites per chain and stronger PE–PE complexation at first contact123 like the case of alginate/poly lysine complexes forming very compact PEMs.
Besides the association with biopolymers, fundamental studies of chitosan with synthetic polymers, for instance poly(acrylic acid), have been particularly useful.138,139 One of these studies have highlighted another mechanism leading to the exponential growth: the formation of islands by the first deposited layers, which grow laterally and vertically with the number of deposition cycles.119,136
A fundamental aspect for (bio)technological applications of functional coatings is their stability under physiological and harsh conditions. For chitosan-based thin films, it has been observed that post-preparation stabilization via cross-linking is a valuable tool to enhance their mechanical and chemical stability under both acidic and alkaline conditions.140,141 In addition to providing mechanical stability, cross-linking seems to control the extent of protein adsorption onto the modified substrate.112,142 This possibility is not offered by all polymers, and it renders chitosan particularly interesting for post-synthetic modifications. Particularly relevant for regenerative tissue engineering and as antimicrobial surfaces143 are those functional coatings combining chitosan with pectin. In this case, the 1:1 charge ratio achieved at pH 5.6 suppresses the chitosan diffusion and reduces its water uptake, which results in a linear thickness increment with initial mass uptake attachment and a slower chain rearrangement.134 An important prerequisite for the film stability is that both polyelectrolytes are charged, and this corresponds to the pH between 3.6 and 7, and the salt concentration between 0.05 and 0.15 M NaCl.144
Another very promising system for regenerative medicine is represented by chitosan/collagen films. Collagen is a fibrous protein that plays an important role in tissue healing, providing a suitable biological environment for cell growth and attachment, migration, and proliferation.145 Collagen thin films and low or high MW chitosans showed enhanced tensile strength and elongation at break compared to pure collagen films, as the intermolecular interactions within the matrix mitigate the strong intramolecular interaction within collagen chains, which increases the film flexibility.146 Furthermore, chitosan contributes to preserve the native structure of collagen, limit hydrolytic and enzymatic degradation, and reduce the swelling of the collagen film (efficient moisture barrier), which could allow a controlled release of epidermal growth factors, when films are used as wound dressings.146,147
Among the rich variety of combinations of chitosan and other polysaccharides in thin films for biomedicine and biotechnology, it is worth mentioning the use of silk,148 casein,149 fucoidan,111 cellulose,150 and DNA.151 In some cases, highly cross-linked structures, as confirmed by surface chemical characterization through X-ray photoelectron spectroscopy, are formed by reactive side groups. Furthermore, enhanced biofilm stability, like mucin, against degradation in surfactant solutions has been proven in the presence of chitosan, as the interactions with the polysaccharide reduce the hydrophobic interactions with the surfactant molecules and preserve the binding to the solid substrates.152 Finally, the possibility of driving the assembly of inorganic nanoparticles (NPs), e.g. gold (Au)-NPs,153 for the preparation of surface electrodes and impedance spectroscopy studies has also been reported, which is added to the numerous examples of highly relevant applications of chitosan-based thin films for the design of bioengineering surfaces, among which are antimicrobial surface102,103,154 and modulated drug release,102 biosensing,155 anticancer treatment,156 anticoagulant for implants in cardiovascular surgery,157 food preservation,110,158 lubrication,99 release of fertilizers,159 and flame retardants.107
For most of these applications, simple preparation methods, simple to speed and scale up for industrial purposes, are fundamental, and therefore alternate or simultaneous spin-coating or spraying of polyelectrolyte solutions160 is a valid alternative to the LbL deposition (Fig. 5B and C). Spraying polyelectrolyte solutions onto a substrate leads to similar structures as dipped multilayers, with minor differences in the growth kinetics of the very first layers due to the suppression of diffusive events,161,162 which results in thinner and smoother layers, as revealed by atomic force and fluorescence microscopy of synthetic polyelectrolytes,163,164 and it has been successfully applied to prepare chitosan-based multilayers onto flat100,165 or curved surfaces.159 The shape and dimensions of chitosan:polyanion complexes remain crucial parameters for the film buildup by either alternate or simultaneous spraying, and they can be properly tuned by the mixing ratio and physico-chemical bulk properties.
The peculiarity of charge inversion from the cationic to the anionic form by proper chemical derivation has awarded the possibility of self-assembling “one component” multilayers, fully based on chitosan.166 The resulting thin films were very smooth and characterized by a linear layer increment, due to the equal charge density along the chain between polyanionic and polycationic forms. Interestingly, their complexation was more exothermic (lower ΔHmix) and more entropically favored (higher ΔSmix) than that for other chitosan/synthetic PE pair, resulting in an overall enhanced film stability.
Less popular than the LbL deposition, another method for thin film preparation is the Langmuir–Blodgett technique. In this case, a Langmuir monolayer is formed by spreading an amphiphilic (macro)molecule dissolved in a volatile organic solvent on the surface of an aqueous subphase.167 The molecules orient their hydrophilic part in the aqueous subphase and the hydrophobic moiety towards the hydrophobic phase (air). Such a monolayer is then transferred onto a solid substrate by the Langmuir–Blodgett (LB) process168 by immersing (or emerging) a solid support in (or from) the aqueous subphase to recover the monolayer, with the possible formation of multilayers by multiple dipping iterations. LB multilayers have been prepared from amphiphilic chitosan derivatives, as well as their mixtures with phospholipids and cholesterol.169 In this case, chemical modifications are a fundamental prerequisite to render chitosan soluble in organic solvents, and most frequently long alkyl chains are attached to the primary hydroxyl and amino group for this purpose.169,170
The highly tunable structure of chitosan:fatty acid complexes in solution, forming multiwalled vesicles, was prepared in bulk under tailored conditions (pH and mixing ratio) to obtain the desired dimensions and number of layers,31 which have been the key properties for the development of a novel approach for the preparation of chitosan–surfactant multilayers from “one step” deposition, as schematically represented in Fig. 7.33 This method overcomes the limitation of time consuming LbL assembly and offers more control over the internal layer structure than spraying methods. Chitosan:fatty acid complexes were then transferred onto a solid substrate by a single spin coating step, which spontaneously formed multilayers with a high degree of inter-layer segregation. The control of structural key parameters, e.g. thickness and number of layers, from the bulk properties of the mixture ease significantly the preparation of films with tailored properties and functions. Furthermore, the low degree of intermixing between subsequent layers leading to high layer segregation makes this kind of multilayer suitable for selective release/uptake and exclusive response of individual parts to external stimuli. The highly segregated internal structure with individual water uptake of the hydrophilic moieties (chitosan layers) could be revealed only by exploiting the isotopic contrast of neutron reflectometry. In general, both X-ray and neutron reflectometry allow the determination of the internal volume fraction distribution of each component along the axis perpendicular to the surface. Their complementarity is due to the fact that their probes, photons and neutrons, interact with different subatomic elements of an atom (the electron cloud and the nuclei, respectively) and therefore provide a different internal contrast of the same systems. In addition, neutrons interact differently with isotopes of the same nucleus, and this property can be used as a tool for identifying the internal structures of chemically homogeneous materials.
