
The hole in the bucky: structure–property mapping of closed- vs. open-cage fullerene solar-cell blends via temperature/composition phase diagrams†
Giovanni Maria
Matrone
a,
Elizabeth
Gutiérrez-Meza
b,
Alex H.
Balzer
c,
Aditi
Khirbat
d,
Artem
Levitsky
e,
Alexander B.
Sieval
f,
Gitti. L.
Frey
 e,
Lee J.
Richter
g,
Carlos
Silva
b and
Natalie
Stingelin
*cd
e,
Lee J.
Richter
g,
Carlos
Silva
b and
Natalie
Stingelin
*cd
aMicrosystems Group & Institute of Complex Molecular Studies (ICMS), Eindhoven University of Technology, Eindhoven 5600 MP, The Netherlands
bSchool of Chemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA
cSchool of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: natalie.stingelin@gatech.edu
dSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
eDepartment of Material Science and Engineering, Technion Israel Institute of Technology, Haifa 3200003, Israel
fResearch Centre Biobased Economy, Hanze University of Applied Sciences, Groningen 9747AS, The Netherlands
gMaterials Science and Engineering Division, National Institute of Standards and Technology, Gaithersburg, MD 20855-8542, USA
First published on 2nd September 2021
Abstract
The morphology development of polymer-based blends, such as those used in organic photovoltaic (OPV) systems, typically arrests in a state away from equilibrium – how far from equilibrium this is will depend on the materials chemistry and the selected assembly parameters/environment. As a consequence, small changes during the blend assembly alter the solid-structure development from solution and, in turn, the final device performance. Comparing an open-cage ketolactam fullerene with the prototypical[6,6]-phenyl-C61-butyric acid methyl ester in blends with poly[2,5-bis(3-hexadecylthiophen-2-yl)thieno[3,2-b]thiophene] (PBTTT), we demonstrate that experimentally established, non-equilibrium temperature/composition phase diagrams can be useful beyond rationalization of optimum blend composition for OPV device performance. Indeed, they can be exploited as tools for rapid, qualitative structure–property mapping, providing insights into why apparent similar donor:acceptor blends display different optoelectronic processes resulting from changes in the phase-morphology formation induced by the different chemistries of the fullerenes.
Introduction
Solution-processable fullerenes have been key in our understanding of important photophysical processes in organic semiconductors, including in blends with donor polymers; and they were critical in providing the knowledge platform that we now build on toward plastic-based, semi-transparent solar cells and their technological exploitation. Today, in organic photovoltaic systems (OPV)s, fullerene derivatives have generally been replaced with non-fullerene acceptors, however, solution-processable fullerenes, first synthesized and applied in devices by Hummelen et al.,1,2 can still provide unique insights into donor:acceptor blends and enable testing methodologies to characterize them.Here, we selected an open- vs. a closed-cage fullerene derivative, i.e. an azafulleroid often referred to as ketolactam fullerene3 (see Fig. 1; simply called ‘ketolactam’ or ‘keto’ in the below) and the prototypical[6,6]-phenyl-C61-butyric acid methyl ester (PCBM), in blends with poly[2,5-bis(3-hexadecylthiophen-2-yl)thieno[3,2-b]thiophene] (PBTTT)4 to scrutinize the usefulness of non-equilibrium temperature/composition phase diagrams, established via thermal analysis, to provide qualitative understanding of why apparently similar blends of comparable polymer:fullerene compositions can display different optoelectronic properties depending whether the open- or the closed cage fullerene is used.
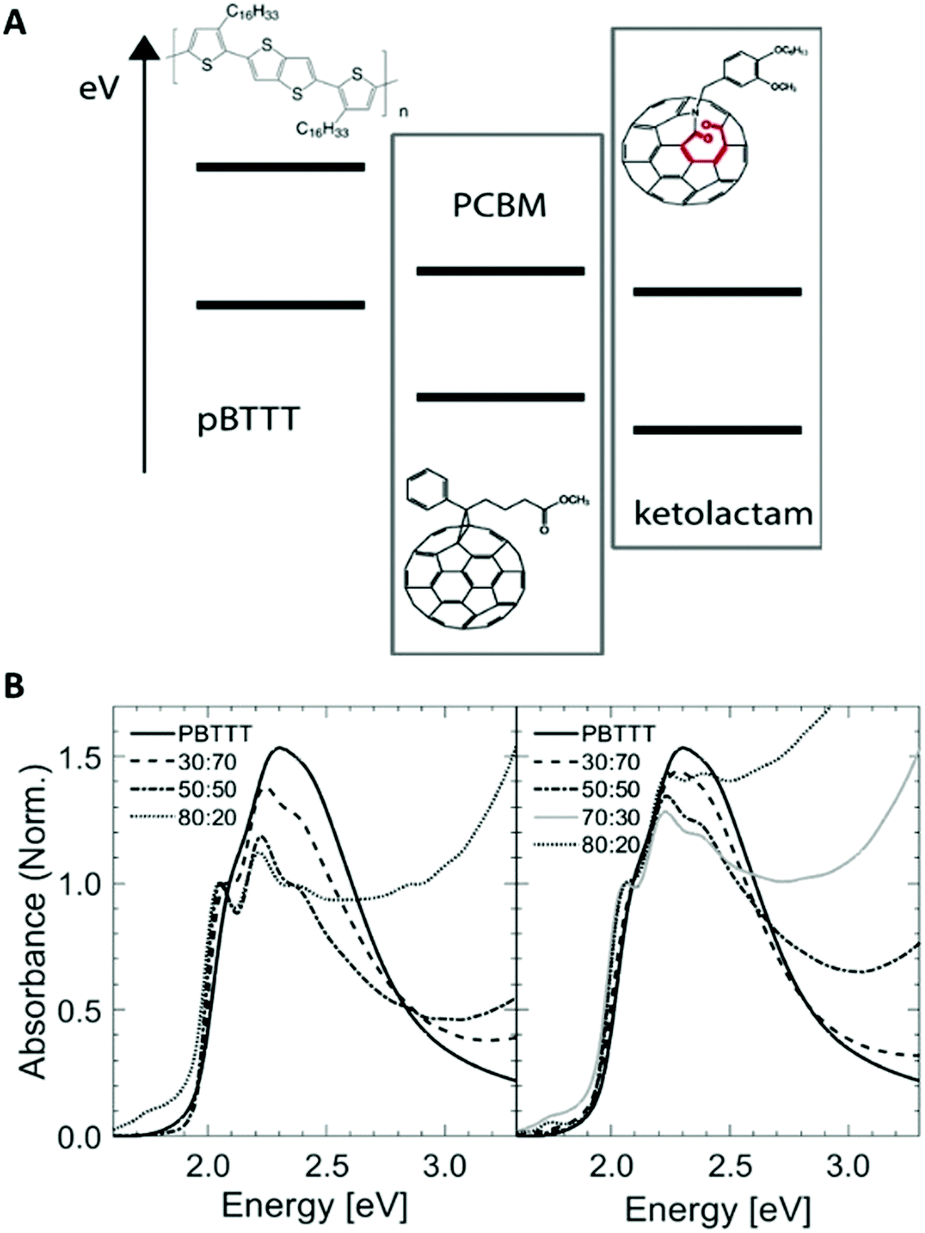 | ||
| Fig. 1 (A) Schematic representation of the energy levels of the polymer (PBTTT) and fullerene acceptors, i.e., PCBM and ketolactam fullerene, used here to scrutinize the power of temperature/composition phase diagrams for property mapping of corresponding blends. Chemical structures are shown as insets. (B) UV-vis spectroscopy data of a range of blends of PCBM:PBTTT (left) and ketolactam:PBTTT (right; compositions given as mass ratio), and for comparison: neat PBTTT. | ||
We selected specific fullerene:PBTTT binaries for which it has been shown that they can form co-crystalline regions, where the fullerene intercalates within the polymer side chains in the solid state, leading to a 1-phase system: i.e., a solid solution.5–9 This can be followed in a relatively straight-forward manner via thermal analysis, linear ultraviolet-visible (UV-vis) absorption spectroscopy as well as vibrational spectroscopy techniques.6 The two fullerene derivatives were chosen because of their similar molecular size and mass, and their rather comparable reduction potentials (1.08 V for PCBM and 0.91 V for ketolactam, corresponding to an ionization potential of −3.75 eV and 3.92 eV, respectively10) and their nearly identical UV-vis absorption behaviour (Fig. 1).
