 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe impact of AlphaFold2 on experimental structure solution
Maximilian
Edich
 a,
David C.
Briggs
b,
Oliver
Kippes
a,
Yunyun
Gao
a and
Andrea
Thorn
*a
a,
David C.
Briggs
b,
Oliver
Kippes
a,
Yunyun
Gao
a and
Andrea
Thorn
*a
aInstitute for Nanostructure and Solid State Physics, Universität Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: andrea.thorn@uni-hamburg.de
bThe Francis Crick Institute, 1 Midland Road, London, NW1 1AT, UK
First published on 24th May 2022
Abstract
AlphaFold2 is a machine-learning based program that predicts a protein structure based on the amino acid sequence. In this article, we report on the current usages of this new tool and give examples from our work in the Coronavirus Structural Task Force. With its unprecedented accuracy, it can be utilized for the design of expression constructs, de novo protein design and the interpretation of Cryo-EM data with an atomic model. However, these methods are limited by their training data and are of limited use to predict conformational variability and fold flexibility; they also lack co-factors, post-translational modifications and multimeric complexes with oligonucleotides. They also are not always perfect in terms of chemical geometry. Nevertheless, machine learning-based fold prediction is a game changer for structural bioinformatics and experimentalists alike, with exciting developments ahead.
Introduction
Cryo-EM structures are an under-determined problem with noisy data
In recent years, cryo-electron microscopy has opened up possibilities to see and understand many large molecular machines and membrane complexes which were previously inaccessible to the structural biology community.1 Single-particle cryo-electron microscopy (Cryo-EM) does not require crystallization, uses very small amounts of material and is applicable to a wide range of macromolecule sizes. It also permits us to study fibrils, membrane proteins and viral assemblies, structures which are typically inaccessible by crystallography. The so-called “resolution revolution”2 (Fig. 1) led to the 2017 Nobel prize for developing cryo-electron microscopy as researchers overcame limitations in sample preparation, detector technology and image processing.3 Very recently, the possibility to determine atomic resolution structures has been experimentally demonstrated,4–6 a very exciting and long predicted development. However, solving an atomic structure from Cryo-EM micrographs remains an under-determined problem with noisy data: molecular particles† are hard to make out with the bare eye in the micrographs as the signal is so weak, and the inherent flexibility of molecules as well as thousands of atomic positions in each particle represent a huge parameter space for the atomic models we utilize for interpretation. For very large structures, computation can be an obstacle. Furthermore, for many structures, only low-resolution data are available, as inherent flexibility and particle heterogeneity blur reconstruction maps. Another kind of revolution, reliable structure prediction software like AlphaFold8 and RoseTTAFold,7 may now enable us to obtain more from such low-resolution data and has many implications for Cryo-EM, as it allows us to add prior knowledge in an unprecedented way to the experimental data.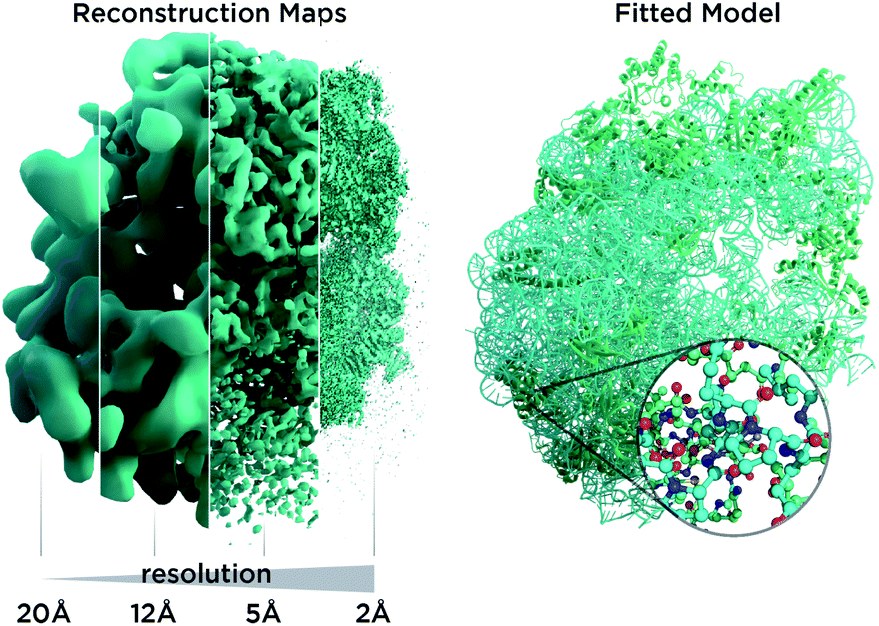 | ||
| Fig. 1 Resolution revolution in Cryo-EM. (Left) Reconstruction maps shown for the 50S ribosome with increasing level of detail from left to right as the field evolved. (Right) A typical atomic model used for interpretation of reconstruction maps. Picture courtesy of Andrea Thorn and Thomas Splettstößer. | ||
AlphaFold
AlphaFold8 is a machine learning (ML)-based program that predicts a protein structure based on the amino acid sequence. Its second generation, AlphaFold2 won the CASP14 challenge in 2020 in predicting protein folds, outperforming all previous algorithms for fold prediction from individual protein sequences. Chemical and Engineering News wrote “At this year’s competition, two-thirds of the protein structures predicted by AlphaFold were within experimental error. Basically, these structures were as good as the ones researchers could obtain through their laboratory techniques.”9These experimental techniques, such as Cryo-EM, are currently the main method for structure solution, and will certainly remain in use, in particular in studies focussing on large macromolecular assemblies with complex interactions. However, some structure determinations can now be replaced by ML-based fold prediction. AlphaFold2, and its alternative RoseTTAFold which is also ML-based, are already used to solve, model and improve protein structures,10–13 and ab initio model building in electron density or Cryo-EM reconstruction maps will no longer be a major challenge for experimental structural biology. This will have significant impact, not only for single-particle cryo-electron microscopy and crystallography, but also on NMR, electron tomography and small angle X-ray scattering. Most developments detailed here are very recent and ongoing, with many references being not yet peer-reviewed.
