
Unconventional aliphatic fluorophores discovered as the luminescence origin in citric acid–urea carbon dots†
Xiaoxiao
Yao
 a,
Yinhan
Wang
b,
Fangjia
Li
c,
Joseph J.
Dalluge
a,
Galya
Orr
c,
Rigoberto
Hernandez
b,
Qiang
Cui
de and
Christy L.
Haynes
*a
a,
Yinhan
Wang
b,
Fangjia
Li
c,
Joseph J.
Dalluge
a,
Galya
Orr
c,
Rigoberto
Hernandez
b,
Qiang
Cui
de and
Christy L.
Haynes
*a
aDepartment of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant Street SE, Minneapolis, Minnesota 55455, USA. E-mail: chaynes@umn.edu
bDepartments of Chemistry, Chemical and Biomolecular Engineering, and Materials Science and Engineering, Johns Hopkins University, Baltimore, Maryland 21218, USA
cEnvironmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99354, USA
dDepartment of Chemistry, Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, USA
eDepartments of Physics and Biomedical Engineering, Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, USA
First published on 17th June 2022
Abstract
Carbon dots (CDs) are emerging as the material of choice in a range of applications due to their excellent photoluminescence properties, ease of preparation from inexpensive precursors, and low toxicity. However, the precise nature of the mechanism for the fluorescence is still under debate, and several molecular fluorophores have been reported. In this work, a new blue fluorophore, 5-oxopyrrolidine-3-carboxylic acid, was discovered in carbon dots synthesized from the most commonly used precursors: citric acid and urea. The molecular product alone has demonstrated interesting aggregation-enhanced emission (AEE), making it unique compared to other fluorophores known to be generated in CDs. We propose that this molecular fluorophore is associated with a polymer backbone within the CDs, and its fluorescence behavior is largely dependent on intermolecular interactions with the polymers or other fluorophores. Thus, a new class of non-traditional fluorophores is now relevant to the consideration of the CD fluorescence mechanism, providing both an additional challenge to the community in resolving the mechanism and an opportunity for a greater range of CD design schemes and applications.
Introduction
The desirable fluorescence properties of carbon dots (CDs) have been applied in a variety of fields such as bioimaging, LEDs, sensing etc.1,2 Their excellent photophysical properties,2,3 limited toxicity,4 and the potential for large scale production5 have made them a promising replacement for semiconductor quantum dots and molecular fluorophores. While their fluorescence properties have attracted great attention, the fluorescence mechanism of CDs has remained unclear, and a few hypotheses are under debate, including mechanisms that invoke quantum confinement effects, surface states, and molecular states.6 First, the quantum confinement effect is well understood for semiconductor quantum dots, but appears less applicable for CDs. For example, Yuan et al. have synthesized triangular CDs with an unprecedentedly narrow bandwidth via solvothermal treatment of phloroglucinol; the measured increase in size for a series of these CDs matched with the red-shift in the CD emission.7 However, other studies have found out that CD size is not related to emission wavelength.8 Second, the surface state hypothesis for CD emission is usually related to surface functional groups9 or nitrogen/oxygen doping,10 which are deemed to be different from the sp2 carbonized core structure.11 Nguyen et al. used scanning tunneling microscopy to image individual CDs with subparticle resolution and found that the localized surface defects have electronic gaps that align with the CD emission.12 Third, in some cases, molecular fluorophores have been found to be fluorescent origin for CDs, especially for CDs that are made using bottom-up routes.13 For example, Song et al. have isolated 5-oxo-1,2,3,5-tetrahydroimidazo[1,2-a]pyridine-7-carboxylic acid (IPCA) from citric acid and ethylenediamine carbon dots.14 Fluorophores with similar structures to IPCA were also found in the reaction products of citric acid and other α,β-diamines.15,16 Kasprzyk et al. identified the structure of a green fluorophore, 4-hydroxy-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-trione (HPPT), from citric acid and urea carbon dots.17 However, to our best knowledge, the blue fluorophore in citric acid and urea CDs is yet to be determined. Some studies have suggested citrazinic acid is a good candidate for the blue fluorophore;17,18 however, it hasn't been isolated from a CD synthesis or proven with structural characterization such as nuclear magnetic resonance (NMR).In this work, a blue fluorophore 5-oxopyrrolidine-3-carboxylic acid (1) has been fully isolated from a citric acid and urea CD synthesis and characterized with NMR, electrospray ionization-mass spectrometry (ESI-MS), and Fourier-transform infrared spectroscopy (FTIR). The isolated fluorophore has been compared with a commercially available chemical, and their spectral characteristics align very well. The fluorophore 1 shows maximum emission at 420 nm, with interesting AEE properties. In addition, pyrrolidone-related structures have been reported to be fluorescent, especially polymers synthesized from monomers that contain pyrrolidone.19–21 This type of compound lacks aromatic rings which are usually found in traditional fluorophores, and thus they have non-traditional intrinsic luminescence (NTIL).21 The fluorescence of NTIL usually comes from confinement through immobilization or rigidification of moieties that are electron rich and heteroatomic, which associate with one another to form highly emissive units.21 We hypothesize that CDs are made of polymer structures such as polyamide, and molecular fluorophores such as 1 are attached to the polymer structure. The fluorescence of the CDs largely depends on the intermolecular interaction of the fluorophores with its local environment. To test this hypothesis computationally, time-dependent density functional theory (TD-DFT) calculations are performed for a single molecule 1 in both vacuum and implicit water solvent, and both models show that molecule 1 has fluorescence in blue region. Furthermore, we have investigated the stability of the CDs, fluorophore 1, and citrazinic acid. The instability of citrazinic acid in water makes it less likely to be responsible for the fluorescence properties of the citric acid and urea-derived CDs.