Results and discussions
We started with establishing temperature/composition phase diagrams using differential scanning calorimetry (DSC)8 and measuring blend films cast from 20 mg mL−1 solutions at 40 °C. The first heating thermograms were utilised for this purpose to assess the thermal phase behaviour of films with an essentially identical processing history as used in linear UV-vis and photoluminescence spectroscopy, transient absorption spectroscopy, as well as IR vibrational spectroscopy. Various blend compositions were thereby tested, both for blends of PBTTT with the open-cage ketolactam fullerene derivative and with PCBM.Comparing the DSC data of both systems, a few observations can immediately be made (Fig. 2, top panels). The melting temperature, Tm (≈255 °C), of the open cage ketolactam fullerene is very ill defined compared to PCBM (Tm ≈ 275 °C), indicating low molecular order. It, however, evolves upon addition of the polymer. Indeed, both the melting endotherms of the PCBM and the ketolactam become sharper and easier to identify with increasing PBTTT content. Moreover, a second endotherm around 240–250 °C is readily observed in the PCBM:PBTTT system at PBTTT content below 60% (all compositions are mass fraction unless otherwise noted), Fig. 2A, top panel, while only some broadening in melting endotherms of specific blend compositions is found for the ketolactam:PBTTT system (e.g., 5% and 15% PBTTT; Fig. 2B, top panel). Increasing the polymer content further (above ≈60%), a low-temperature shoulder evolves in both systems. These endothermic features are relatively weak in the PCBM:PBTTT system but well resolved in the ketolactam:PBTTT binary.
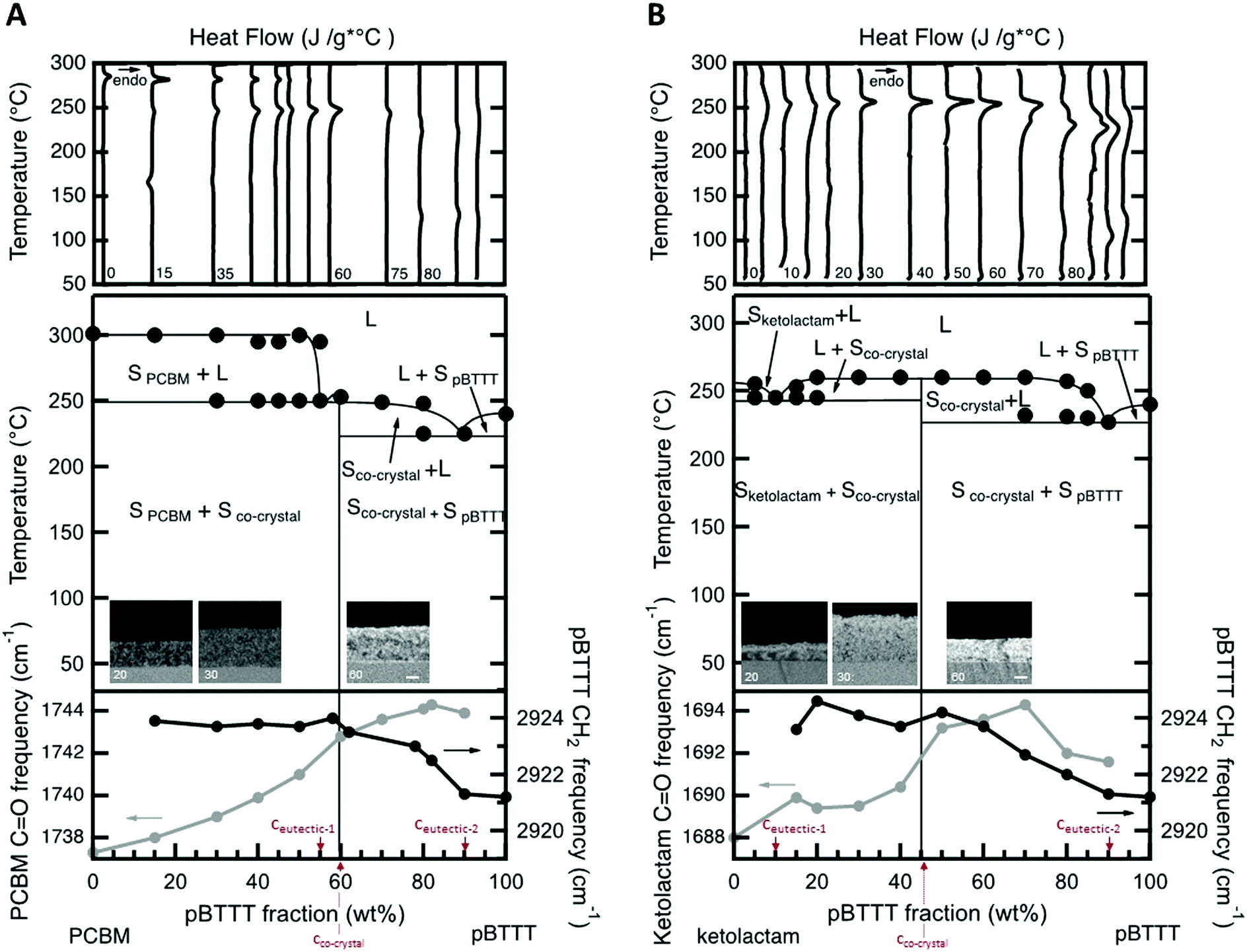 | ||
| Fig. 2 Top: Differential scanning calorimetry (DSC) first heating thermograms of PCBM:PBTTT (A) and ketolactam:PBTTT (B) blends, recorded at 20 °C min−1 in N2-atmosphere on films drop-cast at 40 °C. {Note: the thermograms obtained for the PBTTT:ketolactam system were magnified by a factor 5 so that specific features are better discernable.} Middle: Experimentally established temperature/composition phase diagrams of fullerene:PBTTT blends, using the DSC data displayed in the top panel. A double eutectic system is identified for both binaries, with one eutectic point of composition, ceutectic-1 at 55% and 10% PBTTT for, respectively, PCBM:PBTTT and ketolactam:PBTTT, and a single co-crystalline phase at 60% and 45% PBTTT (all compositions are mass fraction). In the inset, cross-section BSE HRSEM micrographs of PCBM:PBTTT and ketolactam:PBTTT films after 30 cycles of VPI at 60 °C. The bright contrast in the film represent areas rich in ZnO, indicating the PBTTT-rich phase and the fullerene:PBTTT intercalated structure, while the dark areas represent the organic phase, indicating the fullerene-rich phase. Scale bar is 100 nm for all the micrographs. Bottom: PCBM carbonyl and PBTTT d-center frequencies as a function of composition allows one to probe, respectively, the local environment of the fullerene molecules and PBTTT side-chain order via IR vibrational spectroscopy. | ||
These observations can be explained with a double eutectic behaviour for both binaries (as previously postulated for PCBM:PBTTT),8,11i.e. systems with two eutectic points, which are binaries that feature two low-melting compositions. One eutectic point is observed at a eutectic composition, ceutectic-1, and forms between the fullerene and the fullerene:polymer co-crystalline structure; a second eutectic is found at ceutectic-2 and develops between the co-crystalline phase and the polymer (Fig. 2, middle panels). Thereby, ceutectic-1PCBM:PBTTT ≈ 55% PBTTT, ceutectic-2PCBM:PBTTT ≈ 90% PBTTT; while ceutectic-1ketolactam:PBTTT ≈ 10% PBTTT, and ceutectic-2ketolactam:PBTTT ≈ 90% PBTTT. The co-crystal compositions, where essentially all fullerene is intercalated in the polymer, without excess fullerene or excess polymer being present (i.e., a 1-phase system is formed5,6), are identified as 60% PBTTT for the PCBM:PBTTT binary and 45% PBTTT for the ketolactam-based system.