Therefore, selected examples from our work as part of the Coronavirus Structural Task Force will be used to highlight major points. This task force is an initiative of 26 structural biologists and students who focus on evaluating, improving and consolidating our knowledge of SARS-CoV-2 by critically assessing and disseminating structural information about SARS-CoV-2 proteins,14 which occasionally involves machine learning-based fold prediction as well. For the sake of simplicity, we focus here on AlphaFold2 and do not compare it with RoseTTAFold, as both tools face similar challenges.
Applications in experimental structural biology
Machine learning-based fold prediction is an extremely valuable tool for structural biologists. We will highlight how it can be used in connection with experimental work, but also which limitations apply. AlphaFold2 estimates per-residue confidence on a scale from 0–100, with higher values being better; this score is called predicted Local Distance Difference Test (pLDDT) and is stored in the B-factor fields of the model file. The AlphaFold2 pipeline begins with the generation of a deep multiple sequence alignment and a matrix of distance restraints derived from covariant evolutionary differences in the amino acid sequences. These inputs are then passed through a neural network block called “Evoformer” that produces processed multi-sequence alignments and an array of pair–pair distances. The following structure block uses these inputs to position and refine the relative location of each amino acid in 3D space. This whole process is then recycled several times to produce the final model.Construct design
Perhaps the first application that comes to mind for usage of AlphaFold2 for experiments is the design of expression constructs. Both the actual geometry of a predicted fold as well as the pLDDT for each residue can be utilized to get an idea about the less compact or ordered regions and the more ordered, or folded, parts of a given protein sequence, where backbone hydrogen bonds are saturated. Often, omitting less ordered regions from a protein sequence is beneficial to design well behaved recombinant proteins for structural study. In order to ensure solubility, ML-based fold prediction may also give information on surface residues of a given construct, and can permit biologists to better decide where a domain starts and ends in the sequence.One example for this is the betacoronavirus-specific marker domain of the protein nsp3. The roughly 200 residues long domain had previously been predicted to be mostly disordered.15 However, an AlphaFold2 prediction in our lab revealed a stable folded region of ∼80 residues surrounded by large disordered linkers, and recently, this region was experimentally confirmed to be folded (PDB ID 7T9W). Although it is not clear if this structure was solved with assistance from structure prediction, it is a clear example on how AlphaFold2 could be utilized for the identification and design of stable constructs.
De novo protein design
De novo protein design has been a goal of the structural biology community for decades, but was limited by the need to experimentally validate each and every new sequence in order to test hypotheses. With more reliable fold prediction available, this has now changed.16,17 However, at the time of writing this article, no experimentally validated folds designed by AlphaFold2 have yet been published.An obvious translational outlet from de novo protein design would be the creation of “biologics”, such as antibodies18 or DARPins.19 To date, creation of such molecules has favoured time- and resource-intensive experimental techniques such as ribosome-scanning.20 If the design of biologic therapeutics could be carried out computationally, this would represent a huge advance for potential therapeutics but also research and diagnostic tools.