Experimental
All the materials information is included in the supplemental materials.Methods
CD synthesis and fluorophore purification: 0.40 g citric acid and 0.36 g urea were weighed and dissolved in 10 ml Milli-Q water. Hydrothermal treatment in a CEM® microwave reactor was applied at 160 °C for 4 h. The raw product was filtered through a 0.22 μm filter and dialyzed with a molecular weight cut-off = 3500 Da membrane for 3 days. The solution inside the dialysis bag was collected as CDs. After the purification with dialysis, there were minimal free fluorophores in the CD sample. The dialysate was collected and concentrated for fluorophore analysis. The dialysate was purified using a Teledyne ISCO CombiFlash® automated column chromatograph, using reversed phase C18 columns and water/acetonitrile as the mobile phase. Multiple runs of column chromatography and/or extraction were utilized to obtain a fluorophore fraction with high purity for subsequent analysis.Stability test
CDs, citrazinic acid, and 5-oxopyrrolidine-3-carboxylic acid solutions were tested for room temperature stability, UV stability, heat stability, and pH stability. There are 3 materials replicates for each sample in every test. For room temperature stability, the samples were dissolved in MilliQ water and sat at room temperature in a dark environment. The fluorescence of the samples were measured every day for 7 days. For UV stability, the samples were placed in a box with 365 nm UV lamp illumination (23 Watts). The fluorescence of samples was measured after 30 min, and every hour from 1 h to 8 h. For heat stability, the solutions were in an oven at 50 °C for 3, 18, 24, 48, or 72 h. Their fluorescence was measured at each of those time points. For pH stability, the solutions were adjusted with HCl or NaOH addition to reach pH 2, 4, 6, 8 or 10. The fluorescence was measured and recorded at each pH. The fluorescence was normalized to the average of the initial fluorescence value, except in pH variation experiments where the fluorescence was normalized to the highest value across all pHs. The statistical analyses were done with ANOVA in GraphPad.Material characterizations
NMR was conducted on Bruker Avance III series spectrometers (400 MHz and 500 MHz). NMR spectra were analyzed with MestreNova software and referenced using the solvent signal. ESI-MS was conducted on a Bruker BioTOF II ESI/TOF-MS. Accurate mass ESI-MS/MS was carried out on a Sciex ExionLC UHPLC system couple to a Sciex X500R quadrupole-time-of-flight mass spectrometer (UHPLC/QTOF-MS). FTIR spectra were obtained on a Thermo Scientific Nicolet iS5 Fourier Transform Infrared Spectrometer. Samples for FTIR analysis were prepared by dissolving 1 (separated from the CD synthesis or commercially purchased) in methanol. Then some aliquots were dropped on a KBr pellet and let dry to form a thin film. The FTIR spectra were averaged for 16 scans. UV-vis spectra were obtained using a Mikropack DH-2000 UV-Vis-NIR spectrometer. Fluorescence spectra were measured with a PTI QuantaMaster™ 400 fluorometer. Transmission electron microscopy (TEM) was taken on a JOEL-T12 TEM. Bright field and fluorescence macroscopic images of solid 1 particles were obtained on a Nikon AZ100M macroscope. Imaging of single molecule 1 was performed using Olympus IX-71 inverted microscope. Experimental details of ESI-MS/MS and single molecule imaging can be found in the ESI.†Computational methods
The simulations were performed using Gaussian 09.22 The molecules were prepared with Gaussview5.23 The molecule was first optimized in the ground state using the B3LYP/Def2-TZVPP24 method, and then the UV-vis spectrum was calculated using the CAM-B3LYP25 functional and the same basis set. For the implicit solvent calculations, the PCM model26 was used with water as the solvent.Results and discussion
Carbon dots are synthesized by reacting 2.1 mmol citric acid with 6.0 mmol urea at 160 °C for 4 h. The raw product was dialyzed in MilliQ water, and the dialysate has bright blue fluorescence (Fig. S1†). The NMR spectra of the dialysate show that a number of molecules were formed, and the majority of the spectral bands are in the aliphatic region. Characteristic peaks of 1 can be found in the unseparated dialysate and seem to be one of the major components (Fig. S2†). Preliminary ESI-MS of the CD dialysate show that the peak at m/z = 130 represents the protonated molecular ion [1 + H]+. It is interesting to note here that one substance with molecular weight (MW) 155 Da appeared in the CD dialysate; in other work, this peak has been suggested as evidence of the presence of citrazinic acid.17 However, signature citrazinic acid peaks do not appear in the NMR of the CD dialysate from this synthesis condition. Fig. S4† shows that citrazinic acid has proton features at 6.2 or 3.6 and 6.6 ppm for its isomer, while the CD dialysate here has a singlet at 5.7 ppm (Fig. S2†). Thus, this substance of MW 155 Da is not likely to be citrazinic acid. Citrazinic acid has been reported to be synthesized from citric acid and urea;27 however, it is not generated in a significant amount in these synthesis conditions, thus, it is unlikely to be the origin of blue fluorescence from the CDs. After the raw dialysate was purified with reversed phase column chromatography, 1 was found as the first and most abundant blue fraction coming off the column. High resolution ESI-MS (spectrum not shown) and ESI-MS/MS (Fig. S11†) detect the protonated molecular ion, [1 + H]+ at m/z 130.0497. This corresponds to the theoretical molecular formula of 1 (C5H7NO3, theoretical [1 + H]+ = 130.0499) with a measurement error of 1.5 ppm.The structure of 1 was analyzed by multiple 1D and 2D NMR experiments in either D2O or 90% H2O+ 10% D2O. 1H–13C heteronuclear multiple bond correlation (HMBC) spectra showed five non-labile protons (δ 2.6, 3.3, 3.5, and 3.7 ppm) and five carbons (δ 34, 41, 46, 180, and 181 ppm) (Fig. 1). The multiplicity of the protons showed that one proton is in between the other 4 protons (CH2–CH–CH2), as it is pentet splitting and the other protons are d–d splitting. The d–d splitting also indicates that these protons are probably on a ring structure, putting the CH2 protons in different chemical environments. The protons at 3.5 ppm and 3.7 ppm showed second order effects where the peaks are leaning toward each other. These two protons are probably derived from citric acid, as citric acid also has protons showing the second order effect (Fig. S5 and S10†). The two carbons at 180 ppm indicate that they are C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. One nitrogen is at 120 ppm, which shows that it is probably an amide nitrogen. 1H and 13C NMR spectra, 1H–1H homonuclear correlated spectroscopy (COSY), 1H–13C heteronuclear single quantum coherence (HSQC) and 1H–15N HMBC spectra of 1 further confirmed its structure (Fig. S5–9†). It's interesting to note that the CH proton shifts from 3.3 ppm to 3.4 ppm when the carboxylate group changes from the protonated to ionized state. ESI-MS/MS shows that the major fragment ion arising from the precursor ion [1 + H]+ is m/z 84.0444 Da. This is because 1 loses the carboxylate group and forms the positively charged 3-pyrroline-2-one [C4H6NO]+, which has a theoretical m/z of 84.0443 Da (Fig. S11†).
O. One nitrogen is at 120 ppm, which shows that it is probably an amide nitrogen. 1H and 13C NMR spectra, 1H–1H homonuclear correlated spectroscopy (COSY), 1H–13C heteronuclear single quantum coherence (HSQC) and 1H–15N HMBC spectra of 1 further confirmed its structure (Fig. S5–9†). It's interesting to note that the CH proton shifts from 3.3 ppm to 3.4 ppm when the carboxylate group changes from the protonated to ionized state. ESI-MS/MS shows that the major fragment ion arising from the precursor ion [1 + H]+ is m/z 84.0444 Da. This is because 1 loses the carboxylate group and forms the positively charged 3-pyrroline-2-one [C4H6NO]+, which has a theoretical m/z of 84.0443 Da (Fig. S11†).
 | ||
| Fig. 1 1H–13C HMBC spectra taken in D2O and the chemical structure with NMR assignments of 1 (5-oxopyrrolidine-3-carboxylic acid) isolated from CD dialysate. Chemical shifts of proton, carbon and nitrogen are labeled in blue, red and green, respectively. | ||
The FTIR spectrum (Fig. 2) shows the N–H stretching of the amide is at 3200 cm−1 and O–H stretching of the carboxylic acid is 2950 cm−1, while the C–H stretching of the alkanes is at 2550 cm−1. The CO stretching of the lactam and carboxylic acid group overlap at around 1707 cm−1. The band at 1414 cm−1 is due to the C–H bending vibration, and the band at 1191 cm−1 is from the C–N stretching vibration. The C–O stretching of the carboxylic acid group occurs at 1021 cm−1. Additionally, a commercial product matching the putative structure of 1 was purchased from Combi-blocks with 97% purity. Its 1H NMR and FTIR spectra were measured and stacked with 1 isolated from CDs (Fig. S12 and S13†). The spectra of the isolated product 1 match perfectly with the commercial product, which firmly proves that the identity of 1 is 5-oxopyrrolidine-3-carboxylic acid.
