
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis(di-tert-butylindenyl)tetrelocenes†
Liane Hildegard
Staub‡
a,
Jessica
Lambert‡
a,
Carsten
Müller
a,
Bernd
Morgenstern
a,
Michael
Zimmer
a,
Joshua
Warken
a,
Aylin
Koldemir
b,
Theresa
Block
b,
Rainer
Pöttgen
 b and
André
Schäfer
*a
b and
André
Schäfer
*a
aDepartment of Chemistry, Faculty of Natural Sciences and Technology, Saarland University, Campus Saarbrücken, 66123 Saarbrücken, Germany. E-mail: andre.schaefer@uni-saarland.de
bInstitute of Inorganic and Analytical Chemistry, Westfälische Wilhelms-Universität Münster, Corrensstrasse 30, 48149 Münster, Germany
First published on 14th April 2022
Abstract
The synthesis and characterization of bis(di-tert-butylindenyl) germanium(II), tin(II) and lead(II) complexes are reported, which includes the first structurally authenticated example of a bis(indenyl)germanocene. The species were studied in detail in solution and in the solid, which includes single crystal X-ray diffraction and NMR spectroscopy, as well as Mössbauer spectroscopy of the tin compound.
Introduction
Since the report of bis(indenyl)ferrocene, Ind2Fe, and bis(indenyl)cobaltocene, Ind2Co, in 1954,1 just a few years after the discovery of its parent compound, Cp2Fe,2 indenyl has developed into an important ligand in transition metal chemistry and many indenyl-based metallocenes have been reported, ever since.3 On the other hand, although group 14 metallocenes (tetrelocenes) have been known for many decades, it is rather surprising that bis(indenyl)tetrelocenes are almost unexplored.4 This might be related to the tendency of the indenyl ligand to adopt an η3 rather than an η5 coordination mode in some cases – often referred to as the indenyl effect5 – presumably making these compounds less stable than their cyclopentadienyl counterparts. To date, only a handful of bis(indenyl)tetrelocenes have been reported and only two of them have been structurally authenticated, namely a bis(bis(trimethylsilyl)indenyl)stannocene and a bis(bis(trimethylsilyl)indenyl)plumbocene. In addition, bis(bis(trimethylsilyl)indenyl)germanocene and bis(diisopropylindenyl)plumbocene have been characterized by NMR spectroscopy in solution, even though the latter was described to be unstable at ambient conditions, decomposing under the formation of elemental lead after just a few minutes. Furthermore, some related mixed heteroleptic indenyl cyclopentadienyl derivatives have also been described.6Following our group's continued interest in main group metallocene chemistry, we investigate the applicability of diisopropylindenyl and di-tert-butylindenyl as ligands for heavier group 14 elements and herein report the synthesis and structural characterization of a series of bis(di-tert-butylindenyl)tetrelocenes of germanium, tin and lead.
Results and discussion
We started our investigation by preparing diisopropylindene and di-tert-butylindene, followed by metalations with n-butyllithium and sodium amide, as described in the literature (Scheme 1).7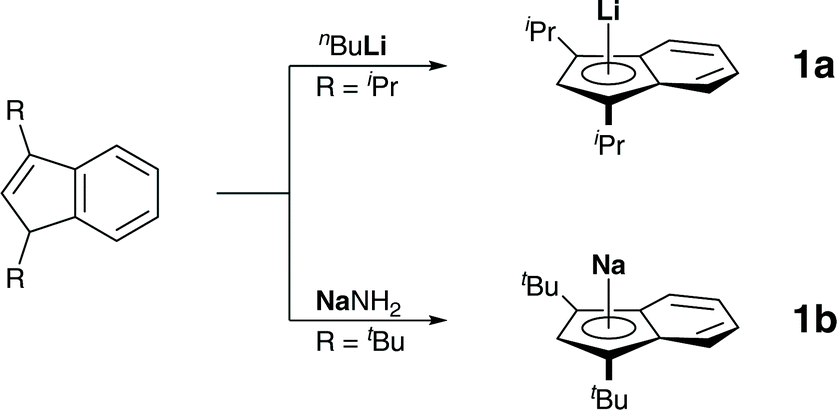 | ||
| Scheme 1 Synthesis of lithium diisopropyl indenide, 1a, and sodium di-tert-butyl indenide, 1b. | ||
Crystals of these alkaline metal indenyl complexes, 1a,b, suitable for X-ray diffraction, could be obtained from dimethoxyethane solutions at 273 K and allowed for structural characterizations (Fig. 1). 1a·(dme) crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , while 1b·(dme)3 crystallized in the monoclinic space group P21/c, both with one formular unit per asymmetric unit (Z = 1). Interestingly, while both compounds crystallize as dimethoxyethane complexes, lithium diisopropylindenide 1a forms a mono(dimethoxyethane) contact ion pair complex, while sodium di-tert-butylindenide 1b forms solvent separated ion pairs with three dimethoxyethane molecules coordinated to the sodium ion. This might be related to the increased steric demand of the di-tert-butylindenyl ligand compared to diisopropylindenyl and to a more ionic sodium indenyl bonding interaction compared to the lithium indenyl bonding.
, while 1b·(dme)3 crystallized in the monoclinic space group P21/c, both with one formular unit per asymmetric unit (Z = 1). Interestingly, while both compounds crystallize as dimethoxyethane complexes, lithium diisopropylindenide 1a forms a mono(dimethoxyethane) contact ion pair complex, while sodium di-tert-butylindenide 1b forms solvent separated ion pairs with three dimethoxyethane molecules coordinated to the sodium ion. This might be related to the increased steric demand of the di-tert-butylindenyl ligand compared to diisopropylindenyl and to a more ionic sodium indenyl bonding interaction compared to the lithium indenyl bonding.