Another related use of protein design technology would be vaccinology. At the beginning of the COVID-19 pandemic, Pfizer/BioNTech and Moderna vaccines included Spike-protein stabilising mutations to increase the half-life and thus the efficacy of their vaccines.21 Mutations to SARS-CoV-2 Spike-protein were introduced based upon knowledge of structures of the related SARS-CoV-1 and MERS Spike proteins, but it is clear that de novo design of stabilised immunogens would be beneficial to future vaccine-design work. Predicting such stabilizing mutations would also be beneficial elsewhere, for example for membrane proteins.
Structure solution
ML-based fold prediction can be used to solve structures in crystallography by molecular replacement,12 and to fit electron density and Cryo-EM reconstruction maps. Terwilliger et al. have designed an iterative AlphaFold2 modelling pipeline that iterates between AlphaFold2 modelling and refinement against experimental data (either Cryo-EM or macromolecular crystallography) to yield improved models.13 Machine learning-based fold prediction has not been used for validation yet, but it is being used to actively improve and correct experimental structure determination.10–12,22Limitations of machine learning based fold predictions
Impact of training data
AlphaFold2 is a supervised machine learning method, hence its neural networks are trained with data for which the fold is known, allowing its constituent neural networks to learn how to derive the fold from the sequence. The correct choice of training data is crucial to any machine learning project.23 In the case of AlphaFold2, the training data are from the world-wide Protein Data Bank (PDB),24 supplemented with selected “self-training” predictions25 the source of which remains somewhat vague.8 However, the PDB data have some very specific properties which affect the performance of AlphaFold2, mainly the lack of intrinsically disordered structures and a very limited sampling of conformational space.26Dark proteome
The “dark proteome” is comprised of proteins with no stable fold that represents a well-defined three-dimensional structure, an estimated 44% of proteins in eukaryotes and viruses.27,28 These are predominantly intrinsically disordered proteins which are thought to contribute to defence and signalling, sometimes becoming ordered when interacting with other macromolecules. As we have very little data about these proteins in a structural sense, besides their sequence, their structures as well as function cannot (yet) be modelled by machine learning. However, to an extent AlphaFold2 is able to predict where disorder occurs, as the pLDDT in such regions will be low.29 Williams et al.30 differentiate structures which AlphaFold2 cannot predict well into those which still look “like” proteins and so-called “barbed wire” folds where residues are just lined up next to each other in a nonsensical conformation with little hydrogen bonding. They speculate that in the latter case, there was little evolutionary covariance in that region.One exciting example of such a protein is the SARS-CoV-2 nucleocapsid, the complete structure of which is still unknown, but which is essential for the viral infection cycle, and hence an important drug target against COVID-19.31 The nucleocapsid protein has two ordered domains (which were experimentally determined32) and three intrinsically disordered regions: the N-terminal (residues 1–47), C-terminal (365–419) and linker (175–247) domains. These regions may order upon binding of RNA, when nucleocapsid protects the RNA as part of the virion.33 Nucleocapsid has also been implicated in RNA regulation. AlphaFold2 simulations of the entire sequence are a very typical example of such a prediction, where ordered compact domains are clearly separated from low confidence disordered areas which show mostly “barbed wire” – with the exception of a leucine-rich helix inside the conserved flexible linker (Fig. 2) and a lower confidence helix in the C terminus. Interestingly, the predicted helix in the linker domain is the site of the G215C mutation in the delta variant of SARS-CoV-2. This mutation is likely to stabilize conserved transient helices.34 If machine learning-based fold prediction could give us more insight about what happens when RNA is bound, this may shed light on nucleocapsid’s role in the cycle of infection.