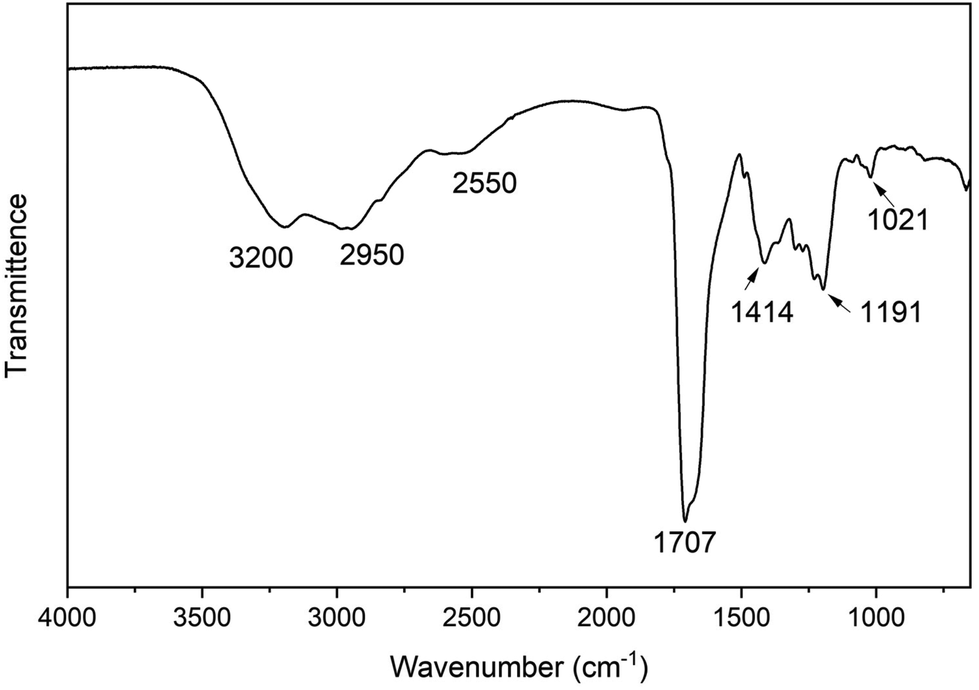 | ||
| Fig. 2 FTIR spectrum of 1, isolated from the CD synthesis. | ||
The formation mechanism of 1 is proposed in Fig. 3. In this scheme, citric acid undergoes multiple routes of dehydration and decarboxylation at high temperature. Itaconic acid has previously been shown to be one of the decomposition products of citric acid.28 Additionally, it is well-known that urea decomposes into ammonia at high temperatures. Finally, itaconic acid has been shown to react with amine groups on chitosan to form structure 1.29,30 Thus, it is reasonable that in the synthesis of carbon dots, citric acid and urea form decomposition products and then itaconic acid and ammonia react to form 1. Since previous work has shown 1 can be attached to chitosan, it is possible that in this case, 1 is attached to the polymer structure of carbon dots, acting as the luminescence source in CDs. To verify the proposed mechanism, itaconic acid and urea underwent hydrothermal treatment at 150 °C for 2 h. The ESI-MS and NMR spectra of the raw product showed that 1 can be synthesized from itaconic acid and urea (Fig. S14 and S15†). The raw product of itaconic acid and urea showed blue fluorescence, and its fluorescence spectrum is in Fig. S16.† As a ternary acid, citric acid has been used as a precursor for various polymer synthesis.31,32 Thus citric acid instead of itaconic acid is used most often in CD synthesis since it is more likely to form both molecular fluorophore and the polymer backbone of CDs.
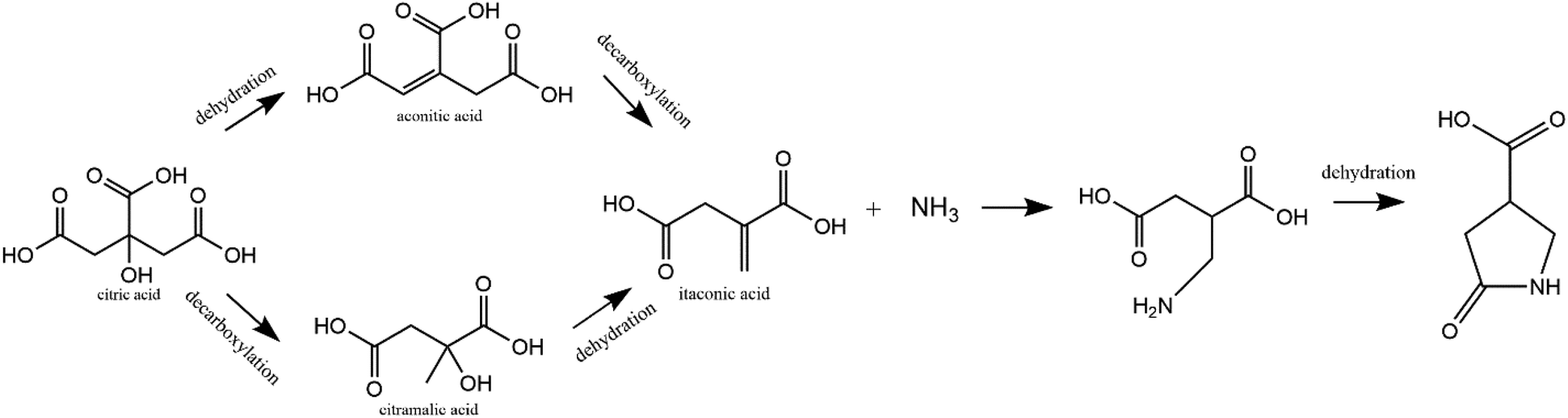 | ||
| Fig. 3 A scheme for the formation of 1 (5-oxopyrrolidine-3-carboxylic acid) from hydrothermal reaction of citric acid and urea. | ||
In addition to 1, other fluorophores were also found in CD dialysate. The amide form of 1, 5-oxopyrrolidine-3-carboxyamide (2) appeared. The spectra of 2 also matched well with a commercial product (Fig. S17†). Compared with 1, the CH proton shifted further to 3.45 ppm, which seems to be impacted most when the COOH group changed to CONH2. The fluorescence of 2 is weaker and blue shifted to 400 nm compared with 1, which is at 420 nm (Fig. S18A†). Other fluorophores, including blue, green and red fluorophores and their possible molecular weight information are listed in Table S1.† It should be noted that even though a number of fluorophores were formed in the CD synthesis, 1 is the most abundant blue fluorophore and thus is most likely to account for the blue fluorescence in CDs.