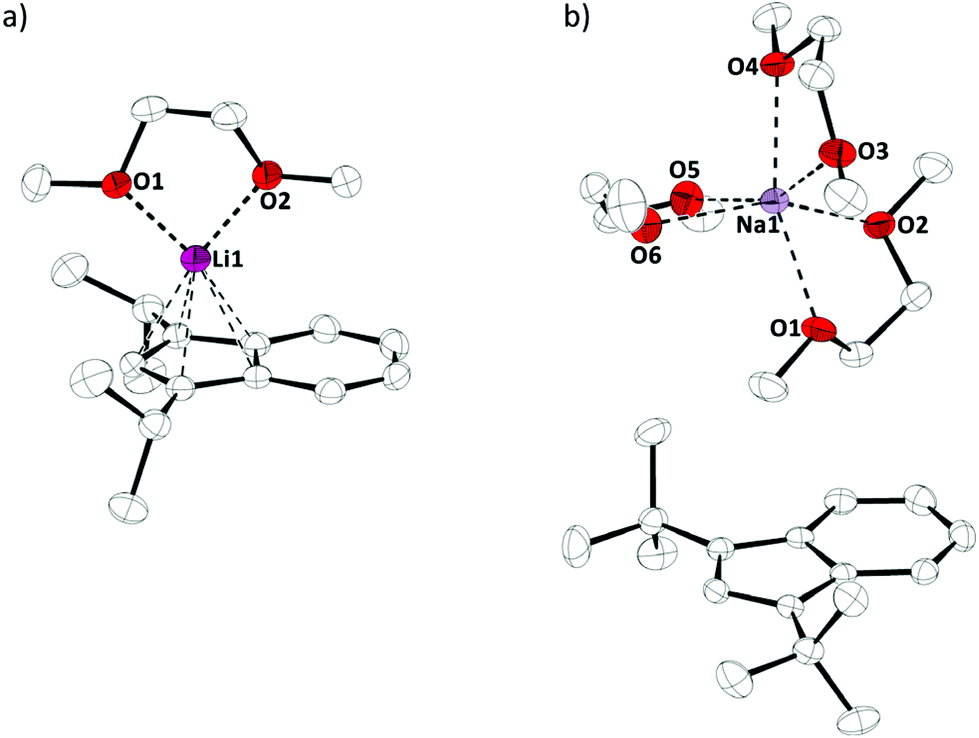 | ||
| Fig. 1 Molecular structure of (a) 1a·(dme) and (b) 1b·(dme)3 in the crystal (displacement ellipsoids at 50% probability level, H-atoms omitted for clarity). | ||
The Li–Indcentroid bonding distance in 1a·(dme) is 194.35(43) pm, which is slightly shorter than in lithium bis(tetrahydrofuran) bis(trimethylsilyl)indenide6c (199.79(62) pm), but longer than in lithium dimethoxyethane cyclopentadienide8 (190.98(29) pm). Characterization of lithium dimethoxyethane diisopropyl-indenide, 1a·(dme), by 7Li NMR spectroscopy in solution (C6D6) and in the solid state revealed quasi-identical 7Li NMR chemical shifts (δ7Li(C6D6) = −8.8; (δ7Li(CP-MAS) = −8.2), indicating that the complex is stable in benzene solutions (Fig. 2).
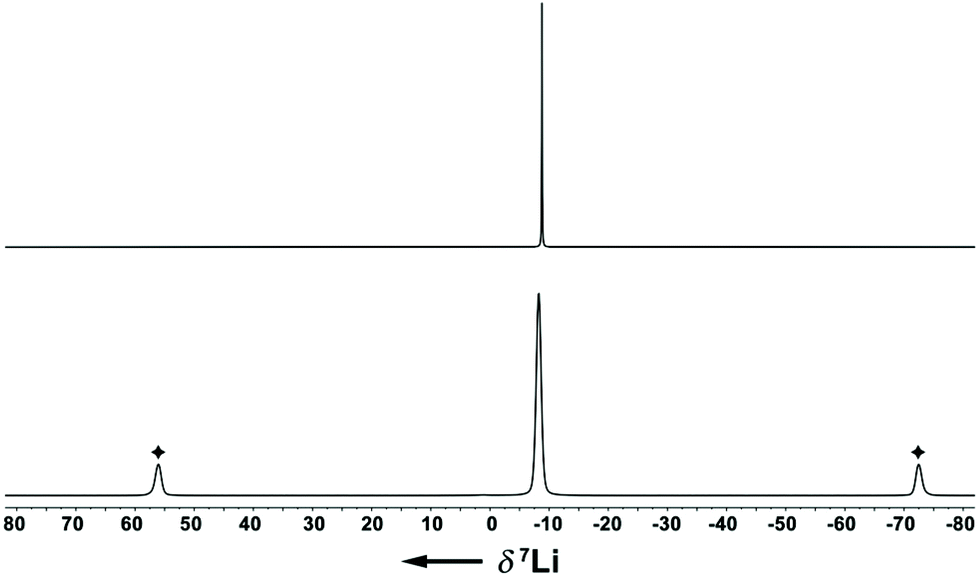 | ||
| Fig. 2 7Li{1H} NMR spectra of 1a·(dme) (upper trace: C6D6 solution, 156 MHz, 295 K; lower trace: CP-MAS(10 kHz), 156 MHz, 297 K; ✦ spinning sidebands). | ||
Following the successful synthesis of lithium diisopropyl-indenide 1a and sodium di-tert-butylindenide 1b, we investigated the reactivity of these compounds toward group 14 dihalides. Hanusa et al. had already reported that the reaction of potassium indenide or potassium diisopropylindenide with lead(II) iodide did not yield stable complexes, but resulted in the formation of lead metal.6b Likewise, an attempt by us to react bis(indenyl)magnesocene9 with germanium(II) chloride dioxane only yielded a complex mixture of unidentifiable products, indicating that the unsubstituted bis(indenyl)tetrelocenes are not stable. As stated before, this might be related to the indenyl effect,5 as the associated tendency of η5 to η3 ring-slippage may result in weaker E–Ind bonds compared to E–Cp bonds. However, it must also be taken into consideration that electronic effect can play a role here, as 1,3-di-tert-butylindenide is more electron rich than unsubstituted indenide, which promotes stronger bonding. Nevertheless, we attempted the synthesis of the bis(diisopropylindenyl)tetrelocenes, starting from 1a and germanium(II) chloride, tin(II) chloride and lead(II) chloride, but in all cases only obtained complex mixtures of products, in case of tin and lead containing large amounts of diisopropylindene along with metallic tin and lead. Thus, we turned to the sterically more demanding di-tert-butylindenyl ligand. Indeed, the reactions of sodium di-tert-butylindenide, 1b, with germanium(II) chloride, tin(II) chloride and lead(II) bromide yielded the corresponding tetrelocenes as air and moisture sensitive, but somewhat stable and isolatable solids (Scheme 2). Noteworthy, we found that shorter reaction times in case of the synthesis of plumbocene 2c were extremely beneficial. Stirring the reaction mixture over night yielded almost no product and large amount of lead metal, while rapid workup after just 15 minutes gave plumbocene 2c in good yields, indicating that 2c exhibits only limited stability in solution.