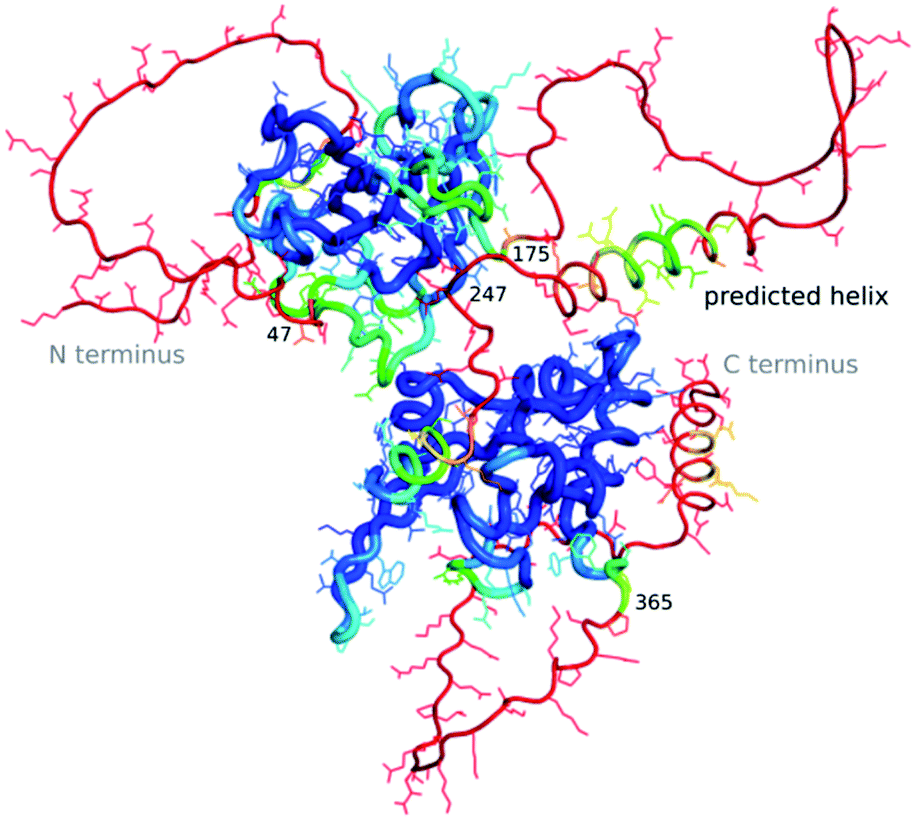 | ||
| Fig. 2 AlphaFold2 prediction for SARS-CoV-2 nucleocapsid. High pLDDT in blue, low pLDDT in red. While the two known domains are predicted with a high confidence (blue), the N-terminus, C-terminus and linker domain are showing a so-called “barbed wire” structure where AlphaFold2 could not find a solution and has low confidence (red), with the exception of two helices in the C-terminus and the linker domain. Picture courtesy of Lisa Schmidt and Andrea Thorn. | ||
Sampling conformational space
Many macromolecules exist in multiple conformations and are required to adopt these conformations as part of their biological function, particularly enzymes and membrane transporters.Even when the structural biologists report only one structure from a Cryo-EM experiment there is a wealth of hidden biological information in the Cryo-EM dataset, which can, in principle, be used to observe structural changes and determine free-energy landscapes.35 Currently, if structures of different states of a molecular machine are needed, the usual approach is to collect and process enough data to determine individual high-resolution reconstruction maps for each state, which are then interpreted individually.36 However, the fact that many molecular movements are more of a continuous process than jumps between discrete states can make this classical approach problematic.37
Indeed, the PDB arguably contains mostly stable conformations, as the majority of structures are derived from vitrified particles (in the case of Cryo-EM) or readily crystallizable proteins (in the case of crystallography). As AlphaFold2 was trained on this inherently biased training data, it could be assumed that, as a consequence, it can tell us little about different conformations and the inherent flexibility or movement of proteins. There are occasional exceptions, for example, when there is a homomultimer as opposed to a monomer38 (see section on “complexes with other proteins”, below). A recent study by del Alamo and colleagues39 showed that by reducing the depth of the multiple sequence alignment and limiting recycling of models within the AlphaFold2 pipeline, they were able to produce multiple fold predictions that appeared to bridge experimentally-determined conformations of the same protein, demonstrating that this technique samples conformational space for a range of G-protein coupled receptors and transmembrane transport proteins. As both these classes of protein are important drug targets,40 this is an important step towards leveraging AlphaFold2 for translational applications.
It would be highly desirable to get a better grip on the conformational variability information which is present in crystallographic and Cryo-EM data, but not currently really meaningfully used,37,41 and then to utilize these data to enhance the training data for neural networks like AlphaFold2.