According to this assignment, the two high-temperature endotherms at ≈300 °C and ≈250 °C in the fullerene-rich PCBM:PBTTT binaries (PBTTT content of ≤50%) can be attributed to the liquidus transition (i.e. the transition to a fully liquid state) and, respectively, the eutectic temperature of the PCBM/PCBM:PBTTT co-crystal system (i.e. the transition from solid PCBM and a solid co-crystalline phase to a 2-phase system comprised of solid PCBM and a melt composed of PBTTT- and PCBM molecules11). Note: this behaviour is somewhat more difficult to discern for the ketolactam:PBTTT system, though, the broadening of the endotherms at 5 and 15% PBTTT indicate a similar behaviour. In addition, we see clear fullerene-rich domains (dark contrast) at low polymer content (20%) in back-scattered electron (BSE) high-resolution electron microscraphs (HRSEM) of vapour-phase infiltrated (VPI) samples (insets in Fig. 2, middle panel; see the ESI† for details on the VPI process as well as ref. 12–14), indicating presence of fullerene-rich domains. Relatively, homogenous samples are found for compositions with predominant co-crystalline phase content (micrographs for a polymer content of 30% and 70% are shown).
On the PBTTT-rich side of the phase diagrams, a similar behaviour is observed; which, this time, is more clear in the ketolactam:PBTTT system. More specifically, in the ketolactam binary, at PBTTT content ≥50%, initially, a broadening of the liquidus endotherm is recorded, which evolves into two clearly distinguishable endotherms, one of which can be assigned to the eutectic temperature (≈230 °C; i.e. the transition from solid PBTTT and solid ketolactam:PBTTT co-crystalline domains to a two-phase system with solid co-crystal being in co-existence with a melt composed of PBTTT and ketolactam), and the other to the liquidus transition above which all material is in the melt (around 240 °C to 260 °C, depending on composition). A similar trend is found for the PCBM:PBTTT binary although the endotherms at polymer-rich compositions are less well resolved.
Such a phase behaviour (formation of two eutectics) implies that we have distinct solid-state phase morphologies in these fullerene:PBTTT systems.11 Solid fullerene- and co-crystalline phases co-exist at room temperature for PBTTT contents of ≤60% and ≤45% for PCBM:PBTTT and ketolactam:PBTTT binaries, respectively. Solid polymer and co-crystalline phase are present at higher polymer contents (i.e. above 60% and 45%). Thereby, fullerene-rich domains, so-called primary domains of fullerene,11,12 form at compositions of ≤55% PBTTT in the PCBM-based binary, while for ketolactam:PBTTT, they only form over a very limited composition regime (≤ceutectic-1 ≈ 10%). In contrast, polymer-rich primary domains develop in a similar composition regime for both systems: at fullerene content of ≤10%; i.e. above ceutectic-2 (≈90% PBTTT). In many cases, these primary regions are embedded in a eutectic morphology (finely-phase separated structures of the co-crystal and, respectively, polymer- or fullerene-rich regions11).
This morphology development can be followed by IR vibrational spectroscopy at room temperature (Fig. 2, bottom panels) using the CH2-assymetric stretch d-frequency in PBTTT, which can be used to probe the side chain order of the polymer (black circles), and the carbonyl stretching frequency of the fullerene (grey circles). We focus on these vibrations because intercalation of the fullerenes within the polymer side-chains (i.e., co-crystalline phase formation) disrupts the nearly all-trans configuration of the interdigitated side chains of the neat polymer regions,6 resulting in an increase of the d-frequency. Similarly, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O frequency of the fullerene provides useful information of the fullerene molecules environment, decreasing upon aggregation due to the high dielectric environment of the neat fullerene regions.
O frequency of the fullerene provides useful information of the fullerene molecules environment, decreasing upon aggregation due to the high dielectric environment of the neat fullerene regions.
For both systems, we observe the CH2-asymmetric stretch d-frequency to increase with fullerene content till a blend composition of 40% fullerene (60% PBTTT) is reached for PCBM:PBTTT, and 55% fullerene (45% PBTTT) for ketolactam:PBTTT. At these compositions, and for more fullerene-rich blends, the CH2-asymmetric stretch d-frequency stabilizes around 2924 cm−1, implying that maximum side-chain disorder is achieved, in agreement with fast calorimetry data presented in the ESI.† We attribute these observations to the fact that at low fullerene content, the PCBM/ketolactam molecules begin to intercalate in the polymer side chains; however, only when sufficient amount of fullerene is added (≥40% PCBM and ≥55% ketolactam), complete intercalation is reached with a composition independent polymer structure.