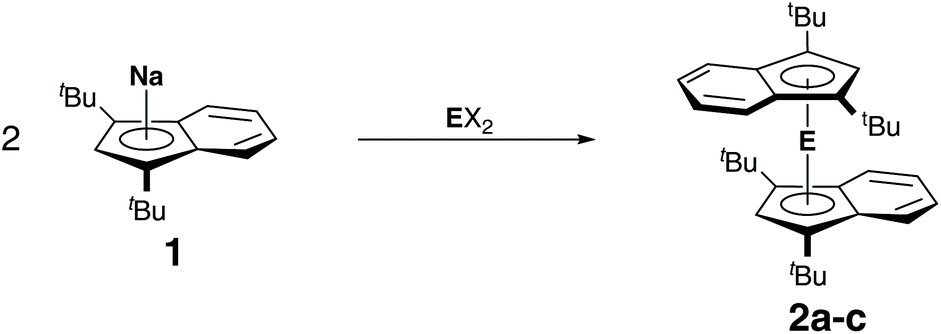 | ||
| Scheme 2 Synthesis of indenyl tetrelocenes 2a–c (a: EX2 = GeCl2; b: EX2 = SnCl2; c: EX2 = PbBr2). | ||
Crystals of tetrelocenes 2a–c, suitable for X-ray diffraction, were obtained at low temperatures and allowed for structural characterization of these species (Fig. 3). 2a–c all crystallized in the same monoclinic space group P21/n, each with one molecule per asymmetric unit (Z = 1). Interestingly, the previously reported solid state structures of bis(bis(trimethylsilyl)indenyl)stannocene6c and bis(bis(trimethylsilyl)indenyl)plumbocene6b showed tilted geometries in the solid state (α = 17.1° to 20.9°), analogous to many other tetrelocenes, while almost parallel arrangements are observed in 2a–c (α = 3.1° to 4.4°). The tetrel indenyl–centroid bonding distances are similar between 2b,c and their trimethylsilyl derivatives, only marginally elongated compared to their unsubstituted Cp2E-type parent compounds, and – as to be expected – increase going from germanium to lead. Furthermore, the two indenyl ligands are twisted against each other, with torsions angles of 83.2° to 92.2° (Table 1 and Fig. 3). Due to this twisting, two different rotamers exist and 2a–c are formally axial chiral in the solid state and crystallize as racemates, thus both rotamers/enantiomers are observed in the crystals.10
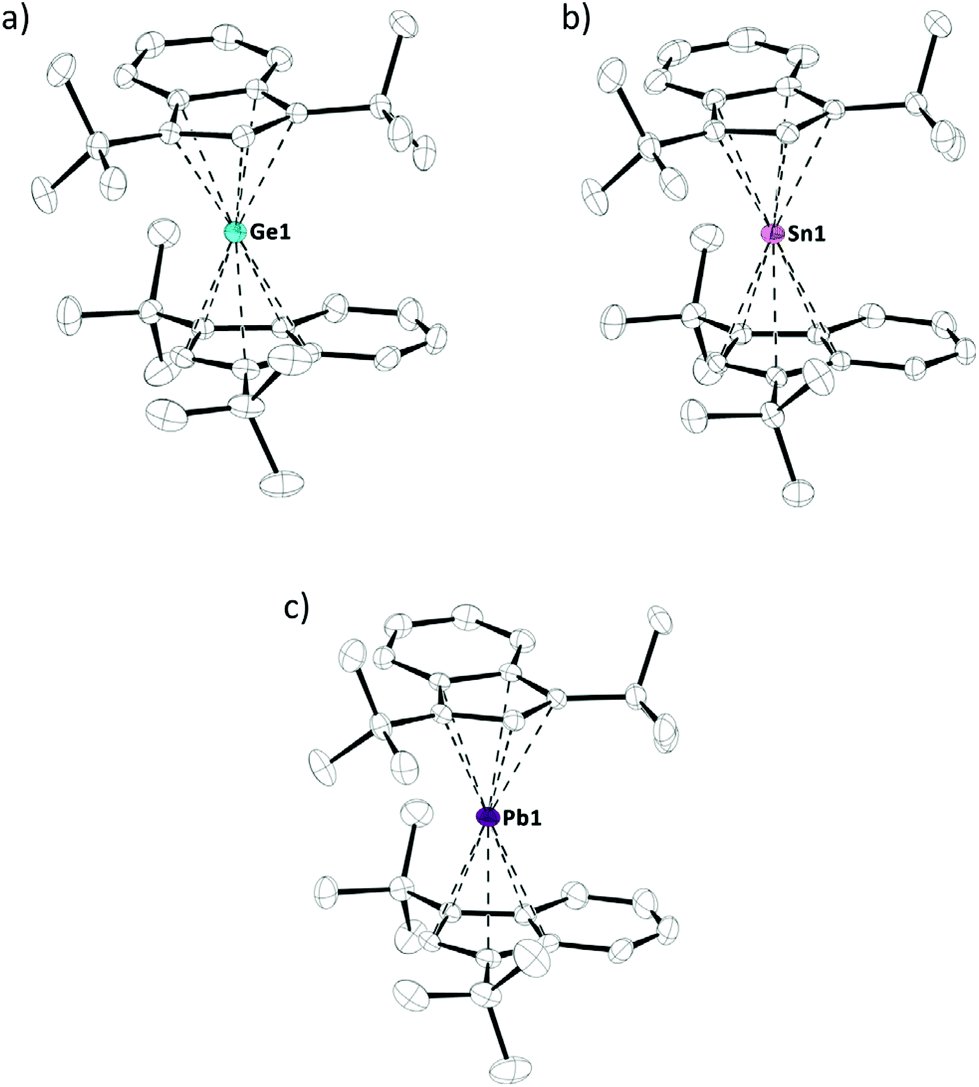 | ||
| Fig. 3 Molecular structure of indenyl tetrelocenes (a) 2a, (b) 2b and (c) 2c in the crystal (displacement ellipsoids at 50% probability level, H-atoms omitted for clarity, only one of two rotamers shown). | ||
| Compound | E–Ind/Cpa [pm] | α [°] | θ [°] | δ 119Sn/δ207Pb |
|---|---|---|---|---|
| a Relative to centroid of C5 moiety. b Dihedral angle between cyclopentadienide or indenide planes. c CInd–centroidInd–centroidInd–CInd torsion angle. d 149 MHz, C6D6, 295 K. e 149 MHz, CP-MAS (13 kHz). f 83 MHz, C6D6, 294 K. g 83 MHz, CP-MAS (13 kHz). | ||||
| 2a | 225.21(5) | 4.4 | 83.2 | — |
| 225.41(5) | ||||
| 2b | 241.08(3) | 3.8 | 91.8 | −2272d |
| 242.13(3) | −2335e | |||
| 2c | 247.72(3) | 3.1 | 92.2 | −4503f |
| 248.55(3) | −4734g | |||
| (2SiInd)2Sn6c | 241.24(7) | 20.3 | 79.2 | −21216c |
| 241.94(7) | 20.9 | 83.4 | ||
| 242.84(7) | ||||
| 242.95(7) | ||||
| (2SiInd)2Pb6b | 248.34(7) | 17.1 | 79.4 | — |
| 248.50(7) | 18.1 | 83.1 | ||
| 248.51(7) | ||||
| 248.86(7) | ||||
| Cp2Ge11 | 223.37(43) | 50.2 | — | — |
| Cp2Sn12 | 237.02(8) | 45.9 | −219913 | |
| 241.04(9) | 46.7 | |||
| 243.69(8) | ||||
| Cp2Pb14 | 246.55(5) | 57.5 | −503015 | |