Effects of mutations
It is still being debated whether AlphaFold2 is suitable42,43 or not44,45 to predict the outcomes of point mutations, with the EBI advising on its homepage that the latter is the case. A preprint by Zhang and co-workers42 suggests that, whilst there is no correlation between pLDDT and the destabilising effect of a point mutation, there is however a correlation when looking at ΔΔG or ΔΔTM values derived from the AlphaFold2 models, which would allow prediction of the destabilising effects of point mutations on protein structure.Ideal geometry
Currently, AlphaFold2 does not produce geometrically perfect structures, even if there are good reference structures in the PDB. SARS-CoV-2 main protease is arguably the most structurally researched drug target for COVID-19, with 485 experimentally determined structures deposited in the PDB. However, when comparing the PDB entry 6YB7 for the viral main protease with the AlphaFold2 prediction of the same sequence, AlphaFold2 produces four additional Ramachandran outliers and a higher number of atoms that come too close to each other (with a Clashscore of 16 instead of 2.5 for the experimentally measured and manually built structure, respectively). The predicted fold also has 88 bad bonds and angles as opposed to just 2 in the PDB entry. Some of these are in the C-terminal region which has a low pLDDT, but they also occur in regions with higher confidence values. It is yet unclear if the deviations from ideal geometry produced by AlphaFold2 differ from those a human would introduce when modelling a fold to a reconstruction map, and how representative such results are. It might be interesting to investigate this, for example by training a so-called adversarial network46 that differentiates between AlphaFold2 and human-made structures.Complexes with other proteins
AlphaFold-Multimer47 is a modification of AlphaFold2 to allow modelling of protein oligomers. The adaptations include pairing the multi-sequence alignments either over n copies of the monomer for homo-oligomeric cases, or all sequences for the hetero-oligomeric cases, and considering permutation symmetry of homo-oligomeric sequences. Currently this methodology appears to have more success with homo-oligomers than hetero-oligomers, and the authors assume that this is because the alignment of the homo-oligomer contains more evolutionary information about the homo-typic interfaces than for hetero-typic interfaces.47 The authors of AlphaFold-Multimer also note that it is not yet capable of accurately predicting antibody epitopes.47 Further development of this technology is highly desirable of course, as there would be considerable synergy with subtomogram averaging and single-particle Cryo-EM structure determinations of large multimeric complexes. Predictions could be used to estimate the mass of a complex and its projections, enabling a more target-oriented approach during particle picking. Nevertheless, the users of such tools should be careful not to rely on predictions only and therefore risk biased results. Combining AlphaFold predictions of single components and maps from tomography in integrative modelling can already reveal structures of large complexes at atomic resolution, as Mosalganti et al. demonstrated with the human nuclear pore complex, where the greatest limit is the resolution of the tomography map.48Complexes with ligands or oligonucleotides
AlphaFold2 is not designed to predict the interactions between proteins and other molecules such as RNA, DNA or smaller ligands and co-factors, or of post-translational modifications, glycosides chief among them.49 As a consequence, single-chain prediction may or may not correspond to the structure adopted in a complex, be it ligand-induced or through interactions with another macromolecule. Some fold predictions have “holes” where there should be a co-factor or a coordinated metal ion.50 Efforts to overcome this limitation are being made in the form of adding ligands geometrically to predicted structures51 and neural network based methods may soon supplement them. Since information on post-translational modification is readily available from UniProt, some of these such as glycosylation and phosphorylation may be a very viable first target for modelling.Experimental
For our research we used the Google Colab52 “Colabfold: AlphaFold2 using MMseqs2” with the AlphaFold8,53–55 version v2.2.0 and non-premium access.Clashscores, Ramachandran outliers and so-called “bad” bond lengths and angles were calculated with Molprobity.56
Conclusions
The Protein Data Bank24 represents the most important resource for structural biology, and machine learning-based fold prediction is now taking full advantage of this resource. In exchange, AlphaFold2 and more recently, RoseTTAFold, can aid experimental design, facilitate structure solution and interpretation of maps, and help to identify which part of a protein sequence may be intrinsically disordered. AlphaFold2 and RoseTTAFold are exciting new tools which will allow us to better understand macromolecular structures. Their shortcomings shine a spotlight on the shortcomings of our current experiments and how we interpret them with models. In order to push the boundaries of machine learning-based fold prediction, we will need better training data. And this means that we need experiments and modelling methods that sample, for example, the entire conformational space of proteins. The machine learning methods themselves will also have to evolve to include ligands, post-translational modifications and complexes of different types of molecules. Nevertheless, machine learning-based fold predictions are a game changer for structural bioinformatics and experimentalists alike, with exciting possibilities ahead.Author contributions
Andrea Thorn: supervision, conceptualization, formal analysis, visualization, investigation, writing – original draft, review & editing, project administration, funding acquisition. Dave Briggs: writing – original draft, review & editing. Maximilian Edich: methodology, investigation, formal analysis, writing – editing. Oliver Kippes: investigation, writing – original draft. Yunyun Gao: data curation & resources, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Christopher Williams, Jane Richardson, Gianluca Santoni and Arwen Pearson for discussion. This work was supported by the German Federal Ministry of Education and Research (grant nos. 05K19WWA and 05K22GU5) and the Deutsche Forschungsgemeinschaft (grant no. TH2135/21), D. C. B. acknowledges that this work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (CC2068), the UK Medical Research Council (CC2068) and the Wellcome Trust (CC2068).References
- X. Bai, G. McMullan and S. H. W. Scheres, Trends Biochem. Sci., 2015, 40, 49–57 CrossRef CAS PubMed
.