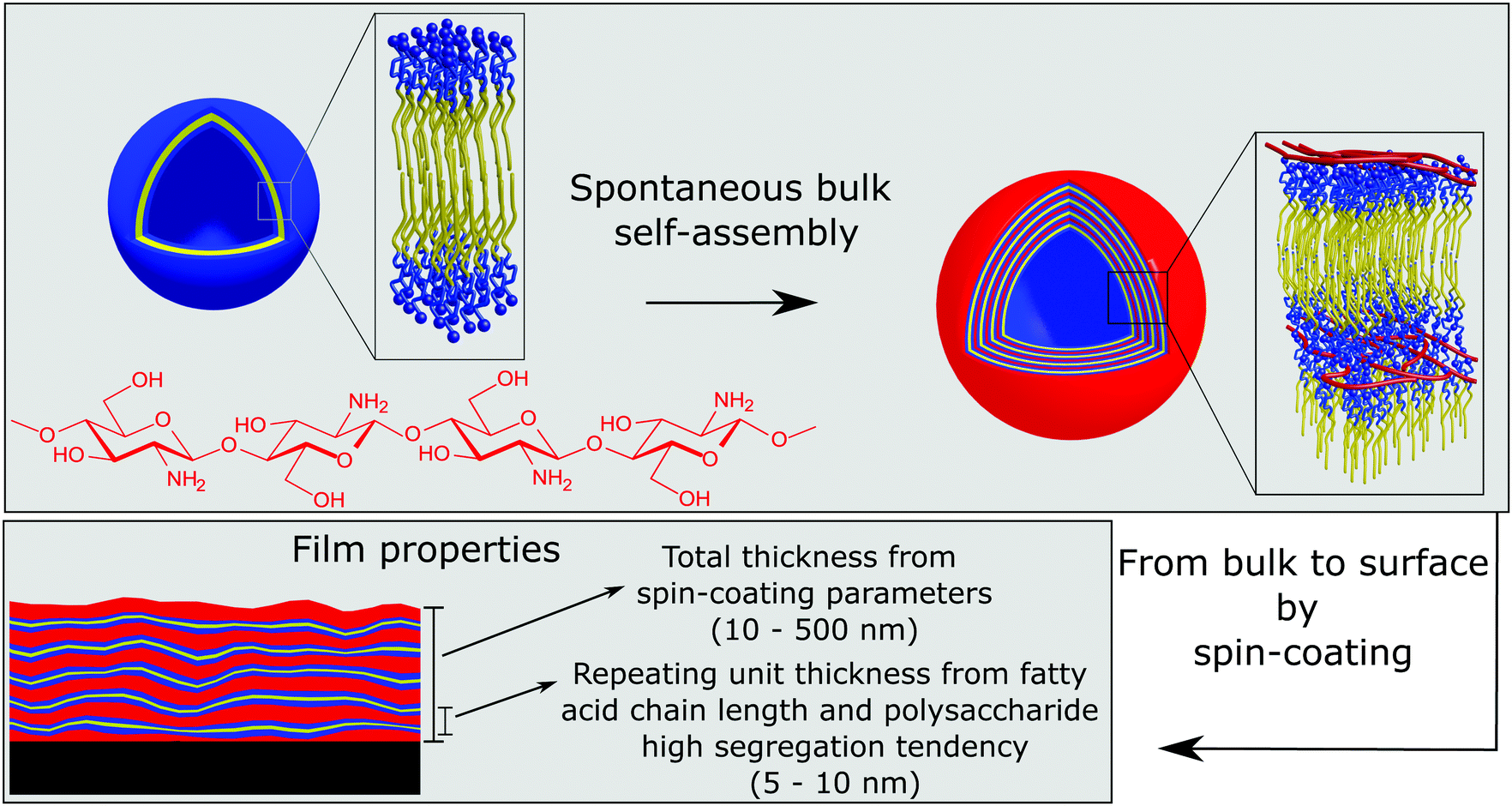 | ||
| Fig. 7 Schematic representation of the formation of chitosan/fatty acid multilayered thin films via a one-step procedure which exploits the spontaneous association of the components into multilayered vesicles in solution. Adapted with permission from ref. 33. | ||
Our results showed that the morphology of coatings produced by this method is macroscopically highly homogeneous, but it phase separates microscopically on a length scale of 5 nm. The electrostatic interaction between the amino-group of chitosan and the carboxylic termination of the fatty acids and the very low miscibility of chitosan with both hydrophilic and hydrophobic materials are at the origin of the very small-scale segregation. Since the size of the microseparated domains depends on the degree of polymerization of the polymer and on the Flory–Huggins interaction parameter(s) χ with different film components, a common strategy to prepare thin films with sub-10 nm structural features, essential for novel applications in nanolithography, is to use oligosaccharide based block copolymers.171,172
Finally, the preparation of chitosan thin films from end-tethering polymer chains onto a solid surface, a geometry known as polymer brush,173,174 is worth mentioning. Either neutral or modified chitosan chains by quaternary ammonium salts, CHI-Qx, were grafted to the epoxide derivatized silicon oxide surface,175,176 with characteristic dry thickness from 5 to 50 nm from fully charged (CHI-Q100) to partial (CHI-Q50, CHI-Q25) to neutral brushes, respectively, due to a decreased grafting density for a higher charge density. An interesting property of polymer brushes is their swelling behavior, which can be exploited to design (bio)sensors and microactuators. These chitosan-based brushes have shown a variable swelling behavior over a broad pH range as a function of their quaternization form, with CHI-Q100 swelling up to 5 times their dry thickness at pH 5 and with an intermediate swelling degree symmetrically around this pH. Partially modified CHI-Q50 brushes were swelling symmetrically around pH 4.5, which is likely due to the balance between increasing protonation of primary amines below pH 6.5 and quaternary ammonium salts above pH 6.5. In contrast, the swelling of native chitosan and CH-Q25 was constant from pH 8.2 to 5.5 and increases near pH 4. The swelling behavior could be rationalised in terms of the size of the counterions condensed around the charged groups: for fully protonated brushes, the swelling clearly increases with the size of counterions, while for partially protonated brushes the influence of the ions is evident at high pH, where there is a complete exchange of counterions from chloride to hydroxide, which have larger hydration shells. Such an effect is not visible for conditions of low pH and a low degree of quaternization, where the ammonium cation content is below a critical value.
Chitosan brushes also exhibited a reduced bacterial attachment/growth of about 30 times compared to the silane (APTES)-modified surface, which has been explained by the capability of the quaternary salt of disrupting the bacterial cell membrane, as well as by the flexible nature of polymer brushes.177 In fact, S. aureus biofilms adhered strongly to silicon oxide and CH surfaces even at a high shear stress (up to 12 dyne per cm2), whereas they detached at a low shear stress (1.5 dyne per cm2).
Brushes were prepared also from chitosan-grafted-poly(ethylene glycol) (PEG) copolymers,178,179 reaching a very high degree of substitution, and adsorbed onto a thiol-modified gold substrate by microcontact printing, covalent grafting and solution adsorption, with the latter leading to the highest polymer adsorption. The presence of PEG units has been crucial to reach high surface adsorption, as revealed by QCM-D studies, and it enhances the hydration degree of chitosan thin films.
Chitosan-based nanocomposites and solid films
The combination of polymers and inorganic nanoparticles represents a well-known strategy to obtain hybrid materials with unique performances as well as specific functionalities. Among sustainable polymers, chitosan was largely employed as a matrix for the fabrication of bionanocomposites suitable for several applications, such as tissue engineering,180,181 drug delivery,182,183 gas sensors,184 packaging,185,186 remediation187,188 and cultural heritage.189,190 Such a wide industrial interest is related to some interesting features of chitosan itself in the solid state. In particular, chitosan and its phosphorylated derivative are flame retardant and therefore they are perspective additives to control the flammability properties of polyethylene or to produce self-extinguishing cotton fabrics.191–193 Additionally, chitosan, being a polycation, has a broad-spectrum antimicrobial activity against both Gram-positive and Gram-negative bacteria as well as fungi that can be further enhanced by transforming the primary amine groups into quaternary salts with permanent positive charge.194The mechanical performance of chitosan composites is affected by the crystallinity that has a great influence on tensile strength. Therefore, chitosan films fabricated through solvent casting from acetic acid may have significant differences in mechanical properties if DDA, pH and water content in the final composite (relative humidity), which highly influence the polymer crystallinity, are altered.195,196
Besides the chemical modification of the chitosan structure, both natural and synthetic nanoparticles were successfully filled within the chitosan matrix to tune its properties. The nanocomposite preparation includes physical and chemical methods.198 Metal nanoparticles (Cu, Ag and Au) were embedded in chitosan through the following subsequent steps:199 (1) metal vapour synthesis for the preparation of the metal nanoparticle sols and (2) deposition of the metal nanoparticle sols on chitosan supports. Chitosan/ZnO nanocomposites were fabricated by the microwave heating technique,187 which reduced the reaction time for the ligand substitution occurring between the functional groups of the biopolymer and the zinc cations of ZnO nanoparticles. The addition of ZnO nanoparticles improved the removal capacity of chitosan towards methylene blue.187 The freeze-drying process was employed for the filling of nano-hydroxyapatite particles within the chitosan matrix,200 while the ultrasonic-assisted method was used for the fabrication of composite scaffolds based on the chitosan hydrogel and multiwalled carbon nanotubes (MWCNTs).181 The addition of nano-hydroxyapatite improved the compression behaviour of the chitosan scaffold in terms of elasticity and flexibility.200 A multifunctional hybrid material composed by chitosan, graphene oxide (GO) and iron oxide (IO) was obtained by the hydrothermal method exploiting the 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) reaction chemistry.201 The chitosan–GO–IO nanocomposite revealed an efficient antimicrobial ability towards both Gram-positive (Staphylococcus aureus) and Gram-negative (Escherichia coli) bacteria.201 Due to its super-paramagnetic properties, the chitosan–GO–IO hybrid can be easily separated from the bacteria and reutilized for subsequent biocide applications.201
The aqueous casting method was largely employed in the fabrication of chitosan based nanocomposites containing natural clay nanoparticles, such as kaolinite nanosheets202,203 and halloysite nanotubes (HNTs).203–205 A previous study180 reports that the amino-modification of the halloysite outer surface can favour the chitosan/HNT interfacial interactions allowing hybrid films with excellent tensile and thermal properties to be obtained. Additionally, the amino-modified clay nanotubes strongly improved the water vapour transmission rate of chitosan, making the bionanocomposite films promising for biomedical purposes.180 Ethylene glycol diglycidyl ether (EGDE) was used as a cross-linker for the preparation of chitosan/amino-modified halloysite composite films. In the process, the hollow tubular shape of halloysite HNTs-NH2 was not altered by the amino functionalization, and the composite film exhibits a porous structure, as shown by scanning electron microscopy (Fig. 8a). It was observed that the swelling ratio of chitosan based nanocomposites decreases with the HNTs-NH2 content (Fig. 8b).