This picture is supported by the evolution of the CO frequency upon addition of the polymer to the fullerenes. At low fullerene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :polymer ratios (i.e., low fullerene content), the CO frequency is relatively independent of loading, due to the homogeneous environment of the cocrystal. At high fullerene:polymer ratios, the frequency shifts as we enter a phase region where fullerene-intercalated PBTTT co-exists with eutectic co-crystal/fullerene regions (ketolactam:PBTTT), or a fullerene-rich phase co-exists with eutectic co-crystal/fullerene domains (PCBM:PBTTT). The transition from polymer:co-crystal co-existence to fullerene:co-crystal coexistence is clear at ≈40% PCBM and ≈55% ketolactam.
:polymer ratios (i.e., low fullerene content), the CO frequency is relatively independent of loading, due to the homogeneous environment of the cocrystal. At high fullerene:polymer ratios, the frequency shifts as we enter a phase region where fullerene-intercalated PBTTT co-exists with eutectic co-crystal/fullerene regions (ketolactam:PBTTT), or a fullerene-rich phase co-exists with eutectic co-crystal/fullerene domains (PCBM:PBTTT). The transition from polymer:co-crystal co-existence to fullerene:co-crystal coexistence is clear at ≈40% PCBM and ≈55% ketolactam.
Having established critical compositions in the phase diagrams of fullerene:PBTTT blends, we went on to scrutinize the allocation of the eutectic temperatures and liquidus lines based on the DSC data shown in Fig. 2, top panels, using temperature resolved absorption and photoluminescence (PL) spectroscopy (Fig. 3), with cuts taken at 30 °C (black dashed lines), at close to the full melting of the blend (270 to 315) °C (depending on the system; red lines) and then after cooling back to 50 °C (blue lines). Thereby we focus on 50:50 and 40:60 fullerene:polymer blends.
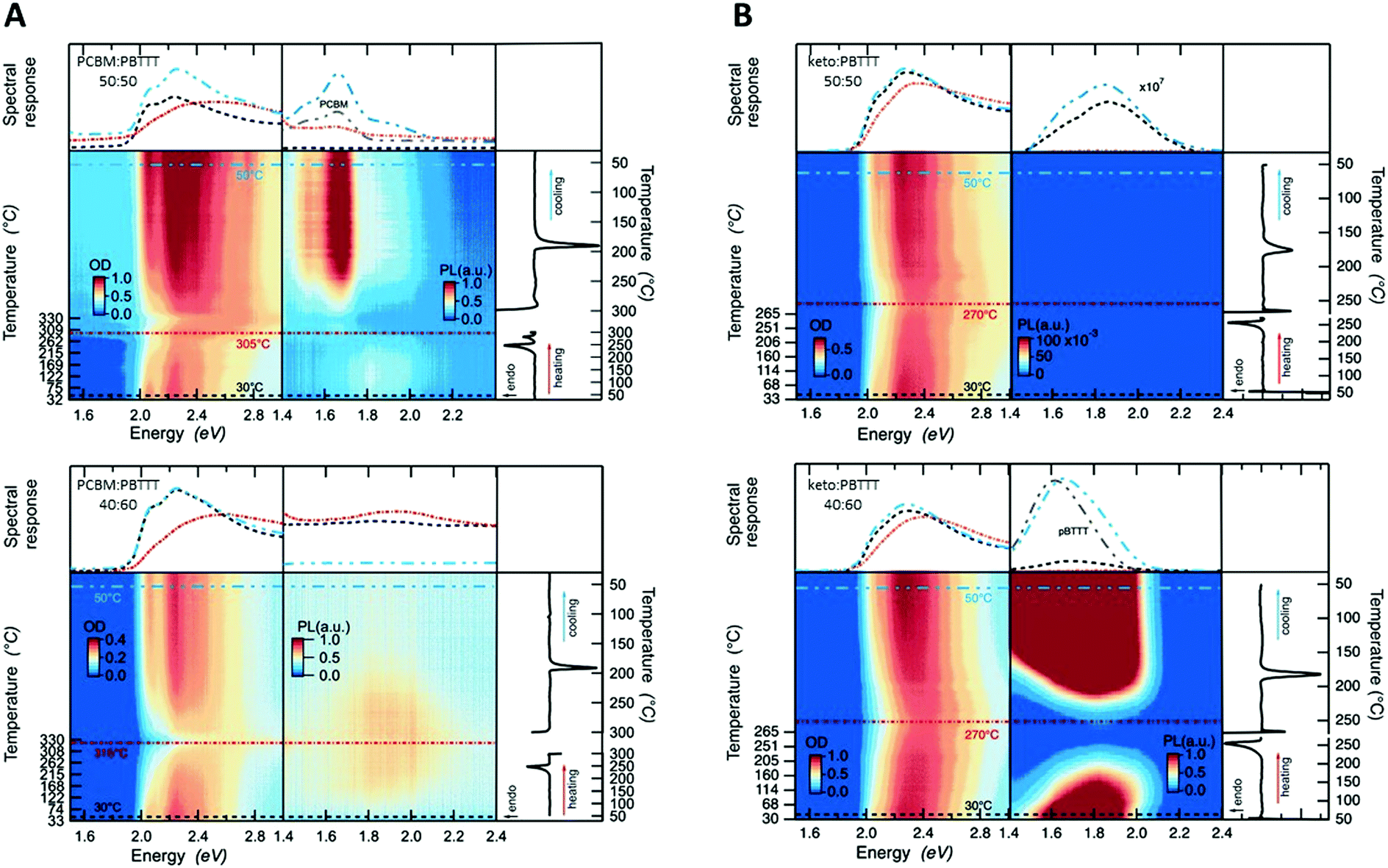 | ||
| Fig. 3 Temperature-resolved steady-state absorption (left) and photoluminescence (middle) spectra shown along with DSC thermograms (right), obtained at 20 °C min−1 heating/cooling for DSC, and at 20 °C min−1 heating (10 °C min−1 cooling) for absorption and PL for (A) 50:50 and 40:60 PCBM:PBTTT (top and bottom, respectively), and (B) the equivalent ketolactam:PBTTT binaries. | ||
We first discuss 50:50 fullerene:PBTTT binaries. While for the PCBM-based binaries the critical absorption features between (2.0 and 2.5) eV fade only around 305 °C (Fig. 3A, top), for the ketolactam blends this occurs already around 270 °C (Fig. 3B, top) in agreement with full melting occurring around these temperatures. For both systems, the line shape becomes, however, less defined at lower temperatures: around 250 °C for PCBM:PBTTT, and ≈230 °C for ketolactam:PBTTT, which we assign to the fact that, at the eutectic temperature, the co-crystalline phase and some PCBM melt (in case of PCBM:PBTTT), while in the ketolactam:PBTTT system, PBTTT and some co-crystalline domains melt. Tellingly, the line-shape for 40:60 ketolactam:PBTTT is relatively ill-defined as cast (prior to heating), as can be better seen in Fig. 1B (right), suggesting incomplete fullerene intercalation at that stage (complete intercalation leads to a structured line-shape with well-resolved 0–0, 0–1 and 0–2 transitions7). Moreover, upon heating, the absorption rapidly decreases above ≈230 °C, i.e., above temperatures where we have identified the eutectic temperature of the co-crystal:PBTTT system to occur.