- W. Kuhlbrandt, Science, 2014, 343, 1443–1444 CrossRef PubMed
-
P. Brzezinski, Chemistry 2017 Nobel Price Announcement: Scientific Background, 2017 Search PubMed
- T. Nakane, A. Kotecha, A. Sente, G. McMullan, S. Masiulis, P. M. G. E. Brown, I. T. Grigoras, L. Malinauskaite, T. Malinauskas, J. Miehling, T. Uchański, L. Yu, D. Karia, E. V. Pechnikova, E. de Jong, J. Keizer, M. Bischoff, J. McCormack, P. Tiemeijer, S. W. Hardwick, D. Y. Chirgadze, G. Murshudov, A. R. Aricescu and S. H. W. Scheres, Nature, 2020, 587, 152–156 CrossRef CAS PubMed
- K. M. Yip, N. Fischer, E. Paknia, A. Chari and H. Stark, Nature, 2020, 587, 157–161 CrossRef CAS PubMed
- K. Zhang, G. D. Pintilie, S. Li, M. F. Schmid and W. Chiu, Cell Res., 2020, 30, 1136–1139 CrossRef PubMed
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millán, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed
-
DeepMind AI predicts protein structures, https://cen.acs.org/physical-chemistry/protein-folding/DeepMind-AI-predicts-protein-structures/98/web/2020/12, accessed May 7, 2021 Search PubMed
-
M. Gupta, C. M. Azumaya, M. Moritz, S. Pourmal, A. Diallo, G. E. Merz, G. Jang, M. Bouhaddou, A. Fossati, A. F. Brilot, D. Diwanji, E. Hernandez, N. Herrera, H. T. Kratochvil, V. L. Lam, F. Li, Y. Li, H. C. Nguyen, C. Nowotny, T. W. Owens, J. K. Peters, A. N. Rizo, U. Schulze-Gahmen, A. M. Smith, I. D. Young, Z. Yu, D. Asarnow, C. Billesbølle, M. G. Campbell, J. Chen, K.-H. Chen, U. S. Chio, M. S. Dickinson, L. Doan, M. Jin, K. Kim, J. Li, Y.-L. Li, E. Linossi, Y. Liu, M. Lo, J. Lopez, K. E. Lopez, A. Mancino, F. R. Moss, M. D. Paul, K. I. Pawar, A. Pelin, T. H. Pospiech, C. Puchades, S. G. Remesh, M. Safari, K. Schaefer, M. Sun, M. C. Tabios, A. C. Thwin, E. W. Titus, R. Trenker, E. Tse, T. K. M. Tsui, F. Wang, K. Zhang, Y. Zhang, J. Zhao, F. Zhou, Y. Zhou, L. Zuliani-Alvarez, D. A. Agard, Y. Cheng, J. S. Fraser, N. Jura, T. Kortemme, A. Manglik, D. R. Southworth, R. M. Stroud, D. L. Swaney, N. J. Krogan, A. Frost, O. S. Rosenberg and K. A. Verba, QCRG Structural Biology Consortium, 2021 Search PubMed
- T. G. Flower and J. H. Hurley, Protein Sci., 2021, 30, 728–734 CrossRef CAS PubMed
- A. J. McCoy, M. D. Sammito and R. J. Read, Acta Crystallogr., Sect. D: Struct. Biol., 2022, 78, 1–13 CrossRef PubMed
- T. C. Terwilliger, B. K. Poon, P. V. Afonine, C. J. Schlicksup, T. I. Croll, C. Millán, J. S. Richardson, R. J. Read and P. D. Adams, bioRxiv, 2022, preprint, DOI:10.1101/2022.01.07.475350.
- T. I. Croll, K. Diederichs, F. Fischer, C. D. Fyfe, Y. Gao, S. Horrell, A. P. Joseph, L. Kandler, O. Kippes, F. Kirsten, K. Müller, K. Nolte, A. M. Payne, M. Reeves, J. S. Richardson, G. Santoni, S. Stäb, D. E. Tronrud, L. C. von Soosten, C. J. Williams and A. Thorn, Nat. Struct. Mol. Biol., 2021, 28, 404–408 CrossRef CAS PubMed
- J. Lei, Y. Kusov and R. Hilgenfeld, Antiviral Res., 2018, 149, 58 CrossRef CAS PubMed
- M. Jendrusch, J. O. Korbel and S. K. Sadiq, bioRxiv, 2021, preprint, DOI:10.1101/2021.10.11.463937.