Material characterization
The synthesized CDs were imaged with transmission electron microscopy (TEM) and have an average size of 10.0 ± 0.9 nm (n = 231) (Fig. 4A and Fig. S19†). The fluorescence spectra show that CDs have optimum emission at 448 nm when excited at 370 nm (Fig. 4B). The fluorescence of molecule 1, however, has emission maximum at 420 nm when excited at 330 nm (Fig. 4C). The UV-vis absorption of purified CDs at around 235 nm is probably due to π–π* transitions within the carbonized cores, while the peak at 330 nm is from n–π* transitions (Fig. 4E).33 For 1 dispersed in water, it has strong absorption below the 300 nm range; this is attributed to n–π* and π–π* transitions of the CO.
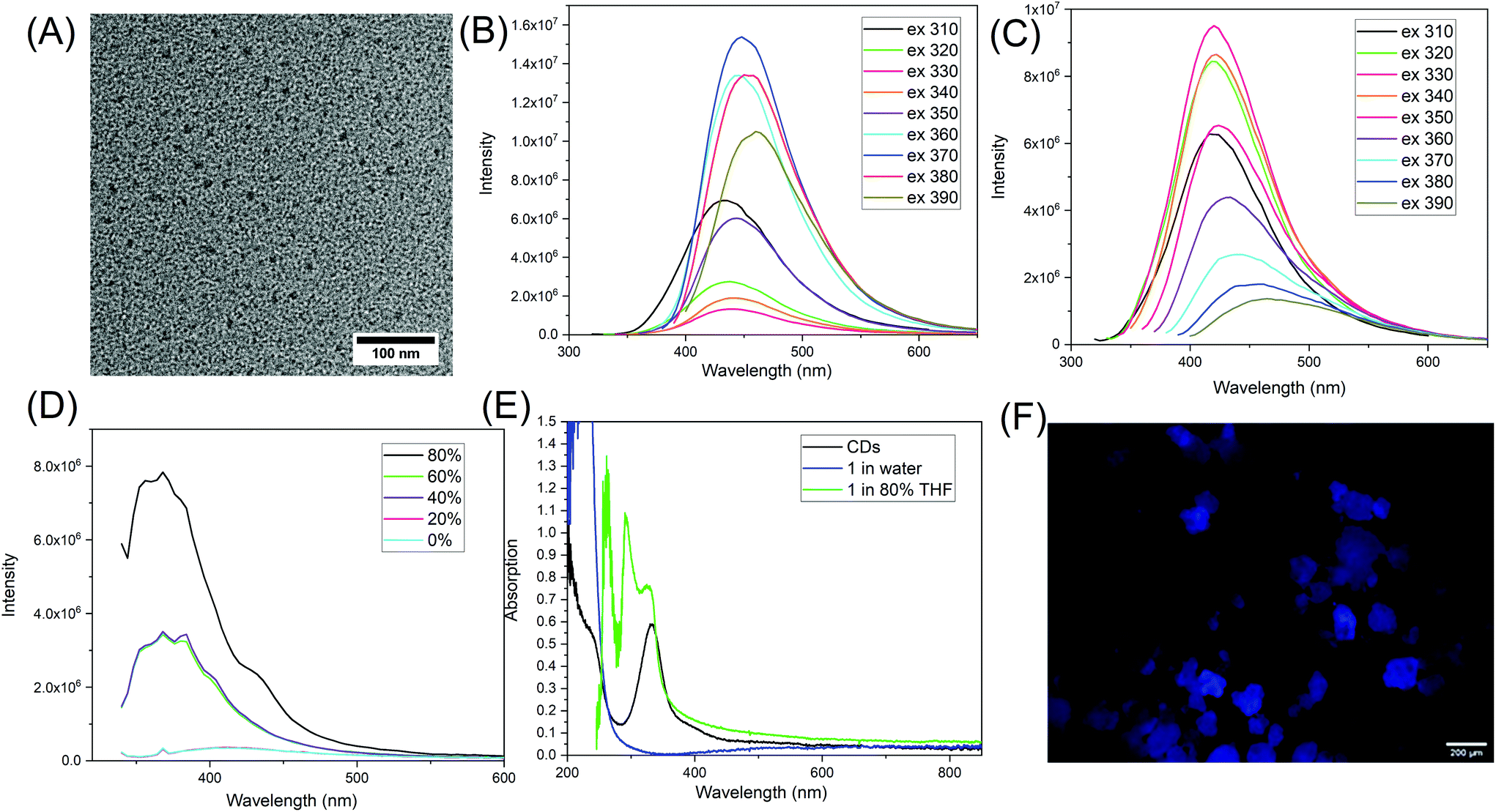 | ||
| Fig. 4 (A) A representative TEM image of the CDs; (B) fluorescence spectra of CDs; (C) fluorescence spectra of isolated 1; (D) fluorescence spectra of 1 excited at 330 nm in mixed solvents of DMSO and THF, with the percentage indicating the THF%; (E) UV-vis absorption of CDs as well as 1 in water and in a mixed solvent of 80% THF and 20% DMSO; (F) a fluorescent macroscopic image of solid 1 particles (excitation 360–380 nm; emission 415 nm with longpass filter, scale bar is 200 μm). | ||
The fluorescence of similar compounds, such as vinyl pyrrolidone has been reported by Song et al.19 They mentioned that the monomer N-vinyl pyrrolidone has low fluorescence at 350 nm, but that the fluorescence increases dramatically with the peak shifted to 380 nm when it forms the poly(vinyl pyrrolidone). This increase and shift was attributed to aggregation-induced emission (AIE).19,34 It seems for these type of unconventional fluorophores, that the intermolecular interactions of the pyrrolidone ring with other electron-rich groups such as CO or nitrogen, have a significant impact on the fluorescence properties. To investigate this possibility, we increased the intermolecular interactions by inducing aggregation of 1 in mixed solvents of dimethyl sulfoxide (DMSO) and tetrahydrofuran (THF). 1 is more soluble in DMSO and less soluble in THF. Increased THF leads to the formation of molecular clusters and then at 80% volume ratio, white precipitate forms. Interestingly, the fluorescence of 1 increased dramatically with increased THF. Its fluorescence was enhanced 22.5 times when the THF was at 80%. The emission also shifted from 420 nm to 400 nm (Fig. 4D). This is a strong evidence indicating that 1 demonstrates AEE. Meanwhile, there are noticeable changes in the absorption spectrum of 1 once it starts to aggregate. In the mixed solvent of 80% THF and 20% DMSO, a few new peaks at 260 nm, 293 nm, and 327 nm appear (Fig. 4E). This indicates that 1 probably forms some new structures where intermolecular interactions play a role in photon absorption. A similar AEE phenomenon was also observed when 1 was in mixed solvents of DMSO and ethyl acetate, when ethyl acetate made up 90% of the volume. In addition, we found that 1 is also fluorescent in the solid state, as shown by the macroscopic images in Fig. 4F and others in Fig. S20.† Similarly, the amide form of 1, 2 was also dissolved in a series of mixed solvents of DMSO and THF. The emission of 2 in DMSO centers at 380 nm, and AEE was also observed with increasing THF% as shown in Fig. S21.† From here, we can see that 1 and 2 are different from traditional aromatic fluorophores where the fluorescence comes from electron delocalization in π–π bonds, and once the molecule aggregates, there is intense intermolecular stacking which usually results in fluorescence quenching. It seems 1 and 2 demonstrate AEE and that intermolecular interactions play a key role in enhancing their fluorescence properties. The intermolecular interactions include the physico-chemical confinement of the moieties that are normally non-emissive or low emissive, electron rich, and heteroatomic.21 Here, the pyrrolidone structure is one of the hetero-atomic sub-luminophores and thus the compound 1 has non-traditional intrinsic luminescence (NTIL).21 A similar phenomenon has been summarized by Tang and coworkers as clusteroluminescence.35
The fluorescence from nonconjugated systems was first found with aliphatic amines,36,37 and later some aliphatic polymers such as poly(amidoamine) (PAMAM) were also found to have blue fluorescence.38,39 The blue fluorescence is thought to originate from n–π* transitions from the amide group inside the PAMAM dendrimer.21,38 The fluorescence of PAMAMs can be enhanced by surface modification with N-(4-carbomethoxy)pyrrolidone.40–42 The pyrrolidone shell is suggested to play multiple roles including stabilization of the PAMAMs sterically to prevent access by external solutes and the impact of protonation.40 It is interesting to note that a prior report on polyamide simulation of CDs showed that the blue fluorescence comes from charge transfer of the amide and carboxylate groups of the polyamide backbone;43 this would also be a form of NTIL.
Single molecule fluorescence of 1 was also measured and found to be intermittent, which is usually called “blinking”. Blinking is common among traditional fluorophores44 and nanoscale particles such as quantum dots or carbon dots,45,46 though there is little investigation on the blinking properties of non-traditional fluorophores. We found that under 405 nm excitation, most of 1 molecules emitted fluorescence, as shown in Movie S1.† The blinking rate of 1 under 405 nm is 5.48%. The blinking rate is defined as the ratio between the on-time of a molecule and the acquisition time that is 100 seconds. With longer excitation wavelengths, 488 nm, 542 nm, or 594 nm, a smaller number of molecules in the same fields of view emitted light (Movie S2–S4†). Under longer excitation wavelengths, blinking rates were slightly higher, from 8.34%–10.02%. Representative emission traces and blinking rate histograms are shown in Fig. S22.† At the moment, the blinking mechanism for non-traditional fluorophores is not clear and merits further investigation in future studies.
To probe the intrinsic photophysical properties of 1, TD-DFT calculations are performed in both vacuum and in an implicit water solvent using the polarizable continuum (PCM) model. The simulated absorption and emission spectra are shown in Fig. 5A and B, respectively. The absorption spectrum peaks are at ∼229 nm in vacuum and ∼228 nm with the implicit solvent. The emission peaks are at 480.7 nm in vacuum and 473.3 nm with the implicit solvent model. The computed natural transition orbitals (NTOs) with optimized structure for the first excited state indicate that the transition involves n to π* character in the carboxyl group.
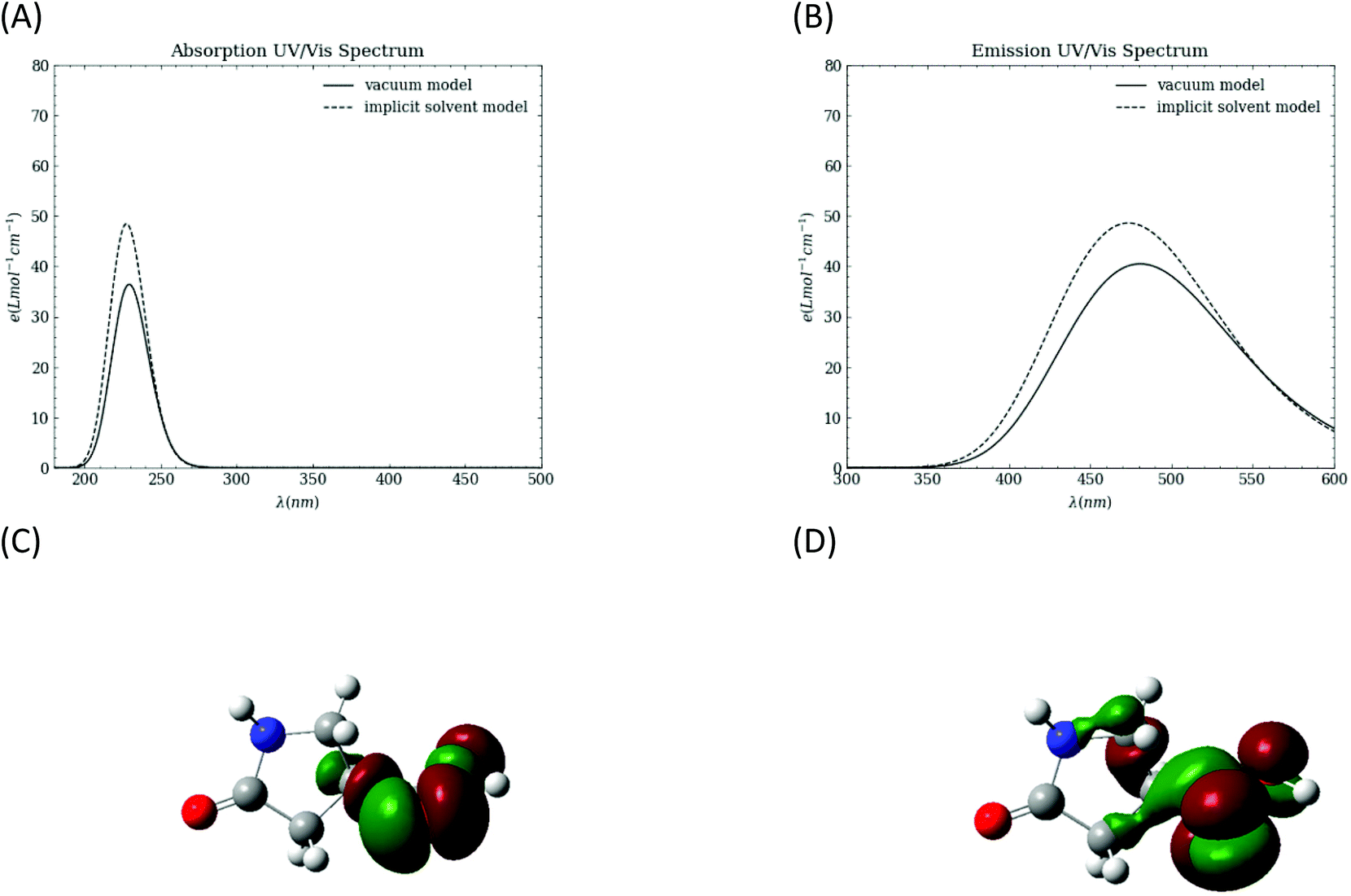 | ||
| Fig. 5 TD-DFT results for molecule 1. (A) simulated absorption UV-vis spectrum; (B) simulated emission UV-vis spectrum; (C) and (D) NTOs calculated for molecule 1 in vacuum, using an optimized structure for the excited state. The NTOs suggest that the excitation involves n (shown in panel C) to π* orbitals (shown in panel D) in the carboxyl group. | ||
We propose here that small molecule 1 is connected to the polymer structure of CDs through either the carboxylate group or the nitrogen. To assess this possibility, we considered two representative compounds: ethyl 5-oxopyrrolidine-3-carboxylate (3) and N-ethyl-5-oxopyrrolidine-3-carboxylic acid (4), where an ethyl group is acting as a simplified model of the proposed polymer. The fluorescence spectra of 3 and 4 are shown in Fig. S18 B and C.† The fluorescence of 3 is more similar to 1 in that the maximum emission is at 420 nm. The fluorescence peak for 4 is much broader than other fluorophores in this system, extending from 350 nm to 600 nm. It is interesting to note that compound 4 shows excitation-dependent emission, which has been widely reported for CDs. This has been typically thought to originate from the inhomogeneity of the fluorophores or CD structures; however, these data show that a single fluorophore can demonstrate excitation-dependent emission as well. The excitation-dependent fluorescence has been reported to be a general feature for sub-fluorophores which have shown clusteroluminescence.35 It was attributed to different extents of electron delocalization in the molecular clusters.351 also shows excitation-dependent emission in the solid state (Fig. S20†). Taken together, these measurements show that modification of the structure of 1 alters its fluorescence, including emission wavelength and peak width. Considering AEE, crosslinking of 1 rigidifies its structure and embedding it into a supramolecular structure such as CDs probably enhance its fluorescence. Given that the polymer backbone structure of CDs is complex and possibly contains multiple functional groups such as CO, N–H etc., the final fluorescence of CDs is dependent on the local environment of the fluorophore 1, such as its interaction with the polymer backbone and with other fluorophores. Further computational simulations in future work will likely be very helpful in visualizing the local structures and predicting the fluorescence of CDs.