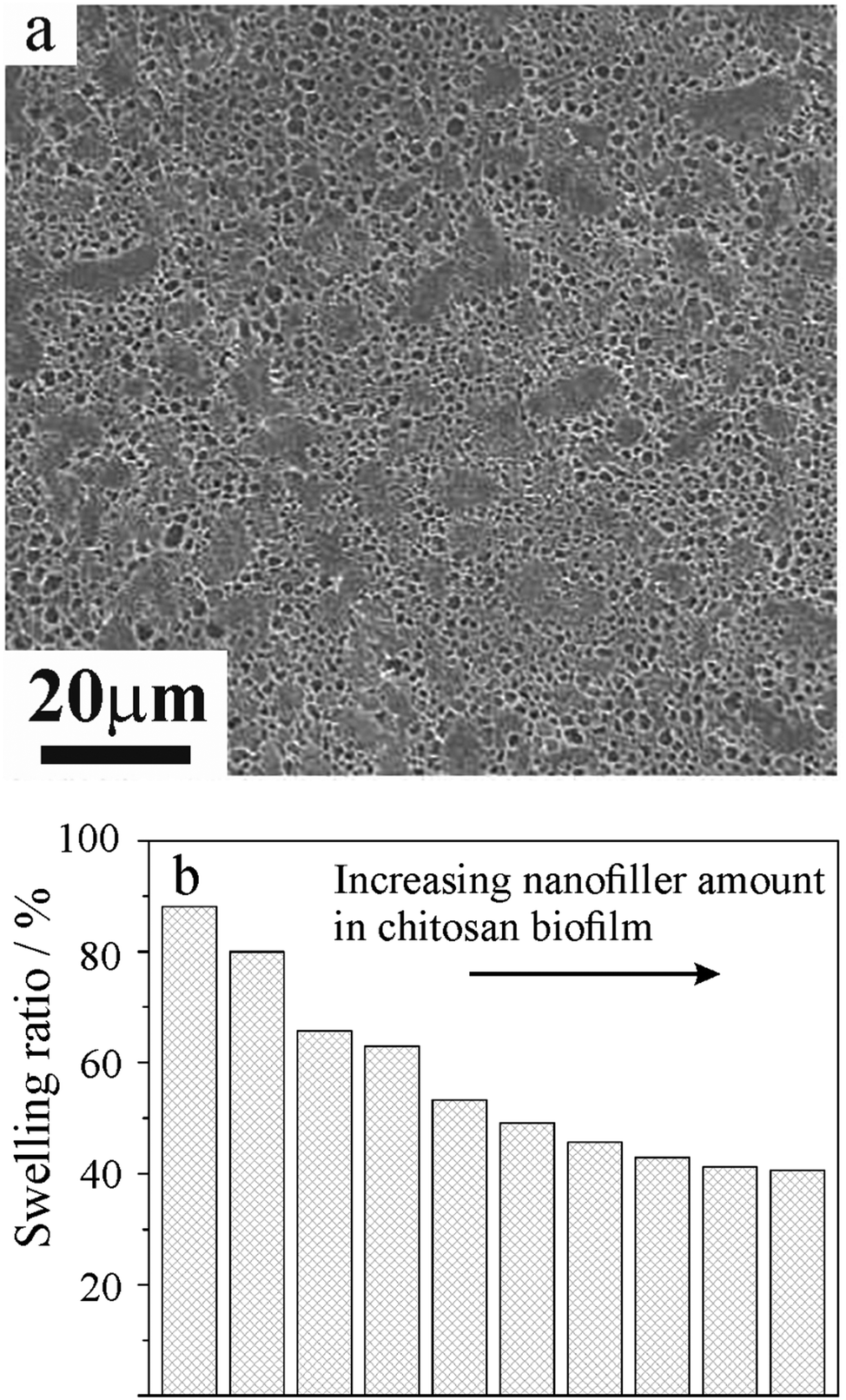 | ||
| Fig. 8 (a) SEM image of the chitosan/HNTs-NH2 nanocomposite film containing ethylene glycol diglycidyl ether (EGDE) as the cross-linking agent. (b) The swelling ratio of chitosan/HNTs-NH2 nanocomposite films. Adapted from ref. 180, coryright 2020, with permission from Elsevier | ||
Within tissue engineering applications, a chitosan/halloysite composite scaffold was fabricated by the combination of solution-mixing and freeze-drying techniques.206 The presence of clay nanotubes induced an improvement of both the compressive behaviour (in terms of strength and Young modulus) and the thermal stability with respect to the scaffold based on pristine chitosan.206 Nanoclays with variable morphology (bentonite, sepiolite, and montmorillonite) were successfully filled into chitosan blended with glycerol by using the casting technique from water.207 Similarly, the chitosan/polyvinyl alcohol (PVA) blend was reinforced with different concentrations of bentonite nanoparticles combined with anthocyanin in order to obtain antibacterial films with improved thermo-mechanical performances.183 The casting procedure was effective in the preparation of films composed by copper oxide (CuO) nanoparticles and chitosan doped with glycerol ionic liquid.184
Recently, chitosan/halloysite nanocomposite films with a sandwich-like structure, as sketched in Fig. 9a, were fabricated by using a sequential casting method.197 The preparation protocol is based on the sequential deposition of chitosan and halloysite aqueous suspensions under controlled pH conditions. SEM images (Fig. 9b) showed that the nanocomposite possesses a multilayer morphology being that halloysite nanotubes are confined between the outer chitosan layers. Compared to pure chitosan, a significant increase (up to ca. 150 °C) of the ignition temperature, as well as the enthalpy of the oxidation (Fig. 9c), was detected in the hybrid films as a consequence of their layered structure.197 The flame retardant features of materials where the polymer and inorganic particles, typically clays, are alternated are well known but a large number of multilayers with micro/nanosized thickness is required;208 on the other hand, the flame retardant features of the chitosan itself combined with the peculiar pH dependent solubility endow the reduction of flammability in more simple layered structures that are easy to generate. Accordingly, the sequential casting procedure can be considered a successful protocol to fabricate chitosan based nanocomposites with flame retardant properties.