- J. G. Greener, S. M. Kandathil, L. Moffat and D. T. Jones, Nat. Rev. Mol. Cell Biol., 2022, 23, 40–55 CrossRef CAS PubMed
- R.-M. Lu, Y.-C. Hwang, I.-J. Liu, C.-C. Lee, H.-Z. Tsai, H.-J. Li and H.-C. Wu, J. Biomed. Sci., 2020, 27, 1 CrossRef CAS PubMed
- Y. L. Boersma, G. Chao, D. Steiner, K. D. Wittrup and A. Plückthun, J. Biol. Chem., 2011, 286, 41273–41285 CrossRef CAS PubMed
- A. Kunamneni, C. Ogaugwu, S. Bradfute and R. Durvasula, Antibodies, 2020, 9, 28 CrossRef CAS PubMed
- M. K. Higgins, J. Mol. Biol., 2021, 433, 167093 CrossRef CAS PubMed
- I. Barbarin-Bocahu and M. Graille, Acta Crystallogr., Sect. D: Struct. Biol., 2022, 78, 517–531 CrossRef CAS PubMed
- A. Thorn, Curr.Opin. Struct.
Biol., 2022, 74, 102368 CrossRef CAS PubMed
- H. Berman, K. Henrick and H. Nakamura, Nat. Struct. Mol. Biol., 2003, 10, 980 CrossRef CAS PubMed
-
Q. Xie, M.-T. Luong, E. Hovy and Q. V. Le, in Proceedings of the IEEE/CVF conference on computer vision and pattern recognition, 2020, pp. 10687–10698 Search PubMed
- C. Marino-Buslje, A. M. Monzon, D. J. Zea, M. S. Fornasari and G. Parisi, Briefings Bioinf., 2019, 20, 356–359 CrossRef CAS PubMed
- N. Perdigão, J. Heinrich, C. Stolte, K. S. Sabir, M. J. Buckley, B. Tabor, B. Signal, B. S. Gloss, C. J. Hammang, B. Rost, A. Schafferhans and S. I. O’Donoghue, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15898–15903 CrossRef PubMed
- N. Perdigão and A. Rosa, High-Throughput, 2019, 8, 8 CrossRef PubMed
- K. M. Ruff and R. V. Pappu, J. Mol. Biol., 2021, 433, 167208 CrossRef CAS PubMed
- C. Williams, D. C. Richardson and J. S. Richardson, Comp. Cryst. Newsl., 2022, 13, 7–12 Search PubMed
- Y. Peng, N. Du, Y. Lei, S. Dorje, J. Qi, T. Luo, G. F. Gao and H. Song, EMBO J., 2020, 39, e105938 CrossRef CAS PubMed
- Q. Ye, S. Lu and K. D. Corbett, Front. Immunol., 2021, 12, 719037 CrossRef CAS PubMed
- R. McBride, M. van Zyl and B. Fielding, Viruses, 2014, 6, 2991–3018 CrossRef PubMed
- H. Zhao, A. Nguyen, D. Wu, Y. Li, S. A. Hassan, J. Chen, H. Shroff, G. Piszczek and P. Schuck, PNAS Nexus, 2022, 1, pgac049 CrossRef PubMed
- D. Haselbach, J. Schrader, F. Lambrecht, F. Henneberg, A. Chari and H. Stark, Nat. Commun., 2017, 8, 15578 CrossRef CAS PubMed
- N. Fischer, A. L. Konevega, W. Wintermeyer, M. V. Rodnina and H. Stark, Nature, 2010, 466, 329–333 CrossRef CAS PubMed
- E. D. Zhong, T. Bepler, B. Berger and J. H. Davis, Nat. Methods, 2021, 18, 176–185 CrossRef CAS PubMed
- M. C. Cummins, T. M. Jacobs, F. D. Teets, F. DiMaio, A. Tripathy and B. Kuhlman, Protein Sci., 2022, 31, e4368 CrossRef CAS PubMed
- D. del Alamo, D. Sala, H. S. Mchaourab and J. Meiler, eLife, 2022, 11, e75751 CrossRef CAS PubMed
- R. Aguayo-Ortiz, J. Creech, E. N. Jiménez-Vázquez, G. Guerrero-Serna, N. Wang, A. M. da Rocha, T. J. Herron and L. M. Espinoza-Fonseca, Sci. Rep., 2021, 11, 16580 CrossRef CAS PubMed
- N. M. Pearce and P. Gros, Nat. Commun., 2021, 12, 5493 CrossRef CAS PubMed
- Y. Zhang, P. Li, F. Pan, H. Liu, P. Hong, X. Liu and J. Zhang, bioRxiv, 2021, preprint, DOI:10.1101/2021.11.03.467194.