Stability test
One way to characterize a fluorophore (and, in this case, compare it to other fluorophores) is to monitor luminescent stability in various circumstances. The stability of CDs, citrazinic acid, and 1 were evaluated as the fluorescence intensity variation in four different conditions: (1) dispersing in water at room temperature, (2) under UV illumination, (3) after 50 °C oven treatment, and (4) with variation in solution pH. Although citrazinic acid has been thought to be the origin of the blue fluorescence from CDs, it was found to be very unstable when dispersed in water. It changed from colorless to purple after it was dispersed in water for 7 days, while there is no obvious difference for CDs or 1 over the same time scale (Fig. S23†). Its fluorescence also decreased to 80% of the initial fluorescence after 4 days (p ≤ 0.05), and after 7 days, its fluorescence was only 24% of the original (p ≤ 0.0001). This is likely due to the imine structure which is prone to hydrolysis.47 The NMR spectra of citrazinic acid just dissolved in water and after 7 days are in Fig. S4 and S24.† Characteristic citrazinic acid NMR peaks at 6.2 ppm are absent, and a new peak at 2.7 ppm appears. This indicates structural rearrangement, and new substances were formed from the hydrolysis of citrazinic acid. In comparison, CDs and 1 were very stable in water at room temperature, as there is no statistical difference in their fluorescence across 7 days (p > 0.05) in Fig. 6A.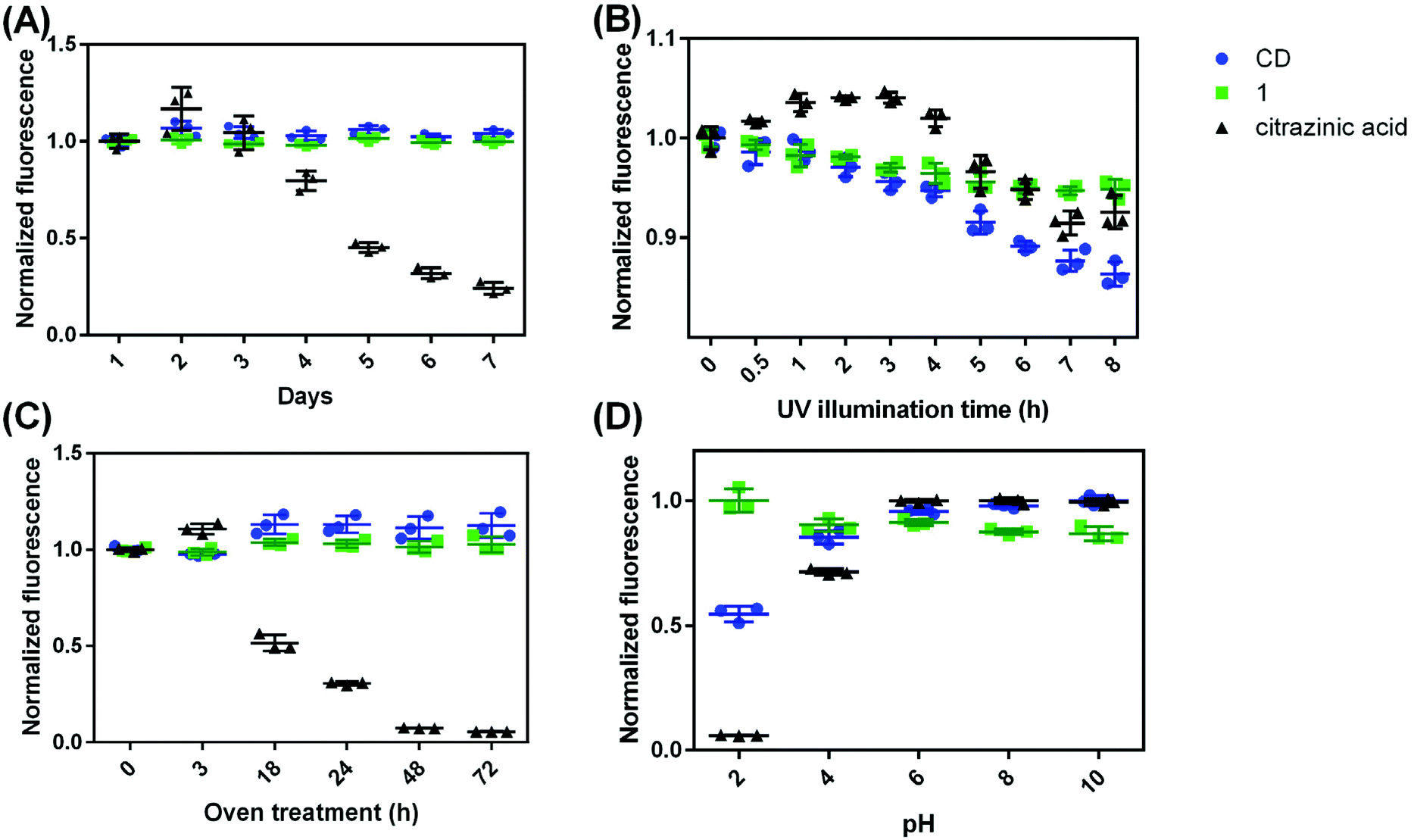 | ||
| Fig. 6 Normalized fluorescence of CDs, 1, and citrazinic acid. (A) samples were dissolved in water at room temperature; (B) underwent 365 nm UV illumination; (C) underwent 50 °C oven treatment; and (D) in a series of pH conditions. Each treatment has 3 materials replicates, and the error bars show the standard deviation. | ||
The photostability of these substances were compared after they were treated under 365 nm UV light (Fig. 6B). The fluorescence of the CDs was found to decrease gradually, retaining 86% of the original intensity after 10 h UV illumination, while the fluorescence of 1 decreased a bit to 95%, and the fluorescence of citrazinic acid was at 93% of the original (p ≤ 0.0001 for all three). It seems that all samples were impacted similarly by UV illumination, though the fluorescence of the CDs decreased more than 1, which is likely attributed to the disintegration of 1 from the polymer backbone of CDs. Free 1 would no longer benefit from the AEE effect and then leads to the observed decrease in fluorescence. Treatment at elevated temperatures showed CDs and 1 to be stable, with their fluorescence increased to 113% (p ≤ 0.05) and remaining at 103% (p > 0.05), respectively. However, citrazinic acid was very unstable at the 50 °C temperature, and its fluorescence decreased to only 5% after 72 h in the oven (Fig. 6C). It was also observed that its color changed from colorless to purple after 18 h in the oven and finally turned brown. High temperature probably accelerates the hydrolysis of citrazinic acid. Considering CD syntheses are conducted under high temperatures for hours in most cases, even if citrazinic acid did form from a citric acid and urea reaction, the chance of it surviving to the end of the hydrothermal treatment is low. The solution pH of all three substances were adjusted to 2, 4, 6, 8, or 10 with either HCl or NaOH, and their fluorescence is shown in Fig. 6D. Again, the fluorescence of 1 seems to be the most stable, as there is no statistically significant difference measured across the pHs considered (p > 0.05). The fluorescence of CDs is lower at acidic pHs, 55% at pH 2, and 85% at pH 4, and retains almost 100% fluorescence at pH 6, 8, and 10. Citrazinic acid seems to be impacted most by pH, as its fluorescence was only 6% at pH 2. This shows that the protonation of the carboxylate group probably affects the delocalization of the electrons in the dihydropyridine ring. Literature also suggests that citrazinic acid will form tautomers in acidic pHs.48