Also in agreement with the established phase diagrams, for 40:60 PCBM:PBTTT there is nearly complete quenching of the PL, due to nearly comprehensive formation of the co-crystalline phase. The quenching is persistent upon cooling from the melt, in accord with expectations for soldification at the co-crystal composition with no excess PCBM or PBTTT. In contrast, while the as cast 50:50 PCBM:PBTTT again has minimal PL, indicating dominant co-crystal formation, significant PCBM PL appears after cooling from the melt, due to the formation of more phase-pure primary PCBM through the more equilibrium (melt) processing (cooling was performed at 10 °C min−1).
Similar behaviour is observed for the ketolactam:PBTTT blends. At 50:50 ketolactam:PBTTT, the system exhibits nearly complete PL quenching, both in the as-cast and melt-solidified states, consistent with the near co-crystal composition. For 40:60 ketolactam:PBTTT, the system is polymer rich, with the presence of primary polymer domains being inferred from the recorded PBTTT PL both in the as-cast and melt-solidified states. We note that the PL is maximally quenched in the intimately mixed melt, for both the hypo-co-crystalline (i.e. the PCBM-rich) 50:50 PCBM:PBTTT and the hyper-co-crystalline (i.e., polymer-rich) 40:60 ketolactam:PBTTT.
Having established the phase morphology of PCBM:PBTTT and ketolactam:PBTTT blends via the temperature/composition phase diagrams in Fig. 2, middle panels, we now show that this information aids in explaining some of the photo-physical behaviour of such binaries. We use for this purpose transient absorption spectroscopy data taken at a photoexcitation of 2.29 eV (exciting the polymer and partly the fullerene) and 3.10 eV (exciting predominantly the fullerene), Fig. 4 and 5.
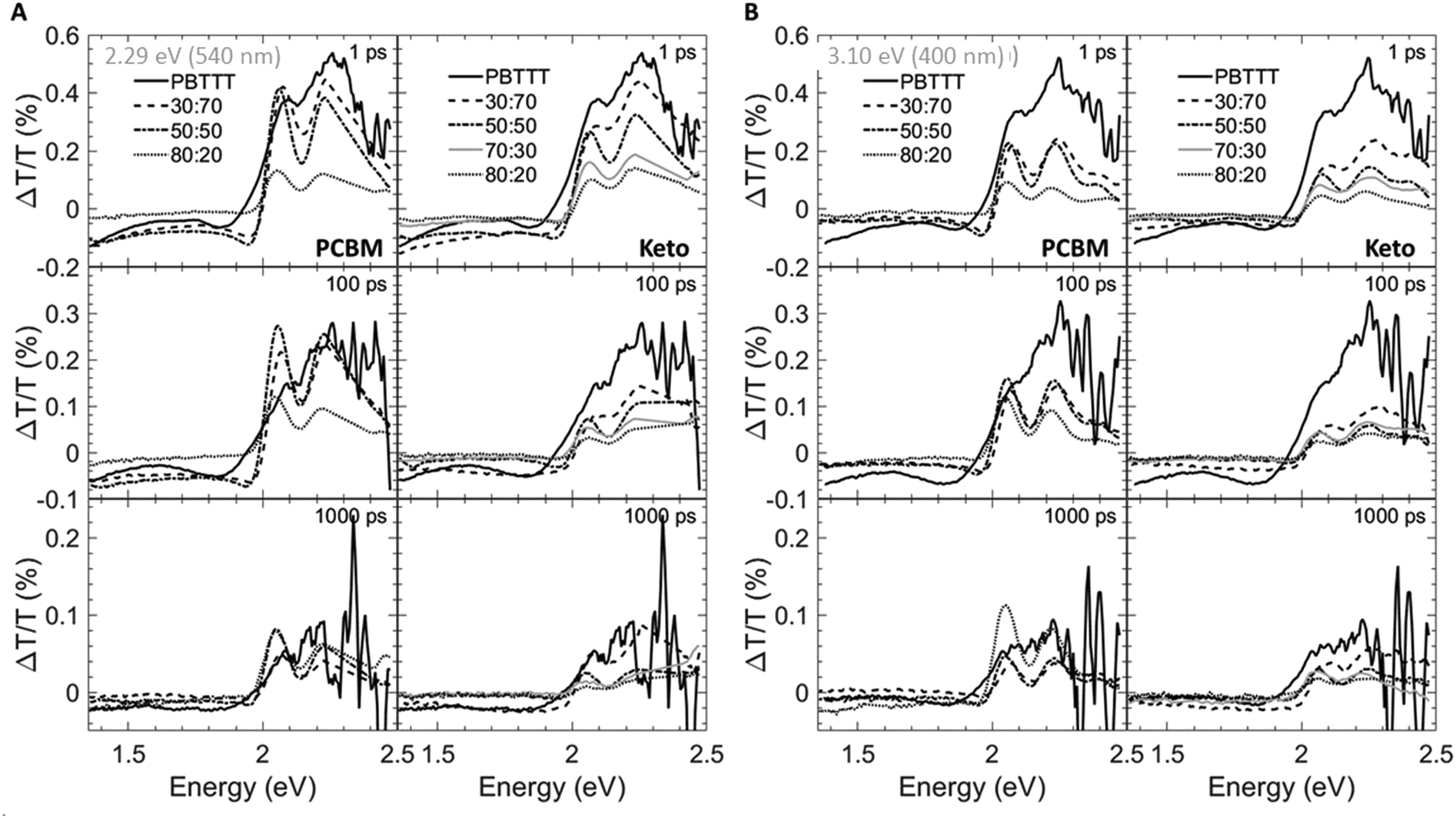 | ||
| Fig. 4 Spectral transient-absorption cuts taken at different delay times (1 ps: top panels; 100 ps: middle panels; 1000 ps: bottom panels) for fullerene:PBTTT blends photoexcited at 2.29 eV (540 nm) (A) and 3.1 eV (400 nm) (B). Data for PCBM-based binaries are shown in the left panels; those for ketolactam systems on the right. | ||
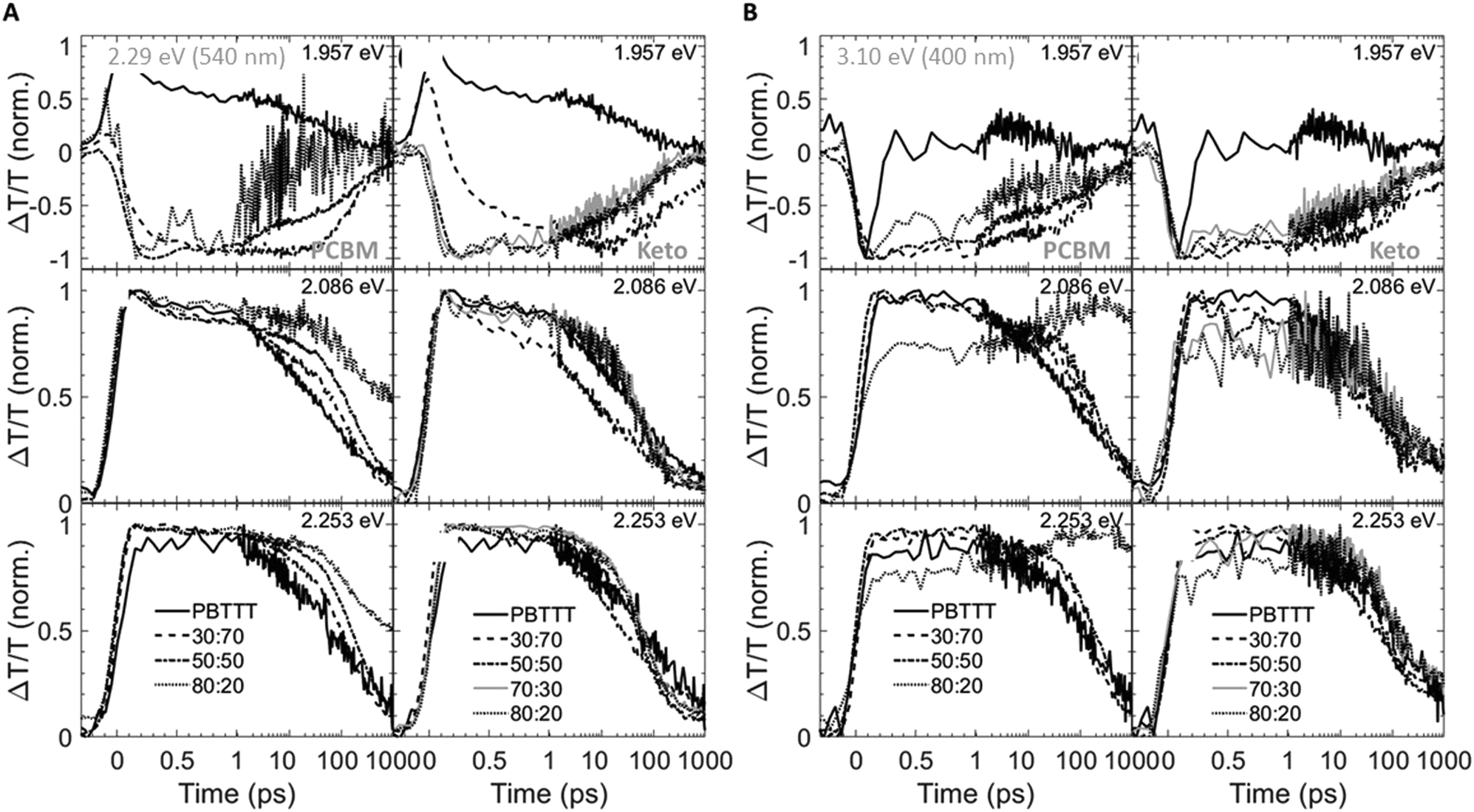 | ||
| Fig. 5 Spectral transient absorption cuts taken at different energies (1.957 eV: top panels; 2.086 eV: middle panels; 2.253 eV: bottom panels), photoexcited at 2.29 eV (540 nm) (A) and 3.1 eV (400 nm) (B). Data for PCBM-based binaries are shown in the left panels; those for ketolactam systems on the right. | ||
We observe that the photoexcitation bleaches the ground state absorption (see the regions around 2.086 eV and 2.253 eV) for all blends. Moreover, a distinct negative differential transmission around 1.957 eV, independent of excitation and with spectral features consistent with electro-absorption that is indicative of charge generation,15–17 is found for 30:70 and 50:50 PCBM:PBTTT binaries (Fig. 4A and B, left), i.e. where the solid-state structure is dominated by co-crystalline domains. However at excess fullerene (80: 20 PCBM:PBTTT), this feature is lost, likely because of the dominance of fullerene-rich domains. For the ketolactam-based system (Fig. 4A and B, right), similar observations are made, though the effect on the electroabsorption lineshape is more clearly discernible when exciting at 3.10 eV (400 nm; Fig. 4B, right).