- M. Akdel, D. E. V. Pires, E. Porta Pardo, J. Jänes, A. O. Zalevsky, B. Mészáros, P. Bryant, L. L. Good, R. A. Laskowski, G. Pozzati, A. Shenoy, W. Zhu, P. Kundrotas, V. Ruiz Serra, C. H. M. Rodrigues, A. S. Dunham, D. Burke, N. Borkakoti, S. Velankar, A. Frost, K. Lindorff-Larsen, A. Valencia, S. Ovchinnikov, J. Durairaj, D. B. Ascher, J. M. Thornton, N. E. Davey, A. Stein, A. Elofsson, T. I. Croll and P. Beltrao, bioRxiv, 2021, preprint, DOI:10.1101/2021.09.26.461876.
- M. A. Pak, K. A. Markhieva, M. S. Novikova, D. S. Petrov, I. S. Vorobyev, E. S. Maksimova, F. A. Kondrashov and D. N. Ivankov, bioRxiv, 2021, preprint, DOI:10.1101/2021.09.19.460937.
- G. R. Buel and K. J. Walters, Nat. Struct. Mol. Biol., 2022, 29, 1–2 CrossRef CAS PubMed
-
I. Goodfellow, Y. Bengio and A. Courville, Deep Learning, https://www.deeplearningbook.org, accessed April 29, 2022 Search PubMed
- R. Evans, M. O’Neill, A. Pritzel, N. Antropova, A. Senior, T. Green, A. Žídek, R. Bates, S. Blackwell, J. Yim, O. Ronneberger, S. Bodenstein, M. Zielinski, A. Bridgland, A. Potapenko, A. Cowie, K. Tunyasuvunakool, R. Jain, E. Clancy, P. Kohli, J. Jumper and D. Hassabis, bioRxiv, 2022, preprint, DOI:10.1101/2021.10.04.463034.
- S. Mosalaganti, A. Obarska-Kosinska, M. Siggel, R. Taniguchi, B. Turoňová, C. E. Zimmerli, K. Buczak, F. H. Schmidt, E. Margiotta, M.-T. Mackmull, W. J. H. Hagen, G. Hummer, J. Kosinski and M. Beck, Science, 2022, 376, eabm9506 CrossRef CAS PubMed
- H. Bagdonas, C. A. Fogarty, E. Fadda and J. Agirre, Nat. Struct. Mol. Biol., 2021, 28, 869–870 CrossRef CAS PubMed
- A. Kryshtafovych, J. Moult, R. Albrecht, G. A. Chang, K. Chao, A. Fraser, J. Greenfield, M. D. Hartmann, O. Herzberg, I. Josts, P. G. Leiman, S. B. Linden, A. N. Lupas, D. C. Nelson, S. D. Rees, X. Shang, M. L. Sokolova and H. Tidow, AlphaFold2 team, Proteins, 2021, 89, 1633–1646 CrossRef CAS PubMed
- M. L. Hekkelman, I. de Vries, R. P. Joosten and A. Perrakis, bioRxiv, 2021, preprint, DOI:10.1101/2021.11.26.470110.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed
- A. L. Mitchell, A. Almeida, M. Beracochea, M. Boland, J. Burgin, G. Cochrane, M. R. Crusoe, V. Kale, S. C. Potter, L. J. Richardson, E. Sakharova, M. Scheremetjew, A. Korobeynikov, A. Shlemov, O. Kunyavskaya, A. Lapidus and R. D. Finn, Nucleic Acids Res., 2020, 48, D570–D578 CAS
- M. Mirdita, M. Steinegger and J. Söding, Bioinformatics, 2019, 35, 2856–2858 CrossRef CAS PubMed
- M. Mirdita, L. von den Driesch, C. Galiez, M. J. Martin, J. Söding and M. Steinegger, Nucleic Acids Res., 2017, 45, D170–D176 CrossRef CAS PubMed
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed
Footnote |
| † In this context, a particle can be both a single molecule or a distinct assembly of molecules, such as a virus. |
| This journal is © The Royal Society of Chemistry 2022 |