 | ||
| Fig. 9 Schematic representation of the chitosan/HNT nanocomposites with a layered structure. (a and b) SEM micrographs at different magnifications of the cross section of chitosan/HNT nanocomposites with a mass ratio of 0.62. (c) Enthalpy and onset temperature of oxidative degradation as functions of the HNT/chitosan mass ratio for the layered nanocomposites. Adapted with permission from ref. 197. | ||
Nanocomposite films formed using the chitosan/polyvinyl alcohol (PVA) blend as the matrix and graphene oxide/hydroxyapatite/gold nanoparticles as fillers were prepared by the gel casting method using glutaraldehyde as the crosslinker.209 These bionanocomposite films are promising for bone tissue regeneration as evidenced by the MTT assays and ALP straining results, which evidenced their capacity to enhance the osteoblast differentiation.209 Within biomedical applications, the addition of rectorite clay particles into chitosan allowed a composite viscous mucus with injectable properties for skin hemostasis to be obtained,210 whereas montmorillonite clay was introduced to the methacrylated glycol chitosan (MeGC) hydrogel, which was obtained by using riboflavin as a photoinitiator.211 The composite hydrogel evidenced a well interconnected microporous structure promoting the cell infiltration, proliferation, and in situ differentiation.211 Hybrid gel beads based on chitosan and halloysite were prepared through the dropping and pH-precipitation method, which is based on the drop-wise addition of chitosan/halloysite dispersion into an aqueous NaOH solution.182,188 The chitosan/HNT gel beads exhibited higher adsorption capacities towards dyes (methylene blue and malachite green) with respect to those of chitosan gel beads.188 As concerns pharmaceutical purposes, chitosan/HNT gel beads were shown to be efficient in the controlled release of doxycycline (an antibiotic of the tetracycline class), highlighting their suitability as a drug delivery system.188 Interestingly, drug release can be extended by covering the surface of the chitosan/HNT gel beads with alginate exploiting the electrostatic attractions occurring between the biopolymers, which are oppositely charged.188 Regarding Cultural Heritage, hybrid gels with surface cleaning ability were fabricated by mixing a chitosan aqueous solution with a Pickering emulsion based on HNTs and n-decane.189 It should be noted that a subsequent drop-wise addition of NaOH solution was conducted to obtain the gel phase from the chitosan/HNTs/n-decane mixture.189
Summary and perspectives
Chitosan exhibits a unique set of physico-chemical characteristics, most notably its low solubility, high intrinsic rigidity, large charge separation, and strong tendency to form intra- and intermolecular hydrogen bonds. In this contribution, an overview of recent advances in chitosan-based materials is presented, whereby attention has been paid to clarifying how the peculiarities of chitosan affect the physico-chemical properties of the resulting materials.The focus of the review was placed on chitosan-based aqueous systems, thin films, and composite materials. Few polymers have attracted comparable attention and have been used for the design of so different systems. Accordingly, an overwhelming amount of literature appears each year on the topic. Similarly, the number of patents involving chitosan is continuously growing, indicating that chitosan not only attracts the interest of the scientific community but also finds wide practical application. Chitosan is abundant (second to cellulose among biopolymers), competitive for physico-chemical properties and its use fits the idea of a circular economy as it is a byproduct of the food industry.
A contrast, commonly found in the field of materials science, is that the rapid development of applications goes with a comparably slow progress in the understanding of the fundamental properties of the investigated system. Chitosan makes no exception to this rule. In fact, very fundamental questions about chitosan remain unanswered. For instance, a random distribution of N-acetylamine units along the polymer backbone is assumed, despite the fact that no evidence for chitosan being a random copolymer was ever presented. From a biological viewpoint, the mechanisms of interaction of chitosan with cell, bacteria, and plants are not well understood and therefore a discrepancy in the reproducibility of bioactivity is observed. It seems clear that although several mechanisms of action are reported, the most established idea is that electrostatic interactions between chitosan and the anionic molecules of cells/DNA may control the bioactivity instead of interactions with a specific receptor.
Moreover, the interactions determining the behavior of hybrid systems can strongly vary: from non-specific, long range, electrostatic interactions, through more specific hydrogen bonds, to short-range and highly directional ionic bridges. Such a diversity makes systematic studies difficult to perform and predictions hardly apply to a broad variety of systems. Clearly, a better understanding of the behavior of this macromolecule is needed to improve our capacity to design chitosan-based materials. In conclusion, it is generally true that the physical and chemical properties of chitosan-based materials can be rationalized on the basis of the physico-chemical properties of this important bio-macromolecule. It is equally true that further fundamental and applied studies are required to improve our capacity to predict the properties of highly complex, multi-component, chitosan based materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by Progetto di ricerca e sviluppo “AGM for CuHe” (ARS01_00697) and University of Palermo. The experimental work performed on chitosan–fatty acid complexes and thin films has profited from the laboratory infrastructure provided by the Partnership of Soft Condensed Matter (PSCM) at the Institut Laue-Langevin.References
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- C. Tang, N. Chen, Q. Zhang, K. Wang, Q. Fu and X. Zhang, Polym. Degrad. Stab., 2009, 94, 124–131 CrossRef CAS.
- K. B. Mukhtar Ahmed, M. M. A. Khan, H. Siddiqui and A. Jahan, Carbohydr. Polym., 2020, 227, 115331 CrossRef CAS.
- M. Rinaudo, M. Milas and P. Le Dung, Int. J. Biol. Macromol., 1993, 15, 281–285 CrossRef CAS.
- C. Schatz, C. Viton, T. Delair, C. Pichot and A. Domard, Biomacromolecules, 2003, 4, 641–648 CrossRef CAS.
- H. Wang, J. Qian and F. Ding, J. Mater. Chem. B, 2017, 5, 6986–7007 RSC.
- Y. P. Singh, J. C. Moses, N. Bhardwaj and B. B. Mandal, J. Mater. Chem. B, 2018, 6, 5499–5529 RSC.
- F. Khan, D. T. N. Pham, S. F. Oloketuyi, P. Manivasagan, J. Oh and Y.-M. Kim, Colloids Surf., B, 2020, 185, 110627 CrossRef CAS.
- W. Zhang, H. Wang, X. Hu, H. Feng, W. Xiong, W. Guo, J. Zhou, A. Mosa and Y. Peng, J. Cleaner Prod., 2019, 231, 733–745 CrossRef CAS.
- L. Xing, Y.-T. Fan, L.-J. Shen, C.-X. Yang, X.-Y. Liu, Y.-N. Ma, L.-Y. Qi, K.-H. Cho, C.-S. Cho and H.-L. Jiang, Int. J. Biol. Macromol., 2019, 141, 85–97 CrossRef CAS.
- M. Nasrollahzadeh, N. Shafiei, Z. Nezafat, N. S. Soheili Bidgoli and F. Soleimani, Carbohydr. Polym., 2020, 241, 116353 CrossRef CAS.
- L. Xu, C. Wang, Y. Cui, A. Li, Y. Qiao and D. Qiu, Sci. Adv., 2019, 5, eaau3442 CrossRef.
- M. Rinaudo, N. R. Kil'deeva and V. G. Babak, Russ. J. Gen. Chem., 2008, 78, 2239–2246 CrossRef CAS.
- L. Chiappisi, I. Hoffmann and M. Gradzielski, Soft Matter, 2013, 9, 3896–3909 RSC.
- L. Chiappisi and M. Gradzielski, Adv. Colloid Interface Sci., 2015, 220, 92–107 CrossRef CAS.
- C. Onesippe and S. Lagerge, Carbohydr. Polym., 2008, 74, 648–658 CrossRef CAS.
- Y. C. Wei and S. M. Hudson, Macromolecules, 1993, 26, 4151–4154 CrossRef CAS.
- L. Petrović, J. Milinković, J. Fraj, S. Bučko, J. Katona and L. Spasojević, Colloid Polym. Sci., 2017, 295, 2279–2285 Search PubMed.
- S. Peretz, M. Florea-Spiroiu, D.-F. Anghel, C. Munteanu, D. Angelescu, C. Stoian and G. Zgherea, J. Appl. Polym. Sci., 2014, 131, 40059 CrossRef.
- P. Pal and A. Pal, Int. J. Biol. Macromol., 2017, 104, 1548–1555 CrossRef CAS.
- S. Demarger-André and A. Domard, Carbohydr. Polym., 1994, 23, 211–219 CrossRef.
- M. C. Bonferoni, G. Sandri, E. Dellera, S. Rossi, F. Ferrari, M. Mori and C. Caramella, Eur. J. Pharm. Biopharm., 2014, 87, 101–106 CrossRef CAS.
- M. Vargas, A. Albors, A. Chiralt and C. González-Martínez, Food Hydrocolloids, 2009, 23, 536–547 CrossRef CAS.
- E. Dellera, M. C. Bonferoni, G. Sandri, S. Rossi, F. Ferrari, C. Del Fante, C. Perotti, P. Grisoli and C. Caramella, Eur. J. Pharm. Biopharm., 2014, 88, 643–650 CrossRef CAS.
- M. V. Shamov, S. Y. Y. Bratskaya and V. A. Avramenko, J. Colloid Interface Sci., 2002, 249, 316–321 CrossRef CAS.