In addition to a NMR characterization of 2a–c in solution, we also studied stannocene 2b and plumbocene 2c by CP/MAS NMR spectroscopy in the solid state (Fig. 5). The 119Sn NMR chemical shift of 2b is only slightly shifted between solution and solid state, with Δδ119Sn = 63, but both resonances (δ119Sn(C6D6) = −2272; δ119Sn(CP/MAS) = −2335) are within the typical range of stannocene-type compounds13 and are similar to the 119Sn NMR chemical shift of bis(bis(trimethylsilyl)indenyl)stannocene, reported by Jones and Cowley (δ119Sn(C6D6) = −2168.6c Furthermore, chemical shift tensors of δ11 = −2216, δ22 = −2318 and δ33 = −2470 are found, whereby the more deshielded component δ11 corresponds to the tensor along the Ind–Sn–Ind axis (Fig. 4). Hence, a chemical shift span of Ω = δ11 − δ33 = 102, which implies a smaller chemical shift anisotropy than in many stannylene-type compounds, is observed.16
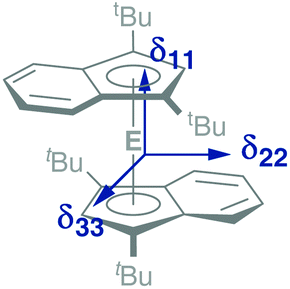 | ||
| Fig. 4 Schematic depiction of the principal axes of the chemical shift tensors δ11, δ22 and δ33 in 2a–c. | ||
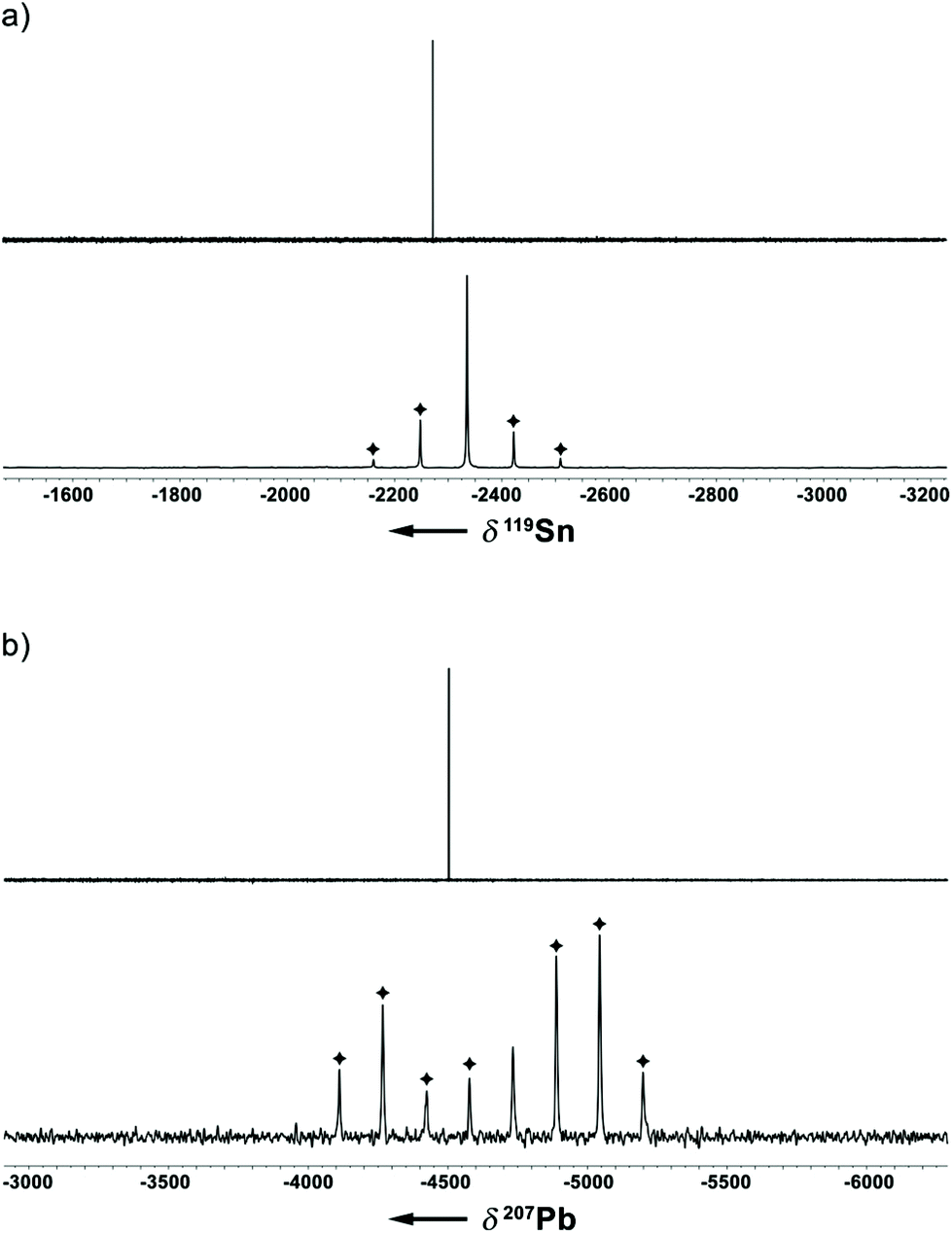 | ||
| Fig. 5 (a) 119Sn{1H} NMR spectra of 2b (upper trace: C6D6 solution, 149 MHz; lower trace: CP-MAS (13 kHz), 149 MHz; ✦ spinning sidebands) and (b) 207Pb{1H} NMR spectra of 2c (upper trace: C6D6 solution, 83 MHz, 294 K; lower trace: CP-MAS (13 kHz), 83 MHz; ✦ spinning sidebands). | ||
Noteworthy, no 207Pb NMR chemical shift of a bis(indenyl)plumbocene has been reported yet, as Hanusa et al. did not describe 207Pb NMR chemical shifts for bis(diisopropylindenyl)plumbocene or bis(bis(trimethylsilyl)indenyl)plumbocene.6b Within this work, we can confirm that the 207Pb NMR chemical shifts for 2c in solution and in the solid state (δ207Pb(C6D6) = −4503; δ207Pb(CP/MAS) = −4734) are within the typical range of Cp-based plumbocenes (e.g.: δ207Pb(Cp2Pb) = −5030; δ207Pb(Cp*2Pb) = −4384.15,17 Furthermore, the resonance of 2c is shifted by Δδ207Pb = 231, compared to the resonance observed in solution, with chemical shift tensors of δ11 = −3891 and δ22 = δ33 = −5169, resulting in a chemical shift span of Ω = δ11 − δ33 = 1278, which is a similar pattern to what was observed for stannocene 2b.
In addition to characterization by NMR spectroscopy, we also performed 119Sn Mössbauer spectroscopy on stannocene 2b at 78 K (Fig. 6). The corresponding fitting parameters are summarized in Table 2. Noteworthy, to the best of our knowledge, this is the first characterization of an indenyl tin(II) complex by Mössbauer spectroscopy, even though Mössbauer spectroscopy of Cp2Sn-type stannocenes has been reported.18