Taken together, we can see that citrazinic acid is unstable in water as it undergoes hydrolysis, which is accelerated at higher temperatures. It is interesting to note that in several cases, the fluorescence of citrazinic acid increased after it was exposed to water for a short time, such as in Fig. 4B, 3 h (p ≤ 0.01). This could be attributed to new substance formation or because during the degradation of citrazinic acid, the fluorophore concentration achieved an optimum value, and its fluorescence intensity was maximized. Overall, 1 seems to be the most stable and not likely to be impacted by the environment. But to most efficiently utilize its fluorescence, 1 needs to be in a polymer or carbon dot structure or in an aggregated state, so that it can induce the AEE effect. In those circumstances, the local environment would be susceptible to the UV light or pH changes.
Conclusions
In summary, we have discovered a new fluorophore present in CDs made from citric acid and urea, 5-oxopyrrolidine-3-carboxylic acid. The fluorophore 1 has been separated from the raw CD product with reversed phase column chromatography. It was analyzed and compared with a commercial product using NMR, ESI-MS and FTIR, and the molecular identity is clear. The TD-DFT calculated spectra support the observed blue fluorescence. 1 was found to have AEE effect, which makes it have much higher emission in aggregated states. It is proposed that when 1 is incorporated into CDs, its fluorescence properties are altered by the local environment, such as the polymer backbone of CDs, since its fluorescence is largely dependent on the intermolecular interactions. Taken together with literature precedent, we can see that various molecular fluorophores, either traditional fluorophores such as IPCA and HPPT,14,17 or non-traditional fluorophores like 1 can be formed in bottom-up preparations of CDs. It's worth further investigation of how they are incorporated into CDs and how the local environment alters their fluorescence properties. Further computational modeling and ultra-fast spectroscopy techniques will likely be helpful in this respect.49 In addition, the NTIL nature of 1 reveals a new perspective in understanding CD fluorescence. Since many CDs are amorphous and have blue emission, they happen to resemble the characteristic optical profiles of NTIL where the emission is between 400–570 nm and the excitation is 225–400 nm.21 It is possible that other CDs, in addition to the one presented here, are also fluorescent due to non-conventional routes. Overall, this structure-focused study provides a new perspective of the fluorescence origin of CDs.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the National Science Foundation under Grant No. CHE-2001611, the NSF Center for Sustainable Nanotechnology (CSN). The CSN is part of the Centers for Chemical Innovation Program. We thank Yukun Cheng, Dr Yuan Sheng, Dr Thomas R. Hoye, Mengyuan Jin, and Dr Letitia Yao (all from the Department of Chemistry, University of Minnesota) for meaningful discussions in the spectral interpretation and proposing the structures of the fluorophores. The fluorescence macroscopic images were taken with the resources and help from Mary Brown and Guillermo Marques at the University of Minnesota University Imaging Centers (UIC). SCR_020997. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from the NSF through the MRSEC (Award Number DMR-2011401) and the NNCI (Award Number ECCS-2025124) programs. The computing resources necessary for this research were provided in part by the National Science Foundation through XSEDE resources under Grant No. CTS090079 and the Advanced Research Computing at Hopkins (ARCH) facilities supported by the NSF MRI Grant (OAC-1920103). Part of the research was performed in the Environmental Molecular Sciences Laboratory, a DOE Office of Science User Facility sponsored by the Biological and Environmental Research program under Contract No. DE-AC05-76RL01830. Part of this work utilized the CEM Discover SP microwave from Jane Wissinger's group (University of Minnesota) that was purchased with funding provided by the NSF Center for Sustainable Polymers, CHE-1901635. The table of contents image was created with BioRender.com.References
- B. Zhao and Z. A. Tan, Adv. Sci., 2021, 8, 2001977 CrossRef CAS PubMed
.