- S. Demarger-André and A. Domard, Carbohydr. Polym., 1995, 27, 101–107 CrossRef.
- I. Ahmed, L. Dildar, A. Haque, P. Patra, M. Mukhopadhyay, S. Hazra, M. Kulkarni, S. Thomas, J. R. Plaisier, S. B. Dutta and J. K. Bal, J. Colloid Interface Sci., 2018, 514, 433–442 CrossRef CAS.
- M. Hasan, G. Ben Messaoud, F. Michaux, A. Tamayol, C. J. F. Kahn, N. Belhaj, M. Linder and E. Arab-Tehrany, RSC Adv., 2016, 6, 45290–45304 RSC.
- H. W. Tan and M. Misran, J. Liposome Res., 2012, 22, 329–335 CrossRef CAS.
- L. Chiappisi, S. Prévost, I. Grillo and M. Gradzielski, Langmuir, 2014, 30, 1778–1787 CrossRef CAS.
- L. Chiappisi, S. Prévost, I. Grillo and M. Gradzielski, Langmuir, 2014, 30, 10608–10616 CrossRef CAS.
- L. Chiappisi, S. David Leach, M. Gradzielski, S. D. Leach and M. Gradzielski, Soft Matter, 2017, 13, 4988–4996 RSC.
- S. Micciulla, D. W. Hayward, Y. Gerelli, A. Panzarella, R. von Klitzing, M. Gradzielski and L. Chiappisi, Commun. Chem., 2019, 2, 61 CrossRef.
- L. Chiappisi, M. Simon and M. Gradzielski, ACS Appl. Mater. Interfaces, 2015, 7, 6139–6145 CrossRef CAS.
- M. Valtiner, S. H. Donaldson, M. A. Gebbie and J. N. Israelachvili, J. Am. Chem. Soc., 2012, 134, 1746–1753 CrossRef CAS.
- J. H. J. Hamman, Mar. Drugs, 2010, 8, 1305–1322 CrossRef CAS.
- Y. Luo and Q. Wang, Int. J. Biol. Macromol., 2014, 64, 353–367 CrossRef CAS.
- P. Pakornpadungsit, T. Prasopdee, N. M. Swainson, A. Chworos and W. Smitthipong, Polym. Test., 2020, 83, 106333 CrossRef CAS.
- L. M. Bravo-Anaya, K. G. Fernández-Solís, J. Rosselgong, J. L. E. Nano-Rodríguez, F. Carvajal and M. Rinaudo, Int. J. Biol. Macromol., 2019, 126, 1037–1049 CrossRef CAS.
- P. L. Ma, M. Lavertu, F. M. Winnik and M. D. Buschmann, Carbohydr. Polym., 2017, 176, 167–176 CrossRef CAS.
- J.-W. Shen, J. Li, Z. Zhao, L. Zhang, G. Peng and L. Liang, Sci. Rep., 2017, 7, 5050 CrossRef.
- P. L. Ma, M. Lavertu, F. M. Winnik, M. D. Buschmann, L. M. Pei, M. Lavertu, F. M. Winnik and M. D. Buschmann, Biomacromolecules, 2009, 10, 1490–1499 CrossRef CAS.
- H. Ragelle, R. Riva, G. Vandermeulen, B. Naeye, V. Pourcelle, C. S. Le Duff, C. D'Haese, B. Nysten, K. Braeckmans, S. De Smedt, C. Jérôme and V. Préat, J. Controlled Release, 2014, 176, 54–63 CrossRef CAS.
- I. Pilipenko, V. Korzhikov-Vlakh, V. Sharoyko, N. Zhang, M. Schäfer-Korting, E. Rühl, C. Zoschke and T. Tennikova, Pharmaceutics, 2019, 11, 317 CrossRef CAS.
- C. Zandanel, M. Noiray and C. Vauthier, Pharm. Res., 2020, 37, 22 CrossRef CAS.
- P. Holzerny, B. Ajdini, W. Heusermann, K. Bruno, M. Schuleit, L. Meinel and M. Keller, J. Controlled Release, 2012, 157, 297–304 CrossRef CAS.
- S. Reed and B. M. Wu, J. Biomed. Mater. Res., Part B, 2017, 105, 272–282 CrossRef CAS.
- K. Xu, K. Ganapathy, T. Andl, Z. Wang, J. A. Copland, R. Chakrabarti and S. J. Florczyk, Biomaterials, 2019, 217, 119311 CrossRef CAS.
- E. A. Krisanti, G. M. Naziha, N. S. Amany, K. Mulia and N. A. Handayani, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 509, 012100 CAS.
- M. Kuczajowska-Zadrożna, U. Filipkowska and T. Jóźwiak, J. Environ. Chem. Eng., 2020, 103878 CrossRef.
- A. B. Kayitmazer, A. F. Koksal and E. Kilic Iyilik, Soft Matter, 2015, 11, 8605–8612 RSC.
- O. Karabiyik Acar, A. B. Kayitmazer and G. Torun Kose, Biomacromolecules, 2018, 19, 1198–1211 CrossRef CAS.
- C. Schatz, J.-M. Lucas, C. Viton, A. Domard, C. Pichot and T. Delair, Langmuir, 2004, 20, 7766–7778 CrossRef CAS.
- J. Valente, V. Gaspar, B. Antunes, P. Countinho and I. Correia, Polymer, 2013, 54, 5–15 CrossRef CAS.
- P. M. de la Torre, S. Torrado and S. Torrado, Biomaterials, 2003, 24, 1459–1468 CrossRef CAS.
- Q. Chen, Y. Hu, Y. Chen, X. Jiang and Y. Yang, Macromol. Biosci., 2005, 5, 993–1000 CrossRef CAS.
- Y. Hu, X. Jiang, Y. Ding, H. Ge, Y. Yuan and C. Yang, Biomaterials, 2002, 23, 3193–3201 CrossRef CAS.
- R.-Y. Zhang, E. Zaslavski, G. Vasilyev, M. Boas and E. Zussman, Biomacromolecules, 2018, 19, 588–595 CrossRef CAS.
- D. Chuan, T. Jin, R. Fan, L. Zhou and G. Guo, Adv. Colloid Interface Sci., 2019, 268, 25–38 CrossRef.
- S. P. Strand, S. Lelu, N. K. Reitan, C. de Lange Davies, P. Artursson and K. M. Vårum, Biomaterials, 2010, 31, 975–987 CrossRef CAS.
- C. Cui, C. Shao, L. Meng and J. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 39228–39237 CrossRef CAS.
- S. Gokila, T. Gomathi, P. Sudha and S. Anil, Int. J. Biol. Macromol., 2017, 104, 1459–1468 CrossRef CAS.
- D. P. Facchi, A. L. Cazetta, E. A. Canesin, V. C. Almeida, E. G. Bonafé, M. J. Kipper and A. F. Martins, Chem. Eng. J., 2018, 337, 595–608 CrossRef CAS.
- F.-L. Mi, H.-W. Sung and S.-S. Shyu, Carbohydr. Polym., 2002, 48, 61–72 CrossRef CAS.
- K. Baysal, A. Z. Aroguz, Z. Adiguzel and B. M. Baysal, Int. J. Biol. Macromol., 2013, 59, 342–348 CrossRef CAS.
- W. Zhang, X. Jin, H. Li, R.-R. Zhang and C.-W. Wu, Carbohydr. Polym., 2018, 186, 82–90 CrossRef CAS.
- R. Rodríguez-Rodríguez, H. Espinosa-Andrews, C. Velasquillo-Martínez and Z. Y. García-Carvajal, Int. J. Polym. Mater. Polym. Biomater., 2020, 69, 1–20 CrossRef.
- H. Hamedi, S. Moradi, S. M. Hudson and A. E. Tonelli, Carbohydr. Polym., 2018, 199, 445–460 CrossRef CAS.
- P. Mohammadzadeh Pakdel and S. J. Peighambardoust, Carbohydr. Polym., 2018, 201, 264–279 CrossRef CAS.
- N. Sereni, A. Enache, G. Sudre, A. Montembault, C. Rochas, P. Durand, M. H. Perrard, G. Bozga, J. P. Puaux, T. Delair and L. David, Langmuir, 2017, 33, 12697–12707 CrossRef CAS.