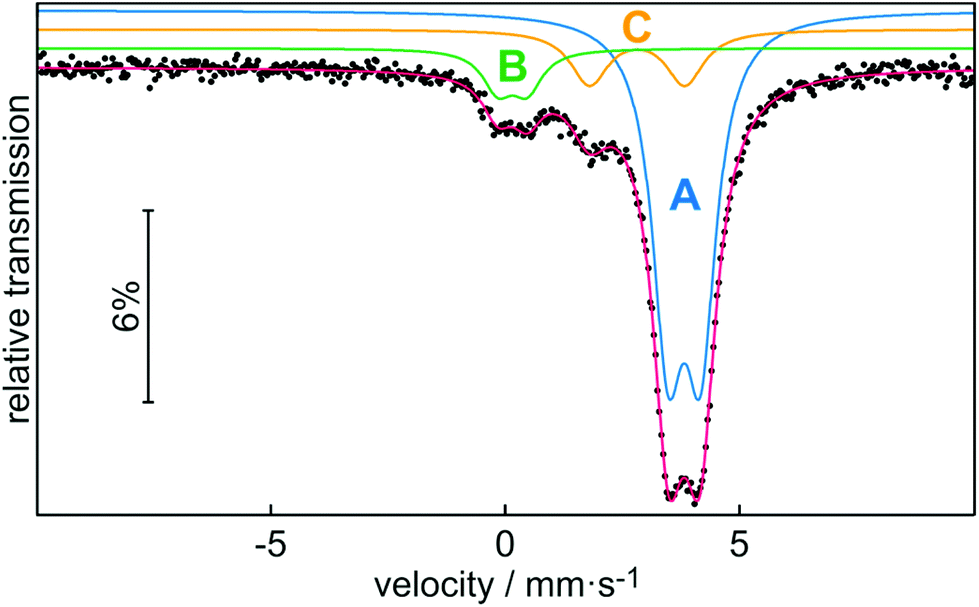 | ||
| Fig. 6 Experimental and simulated 119Sn Mössbauer spectrum of 2b. | ||
| Signal | δ [mm s−1] | ΔEQ [mm s−1] | Γ [mm s−1] | Area [%] |
|---|---|---|---|---|
| A | 3.80(1) | 0.70(1) | 0.81(1) | 75(1) |
| B | 0.14(2) | 0.63(3) | 0.78(6) | 9(1) |
| C | 2.80(1) | 2.03(3) | 0.98(6) | 16(1) |
The experimental Mössbauer spectrum is well reproduced with three subsignals. Signal A with the highest signal intensity can be assigned to stannocene 2b, while signal B and C can be attributed to decomposition products of this compound. Because of the identical chemical environment for all tin(II) atoms in stannocene 2b, a single signal (A) is expected. The isomer shift of δ = 3.80(1) mm s−1 is close to isomer shifts of Cp-based stannocenes (δ(Cp2Sn) = 3.73(6) mm s−1; δ(Me4Si2[2]Cp2Sn) = 3.734(6) mm s−1.18a,c Thus, there is no strong effect of the indenyl ligand on the isomer shift compared to cyclopentadienyl. This means that s electron density of tin(II) is predominantly affected by the C5 moiety of the di-tert-butylindenyl ligand. The coplanar (linear with regards to the centroids) coordination of tin(II) in 2b causes the small electric quadrupole splitting of ΔEQ = 0.70(1) mm s−1, which is also close to the reported one for Cp2Sn (ΔEQ = 0.65(6) mm s−1 (ref. 18a)). Signal B with an isomer shift of δ = 0.14(2) mm s−1 and a quadrupole splitting of ΔEQ = 0.63(3) mm s−1 is within the typical range of Sn(IV) compounds. Therefore, we assume that signal B can be assigned to SnO2, or, respectively, organotin oxygen derivates resulting from hydrolysis and/or oxidation of 2b.19–21 Signal C shows an isomer shift of δ = 2.80(1) mm s−1 and a quadrupole splitting of ΔEQ = 2.03(3) mm s−1 which is comparable to the Mössbauer spectroscopic data of Sn(OH)2 (δ = 2.83(6) mm s−1; ΔEQ = 2.20(6) mm s−1 or other organotin hydroxide byproducts,20–22 which may also result from hydrolysis of 2b. All observed line widths are in the usual range for 119Sn Mössbauer spectroscopic investigations.19,20
Following our experimental investigation, we also conducted DFT calculations at the PBE0-D3/def2-TZVP level of theory,23 choosing germanocene 2a as a model compound, since little was known about the frontier orbitals of bis(indenyl)-tetrelocenes. The calculated structure is in good agreement with the experimentally determined solid-state structure, with the Ge–Indcentroid bonds being marginally shorter by ca. 2 pm in the solid-state structure. Inspection of the frontier orbitals of germanocene 2a reveals a picture typical for tetrelocenes (Fig. 7).