- B. Zhi, X. Yao, Y. Cui, G. Orr and C. L. Haynes, Nanoscale, 2019, 11, 20411–20428 RSC
- C.-X. Li, C. Yu, C.-F. Wang and S. Chen, J. Mater. Sci., 2013, 48, 6307–6311 CrossRef CAS
- B. Zhi, X. Yao, M. Wu, A. Mensch, Y. Cui, J. Deng, J. J. Duchimaza-Heredia, K. J. Trerayapiwat, T. Niehaus, Y. Nishimoto, B. P. Frank, Y. Zhang, R. E. Lewis, E. A. Kappel, R. J. Hamers, H. D. Fairbrother, G. Orr, C. J. Murphy, Q. Cui and C. L. Haynes, Chem. Sci., 2021, 12, 2441–2455 RSC
- L. Li, Y. Li, Y. Ye, R. Guo, A. Wang, G. Zou, H. Hou and X. Ji, ACS Nano, 2021, 15, 6872–6885 CrossRef CAS PubMed
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8, 355–381 CrossRef CAS
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. a. Tan, A. Chen, M. Jin and S. Yang, Nat. Commun., 2018, 9, 2249 CrossRef PubMed
- S. K. Misra, I. Srivastava, J. S. Khamo, V. V. Krishnamurthy, D. Sar, A. S. Schwartz-Duval, J. A. N. T. Soares, K. Zhang and D. Pan, Nanoscale, 2018, 10, 18510–18519 RSC
- F. Yang, G. E. LeCroy, P. Wang, W. Liang, J. Chen, K. A. S. Fernando, C. E. Bunker, H. Qian and Y.-P. Sun, J. Phys. Chem. C, 2016, 120, 25604–25611 CrossRef CAS
- H. Ding, S.-B. Yu, J.-S. Wei and H.-M. Xiong, ACS Nano, 2016, 10, 484–491 CrossRef CAS PubMed
- H. Ding, X.-H. Li, X.-B. Chen, J.-S. Wei, X.-B. Li and H.-M. Xiong, J. Appl. Phys., 2020, 127, 231101 CrossRef CAS
- H. A. Nguyen, I. Srivastava, D. Pan and M. Gruebele, ACS Nano, 2020, 14, 6127–6137 CrossRef CAS PubMed
- J. B. Essner, J. A. Kist, L. Polo-Parada and G. A. Baker, Chem. Mater., 2018, 30, 1878–1887 CrossRef CAS
- Y. Song, S. Zhu, S. Zhang, Y. Fu, L. Wang, X. Zhao and B. Yang, J. Mater. Chem. C, 2015, 3, 5976–5984 RSC
- W. Kasprzyk, S. Bednarz, P. Żmudzki, M. Galica and D. Bogdał, RSC Adv., 2015, 5, 34795–34799 RSC
- Y. Xiong, J. Schneider, E. V. Ushakova and A. L. Rogach, Nano Today, 2018, 23, 124–139 CrossRef CAS
- W. Kasprzyk, T. Świergosz, S. Bednarz, K. Walas, N. V. Bashmakova and D. Bogdał, Nanoscale, 2018, 10, 13889–13894 RSC
- C. J. Reckmeier, J. Schneider, Y. Xiong, J. Hausler, P. Kasak, W. Schnick and A. L. Rogach, Chem. Mater., 2017, 29, 10352–10361 CrossRef CAS
- G. Song, Y. Lin, Z. Zhu, H. Zheng, J. Qiao, C. He and H. Wang, Macromol. Rapid Commun., 2015, 36, 278–285 CrossRef CAS PubMed
- C. Shang, N. Wei, H. Zhuo, Y. Shao, Q. Zhang, Z. Zhang and H. Wang, J. Mater. Chem. C, 2017, 5, 8082–8090 RSC
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, R. D. Gaussian 09, Gaussian, Inc., Wallingford CT, 2013 Search PubMed
-
R. Dennington, T. A. Keith and J. M. Millam, V. GaussView, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef
- W. J. Sell and T. H. Easterfield, J. Chem. Soc., Dalton Trans., 1893, 63, 1035–1051 RSC
- T. Ye, B. Wang, J. Liu, J. Chen and Y. Yang, Carbohydr. Polym., 2015, 121, 92–98 CrossRef CAS PubMed
- J. A. Sirviö, A. M. Kantola, S. Komulainen and S. Filonenko, Biomacromolecules, 2021, 22, 2119–2128 CrossRef PubMed
- P. L. Paytash, E. Sparrow and J. C. Gathe, J. Am. Chem. Soc., 1950, 72, 1415–1416 CrossRef CAS
- D. Gyawali, P. Nair, Y. Zhang, R. T. Tran, C. Zhang, M. Samchukov, M. Makarov, H. K. W. Kim and J. Yang, Biomaterials, 2010, 31, 9092–9105 CrossRef CAS PubMed
- J. Yang, A. R. Webb and G. A. Ameer, Adv. Mater., 2004, 16, 511–516 CrossRef CAS
- Y. Zhou, A. Desserre, S. K. Sharma, S. Li, M. H. Marksberry, C. C. Chusuei, P. L. Blackwelder and R. M. Leblanc, ChemPhysChem, 2017, 18, 890–897 CrossRef CAS PubMed
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed
- H. Zhang and B. Z. Tang, JACS Au, 2021, 1, 1805–1814 CrossRef CAS PubMed
- A. M. Halpern, Chem. Phys. Lett., 1970, 6, 296–298 CrossRef CAS
- E. M. Arthur and M. Halpern, J. Am. Chem. Soc., 1972, 94, 8273–8274 CrossRef
- C. L. Larson and S. A. Tucker, Appl. Spectrosc., 2001, 55, 679–683 CrossRef CAS
- O. Varnavski, R. G. Ispasoiu, L. Balogh, D. Tomalia and T. Goodson III, J. Chem. Phys., 2001, 114, 1962–1965 CrossRef CAS
- M. Studzian, Ł. Pułaski, D. A. Tomalia and B. Klajnert-Maculewicz, J. Phys. Chem. C, 2019, 123, 18007–18016 CrossRef CAS
- M. Konopka, A. Janaszewska, K. A. M. Johnson, D. Hedstrand, D. A. Tomalia and B. Klajnert-Maculewicz, J. Nanopart. Res., 2018, 20, 220 CrossRef
- A. Janaszewska, M. Studzian, J. F. Petersen, M. Ficker, V. Paolucci, J. B. Christensen, D. A. Tomalia and B. Klajnert-Maculewicz, Colloids Surf., B, 2017, 159, 211–216 CrossRef CAS PubMed
- L. Vallan, E. P. Urriolabeitia, F. Ruipérez, J. M. Matxain, R. Canton-Vitoria, N. Tagmatarchis, A. M. Benito and W. K. Maser, J. Am. Chem. Soc., 2018, 140, 12862–12869 CrossRef CAS PubMed
- E. K. L. Yeow, S. M. Melnikov, T. D. M. Bell, F. C. De Schryver and J. Hofkens, J. Phys. Chem. A, 2006, 110, 1726–1734 CrossRef CAS PubMed
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2001, 115, 1028–1040 CrossRef CAS
- B. Zhi, Y. Cui, S. Wang, B. P. Frank, D. N. Williams, R. P. Brown, E. S. Melby, R. J. Hamers, Z. Rosenzweig, D. H. Fairbrother, G. Orr and C. L. Haynes, ACS Nano, 2018, 12, 5741–5752 CrossRef CAS PubMed
- C.-J. Li and C. Wei, Chem. Commun., 2002, 268–269, 10.1039/B108851N
- L. Stagi, S. Mura, L. Malfatti, C. M. Carbonaro, P. C. Ricci, S. Porcu, F. Secci and P. Innocenzi, ACS Omega, 2020, 5, 10958–10964 CrossRef CAS PubMed
- Q. Cui, R. Hernandez, S. E. Mason, T. Frauenheim, J. A. Pedersen and F. Geiger, J. Phys. Chem. B, 2016, 120, 7297–7306 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR spectra for 1, additional photographs of experiments and fluorescence spectra and movies of 1. See DOI: https://doi.org/10.1039/d2nr02361j |
| This journal is © The Royal Society of Chemistry 2022 |