- A. A. Enache, L. David, J. P. Puaux, I. Banu and G. Bozga, J. Appl. Polym. Sci., 2018, 135, 1–12 CrossRef.
- P. Sacco, F. Brun, I. Donati, D. Porrelli, S. Paoletti and G. Turco, ACS Appl. Mater. Interfaces, 2018, 10, 10761–10770 CrossRef CAS.
- Y. Huang, Y. Cai and Y. Lapitsky, J. Mater. Chem. B, 2015, 3, 5957–5970 RSC.
- P. Sacco, S. Paoletti, M. Cok, F. Asaro, M. Abrami, M. Grassi and I. Donati, Int. J. Biol. Macromol., 2016, 92, 476–483 CrossRef CAS.
- L. Bugnicourt and C. Ladavière, Prog. Polym. Sci., 2016, 60, 1–17 CrossRef CAS.
- L. Dambies, T. Vincent, A. Domard and E. Guibal, Biomacromolecules, 2001, 2, 1198–1205 CrossRef CAS.
- Azizullah, N. ur Rehman, A. Haider, U. Kortz, S. U. Afridi, M. Sohail, S. A. Joshi and J. Iqbal, Int. J. Pharm., 2017, 533, 125–137 CrossRef CAS.
- X. Z. Shu and K. J. Zhu, Int. J. Pharm., 2002, 233, 217–225 CrossRef CAS.
- S. Saravanan, S. Vimalraj, P. Thanikaivelan, S. Banudevi and G. Manivasagam, Int. J. Biol. Macromol., 2019, 121, 38–54 CrossRef CAS.
- H. Y. Zhou, L. J. Jiang, P. P. Cao, J. B. Li and X. G. Chen, Carbohydr. Polym., 2015, 117, 524–536 CrossRef CAS.
- A. Shi, X. Dai and Z. Jing, Polym. Sci., Ser. A, 2020, 62, 228–239 CrossRef.
- G. A. F. Roberts and K. E. Taylor, Makro, 1989, 190, 951–960 CrossRef CAS.
- P. Hu, C. B. Raub, J. S. Choy and X. Luo, J. Mater. Chem. B, 2020, 8, 2519–2529 RSC.
- L. Gao, H. Gan, Z. Meng, R. Gu, Z. Wu, L. Zhang, X. Zhu, W. Sun, J. Li, Y. Zheng and G. Dou, Colloids Surf., B, 2014, 117, 398–405 CrossRef CAS.
- R. A. Muzzarelli, M. El Mehtedi, C. Bottegoni, A. Aquili and A. Gigante, Mar. Drugs, 2015, 13, 7314–7338 CrossRef CAS.
- G. Tripodo, A. Trapani, A. Rosato, C. Di Franco, R. Tamma, G. Trapani, D. Ribatti and D. Mandracchia, Carbohydr. Polym., 2018, 198, 124–130 CrossRef CAS.
- L. Pérez-Álvarez, L. Ruiz-Rubio, B. Artetxe, M. D. M. Vivanco, J. M. Gutiérrez-Zorrilla and J. L. Vilas-Vilela, Carbohydr. Polym., 2019, 213, 159–167 CrossRef.
- L. Li, N. Wang, X. Jin, R. Deng, S. Nie, L. Sun, Q. Wu, Y. Wei and C. Gong, Biomaterials, 2014, 35, 3903–3917 CrossRef CAS.
- E. A. Kamoun, J. Adv. Res., 2016, 7, 69–77 CrossRef CAS.
- E. Lucas de Lima, N. Fittipaldi Vasconcelos, J. da Silva Maciel, F. Karine Andrade, R. Silveira Vieira and J. P. Andrade Feitosa, J. Mater. Sci.: Mater. Med., 2020, 31, 5 CrossRef CAS.
- J. Berger, M. Reist, J. M. Mayer, O. Felt, N. A. Peppas and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 19–34 CrossRef CAS.
- L. Weng, X. Chen and W. Chen, Biomacromolecules, 2007, 8, 1109–1115 CrossRef CAS.
- P. Sautrot-Ba, N. Razza, L. Breloy, S. A. Andaloussi, A. Chiappone, M. Sangermano, C. Hélary, S. Belbekhouche, T. Coradin and D. L. Versace, J. Mater. Chem. B, 2019, 7, 6526–6538 RSC.
- M. Pei, J. Mao, W. Xu, Y. Zhou and P. Xiao, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1862–1871 CrossRef CAS.
- J. Huang, J. Qin, P. Zhang, X. Chen, X. You, F. Zhang, B. Zuo and M. Yao, Carbohydr. Polym., 2020, 229, 115515 CrossRef CAS.
- A. K. Ospanova, B. E. Savdenbekova, M. K. Iskakova, R. A. Omarova, R. N. Zhartybaev, B. Z. Nussip and A. S. Abdikadyr, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 230, 1–6 Search PubMed.
- M. T. Cook, G. Tzortzis, V. V. Khutoryanskiy and D. Charalampopoulos, J. Mater. Chem. B, 2013, 1, 52–60 RSC.
- K. Tian, C. Xie and X. Xia, Colloids Surf., B, 2013, 109, 82–89 CrossRef CAS.
- J. H. Bongaerts, J. J. Cooper-White and J. R. Stokes, Biomacromolecules, 2009, 10, 1287–1294 CrossRef CAS.
- M. Criado-Gonzalez, M. Fernandez-Gutierrez, J. San Roman, C. Mijangos and R. Hernández, Carbohydr. Polym., 2019, 206, 428–434 CrossRef CAS.
- A. Ali and S. Ahmed, Int. J. Biol. Macromol., 2018, 109, 273–286 CrossRef CAS.
- L. Pérez-Álvarez, L. Ruiz-Rubio, I. Azua, V. Benito, A. Bilbao and J. L. Vilas-Vilela, Eur. Polym. J., 2019, 112, 31–37 CrossRef.
- B. Guan, H. Wang, R. Xu, G. Zheng, J. Yang, Z. Liu, M. Cao, M. Wu, J. Song, N. Li, T. Li, Q. Cai, X. Yang, Y. Li and X. Zhang, Sci. Rep., 2016, 6, 1–12 CrossRef.
- H. Lv, Z. Chen, X. Yang, L. Cen, X. Zhang and P. Gao, J. Dent., 2014, 42, 1464–1472 CrossRef CAS.
- S. Del Hoyo-Gallego, L. Pérez-Álvarez, F. Gómez-Galván, E. Lizundia, I. Kuritka, V. Sedlarik, J. M. Laza and J. L. Vila-Vilela, Carbohydr. Polym., 2016, 143, 35–43 CrossRef CAS.
- V. Nascimento, C. França, J. Hernández-Montelongo, D. Machado, M. Lancellotti, M. Cotta, R. Landers and M. Beppu, Eur. Polym. J., 2018, 109, 198–205 CrossRef CAS.
- L. Maddalena, F. Carosio, J. Gomez, G. Saracco and A. Fina, Polym. Degrad. Stab., 2018, 152, 1–9 CrossRef CAS.
- J. Jung, L. Li, C. K. Yeh, X. Ren and Y. Sun, Mater. Sci. Eng., C, 2019, 104, 109961 CrossRef CAS.
- H. Wang, J. Qian and F. Ding, J. Agric. Food Chem., 2018, 66, 395–413 CrossRef CAS.
- S. Kumar, A. Mukherjee and J. Dutta, Trends Food Sci. Technol., 2020, 97, 196–209 CrossRef CAS.
- N. L. Benbow, J. L. Webber, S. Karpiniec, M. Krasowska, J. K. Ferri and D. A. Beattie, Phys. Chem. Chem. Phys., 2017, 19, 23790–23801 RSC.
- T. I. Croll, A. J. O'Connor, G. W. Stevens and J. J. Cooper-White, Biomacromolecules, 2006, 7, 1610–1622 CrossRef CAS.
- J. J. Richardson, M. Bjornmalm and F. Caruso, Science, 2015, 348, aaa2491 CrossRef.
- H. Kaygusuz, S. Micciulla, F. B. Erim and R. von Klitzing, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 1798–1803 CrossRef CAS.
- J. Huang, S. Zajforoushan Moghaddam and E. Thormann, ACS Omega, 2019, 4, 2019–2029 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- J. B. Schlenoff, A. H. Rmaile and C. B. Bucur, J. Am. Chem. Soc., 2008, 130, 13589–13597 CrossRef CAS.
- R. V. Klitzing, Phys. Chem. Chem. Phys., 2006, 8, 5012 RSC.
- D. Volodkin and R. von Klitzing, Curr. Opin. Colloid Interface Sci., 2014, 1–7 Search PubMed.
- C. Coquery, F. Carosio, C. Negrell, N. Caussé, N. Pébère and G. David, Surf. Interfaces, 2019, 16, 59–66 CrossRef CAS.
- W. Yuan, H. Dong, C. M. Li, X. Cui, L. Yu, Z. Lu and Q. Zhou, Langmuir, 2007, 23, 13046–13052 CrossRef CAS.
- J. M. Silva, A. R. C. Duarte, S. G. Caridade, C. Picart, R. L. Reis and J. F. Mano, Biomacromolecules, 2014, 15, 3817–3826 CrossRef CAS.
- G. Maurstad, Y. A. Mørch, A. R. Bausch and B. T. Stokke, Carbohydr. Polym., 2008, 71, 672–681 CrossRef CAS.
- P. Kujawa, P. Moraille, J. Sanchez, A. Badia and F. M. Winnik, J. Am. Chem. Soc., 2005, 127, 9224–9234 CrossRef CAS.
- T. Tariverdian, T. Navaei, P. B. Milan, A. Samadikuchaksaraei and M. Mozafari, in Advanced Functional Polymers for Biomedical Applications, ed. M. Mozafari and N. P. S. Chauhan, Elsevier, 2019, pp. 323–357 Search PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, 41–56 CrossRef.
- H. H. Trimm and B. R. Jennings, Biochem. J., 1983, 213, 671–677 CrossRef CAS.
- N. Ghavidel Mehr, C. D. Hoemann and B. D. Favis, Polymer, 2015, 64, 112–121 CrossRef CAS.
- J. Choi and M. F. Rubner, Macromolecules, 2005, 38, 116–124 CrossRef CAS.
- M. Elzbieciak, S. Zapotoczny, P. Nowak, R. Krastev, M. Nowakowska and P. Warszyński, Langmuir, 2009, 25, 3255–3259 CrossRef CAS.
- E. Guzmán, J. A. Cavallo, R. Chuliá-Jordán, C. Gómez, M. C. Strumia, F. Ortega and R. G. Rubio, Langmuir, 2011, 27, 6836–6845 CrossRef.
- C. Huang, G. Fang, Y. Zhao, S. Bhagia, X. Meng, Q. Yong and A. J. Ragauskas, Carbohydr. Polym., 2019, 222, 115036 CrossRef CAS.
- U. Voigt, V. Khrenov, K. Tauer, M. Hahn, W. Jaeger and R. V. Klitzing, J. Phys.: Condens. Matter, 2003, 15, S213–S218 CrossRef CAS.
- S. Micciulla, S. Dodoo, C. Chevigny, A. Laschewsky and R. von Klitzing, Phys. Chem. Chem. Phys., 2014, 16, 21988–21998 RSC.
- L. Richert, P. Lavalle, E. Payan, X. Z. Shu, G. D. Prestwich, J. F. Stoltz, P. Schaaf, J. C. Voegel and C. Picart, Langmuir, 2004, 20, 448–458 CrossRef CAS.
- C. Picart, J. Mutterer, L. Richert, Y. Luo, G. D. Prestwich, P. Schaaf, J.-C. C. Voegel and P. Lavalle, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12531–12535 CrossRef CAS.
- M. Zerball, A. Laschewsky and R. Von Klitzing, J. Phys. Chem. B, 2015, 119, 11879–11886 CrossRef CAS.
- C. Liu, E. Thormann, P. M. Claesson and E. Tyrode, Langmuir, 2014, 30, 8866–8877 CrossRef CAS.
- K. Wulf, S. Schünemann, A. Strohbach, R. Busch, S. B. Felix, K. P. Schmitz, K. Sternberg and S. Petersen, BioNanoMaterials, 2015, 16, 265–273 Search PubMed.
- J. M. Silva, S. G. Caridade, N. M. Oliveira, R. L. Reis and J. F. Mano, J. Mater. Chem. B, 2015, 3, 4555–4568 RSC.
- N. M. Alves, C. Picart and J. F. Mano, Macromol. Biosci., 2009, 9, 776–785 CrossRef CAS.
- G. V. Martins, E. G. Merino, J. F. Mano and N. M. Alves, Macromol. Biosci., 2010, 10, 1444–1455 CrossRef CAS.
- A. F. Martins, J. Vlcek, T. Wigmosta, M. Hedayati, M. M. Reynolds, K. C. Popat and M. J. Kipper, Appl. Surf. Sci., 2020, 502, 144282 CrossRef CAS.
- M. Marudova, S. Lang, G. J. Brownsey and S. G. Ring, Carbohydr. Res., 2005, 340, 2144–2149 CrossRef CAS.
- A. E. Sorkio, E. P. Vuorimaa-Laukkanen, H. M. Hakola, H. Liang, T. A. Ujula, J. J. Valle-Delgado, M. Österberg, M. L. Yliperttula and H. Skottman, Biomaterials, 2015, 51, 257–269 CrossRef CAS.
- M. Andonegi, K. L. Heras, E. Santos-Vizcaíno, M. Igartua, R. M. Hernandez, K. de la Caba and P. Guerrero, Carbohydr. Polym., 2020, 237, 116159 CrossRef CAS.
- I. Leceta, P. Arana, P. Guerrero and K. De La Caba, Mater. Lett., 2014, 128, 125–127 CrossRef CAS.
- A. Sionkowska and A. Płanecka, J. Mol. Liq., 2013, 186, 157–162 CrossRef CAS.
- T. Yovcheva, B. Pilicheva, A. Marinova, A. Viraneva, I. Bodurov, G. Exner, S. Sotirov, I. Vlaeva, Y. Uzunova and M. Marudova, J. Phys.: Conf. Ser., 2019, 1186, 1–7 CrossRef.
- K. Junka, O. Sundman, J. Salmi, M. Österberg and J. Laine, Carbohydr. Polym., 2014, 108, 34–40 CrossRef CAS.
- K. Cai, Y. Hu, Y. Wang and L. Yang, J. Biomed. Mater. Res., 2008, 84A, 516–522 CrossRef CAS.
- A. Dedinaite, M. Lundin, L. Macakova and T. Auletta, Langmuir, 2005, 21, 9502–9509 CrossRef CAS.
- H. Huang and X. Yang, Colloids Surf., A, 2003, 226, 77–86 CrossRef CAS.
- A. Valverde, L. Pérez-Álvarez, L. Ruiz-Rubio, M. A. Pacha Olivenza, M. B. García Blanco, M. Díaz-Fuentes and J. L. Vilas-Vilela, Carbohydr. Polym., 2019, 207, 824–833 CrossRef CAS.
- H. Zare, G. D. Najafpour, M. Jahanshahi, M. Rahimnejad and M. Rezvani, Rom. Biotechnol. Lett., 2017, 22, 12611–12619 CAS.
- H. Sun, D. Choi, J. Heo, S. Y. Jung and J. Hong, Cancers, 2020, 12, 1–14 Search PubMed.
- P. Li, Y.-N. Dai, J.-P. Zhang, A.-Q. Wang and Q. Wei, Int. J. Biomed. Sci., 2008, 4, 221–228 CAS.
- I. M. Brasil, C. Gomes, A. Puerta-Gomez, M. E. Castell-Perez and R. G. Moreira, LWT–Food Sci. Technol., 2012, 47, 39–45 CrossRef CAS.
- Y. Kusumastuti, A. Istiani, Rochmadi and C. W. Purnomo, Adv. Mater. Sci. Eng., 2019, 2019, 1–8 CrossRef.
- J. B. Schlenoff, S. T. Dubas and T. Farhat, Langmuir, 2000, 16, 9968–9969 CrossRef CAS.
- K. C. Krogman, N. S. Zacharia, S. Schroeder and P. T. Hammond, Langmuir, 2007, 23, 3137–3141 CrossRef CAS.
- K. C. Krogman, J. l. Lowery, N. S. Zacharia, G. C. Rutledge and P. T. Hammond, Nat. Mater., 2009, 8, 512–518 CrossRef CAS.
- A. Izquierdo, S. S. Ono, J.-C. C. Voegel, P. Schaaf and G. Decher, Langmuir, 2005, 21, 7558–7567 CrossRef CAS.
- M. Kolasinska, R. Krastev, T. Gutberlet and P. Warszynski, Langmuir, 2009, 25, 1224–1232 CrossRef CAS.
- M. Criado, E. Rebollar, A. Nogales, T. A. Ezquerra, F. Boulmedais, C. Mijangos and R. Hernández, Biomacromolecules, 2017, 18, 169–177 CrossRef CAS.
- M. Bulwan, S. Zapotoczny and M. Nowakowska, Soft Matter, 2009, 5, 4726–4732 RSC.
- I. Langmuir, J. Am. Chem. Soc., 1917, 39, 1848–1906 CrossRef CAS.
- K. B. Blodgett, J. Am. Chem. Soc., 1935, 57, 1007–1022 CrossRef CAS.
- F. J. Pavinatto, L. Caseli and O. N. Oliveira, Biomacromolecules, 2010, 11, 1897–1908 CrossRef CAS.
- P. T. Hammond, Adv. Mater., 2004, 16, 1271–1293 CrossRef CAS.
- Y. Sakai-Otsuka, S. Zaioncz, I. Otsuka, S. Halila, P. Rannou and R. Borsali, Macromolecules, 2017, 50, 3365–3376 CrossRef CAS.
- Y. Liao, W.-C. Chen and R. Borsali, Adv. Mater., 2017, 29, 1701645 CrossRef.
- N. Ayres, Polym. Chem., 2010, 1, 769–777 RSC.
- O. Azzaroni, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3225–3258 CrossRef CAS.
- H. S. Lee, D. M. Eckmann, D. Lee, N. J. Hickok and R. J. Composto, Langmuir, 2011, 27, 12458–12465 CrossRef CAS.
- H. S. Lee, M. Q. Yee, Y. Y. Eckmann, N. J. Hickok, D. M. Eckmann and R. J. Composto, J. Mater. Chem., 2012, 22, 19605–19616 RSC.
- P. Kurt, L. Wood, D. E. Ohman and K. J. Wynne, Langmuir, 2007, 23, 4719–4723 CrossRef CAS.
- N. Gorochovceva, A. Naderi, A. Dedinaite and R. Makuška, Eur. Polym. J., 2005, 41, 2653–2662 CrossRef CAS.
- Y. Zhou, B. Liedberg, N. Gorochovceva, R. Makuska, A. Dedinaite and P. M. Claesson, J. Colloid Interface Sci., 2007, 305, 62–71 CrossRef CAS.
- M. Xie, K. Huang, F. Yang, R. Wang, L. Han, H. Yu, Z. Ye and F. Wu, Int. J. Biol. Macromol., 2020, 151, 1116–1125 CrossRef CAS.
- S. Velmurugan, S. Palanisamy, T. C.-K. Yang, M. Gochoo and S.-W. W. Chen, Ultrason. Sonochem., 2020, 62, 104863 CrossRef CAS.
- L. Lisuzzo, G. Cavallaro, F. Parisi, S. Milioto, R. Fakhrullin and G. Lazzara, Coatings, 2019, 9, 70 CrossRef.
- M. Koosha and S. Hamedi, Prog. Org. Coat., 2019, 127, 338–347 CrossRef CAS.
- F. I. Ali, S. T. Mahmoud, F. Awwad, Y. E. Greish and A. F. Abu-Hani, Carbohydr. Polym., 2020, 236, 116064 CrossRef CAS.
- A. Giannakas, P. Stathopoulou, G. Tsiamis and C. Salmas, J. Food Process. Preserv., 2020, 44, e14327 CAS.
- V. Labonté, A. Marion, N. Virgilio and J. R. Tavares, Ind. Eng. Chem. Res., 2016, 55, 7362–7372 CrossRef.
- M. H. Mostafa, M. A. Elsawy, M. S. Darwish, L. I. Hussein and A. H. Abdaleem, Mater. Chem. Phys., 2020, 248, 122914 CrossRef CAS.
- Q. Peng, M. Liu, J. Zheng and C. Zhou, Microporous Mesoporous Mater., 2015, 201, 190–201 CrossRef CAS.
- G. Cavallaro, S. Milioto, L. Nigamatzyanova, F. Akhatova, R. Fakhrullin and G. Lazzara, ACS Appl. Nano Mater., 2019, 2, 3169–3176 CrossRef CAS.
- G. Cavallaro, S. Milioto and G. Lazzara, Langmuir, 2020, 36, 3677–3689 CrossRef CAS.
- G. Cavallaro, G. Lazzara, S. Konnova, R. Fakhrullin and Y. Lvov, Green Mater., 2014, 2, 232–242 CrossRef.
- H. Pan, W. Wang, Y. Pan, L. Song, Y. Hu and K. M. Liew, Carbohydr. Polym., 2015, 115, 516–524 CrossRef CAS.
- M. Hassan, M. Nour, Y. Abdelmonem, G. Makhlouf and A. Abdelkhalik, Polym. Degrad. Stab., 2016, 133, 8–15 CrossRef CAS.
- B.-I. Andreica, X. Cheng and L. Marin, Eur. Polym. J., 2020, 139, 110016 CrossRef CAS.
- L. J. R. Foster, S. Ho, J. Hook, M. Basuki and H. Marçal, PLoS One, 2015, 10, e0135153 CrossRef.
- G. Cavallaro, G. Lazzara, S. Konnova, R. Fakhrullin and Y. Lvov, Green Mater., 2014, 2, 232–242 CrossRef.
- V. Bertolino, G. Cavallaro, G. Lazzara, S. Milioto and F. Parisi, New J. Chem., 2018, 42, 8384–8390 RSC.
- T. C. Yadav, P. Saxena, A. K. Srivastava, A. K. Singh, R. K. Yadav, Harish, R. Prasad and V. Pruthi, Advanced Functional Textiles and Polymers, Wiley, 2019, pp. 365–403 Search PubMed.
- M. Rubina, A. Shulenina, R. Svetogorov and A. Vasilkov, Macromol. Symp., 2020, 389, 1900067 CrossRef CAS.
- R. Ying, H. Wang, R. Sun and K. Chen, Mater. Sci. Eng., C, 2020, 110, 110689 CrossRef CAS.
- X. Jia, I. Ahmad, R. Yang and C. Wang, J. Mater. Chem. B, 2017, 5, 2459–2467 RSC.
- A. B. Neji, M. Jridi, H. Kchaou, M. Nasri and R. Dhouib Sahnoun, Polym. Test., 2020, 84, 106380 CrossRef CAS.
- V. Bertolino, G. Cavallaro, G. Lazzara, M. Merli, S. Milioto, F. Parisi and L. Sciascia, Ind. Eng. Chem. Res., 2016, 55, 7373–7380 CrossRef CAS.
- M. Liu, Y. Zhang, C. Wu, S. Xiong and C. Zhou, Int. J. Biol. Macromol., 2012, 51, 566–575 CrossRef CAS.
- L. Lisuzzo, G. Cavallaro, S. Milioto and G. Lazzara, New J. Chem., 2019, 43, 10887–10893 RSC.
- M. Liu, C. Wu, Y. Jiao, S. Xiong and C. Zhou, J. Mater. Chem. B, 2013, 1, 2078 RSC.
- I. Benucci, K. Liburdi, I. Cacciotti, C. Lombardelli, M. Zappino, F. Nanni and M. Esti, Food Hydrocolloids, 2018, 74, 124–131 CrossRef CAS.
- X. Liu, S. Qin, H. Li, J. Sun, X. Gu, S. Zhang and J. C. Grunlan, Macromol. Mater. Eng., 2019, 304, 1800531 CrossRef.
- J. Prakash, D. Prema, K. Venkataprasanna, K. Balagangadharan, N. Selvamurugan and G. D. Venkatasubbu, Int. J. Biol. Macromol., 2020, 154, 62–71 CrossRef CAS.
- X. Li, Y.-C. Li, M. Chen, Q. Shi, R. Sun and X. Wang, J. Mater. Chem. B, 2018, 6, 6544–6549 RSC.
- Z.-K. Cui, S. Kim, J. J. Baljon, B. M. Wu, T. Aghaloo and M. Lee, Nat. Commun., 2019, 10, 3523 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb01865a |
| This journal is © The Royal Society of Chemistry 2021 |