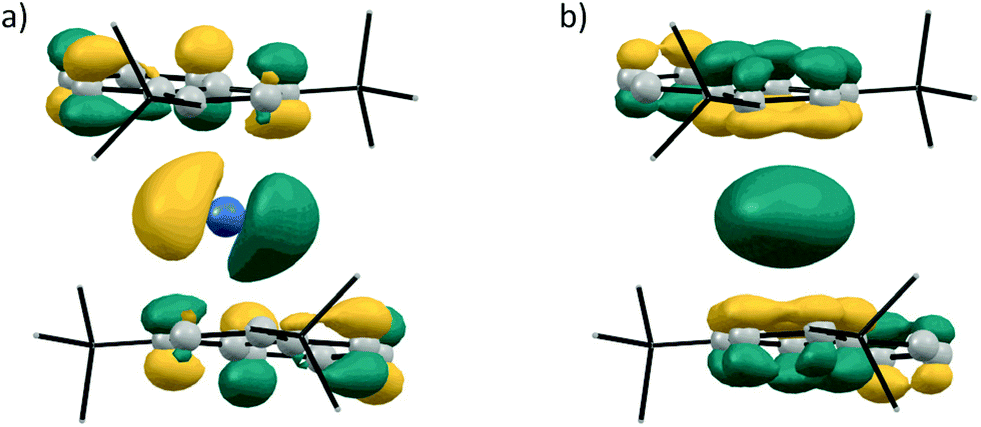 | ||
| Fig. 7 Kohn–Sham molecular orbital contours of (a) LUMO and (b) HOMO−4 of 2a (calculated at PBE0-D3/def2-TZVP; isodensity = 0.045 a.u.). | ||
The LUMO has a large coefficient at the central atom in the shape of a p orbital and is antibonding in nature with regards to the Ge–Ind bonds. As to be expected from the quasi-coplanar geometry of the indenyl ligands, the formal lone pair at the central atom possesses a spherical shape due to its high s character and is also anti-bonding with regards to the germanium indenyl bonds and relatively low in energy, being the HOMO−4.
Conclusions
In this work we studied the underexplored class of bis(indenyl) complexes of group 14 elements in oxidation state +II and found that these bis(indenyl)tetrelocenenes are only stable with a sterically demanding 1,3-substitution pattern. Utilizing the 1,3-di-tert-butylindenyl ligand in the form of its sodium complex, 1b, we were able to prepare the corresponding series of germanocene, 2a, stannocene, 2b, and plumbocene, 2c. The compounds were characterized by multinuclear NMR spectroscopy and single crystal X-ray diffraction, as well as 119Sn Mössbauer spectroscopy in case of 2b. They exhibit 119Sn and 207Pb NMR and 119Sn Mössbauer parameters, which are similar to Cp-based stannocenes and plumbocenes, respectively. Noteworthy, germanocene 2a represents the first X-ray diffraction structurally authenticated example of a bis(indenyl)germanocene.Experimental
All manipulations were carried out under an argon inert gas atmosphere (argon 5.0), using Schlenk techniques and a glovebox. Diisopropyl indene and di-tert-butyl indene were prepared following literature procedures.8NMR spectra were recorded on Bruker Avance III 300 (solution), Bruker Avance III 400 (solution) and Bruker Ascend 400WB (solid state) spectrometers. 1H and 13C NMR spectra were referenced using the solvent signals,247Li, 119Sn and 207Pb NMR spectra were referenced using external standards (δ7Li(LiCl in D20) = 0; δ119Sn(Me4Sn) = 0; δ207Pb(Me4Pb) = 0).
Single crystal X-ray diffraction analysis were carried out on a Bruker D8 Venture diffractometer with a microfocus sealed tube and a Photon II detector and using monochromated MoKα radiation (λ = 0.71073 Å). Corrected for absorption effects was performed with the multi-scan method. The structure was solved by direct methods using SHELXT and was refined by full matrix least squares calculations on F2 (SHELXL 2018) in the graphical user interface ShelXle.25–28 Crystal structure data has been deposited in the Cambridge Structural Database at CCDC 2154213–2154217.†119Sn Mössbauer spectroscopy was performed with a Ca119mSnO3 source in normal transmission geometry for three days, whereby the sample was cooled down at 78 K using a commercial liquid nitrogen cryostat. The sample was prepared in a thin-walled PMMA container. The spectrum was fitted by using the program WinNormos for the Igor6 software package.29 Elemental analyses were carried out on an Elementar vario MICRO cube.