More information can be obtained from the dynamics of the bleach (observed in the ≈2.0 to 2.5 eV region) and the electro-absorption feature (≈1.957 eV; Fig. 5). We focus first on the latter (Fig. 5, top panels). When exciting at 2.29 eV (540 nm), the electro-absorption signature for PCBM:PBTTT blends evolves rapidly for all compositions investigated (i.e., 80:20, 50:50, 30:70 PCBM:PBTTT; Fig. 5A, top left panel), likely because all contain co-crystalline domains. However, the decay is strongly dependent on blend composition. In blends where PCBM-rich phases are present (80:20, 50:50 PCBM:PBTTT), the electro-absorption feature is rapidly lost, which we tentatively assign to the fact that these fullerene-dominated regions enable rapid spatial separation of charges. This is somewhat more pronounced (faster decay) for samples with a larger fraction of PCBM-rich phase; i.e. 80:20 PCBM:PBTTT. In contrast, the decay is drastically slowed down in binaries of incomplete intercalation and lack of PCBM-rich domains (e.g., 30:70 PCBM:PBTTT). A similar trend, but less pronounced is recorded for the PCBM binary when exciting at 3.10 eV (400 nm; Fig. 5B, top left panel).
For 80:20, 70:30 and 50:50 ketolactam:PBTTT blends (Fig. 5A and B, right top panels), the electro-absorption dynamics are comparable to the 50:50 PCBM:PBTTT system (Fig. 5A and B, left top panels), likely because all these blends are dominated by the co-crystalline phase without many fullerene primary domains. No significantly faster decay is observed even for the ketolactam blends of very high fullerene content (80:20), supporting the view that fullerene-rich domains only start to form at ketolactam contents ≥90%, i.e., at PBTTT-contents below ceutectic-1ketolactam:PBTTT compared to the PCBM:PBTTT system where fullerene-rich domains begin evolving at fullerene contents of 45% and more, i.e. at PBTTT-contents below ceutectic-1PCBM:PBTTT.
Another intriguing observation is that the rise in electro-absorption feature is drastically slower for 30:70 ketolactam:PBTTT than for 30:70 PCBM:PBTTT. Considering that the 70:30 ketolactam:PBTTT blend is compositionally less close to the fullerene:polymer ratio where complete intercalation occurs (Fig. 2), these findings emphasise that is not only the phase morphology that matters but also the specific local arrangement of these phases.
The importance of the presence of fullerene-rich domains is also evident from the dynamics of the photo-bleaching at 2.086 eV and 2.254 eV (time cuts were taken when exciting at 2.29 eV; Fig. 5A, middle and bottom panels). In the 80:20 PCBM:PBTTT blend, the bleach displays a long-lived residual component. Conversely, the bleach recovery is nearly complete for the 30:70 PCBM:PBTTT blend, similar to the case of neat PBTTT. This effect is even more pronounced in PCBM-based blends when excited at 3.10 eV (Fig. 5B, left, middle and bottom panels), supporting our hypothesis that rapid charge separation within this timescale is enabled by fullerene-rich primary domains in PCBM:PBTTT blends and geminate recombination is reduced. In contrast, the bleach recovery in the ketolactam blends displays a weaker sensitivity to blend composition at both pump wavelengths (Fig. 5A and B, right, middle and bottom panels), which is attributed to the fact that these blends do not comprise such fullerene-rich primary regions even at relatively high ketolactam content.
Conclusions
We have shown that opening the cage of substituted fullerenes affects the thermal phase behaviour and, as a consequence the structure formation of these acceptor materials when used in blends. More specifically, in the case of ketolactamvs.PCBM, the open cage, perhaps combined with the slightly different substituent, leads to a lower melting temperature compared to PCBM. Accordingly, the eutectic point, here, ceutectic-1, shifts to very low fullerene content similar to the shifts in ceutectic observed for PCBM:poly(3-alkyl thiophene) (P3AT) blends when going from the high-melting poly(3-butyl thiophene) (P3BT; Tm ≈ 284 °C) to the medium-melting poly(3-hexyl thiophene) (P3HT; Tm ≈ 238 °C), followed by the low-melting poly(3-dodecyl thiophene) (P3DDT; Tm ≈ 169 °C).18 In the case of the ketolactam:PBTTT blends, the shift in ceutectic-1 limits the composition range where fullerene-rich primary domains evolve. This can be beneficial for increasing absorption (the co-crystalline phase absorbs more strongly in the visible compared to the neat fullerenes10) but limits charge generation and increases geminate recombination.8 In addition, the open-cage structure leads to more molecular disorder, as deduced from the very weak melting signal in differential scanning calorimetry. This leads to a stronger tendency to vitrify (forming a glass) upon blending, which means that blends are strongly affected by kinetic factors during solidification. This explains why intercalation of ketolactam into the PBTTT is somewhat hindered compared to PCBM. Indeed, PBTTT side-chain disordering sets in only at a larger fullerene fraction (see Fig. 2, bottom panel; Fig. S5–S7 in the ESI†), shifting the composition where essentially only a co-crystalline phase exists to higher fullerene content: 55% fullerene (i.e., 45% PBTTT) for ketolactam, vs. 40% fullerene (i.e., 60% PBTTT) for PCBM. As a consequence, the composition range where co-crystalline regions co-exist with a finely phase-separated eutectic phase, comprising ketolactam-intercalated PBTTT and PBTTT-rich regions, is enlarged. Our work, thus, emphasises the usefulness of the employment of experimentally established temperature/composition phase diagrams. They strengthen the view that relatively phase-pure (primary) domains can function as energetic sinks to create photo-generated charges. They also limit geminate recombination. Most importantly, experimentally established temperature/composition phase diagrams can assist in future materials design. The comparison of blends of PCBM and ketolactam and PBTTT show that relatively slight chemical changes that have no significant effect on molecular mass or energy levels, can affect the materials thermal phase behaviour and, in turn, the blends solid-state structure formation. Our work hence emphasises the importance not only of designing materials with respect to their optoelectronic properties or solution-processability, but also with regard to their thermal phase behaviour.11 This will be of particular importance for higher-efficiency OPV blends.Materials and methods
Materials
PBTTT was supplied by Dr Martin Heeney, Imperial College London, and PCBM was purchased from Solenne. The ketolactam fullerene was prepared according to a literature procedure.3,10,19Solutions
Solutions were prepared by dissolving PBTTT and fullerenes (PCBM; ketolactam) in 1,2-dichlorobenzene (Sigma Aldrich) and stirred on a standard hot plate for a minimum of 1 hour before deposition to ensure complete dissolution. The solutions concentration was 20 mg mL−1 and the standard stirring temperature was 100 °C. Fullerene:PBTTT blends were prepared by adding desired volume of fullerenes solutions to polymer's one.Films
Fullerene:PBTTT blend films were fabricated using a wire-bar coater (K 101 control coater, Printcoat Instruments) connected to a temperature-controlled stage. For all fullerene:PBTTT systems the stage temperature was set at 40 °C while the coating speed use was 8 cm s−1. Blend solutions were directly poured onto the bars, allowing a gap of around 150 μm in between the latter and the substrate.Brewster's angle transmission IR spectroscopy
A custom fixture was used to hold films deposited on double-side polished Si wafers at ≈73°-angle-of-incidence. Spectra were recorded at room temperature with a commercial Fourier transform instrument with a HgCdTe detector. The incident polarization was defined by a wire grid polarizer immediately before the sample. We followed the CH2-asymmetric stretch d-frequency in the polymer, which was previously used to follow disordering of the side chains of PBTTT, in that case to identify the liquid-crystalline transition.20 In the case of the fullerenes, we followed the CO-frequency, which depends on (1) how it is bonded, and (2) what it is embedded in; i.e. the local environment. For the ketolactam, data for the ‘amide’ CO bond is shown. We like to emphasize that a frequency shift of the same magnitude was observed for the ‘ketone’ CO bond (found at ≈1728 cm−1 for the neat ketolactam).
Room-temperature UV-vis absorption spectroscopy
Absorbance of fullerene:PBTTT blends were measured at normal incidence using a double-beam Agilent Cary 5000 UV-Vis-NIR spectrophotometer at 600 nm s−1. All spectra were normalized to the polymer absorbance peak that corresponds to the 0–0 transition, located near 2.0 eV. Spectra for the neat fullerenes are given in the ESI† (Fig. S3).Temperature-resolved UV-vis absorption-/photoluminescence (PL) spectroscopy
A custom setup was used that allows simultaneous p-polarized, near Brewster angle transmission UV-vis spectroscopy, with grazing incidence laser-diode illumination and normal-incidence, high numerical-aperture collection for photoluminescence on a support plate to control the substrate temperature. The setup was in a nitrogen filled glove-box. Films were on glass substrates to allow light transmission in the UV-visible wavelength regime and to suppress the film-thickness modulation in PL. Care was taken to focus the diode laser for PL measurements just beside the spot used for UV-vis absorption measurements, preventing interferences while ensuring that the same material area was probed. The blend films were then heated close or above the melt, followed by cooling to room temperature, while monitoring the linear absorption and photoluminescence spectra. Identical heating rates as used for differential scanning calorimetry (20 °C min−1) were employed. Cooling rates were slower (≈10 °C min−1).Differential scanning calorimetry (DSC)
DSC was performed under N2 atmosphere applying heating/cooling rates of 20 °C min−1 using a Mettler Toledo DSC700 instrument. Powders were produced from films drop cast from 1,2-dichlorobenzene solutions of 5% total material mass onto glass slides at 40 °C. After solvent evaporation at ambient pressure, films were annealed in vacuum for (7 to 8) h before being removed from substrates as scratched flakes/powders, (1.5 to 4) mg, and sealed for testing in aluminium crucibles.Vapour phase infiltration (VPI)
Prior to VPI deposition, samples were held in a vacuum chamber for 8 hours under 10−6 mbar for out-gasing solvent/moisture residues. The VPI deposition was carried out in an Ultratech/Cambridge Nanotech Savannah S200 system. The samples were exposed to 30 alternating cycles of DEZ and H2O at 60 °C. Each precursor pulse followed by 120 s hold step during which the evacuation valve remained close. Purge step with N2 gas of 30 s long was carried out before each exposure of complementary precursor.High-resolution electron microscopy (HRSEM)
For cross-section HRSEM, films were coated on Si substrates, exposed to the VPI process, then immersed in liquid nitrogen and cleaved. Cross-section HRSEM micrographs of the films were taken using a Zeiss Ultra-Plus FEG-SEM operated at 2 KV accelerating voltage with a working distance of 2.7 mm. Backscattered electron signal was collected with an ESB detector.Transient absorption spectroscopy
The experimental set-up uses an ultrafast laser system (Pharos Model PH1-20-0200-02-10, Light Conversion) generating 1030 nm pulses with ≈220 fs pulse duration at 100 kHz repetition rate; 10 W of the output was sent into a commercial optical parametric amplifier (Orpheus, Light Conversion) to produce a pump source covering a spectral range of 360–2600 nm. The probe source was generated by a Sapphire crystal, focused with 2 W of the laser output, to get a single filament white light continuum covering a spectral range of 480–1100 nm. Pump–probe experiments were carried out in a transient absorption commercial set-up (Light Conversion Hera) with the detection system consisting of a multichannel detector (200–1100 nm spectral sensitivity range, 256 pixels) along with an imaging spectrograph (Shamrock 193i, Andor Technology). The samples were photoexcited at 2.29 eV and 3.1 eV, with fluences of 378 nJ cm−2 and 1 μJ cm−2, respectively.Certain commercial equipment, instruments, or materials are identified in this paper in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by NIST, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Marie Skłodowska-Curie Actions Innovative Training Network “H2020-MSCAITN-2014 INFORM—675867”. N. S. and G. L. F thank the NSF-BSF support via the NSF-funded project #1905901 and BSF #2018652. CS acknowledges funding from the National Science Foundation (grant DMR-1729737). We like to thank Profs Natalie Banerji and Brad Chmelka for many valuable discussions and stimulating this work. We like to thank Sebastian Engmann for assistance with temperature dependent spectroscopy.Notes and references
- J. C. Hummelen, B. W. Knight, F. LePeq and F. Wudl, J. Org. Chem., 1995, 60, 532 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CrossRef CAS.
- J. C. Hummelen, M. Prato and F. Wudle, J. Am. Chem. Soc., 1995, 117, 7003 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicious, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. Chabinyc, R. Kline, M. McGehee and M. Toney, Nat. Mater., 2006, 5, 328 CrossRef CAS PubMed.
- N. C. Miller, E. Cho, R. Gysel, C. Risko, V. Coropceanu, C. E. Miller, S. Sweetnam, A. Sellinger, M. Heeney, I. McCulloch, J. L. Bredas, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2012, 2, 1208 CrossRef CAS.
- N. C. Miller, E. Cho, M. J. N. Junk, R. Gysel, C. Risko, D. Kim, S. Sweetnam, C. E. Miller, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, A. Amassian, D. Acevedo-Feliz, C. Knox, M. R. Hansen, D. Dudenko, B. F. Chmelka, M. F. Toney, J. L. Bredas and M. D. McGehee, Adv. Mater., 2012, 24, 6071 CrossRef CAS PubMed.
- E. Buchaca-Domingo, A. J. Ferguson, F. C. Jamieson, T. McCarthy-Ward, S. Shoaee, J. Tumbleston, O. G. Reid, L. Yu, M. B. Madec, M. Pfannmöller, F. Hermmerschmidt, R. R. Schröder, S. Watkins, N. Kopidakis, G. Portale, P. Smith, A. Amassian, M. Heeney, H. Ade, G. Rumbles, J. R. Durrant and N. Stingelin, Mater. Horiz., 2014, 1, 270 RSC.
- F. C. Jamieson, E. Buchaca Domingo, T. McCarthy-Ward, M. Heeney, N. Stingelin and J. Durrant, Chem. Sci., 2012, 3, 485 RSC.
- E. Buchaca-Domingo, K. Vandewal, Z. Fei, S. E. Watkins, F. H. Scholes, J. H. Bannock, J. de Mello, L. J. Richer, D. M. DeLongchamp, A. Amassian, M. Heeney, A. Salleo and N. Stingelin, J. Am. Chem. Soc., 2015, 137, 5256 CrossRef CAS PubMed.
- M. S. Vezie, M. Azzouzi, A. M. Telford, T. R. Hopper, A. B. Sieval, J. C. Hummelen, K. Fallon, H. Bronstein, T. Kirchartz, A. A. Bakulin, T. M. Clarke and J. Nelson, ACS Energy Lett., 2019, 4, 2096 CrossRef CAS.
- I. Botiz, M. M. Durbin and N. Stingelin, Macromolecules, 2021, 54, 5304 CrossRef CAS.
- A. Levitsky, G. M. Matrone, A. Khirbat, I. Bargigia, X. Chu, O. Nahor, T. S. Peretz, A. J. Moule, L. J. Richter, C. Silva, N. Stingelin and G. Frey, Adv. Sci., 2020, 7, 2000960 CrossRef CAS PubMed.
- S. Obuchovsky, M. Levin, A. Levitsky and G. L. Frey, Org. Electron., 2017, 49, 234 CrossRef CAS.
- C. Z. Leng and M. D. Losego, Mater. Horiz., 2017, 4, 747 RSC.
- M. Causa, J. De Jonghe-Risseh, M. Scarongella, J. C. Brauer, E. Buchaca-Domingo, J.-E. Moser, N. Stingelin and N. Banerji, Nat. Commun., 2016, 7, 12556 CrossRef CAS PubMed.
- F. Dou, E. Buchaca-Domingo, M. Sakowicz, E. Rezasoltani, T. McCarthy-Ward, M. Heeney, X. Zhang, N. Stingelin and C. Silva, J. Mater. Chem. C, 2015, 3, 3722 RSC.
- P. Krauspe, D. Tsokkou, M. Causa, E. Buchaca-Domingo, Z. P. Fei, M. Heeney, N. Stingelin and N. Banerji, J. Mater. Chem. A, 2018, 6, 22301 RSC.
- T. Ferenczi, C. Müller, D. D. C. Bradley, P. Smith, N. Stingelin and J. Nelson, Adv. Mater., 2011, 23, 4093 CrossRef CAS PubMed.
- D. F. Kronholm, A. B. Sieval and J. C. Hummelen, US Pat., 9475810B2, 2016 Search PubMed.
- D. M. DeLongchamp, R. J. Kline, Y. Jung, E. K. Lin, D. A. Fischer, D. J. Gundlach, S. K. Cotts, A. J. Moad, L. J. Richer, M. F. Toney, M. Heeney and I. McCulloch, Macromolecules, 2008, 41, 570 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc03082e |
| This journal is © The Royal Society of Chemistry 2021 |