Lithium diisopropyl indenide 1a
Diisopropyl indene (6.0 g, 29.9 mmol) was dissolved in toluene and treated with n-butyl lithium (2.5 M in hexane, 12.0 mL, 29.9 mmol), whereby the colour of the solution turned orange. The mixture was stirred overnight at 353 K. 1a precipitated as a solid and was collected by filtration and dried in vacuo. Recrystallisation from dimethoxyethane at 248 K yielded large crystals of 1a·(dme).Yield: 7.4 g, 25.0 mmol, 84%.
1H NMR (400 MHz, C6D6, 295 K): δ (in ppm) = 1.55 (br s, 12 H, CH(CH3)2), 2.12 (s, 4 H, (CH2OCH3)2), 2.42 (s, 6 H, (CH2OCH3)2), 3.60 (sep, 2 H, CH(CH3)2), 6.58 (br s, 1 H, Ind–H), 7.05–7.09 (m, 2 H, Ind–H), 7.85–7.88 (m, 2 H, Ind–H).
13C{1H} NMR (101 MHz, C6D6, 295 K): δ (in ppm) = 25.8 (br d, CH(CH3)2), 27.5 (CH(CH3)2), 58.7 (dme), 69.4 (dme), 108.3 (Ind), 111.4 (Ind), 115.3 (Ind), 119.5 (Ind), 123.5 (Ind).
7Li{1H} NMR (156 MHz, C6D6, 295 K): δ (in ppm) = −8.8.
13C{1H} CP-MAS (13 kHz) NMR (101 MHz, 297 K): δ (in ppm) = 26.9 (CH(CH3)2), 58.4 (dme), 70.4 (dme), 108.2 (Ind), 111.9 (Ind), 115.1 (Ind), 115.7 (Ind), 117.9 (Ind), 122.9 (Ind).
7Li{1H} CP-MAS(10 kHz) NMR (156 MHz, 297 K): δ (in ppm) = −8.2.
Sodium di-tert-butyl indenide 1b
Di-tert-butyl indene (7.44 g, 32.6 mmol) was added to a suspension of sodium amide (1.33 g, 34.2 mmol) in thf. The mixture was heated to reflux for 7 h and subsequently stirred overnight at ambient conditions, turning from colourless to red brown. After filtration and evaporation of all volatiles, the residue was washed with small portions of hexane, and dried in vacuo.Yield: 6.0 g, 23.8 mmol, 73%.
Recrystallisation from dimethoxyethane at 248 K yielded crystals of 1b·(dme)3.
1H NMR (400 MHz, thf-D8, 295 K): δ (in ppm) = 1.46 (s, 18 H, C(CH3)3), 6.32–6.36 (m, 2 H, Ind–H), 6.46 (s, 1 H, Ind–H), 7.52–7.56 (m, 2 H, Ind–H).
13C{1H} NMR (101 MHz, thf-D8, 295 K): δ (in ppm) = 33.2 (C(CH3)3), 33.8 (C(CH3)3), 110.9 (Ind), 112.9 (Ind), 113.6 (Ind), 120.0 (Ind), 124.9 (Ind).
Germanocene 2a and stannocene 2b
A thf solution of sodium di-tert-butyl indenide 1b (for 2a: 0.25 g, 0.99 mmol; for 2b: 1.00 g, 0.40 mmol) was treated with a thf solution of the corresponding group 14 salt (for 2a: GeCl2·dioxane: 0.12 g, 0.50 mmol; for 2b: SnCl2: 0.38 g, 2.0 mmol) at 193 K. The mixture was allowed to warm to room temperature and stirred overnight. All volatiles were removed in vacuo, the remaining solid residue was taken up in toluene and the resulting suspension was filtered. Subsequent removal of the solvent yielded the title compounds as yellow (2a) or orange (2b) solids. Crystals, suitable for single crystal X-ray diffraction, were obtained from toluene solutions at 248 K.Yield 2a: 153 mg, 0.29 mmol, 58%; 2b: 0.40 g, 0.70 mmol, 35%.
2a: CHN for C34H46Ge: C 76.10 (calc.: 77.44); H 9.10 (calc.: 8.79)%.
1H NMR (400 MHz, C6D6, 295 K): δ (in ppm) = 1.34 (s, 36 H, C(CH3)3), 5.92 (s, 2 H, Ind–H), 6.92–6.97 (m, 4 H, Ind–H), 7.48–7.52 (m, 4 H, Ind–H). 13C{1H} NMR (101 MHz, C6D6, 295 K): δ (in ppm) = 32.3 (C(CH3)3), 32.5 (C(CH3)3), 115.3 (Ind), 121.6 (Ind), 122.5 (Ind), 125.2 (Ind), 129.5 (Ind).
2b: CHN for C34H46Sn: C 71.4 (calc.: 71.2); H 8.3 (calc.: 8.1)%.
1H NMR (400 MHz, C6D6, 295 K): δ (in ppm) = 1.38 (s, 18 H, C(CH3)3), 6.02 (s, 1JH–Sn = 22 Hz, 2 H, Ind–H), 6.89–6.93 (m, 4 H, Ind–H), 7.51–7.56 (m, 4 H, Ind–H).
13C{1H} NMR (101 MHz, C6D6, 296 K): δ (in ppm) = 32.3 (C(CH3)3), 33.2 (C(CH3)3), 114.8 (1JC–Sn = 49 Hz, Ind), 120.7 (Ind), 122.4 (1JC–Sn = 11 Hz, Ind), 124.5 (Ind), 127.9 (Ind).
119Sn{1H} NMR (149 MHz, C6D6, 295 K): δ (in ppm) = −2272.
13C{1H} CP-MAS (13 kHz) NMR (101 MHz): δ (in ppm) = 31, 32, 33, 34 (C(CH3)2), 115 (Ind), 120 (Ind), 122 (Ind), 123 (Ind), 127 (Ind).
119Sn{1H} CP-MAS (13 kHz) NMR (149 MHz): δiso (in ppm) = −2335 (δ11 = −2216 ± 5, δ22 = −2318 ± 5, δ33 = −2470 ± 5).
119Sn Mössbauer: δ = 3.80(1) mm s−1; ΔEQ = 0.70(1) mm s−1; Γ = 0.81(1) mm s−1.
Plumbocene 2c
A dme solution of sodium di-tert-butyl indenide 1b (0.5 g, 2.00 mmol) was treated with a dme solution of PbBr2 (0.367 g, 1.00 mmol) at 273 K. The mixture was allowed to warm to room temperature and stirred for 15 min. All volatiles were removed in vacuo, the remaining solid residue was taken up in toluene and the resulting suspension was filtered. Subsequent removal of the solvent yielded the title compound as a dark red solid. Crystals, suitable for single crystal X-ray diffraction, were obtained from an ortho-difluorobenzene solution at 248 K.Yield: 0.315 g, 0.476 mmol, 48%.
CHN for C34H46Pb: C 59.42 (calc.: 61.69); H 7.10 (calc.: 7.00)%.
1H NMR (400 MHz, C6D6, 294 K): δ (in ppm) = 1.39 (s, 36 H, C(CH3)3), 6.02 (s, 1JH–Pb = 26 Hz, 2 H, Ind–H), 6.88–6.92 (m, 4 H, Ind–H), 7.49–7.53 (m, 4 H, Ind–H).
13C{1H} NMR (101 MHz, C6D6, 294 K): δ (in ppm) = 31.4 (C(CH3)3), 34.2 (C(CH3)3), 112.5 (1JC–Pb = 105 Hz, Ind), 120.2 (Ind), 121.7 (Ind), 127.0 (Ind), 128.8 (Ind).
207Pb{1H} NMR (83 MHz, C6D6, 294 K): δ (in ppm) = −4503.
13C{1H} CP-MAS (13 kHz) NMR (101 MHz): δ (in ppm) = 30, 31, 32, 34, 35 (C(CH3)2), 113 (Ind), 120 (Ind), 121 (Ind), 122 (Ind), 127 (Ind), 129 (Ind), 130 (Ind).
207Pb{1H} CP-MAS (13 kHz) NMR (83 MHz): δiso (in ppm) = −4734 (δ11 = −3891 ± 10, δ22 = −5169 ± 10, δ33 = −5169 ± 10).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Susanne Harling is thanked for elemental analysis. Funding by the Deutsche Forschungsgemeinschaft (Emmy Noether program, SCHA1915/3-1) is gratefully acknowledged. Instrumentation and technical assistance for this work were provided by the Service Center X-ray Diffraction, with financial support from Saarland University and the Deutsche Forschungsgemeinschaft (INST 256/506-1).Notes and references
- P. L. Pauson and G. Wilkinson, J. Am. Chem. Soc., 1954, 76, 2024–2026 CrossRef CAS
.
-
(a) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS
-
(a) V. Cadierno, J. Díez, M. P. Gamasa, J. Gimeno and E. Lastra, Coord. Chem. Rev., 1999, 193–195, 147–205 CrossRef CAS
-
(a)
P. Jutzi and N. Burford, Main Group Metallocenes, in Metallocenes, 1998 Search PubMed
- M. J. Calhorda, C. C. Romão and L. F. Veiros, Chem. – Eur. J., 2002, 8, 868–875 CrossRef CAS
-
(a) A. H. Cowley, M. A. Mardones, S. Avendaño, E. Román, J. M. Manriquez and C. J. Carrano, Polyhedron, 1993, 12, 125–127 CrossRef CAS
-
(a) A. C. Möller, R. Blom, R. H. Heyn, O. Swang, C.-H. Görbitz and T. Seraidaris, Eur. J. Inorg. Chem., 2005, 1759–1769 CrossRef
- R. Michel, R. Herbst-Irmer and D. Stalke, Organometallics, 2011, 30, 4379–4386 CrossRef CAS
- H. Gritzo, F. Schaper and H.-H. Brintzinger, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, E60, m1108–m1110 CrossRef
- Low temperature 1H NMR spectroscopy of 2a at 243 K did not show any signal splitting, suggesting that the Ind–Ge bond rotation has a low energy barrier.
- M. Grenz, E. Hahn, W.-W. du Mont and J. Pickardt, Angew. Chem., Int. Ed. Engl., 1984, 23, 61–63 CrossRef
- J. L. Atwood, W. E. Hunter, A. H. Cowley, R. A. Jones and C. A. Stewart, J. Chem. Soc., Chem. Commun., 1981, 925–927 RSC
- C. Müller, D. M. Andrada, I.-A. Bischoff, M. Zimmer, V. Huch, N. Steinbrück and A. Schäfer, Organometallics, 2019, 38, 1052–1061 CrossRef
- J. S. Overby, T. P. Hanusa and V. G. Young, Inorg. Chem., 1998, 37, 163–165 CrossRef CAS PubMed
- C. Janiak, H. Schumann, C. Stader, B. Wrackmeyer and J. J. Zuckerman, Chem. Ber., 1988, 121, 1745–1751 CrossRef CAS
- B. E. Eichler, B. L. Phillips, P. P. Power and M. P. Augustine, Inorg. Chem., 2000, 39, 5450–5453 CrossRef CAS PubMed
- P. Jutzi, R. Dickbreder and H. Nöth, Chem. Ber., 1989, 122, 865–870 CrossRef CAS
-
(a) P. G. Harrison and J. J. Zuckerman, J. Am. Chem. Soc., 1969, 91, 6885–6886 CrossRef CAS
-
G. K. Shenoy and F. E. Wagner, Mössbauer Isomer Shifts, North-Holland Publishing Company, Amsterdam, 1978 Search PubMed
-
N. N. Greenwood and T. C. Gibb, Mössbauer Spectroscopy, Chapman and Hall Ldt., London, 1971 Search PubMed
- J. J. Zuckermann, Adv. Organomet. Chem., 1971, 9, 21–134 CrossRef
- D. Honnick and J. J. Zuckerman, Inorg. Chem., 1976, 15, 3034–3037 CrossRef
- See ESI† for further details and references.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed
-
R. A. Brand, WinNormos for Igor6 (version for Igor 6.2 or above: 22/02/2017), Universität Duisburg, Duisburg (Germany), 2017 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2154213–2154217. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00582d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |