
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments in enantio- and diastereoselective hydrogenation of N-heteroaromatic compounds
Ramachandran
Gunasekar
 ab,
Ross L.
Goodyear
b,
Ilaria
Proietti Silvestri
b and
Jianliang
Xiao
*a
ab,
Ross L.
Goodyear
b,
Ilaria
Proietti Silvestri
b and
Jianliang
Xiao
*a
aDepartment of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: jxiao@liv.ac.uk
bLiverpool ChiroChem (LCC), Liverpool L69 7ZD, UK
First published on 4th February 2022
Abstract
The enantioselective and diastereoselective hydrogenation of N-heteroaromatic compounds is an efficient strategy to access chirally enriched cyclic heterocycles, which often possess highly bio-active properties. This strategy, however, has only been established in recent times. This is in part due to the challenges of the high stability of the aromatic compounds and the presence of heteroatoms that have the potential to poison the chiral catalysts. Additionally, N-heteroaromatic compounds are a structurally diverse family of substrates, each group showing distinct reactivity in hydrogenation. Advances in recent years have allowed various N-heteroaromatic compounds, including pyridines, indoles, quinolines, isoquinolines, quinoxalines and imidazoles, to be hydrogenated with good to excellent enantioselectivity under appropriate reaction conditions. Transition–metal catalysis, utilising iridium, ruthenium, rhodium, and palladium complexes, has been found to play an important role in this field. More recently, organocatalysis has been shown to be efficient for the hydrogenation of certain N-heteroaromatic compounds. This review provides an analysis of the recent developments in the enantioselective and diastereoselective hydrogenation of N-heteroaromatic compounds. The importance of these molecules and their applications to drug discovery has been highlighted throughout the review.
 Ramachandran Gunasekar | Ramachandran Gunasekar received his PhD from the VIT University, India in 2014. He moved to Japan, as a JSPS postdoctoral Fellow under the supervision of Prof. Masato Kitamura in 2015. He then joined as a KTP Associate at the University of Liverpool collaborating with Liverpool Chirochem (LCC), under the supervision of Prof. Jianliang Xiao in 2019. Recently, he joined the group of Prof. Paul. O'Neill as a Research Associate at the University of Liverpool. His current research interests focus on exploring new chemical space by designing and synthesizing of small molecule toxin inhibitors for the snakebite treatments. |
 Ross L. Goodyear | Ross Goodyear received his PhD from the University of East Anglia under the supervision of Prof. Phil Page. In 2020 he started work as an R&D chemist at LCC specializing on the synthesis of sp3-rich fragments through photochemical C–H activation. His research interests focus on the production of bio-active molecules and natural product analogues. |
 Ilaria Proietti Silvestri | Ilaria Proietti Silvestri is Head of R&D at LCC. Ilaria did her doctoral work at Sapienza University of Rome, where she studied transition metal-catalysed coupling reactions. After post-doctoral research fellowships at the University of Copenhagen and at Politecnico of Milan, in 2015 Ilaria moved to England to join RedX Pharma. In 2017 Ilaria joined LCC, where she is currently leading the R&D team investigating the design and synthesis of novel 3D-rich N-heterocycles specifically designed for leading applications in drug discovery. |
 Jianliang Xiao | Jianliang Xiao is Professor of Catalysis at the University of Liverpool. Prior to joining Liverpool in 1996, he worked with Nobel Laureate Prof. Noyori in Japan and received undergraduate and postgraduate training in China and Canada. His main research interest is in molecular catalysis, asymmetric synthesis and Green chemistry, and he has published ca. 260 papers in these areas with his students and collaborators. For these works, he was awarded the RSC Tilden Prize 2020, Excelling at Innovation Award, University of Liverpool 2015, and UK Prize for Process Chemistry Research 2008 (SCI). |
1. Introduction
Nitrogen heterocycles are highly valuable structural motifs found frequently in bioactive natural products.1,2 In addition to their prevalence in nature, analysis of structurally unique FDA approved small molecule drugs has shown that 59% contain nitrogen heterocycles.3 Among the 640 N-heterocycle-containing drugs, 31% (200 compounds) are saturated N-heterocycles, such as piperidines (72), piperazines (59), pyrrolidines (37), imidazolidines (11), tetrahydroisoquinolines (11) and tetrahydro-2-pyrimidinones (9). The chiral versions of such structures and their substituted derivatives could be potentially accessible directly through the enantioselective hydrogenation (EH) of the corresponding unsaturated N-heterocyclic ring. In the search for unique therapeutic scaffolds, many research groups have realised the potential of EH as a powerful tool for the synthesis of chiral heterocycles.4–14EH of N-heteroaromatic compounds offers a highly useful synthetic protocol for the construction of industrially and pharmaceutically important cyclic heterocycles in a minimal number of steps. However, such EH reactions have been less intensively studied compared to those of other well-known prochiral substrates, such as olefins, ketones, and imines. The key challenges in achieving this transformation include: (1) the high stability of these aromatic compounds, thereby requiring harsh reaction conditions such as high temperature and pressure. This can have a detrimental effect on the resulting enantioselectivities; (2) the poisoning effects of nitrogen or sulphur atoms acting on the chiral catalysts, and (3) the lack of secondary coordinating sites in simple aromatic compounds leading to low selectivities.5,6,10,15 Efficient strategies have been developed to achieve successful EH of N-heteroaromatic compounds through substrate activation, catalyst activation, and relay activation.6 Substrate activation consists of the introduction of a second coordinating group to alleviate the effect of aromaticity and/or assist in coordination with the catalyst. Catalyst activation includes the fine-tuning of steric and electronic effects in the chiral ligands and also the introduction of additives. Relay activation uses a mixed catalyst system where an achiral catalyst is used to partially reduce the substrate, breaking the aromaticity. This is followed by the reduction of a more reactive intermediate in an asymmetric fashion with a second, chiral catalyst. Various N-heteroarenes, like indoles, isoquinolines, pyridines, quinolines and quinoxalines, can be efficiently hydrogenated with good to excellent enantioselectivity based on these approaches.
In 1987, Murata et al. reported the first example of homogeneous EH of 2-methylquinoxaline using a RhH[(S,S)-DIOP] catalyst under H2.16 Later in 1995, Takaya et al. established a method to hydrogenate 2-methylfuran, achieving 50% ee using a chiral Ru complex bearing the (R)-BINAP ligand as the catalyst.17 Bianchini et al. established in 1998 the first EH of 2-methylquinoxaline using an orthometalated iridium dihydride complex to provide the product in 90% ee.18 Thereafter, several efficient and novel strategies have been developed for the successful enantioselective and diastereoselective hydrogenation of N-heteroaromatic compounds. This review focuses on the developments in enantioselective and diastereoselective hydrogenation of N-heteroaromatic compounds since the previous comprehensive report in 2011,6 and where possible, the relevance of the hydrogenation products to pharmaceutical synthesis has been highlighted. While the review was prepared, a perspective article was published on homogeneous catalyst-mediated asymmetric hydrogenation of heteroarenes in late 2020.15
2. Transition metal complex-catalysed asymmetric hydrogenation
Asymmetric hydrogenation of N-heteroaromatic compounds can be achieved using various transition metals, typically bearing chiral phosphorus ligands, as the catalyst. However, the most effective catalysts are based on only a few metals, i.e. Ir, Rh, Ru, and Pd.2.1. Ir-Catalysed enantioselective and diastereoselective hydrogenation
Since Crabtree reported the synthesis and application in catalysis of the complex [Ir(cod)(PCy3)(Py)]PF6, iridium catalysis has been widely utilised in the hydrogenation of olefins.19 A significant further development was brought about by Pfaltz et al. in 1998, in which the two achiral monodentate ligands were replaced with a bulkier chiral bidentate PHOX ligand.20 The new complex allowed for efficient transfer of chirality from the ligand to the product, opening possibilities for asymmetric hydrogenation. The Ir catalysis was initially used to reduce olefins21,22 and imines,23,24 including challenging un-functionalized olefins. Further development of iridium catalysis has enabled the EH of both arenes and heterocyclic compounds. Described below are recent reports on the EH of various N-heteroarene compounds.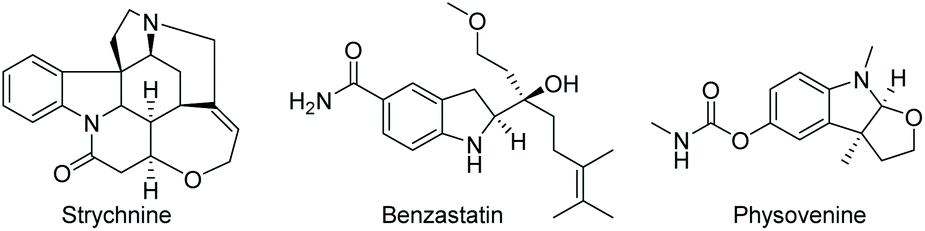 | ||
| Scheme 1 Natural products possessing indoline moiety as a core structure. | ||
Compared to indoles, only a handful of indoline-based drugs are currently on the market. Indapamide as a racemate has been in use since 1977 for the treatment of hypertension. Physostigmine is a natural product with structural similarities to physovenine (Scheme 2). Physostigmine, however, has been approved as a drug. As an enhancer of acetylcholine, its predominant use is as an antidote to various poisons that work by inhibiting acetylcholine.28
 | ||
| Scheme 2 Approved drugs on the market containing indoline moiety. | ||
Despite the current lack of indoline drugs, the biological activities of indoline derivatives are being studied and developed into pharmaceuticals. Relcovaptan, for instance, is being investigated for multiple uses, including the treatment of Raynaud's disease (Scheme 3).29 In addition, WAY-163909 has been shown to have potential uses in anti-obesity drugs and the treatment of certain drug addictions.30
 | ||
| Scheme 3 Highly bio-active indoline derivatives. | ||
To accelerate the development of the indoline moiety in drugs, more methods are required to produce their substituted cores in enantiomerically pure forms. Indoles are a challenging structure for EH to afford indolines. The EH of indole remained unsuccessful for a prolonged time. Since the first report on the EH of indoles using Rh-complexes in 2000,31,32 various other catalytic systems have been investigated. Iridium complexes bearing either bisphosphine ligands or P,N ligands33 have become the most common catalyst used for the EH of substituted indole derivatives.
In 2008 Vidal-Ferran et al. synthesised a highly modular class of P–OP ligands.34 Various metal complexes were formed from the enantiomerically pure ligands and used for the EH of olefins, and later diversely substituted C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond-containing heterocycles.35,36 To develop the method further, the authors began to explore the asymmetric hydrogenation of unprotected indoles. Reusable Brønsted acids together with iridium complexes derived from enantiomerically pure P–OP ligands were explored.37 The study began by using 2-methyl-1H-indole as a model substrate with the Ir–L1 complex as catalyst (Scheme 4). It was found that the lower the pKa of the additive, the higher the conversion. However, enantioselectivities were unaffected by the various Brønsted acid additives. Racemic camphorsulfonic acid (rac-CSA) was found to be the most efficient. Using these optimised conditions, an array of substituted indoles was hydrogenated. It was found that substitution at the C2 position of the indole barely influenced the enantioselectivities which remained high for all 2-substituted indolines reported. Hydrogenation of the 2,3-fused ring derivative provided a cis-indoline in high enantioselectivity. This approach has been extended to the asymmetric hydrogenation of seven-membered CN containing heterocycles.38
N bond-containing heterocycles.35,36 To develop the method further, the authors began to explore the asymmetric hydrogenation of unprotected indoles. Reusable Brønsted acids together with iridium complexes derived from enantiomerically pure P–OP ligands were explored.37 The study began by using 2-methyl-1H-indole as a model substrate with the Ir–L1 complex as catalyst (Scheme 4). It was found that the lower the pKa of the additive, the higher the conversion. However, enantioselectivities were unaffected by the various Brønsted acid additives. Racemic camphorsulfonic acid (rac-CSA) was found to be the most efficient. Using these optimised conditions, an array of substituted indoles was hydrogenated. It was found that substitution at the C2 position of the indole barely influenced the enantioselectivities which remained high for all 2-substituted indolines reported. Hydrogenation of the 2,3-fused ring derivative provided a cis-indoline in high enantioselectivity. This approach has been extended to the asymmetric hydrogenation of seven-membered CN containing heterocycles.38
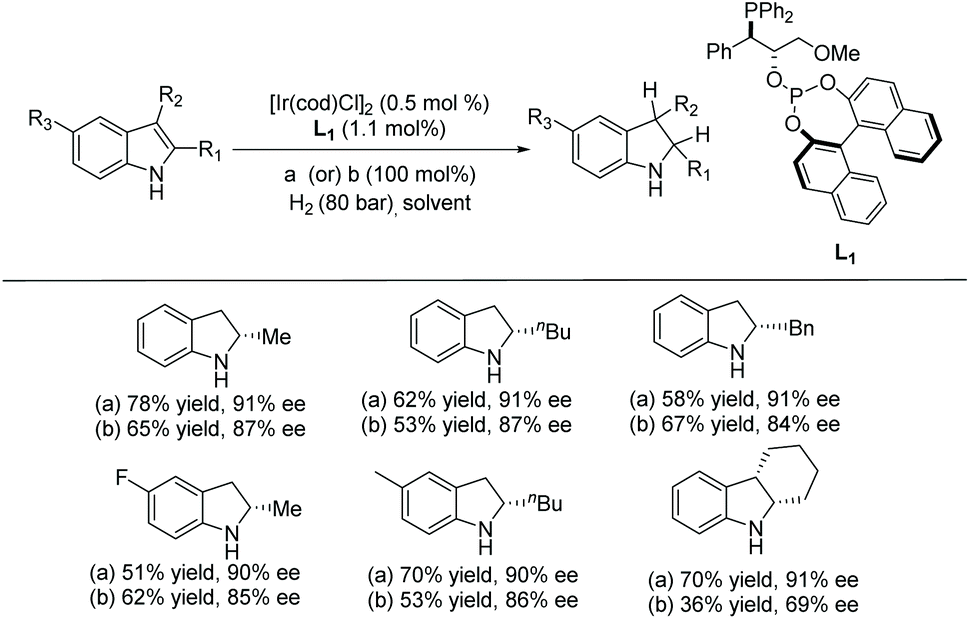 | ||
Scheme 4 EH of unprotected indoles using an Ir–L1 catalyst. Reaction conditions: in situ formed catalyst, [Ir]/L1/rac-CSA (a) (or) OWEX™ 50WX8 (b)/substrate molar ratio = 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1:100:100, H2, rt, 20 h, 0.2 M in 2-Me-THF (or) 0.2 M in 2-Me-THF–DCM (90/10 v/v). :1.1:100:100, H2, rt, 20 h, 0.2 M in 2-Me-THF (or) 0.2 M in 2-Me-THF–DCM (90/10 v/v). | ||
In addition to the use of 1 equivalent of rac-CSA as an additive, ion-exchange resins containing sulfonic acids were also studied. These resins could be recovered and reused for further hydrogenation. The studies revealed that the use of DOWEX™ 50WX8 resin gave the best enantioselectivities, with 87% ee for the 2-methyl indole substrate (Scheme 4).
In 2014, Lyubimov et al. reported an Ir-catalysed EH of indoles using readily available chiral phosphoramidite or chiral phosphite ligands derived from BINOL.39,40 Using 2-methylindole as a model substrate, the phosphite type ligands in the presence of various additives and solvents were tested, leading to the finding that the use of [Ir(cod)2]BARF/L2/I2 could yield excellent conversion (100%) to the corresponding indoline in 80% ee, as shown in Scheme 5.
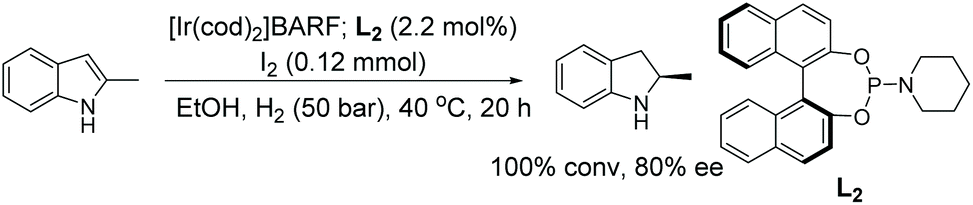 | ||
| Scheme 5 EH of indoles using a phosphoramidite ligand. | ||
 | ||
| Scheme 6 Examples of THIQ-based drugs. | ||
In addition to the approved drugs, there are numerous THIQ-derived compounds in clinical trials. These THIQs contain substitutions in the 1, 3 and 4 positions, showing that all substitutions play an important role in the discovery of new drugs. Examples are found in (+)-SJ733 which is under investigation to treat malaria,45 and Subasumstat which could be used in cancer therapies as an inhibitor of sumoylation (Scheme 7).46
 | ||
| Scheme 7 Examples of THIQ-derived compounds under clinical trial. | ||
The EH of THIQs has been fairly well studied; however, challenges still remain. Despite the biological importance of 4-substituted THIQs, their synthesis through EH remains relatively underdeveloped compared to the 1 and 3 substituted counterparts. Zhou et al. reported in 2006 the first EH of isoquinolines with moderate enantioselectivities.47 Later on, Zhou and Mashima et al. advanced the EH of various mono/di-substituted isoquinolines catalysed with chiral iridium complexes.48 Since then, work has continued to develop this methodology further.
In 2012, Zhou et al. established an enantio- and diastereoselective hydrogenation of isoquinoline derivatives using an iridium-bisphosphine catalyst.49 Previous successful results on the EH of monosubstituted isoquinolines encouraged them to investigate the EH of disubstituted isoquinolines to access tetrahydroisoquinolines diastereoselectively and enantioselectively. Initial screening of various solvents, additives and catalysts showed that [Ir(cod)Cl]2/L3 in combination with an additive, 1,3-dibromo-5,5-dimethylhydantoin (BCDMH), gave high yields and ees (Scheme 8).
 | ||
| Scheme 8 Stereoselective hydrogenation of isoquinolines with an Ir–L3 catalyst. [Ir(cod)Cl]2 (1.0 mol%), L3 (2.2 mol%), BCDMH (10 mol%); d.r. >20:1. | ||
The enantioselectivity of the hydrogenated product was found to arise from a process of dynamic kinetic resolution (DKR) (Scheme 9). DKR is a powerful tool for accessing enantioenriched compounds from racemic substrates/intermediates. During optimisation of the reaction conditions, it was realised that lowering the hydrogen pressure and increasing temperature led to higher enantioselectivities in the cis product. A series of control experiments suggested the formation of the enamine intermediate by 1,2-hydride addition to the CN bond, where there is the least steric hindrance in the initial step. A rapid tautomerisation to the imine is followed by the diastereoselective hydrogenation of int 2. The enantioselectivity of the reaction is controlled by the rate of the isomerization of imine intermediates via the enamine int 1 and that of the hydrogenation of imine int 2. To attain high enantioselectivity, the rate constant associated with the reversible conversion of imine intermediates should be much larger than that of the diastereoselective hydrogenation of int 2. In addition, the rate of diastereoselective reduction of the enantiomer int 2 to II should be faster than that of the other enantiomer. The rate of the isomerization step increased as the reaction temperature was raised, and a low hydrogen pressure decreased the rate of hydrogenation of int 2.50 The reaction was developed further by demonstrating an epimerisation of a cis diastereoisomer to the corresponding trans isomer when treated with LDA, with only minor loss of enantiopurity. trans-Diastereomers are often difficult to achieve via direct asymmetric hydrogenation.
 | ||
| Scheme 9 Mechanistic insights into the EH of isoquinolines with an Ir–L3 catalyst. | ||
In 2013, Mashima et al. developed the EH of isoquinolinium salts using chiral iridium complexes.48 Their previous report on the EH of quinoxalines using halide-bridged dinuclear iridium(III) complexes with aryl amine additives51 led them to expand the methodology to the hydrogenation of isoquinolinium chlorides.
To begin the study, conditions were screened by varying such parameters as solvent and catalyst. Based on the screening results, a 10:1 mixture of 1,4-dioxane/IPA as the solvent and [Ir(H)L4]2(μ-Cl)3Cl as the catalyst were chosen as the optimal conditions. With these in hand, the reaction was carried out on a series of mono-substituted isoquinolinium chlorides, which afforded the corresponding products with high to excellent enantioselectivities regardless of the electronic effect of the substituents (Table 1). To probe the mechanism involved in the reaction, several experiments were carried out, including labelling experiments with D2. Based on these experiments, it was concluded that the reaction proceeds via 1,4 reduction and subsequent CN reduction yields the desired product. Finally, the method was utilised as a key step for the synthesis of the urinary antispasmodic drug (+)-solifenacin, as shown in Scheme 10.
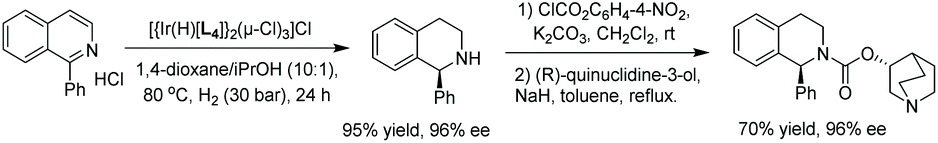 | ||
| Scheme 10 Synthesis of (+)-solifenacin starting from isoquinoline via EH. | ||
|
|
||||
|---|---|---|---|---|
| Entry | R/R′ | T (°C) | Conv. (%) | ee (%) |
| Reaction condition: isoquinoline (0.24 mmol), catalyst (2.4 μmol), solvent (3 mL), 30 °C.a 4.8 μmol of Ir–L4 catalyst was used.b cis:trans >99:1. |
||||
| 1 | Ph/H | 30 | 99 | 96 (+) |
| 2 | 4-MeOC6H4/H | 30 | 99 | 95 (+) |
| 3 | 4-CF3C6H4/H | 30 | 99 | 96 (+) |
| 4 | 2-MeC6H4/H | 30 | 94 | 79 (+) |
| 5 | 2-OMeC6H4/H | 30 | 93 | 81 (+) |
| 6a | H/4-CF3C6H4 | 80 | 99 | 98 (+) |
| 7a | H/4-OMeC6H4 | 80 | 99 | 99 (S) |
| 8a | H/2-BrC6H4 | 80 | 75 | 96 (R) |
| 9a | H/Cy | 80 | 99 | 79 (−) |
| 10b | Ph/Ph | 80 | 98 | 98 (S,S) |

Around the same time, Zhou et al. developed an EH of a variety of 1-substituted isoquinolinium salts using an Ir–L5 catalyst (Table 2). It was expected that activating isoquinoline as the N-benzyl isoquinolinium salt may facilitate hydrogenation, improving both the reactivity and selectivity.52 Initially, reaction conditions including a variety of solvents were screened. The mixed solvent system (THF/DCM (1:1)) was found to provide the highest reactivity and enantioselectivity. Following these findings, various commercially available bisphosphine ligands were investigated. It was found that L5 gave the highest ee and conversion.
|
|
||||
|---|---|---|---|---|
| Entry | R | Ar | Yield (%) | ee (%) |
| Reaction condition: isoquinolinium salt (0.25 mmol), [Ir(cod)Cl]2 (1 mol%), L5 (2.2 mol%), 20 h.a [Ir(cod)Cl]2 (2 mol%), L5 (4.4 mol%). | ||||
| 1 | Ph | Ph | 99 | 93 (R) |
| 2 | 4-MeOC6H4 | Ph | 99 | 94 (−) |
| 3 | 4-CF3C6H4 | Ph | 97 | 92 (−) |
| 4a | iPr | Ph | 99 | 74 (+) |
| 5 | 4-MeC6H4 | 2-(iPrCO2)C6H4 | 99 | 95 (−) |
| 6 | 4-OMeC6H4 | 2-(iPrCO2)C6H4 | 99 | 94 (−) |
| 7 | 4-ClC6H4 | 2-(iPrCO2)C6H4 | 99 | 94 (−) |
| 8 | 4-CF3C6H4 | 2-(iPrCO2)C6H4 | 99 | 94 (−) |
| 9 | 4-FC6H4 | 2-(iPrCO2)C6H4 | 99 | 94 (−) |
| 10 | 3,5-FC6H3 | 2-(iPrCO2)C6H4 | 99 | 94 (−) |

The introduction of a CO2iPr group at the 2-position of the benzyl group led to a slight increase in the enantioselectivity, perhaps due to its steric bulk and/or interaction with the iridium. These optimised conditions were used to explore the scope of 1-substituted isoquinolinium salts. The results show that most of the 1-aryl substituted isoquinolinium salts gave high yields and enantioselectivities, whereas the 1-alkyl-isoquinolinium salts performed well in the transformation but provided only moderate enantioselectivity. To elucidate the reaction mechanism, a series of experiments were carried out. These showed that the reaction is initiated by 1,2-hydride addition with subsequent isomerisation to the iminium salt (Scheme 11). This is followed by rapid hydrogenation to deliver the desired product.
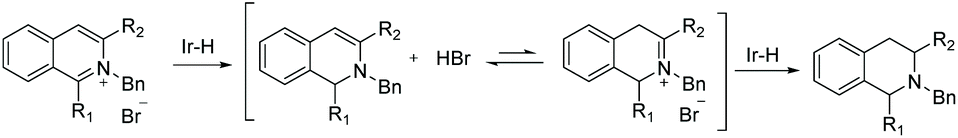 | ||
| Scheme 11 Proposed mechanism for the hydrogenation of isoquinolinium salts. | ||
Expanding further on the EH of isoquinolines, Zhou et al. established an efficient route for the EH of fluorinated isoquinolines (Table 3).8 Fluorine can modify the electronic and physical properties of molecules, making fluorinated analogues highly valuable to medicinal chemists. However, substrates of this nature pose several challenges, such as (i) the highly stable aromatic structure, (ii) hydrodefluorination, and (iii) control of the stereoselectivity. Zhou's strategy includes the use of a catalytic amount of halogenated hydantoin and the hydrochloride salt of the substrates. Optimal conditions were found for the EH of fluorinated isoquinolines, using [Ir(cod)Cl]2–L3 in the presence of DCDMH (1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione), which addressed these limitations. The method showed good to excellent yields (79–97%) with good enantioselectivities (88–93% ee) regardless of the properties of the alkyl chain at the 3-position or the electronic nature of the substituents on the aromatic ring. In general, chiral N-heterocycles bearing a C–F stereogenic centre with high chemo-, diastereo- and enantio-selectivity have been obtained.8 To showcase the practicality of this method, a reaction was carried out on a gram scale, providing 89% ee and 91% yield.
|
|
|||
|---|---|---|---|
| Entry | Isoquinoline salts (R1/R2/R3) | Yield (%) | ee (%) |
| Reaction conditions: isoquinoline salts (0.20 mmol), L3 (2.2 mol%), [Ir(cod)Cl]2 (1.0 mol%), DCDMH (5 mol%).a Gram scale. | |||
| 1 | H/H/n-Bu | 92 | 93 (−) |
| 2 | H/H/Me | 92 | 93 (−) |
| 3 | H/H/Et | 94 | 93 (−) |
| 4 | H/H/nPr | 93 | 92 (R,R) |
| 5 | H/H/cyclopropyl | 79 | 90 (−) |
| 6 | H/H/nBu | 95 | 93 (−) |
| 7 | F/H/nBu | 91 | 91 (−) |
| 8 | H/F/nBu | 93 | 91 (−) |
| 91a | 89 (−) | ||
| 9 | Me/H/nBu | 97 | 91 (−) |
| 10 | Cl/H/nBu | 93 | 90 (−) |

In 2017, Zhou et al. reported the EH of isoquinolines and pyridines by employing trichloroisocyanuric acid (TCCA) as a traceless activation reagent.53 The traceless agent does not require instillation or removal steps. The Lewis basic isoquinolines appear to easily form halogen bond interactions with halogenides, leading to their activation, which could open a new opportunity for the EH of isoquinolines. With this in mind, optimal conditions were searched and found to be [Ir(cod)Cl]2/L6/H2/TCCA. A series of isoquinolines were hydrogenated with high conversion and enantioselectivity, including isoquinolines substituted at the 1 and/or 3 position with some fluorinated at the 4 position (Scheme 12). The practical utility of this method was tested on a gram scale on 1-phenylisoquinoline under the standard conditions, providing 94% yield and 94% ee.
 | ||
| Scheme 12 EH of isoquinolines by traceless activation with TCCA. Reaction condition: [Ir(COD)Cl]2 (1.0 mol%), L6 (2.2 mol%). a[Ir(COD)Cl]2 (2.0 mol%), L6 (4.4 mol%). bTCCA (1.0 equiv.). cDioxane/iPrOH (v/v = 5:1, 3.0 mL), H2 (40 bar), TCCA (1.0 equiv.), 20 h, 30 °C. | ||
In 2020, Stoltz et al. established the enantioselective and diastereoselective hydrogenation of 1,3-disubstituted isoquinolines bearing a hydroxymethyl directing group.54 Optimisation showed that [Ir(cod)Cl]2 and a commercially available chiral xyliphos ligand L7 in the presence of a TBAI additive could provide good yields and selectivity (Scheme 13).
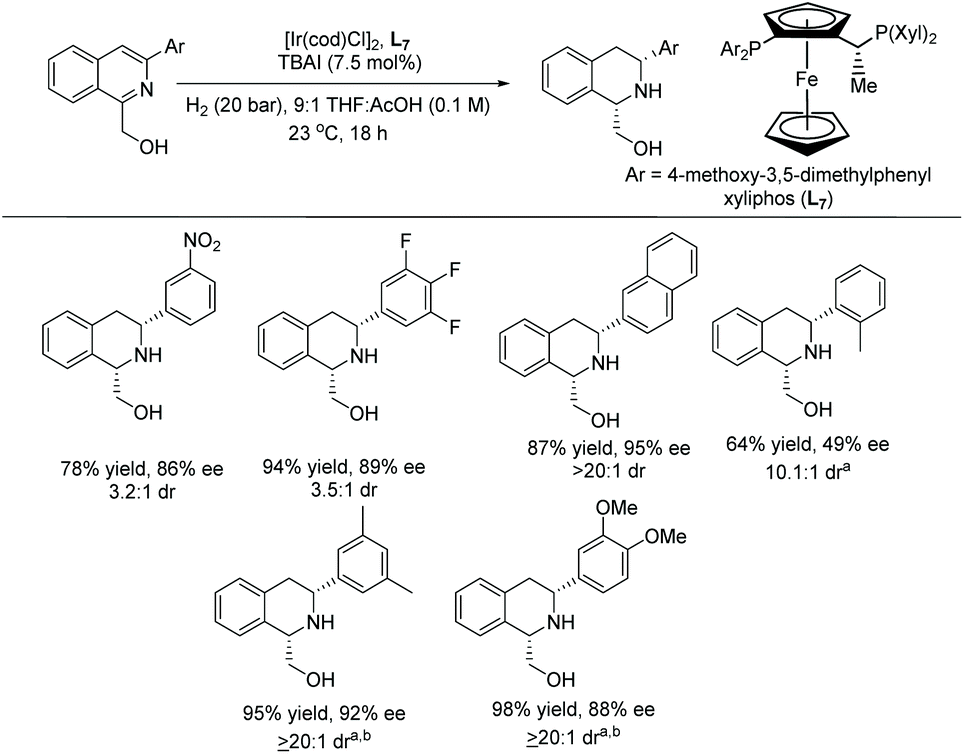 | ||
| Scheme 13 Stereoselective hydrogenation of isoquinolines with an Ir–L7 catalyst. Reactions condition: Substrate (0.2 mmol). a60 °C, H2 (60 bar). bCH2Cl2 cosolvent used to improve substrate solubility. | ||
A broad range of substituted isoquinolines was reduced with high enantioselectivity of up to 95% ee and diastereoselectivity of >20:1 dr. The directing group at the C1 position of the isoquinolines is required to facilitate the hydrogenation, a key strategy for attaining high enantioselectivity and diastereoselectivity. Currently, this report represents the highest tolerance of Lewis basic functionalities of any asymmetric isoquinoline reduction protocols (Schemes 13 and 14).
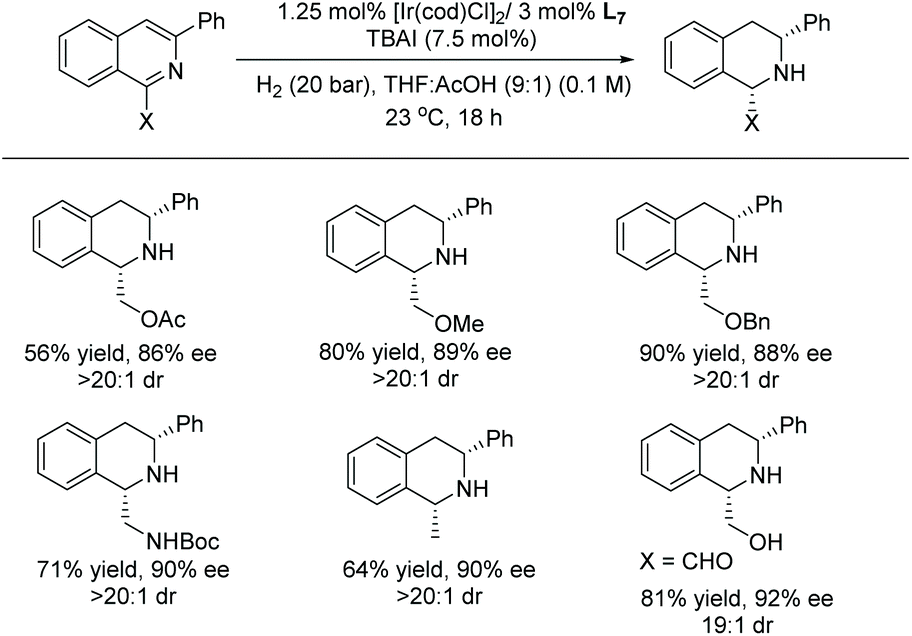 | ||
| Scheme 14 Functional group tolerance of the EH. | ||
This method can be used to produce electron-deficient THIQs. The flexibility of the substrates was showcased by using the hydroxyl directing group as a site for further synthetic manipulations (Scheme 15). Transformations include the elaboration of the synthesised scaffold to various tricyclic and pentacyclic skeletons, which are of potential medicinal interest.
 | ||
| Scheme 15 Derivatization of a hydrogenated product. | ||
Prior to this work, the same group had been able to show the importance of both the transformation and THIQ core through the synthesis of jorumycin, a potential anti-tumour agent with antibiotic character.55 Hydrogenation of a key isoquinoline intermediate furnished the pentacyclic system in jorumycin (Scheme 16). Jorumycin also shares structural features with the experimental drug PM00104, which is under investigation to treat cancer.56
 | ||
| Scheme 16 EH as a key step to synthesise jorumycin. | ||
Current piperidine drugs are used to treat a large array of diseases. Meplvacaine, ropivacaine, bupivacaine and levobupivacaine are all anaesthetics. Paroxetine is an antidepressant and levocabastine is an antiallergic drug (Scheme 17). Perhaps most importantly, the piperidine scaffold has been significant in anti-cancer drugs with a range of different modes of action and structures (Scheme 18). Piperidine drugs have a variety of different substitution patterns. The most common substitutions occur in the N and C4 positions. The most popular number of substitutions on the piperidine scaffold is 2, with the 1,4 disubstituted piperidines the most common, correlating with the most common substitutions. Although of a 3-D structure, a 1,4 disubstituted piperidine would not be chiral. This simplicity could be in part the reason for the substitution pattern's popularity. After the 1,4 positions, position C2 is the third most popular position with almost a third of FDA approved piperidine-based drugs containing this substitution. The synthesis of C2-substituted piperidines is perhaps the most well developed route to chiral piperidines, potentially the reason for its relative popularity. The C3 position is, however, potentially underutilised. In 2014, only 14 of the 72 piperidine-based drugs contained a substitution in the C3 position.3 Further work in the synthesis of enantioenriched piperidines could help unlock previously unknown and increasingly diverse scaffolds with interesting chemical and physical properties.
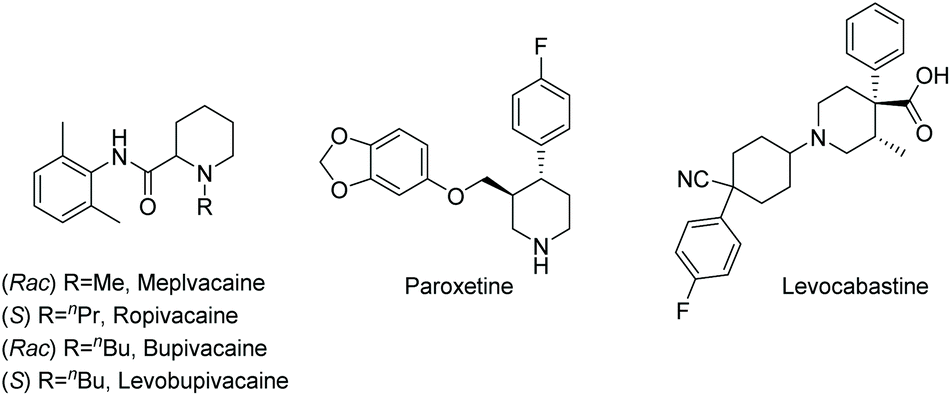 | ||
| Scheme 17 A selection of piperidine-based drugs. | ||
 | ||
| Scheme 18 Piperidines with different substitution patterns showing anti-cancer properties. | ||
Due to the pharmaceutical interest, there have been huge efforts focused on the development of synthetic protocols for such a prevalent motif. EH of various substituted pyridines is the most straightforward and atom-economical route to generate chiral piperidines. However, despite several recent advances in the area, the EH of pyridine derivatives remains a challenging task.
Studer et al. reported in 2000 the first homogeneous asymmetric hydrogenation of an un-activated pyridine using [Rh(nbd)2]BF4/BINAP as a catalyst to yield the piperidine in 25% ee.58 Charette et al. in 2005 established asymmetric hydrogenation of activated pyridine derivatives by introducing N-benzoyliminopyridinium ylides as the substrates.59 Zhou et al. in 2008 discovered the [Ir(cod)Cl]2/L3/I2 catalyst system for the EH of pyridines.60 This work was expanded further in 2012, when Zhou et al. reported the iridium-catalysed EH of 2-substituted pyridinium salts with high enantioselectivity.61 Initial investigations focussed on the hydrogenation of N-benzyl-2-phenylpyridinium bromide using [Ir(cod)Cl]2/L3 as the catalyst. It was found that a 1:1 mixture of PhMe/CH2Cl2 provided the best reactivity and enantioselectivity. Examination of the activating group showed that electron-withdrawing substituents such as CO2Me at the C2-position of the benzyl group on the pyridinium salt led to a considerable increase in enantioselectivity. It was suggested that this group could coordinate with the catalyst, influencing the enantioselectivity. Replacement of the counterion was examined but found to have no impact on reactivity or enantioselectivity. Activation of pyridines to the pyridinium salts was critical to this strategy, as salt formation would avoid the inhibition of the catalyst by the substrate. In addition to this, in situ generated stoichiometric hydrogen bromide would continue to effectively inhibit the coordination ability of the desired product after hydrogenation. Selected results are shown in Table 4.
|
|
|||
|---|---|---|---|
| Entry | Pyridinium salt (R) | Yield (%) | ee (%) |
| Reaction condition: pyridinium salt (0.25 mmol), [Ir(cod)Cl]2 (1 mol%), L3 (2.2 mol%), 24 h. | |||
| 1 | Ph | 99 | 93 (S) |
| 2 | 4-MeC6H4 | 99 | 89 (−) |
| 3 | 3-MeC6H4 | 88 | 86 (−) |
| 4 | 2-MeC6H4 | 82 | 78 (−) |
| 5 | 4-OMeC6H4 | 99 | 92 (−) |
| 6 | 3-OMeC6H4 | 93 | 92 (−) |
| 7 | 4-ClC6H4 | 95 | 92 (−) |
| 8 | 4-FC6H4 | 99 | 93 (−) |
| 9 | 2-Naphthyl | 99 | 87 (−) |
| 10 | 4-CF3C6H5 | 96 | 93 (−) |
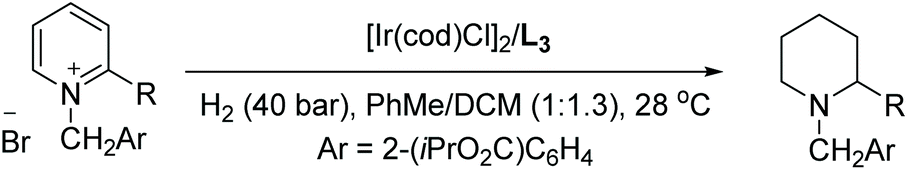
To demonstrate the utility of this methodology, a gram scale reaction was performed, furnishing the protected 2-phenyl piperidine with only a small loss of yield. Changing to a Boc protecting group allowed recrystallisation to give the desired compound in excellent ee. Previous work has shown enantioenriched Boc-protected 2-phenyl piperidine could be elaborated to an NK1 receptor antagonist (Scheme 19).62,63
 | ||
| Scheme 19 Synthesis of NK1 receptor antagonist starting from 2-phenylpyridine. | ||
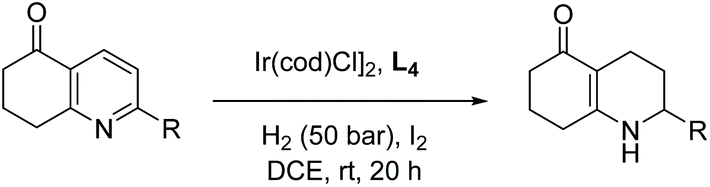
Xu and co-workers reported in 2010 a highly efficient catalytic system comprised of the chiral ligand Difluorphos in combination with [Ir(cod)Cl]2, which allowed for the asymmetric hydrogenation of quinolines to yield high enantioselectivities of up to 96% ee with up to 43000 TON and up to 3510 h−1 TOF.64 The I2 additive played an important role in this reaction, enhancing the catalytic performance by potentially oxidizing Ir(I) to form the highly active Ir(III) species. Under the optimised reaction conditions, a series of 2-substituted quinolines were hydrogenated to display the scope of the reaction at low catalyst loadings (0.05–0.01 mol%). Due to the high activities observed, the EH of trisubstituted pyridines was also investigated. Initially, the reaction was screened with modifications to the solvent, amount of I2 and S/C ratios. A series of fused-ring trisubstituted pyridines were then hydrogenated, providing quantitative yields and good to excellent enantioselectivities (Table 5).
|
|
|||
|---|---|---|---|
| Entry | Substrate (R) | Yield (%) | ee (%) |
| Reaction conditions: [Ir(COD)Cl]2 (0.5 mol%), L4 (1.1 mol%), I2 (15 mol%). | |||
| 1 | Me | 97 | 98 (R) |
| 2 | n C3H7 | 97 | 89 (R) |
| 3 | n C5H11 | 98 | 86 (R) |
| 4 | n C6H13 | 97 | 78 (R) |
| 5 | C6H5 | 99 | 92 (S) |
| 6 | CH2C6H5 | 99 | 92 (S) |
| 7 | CH2CH2C6H5 | 98 | 92 (R) |
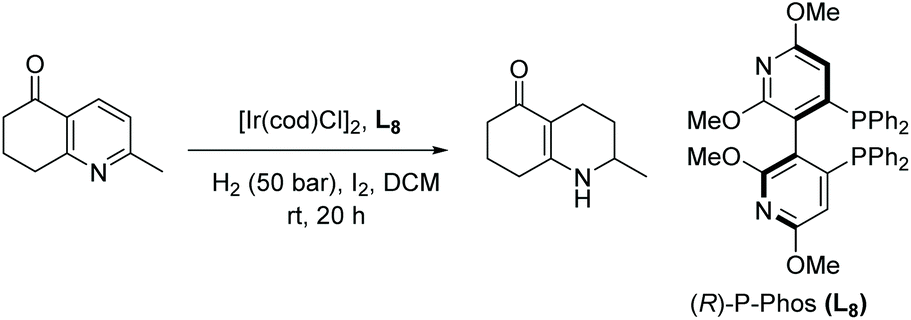
Xu et al. also reported highly enantioselective hydrogenation of quinolines and pyridines using a chiral iridium catalyst generated in situ from [Ir(cod)Cl]2 and a P-Phos ligand in the presence of I2. The EH was carried out at high S/C ratios of 2000–50000, reaching up to 4000 h−1 TOF and up to 43000 TON.65 The catalytic activity was found to be additive-controlled. To maintain a good conversion, decreasing the amount of additive I2 was necessary when lowering the catalytic loading. Initially, hydrogenation was carried out on a series of quinoline derivatives. The same catalytic system was then utilised to enable EH of trisubstituted pyridines, affording the chiral hexahydroquinolinones with quantitative yields and up to 99% ee (Table 6).
|
|
|||
|---|---|---|---|
| Entry | Substrate (R) | Yield (%) | ee (%) |
| Reaction conditions: [Ir(COD)Cl]2 (0.5 mol%), L8 (1.1 mol%), I2 (20 mol%). | |||
| 1 | Me | 98 | 95 (S) |
| 2 | n C3H7 | 96 | 94 (S) |
| 3 | n C5H11 | 97 | 95 (S) |
| 4 | n C6H13 | 97 | 97 (S) |
| 5 | C6H5 | 99 | 81 (R) |
| 6 | CH2C6H5 | 98 | 92 (R) |
| 7 | CH2CH2C6H5 | 99 | 96 (S) |
Andersson and co-workers established a N,P-ligated iridium catalysed EH of ortho-substituted pyridines in 2013 (Table 7).66 The protocol involves the activation of the substrate by forming N-imino-pyridium ylides.59 The use of iodine as an additive was also shown to be highly effective in this method. Although high ees were achieved, the reaction was found to be highly substrate dependant, with the highest ee observed for linear alkyl groups at the C2-position. Bulkier substitutions resulted in a significant drop in selectivity.
|
|
|||
|---|---|---|---|
| Entry | Pyridinium salt (R) | Conversion (%) | ee (%) |
| 1 | Ph | >99 | 86 |
| 2 | 4-MeC6H4 | >99 | 83 |
| 3 | 3-MeC6H4 | >99 | 77 |
| 4 | 2-MeC6H4 | >99 | 77 |
| 5 | 4-MeOC6H4 | >99 | 10 |
| 6 | 3-MeOC6H4 | >99 | 40 |
| 7 | 4-ClC6H4 | >99 | 61 |
| 8 | 4-FC6H4 | >99 | 98 |

In 2014, Zhang et al. described the asymmetric hydrogenation of N-benzyl-2-substituted pyridinium salts. Simple benzyl groups were used to successfully activate the pyridine substrates for hydrogenation.67 The Zhou group had previously shown that EWG substitution at the C2-position of the benzyl activating group could be highly beneficial for EH of the pyridinium salt.61
Using an iridium-phosphole catalyst, simple N-alkyl-2-arylpyridinium salts were hydrogenated with excellent reactivity and enantioselectivity (Table 8).67 An efficient procedure was also developed to overcome the extra steps required for the removal of these activating groups to obtain the desired piperidine product. The nature of the pyridinium counterion had some impact on both the activity and enantioselectivity of the reaction (Scheme 20).
 | ||
| Scheme 20 Effect of the activating group/counterion on ee. | ||
|
|
|||
|---|---|---|---|
| Entry | Pyridinium salt (R) | Yield (%) | ee (%) |
| Reaction conditions: pyridinium salt, 0.025 M, [Ir]/ligand/pyridinium bromide = 0.5:0.55:100, 20 h.a DCE/acetone = 5:1 as solvent.b Acetone as solvent.c DCE as solvent. |
|||
| 1 | Ph | 97 | 96 |
| 2 | 4-MeC6H4 | 93 | 93 |
| 3 | 4-OMeC6H4 | 95 | 94 |
| 4 | 4-Ac(H)NC6H4 | 97 | 95 |
| 5 | 4-ClC6H5 | 96 | 95 |
| 6 | 4-PhC6H5 | 86 | 90 |
| 7a | 4-PhC6H5 | 96 | 95 |
| 8 | 2,4-Cl2C6H5 | 94 | 90 |
| 9 | 3,5-F2C6H5 | 95 | 96 |
| 10 | 2-Naphthyl | 12 | 93 |
| 11b | 2-Naphthyl | 88 | 94 |
| 12b | Me | 81 | 33 |
| 13b | iPr | 24 | 69 |
| 14c | CH2OAc | 92 | 24 |

Mashima et al. in 2014 reported asymmetric hydrogenation of di-substituted pyridinium salts using chiral iridium dinuclear complexes to provide piperidines of high diastereoselectivity.68 The authors had previously exploited the same catalytic strategy for the EH of isoquinolines (see Table 1). This strategy was expanded to include pyridine motifs with only slight modifications. With the iridium dinuclear complex as the catalyst in a mixed solution of 1,4-dioxane/iPrOH at 100 °C for 20 h under H2 (10 bar), various di-substituted pyridinium salts were successfully hydrogenated to afford chiral piperidines (Table 9).
|
|
||||
|---|---|---|---|---|
| Entry | Product | Yield (%) | ee (%) | dr |
| Reaction conditions: pyridine (0.24 mmol), catalyst (12.1 μmol), and solvent (6 mL). | ||||
| 1 |

|
81 | 28 | >95:5 |
| 2 |
|
54 | 55 | >95:5 |
| 3 |
|
>95 | 82 | >95:5 |
| 4 |

|
75 | 73 | >95:5 |
| 5 |

|
66 | 72 | >95:5 |
| 6 |

|
32 | 40 | >95:5 |

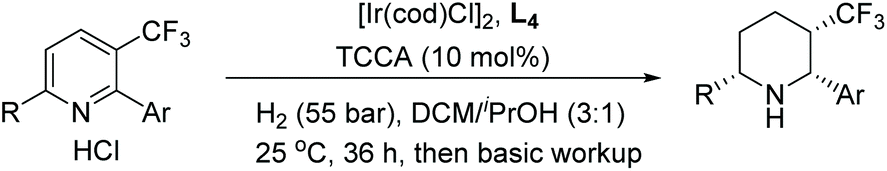
Shortly after, Zhou et al. reported the EH of multi-substituted pyridinium salts to obtain cis-trifluoromethyl substituted piperidines. The trifluoromethyl group is of great interest to medicinal chemists due to its ability to vastly change the chemical and physical properties of molecules. The EH resulted in products with multiple stereogenic centres, with up to 90% ee (Table 10).69 The optimal conditions for this reaction were found to be [Ir(cod)Cl]2/L4/TCCA. The method was further extended by the exploration of 2,6-disubstituted pyridinium hydrochlorides where a trifluoromethyl group was not a substituent (Scheme 21). Moderate reactivity and enantioselectivity were achieved. The relatively harsh conditions (85 bar hydrogen pressure and 80 °C) required for these substrates suggest that the introduction of trifluoromethyl group may increase the substrate reactivity, presumably due to the electron-withdrawing effect.
 | ||
| Scheme 21 EH of 2,6-disubstituent pyridinium hydrochloride salts. Reaction condition: pyridinium HCl (0.125 mmol), L4 (2.2 mol%), [Ir(COD)Cl]2 (1.0 mol%), TCCA (10 mol%), 24 h. | ||
|
|
|||
|---|---|---|---|
| Entry | R/Ar | Yield (%) | ee (%) |
| Reaction condition: pyridinium hydrochloride (0.125 mmol), L4 (5.5 mol%), [Ir(cod)Cl]2 (2.5 mol%). | |||
| 1 | Me/C6H5 | 95 | 90 |
| 2 | Me/4-MeC6H5 | 84 | 89 |
| 3 | Me/3-MeC6H5 | 84 | 88 |
| 4 | Me/4-MeOC6H5 | 94 | 88 |
| 5 | Me/2-Napthyl | 93 | 89 |
| 6 | Me/4-Ph-C6H5 | 90 | 87 (2R,3S,6R) |
| 7 | Me/4-CF3C6H5 | 85 | 86 |
| 8 | Me/3,5-F2C6H5 | 72 | 84 |
| 9 | Et/C6H5 | 82 | 87 |
In 2016, Zhou et al. disclosed a highly EH of pyridinium salts bearing a hydroxyl group, delivering straightforward access to trans 6-substituted piperidin-3-ols with up to 95% ee.70 They began the investigation by screening various reaction parameters, such as solvents and catalysts. Optimal conditions were found to be [Ir(cod)Cl]2/L11/H2 (40 bar)/NaHCO3. With these optimal conditions, various disubstituted pyridinium salts were tested and shown to give the corresponding trans-products with high reactivity and diastereoselectivity (Table 11). The trans-piperidin-3-ols were shown to be convertible to the cis-products by Swern oxidation followed by reduction using K-selectride. Consequently, both the cis & trans-piperidin-3-ols can be easily accessed from the same starting materials.
|
|
||||
|---|---|---|---|---|
| Entry | R | Yield (%) | ee (%) | trans/cis |
| Conditions: substrate (0.20 mmol), [Ir(cod)Cl]2 (1.5 mol%), L11 (3.3 mol%), NaHCO3 (0.20 mmol), THF (3.0 mL), 40 °C, 24 h. | ||||
| 1 | Ph | 98 | 93 (3S,6R) | 89:11 |
| 2 | 3-MeC6H5 | 91 | 94 (+) | 82:18 |
| 3 | 4-MeC6H5 | 93 | 94 (+) | 90:10 |
| 4 | 3,5-Me2C6H5 | 93 | 95 (+) | 83:17 |
| 5 | 3-MeOC6H5 | 95 | 91 (+) | 84:16 |
| 6 | 3,5-F2C6H5 | 97 | 86 (+) | 86:14 |
| 7 | 3-ClC6H5 | 95 | 90 (+) | 88:12 |
| 8 | 4-ClC6H5 | 96 | 92 (+) | 83:17 |
| 9 | 4-CF3C6H5 | 97 | 93 (+) | 83:17 |
| 10 | 4-MeO2CC6H5 | 95 | 90 (+) | 75:25 |
| 11 | 2-Naphthyl | 90 | 81 (+) | 49:51 |
| 12 | n Pr | 81 | 64 (+) | 53:47 |
| 13 | H | 89 | 15 (+) | — |

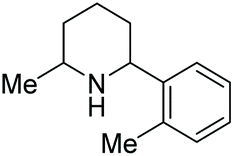
In 2016, Mashima et al. developed the EH of amino pyridines, which gave the corresponding piperidines with an exocyclic nitrogen moiety (Table 12).71 This allowed for a direct synthetic method for the synthesis of neurokinin-1 (NK1) receptor antagonist derivatives. Thus, EH of 3-amido-2-arylpyridinium salts using the dinuclear iridium complex in combination with (S)-SegPhos provided the corresponding chiral piperidines in high cis-diastereoselectivity (>95:5) and moderately high enantioselectivity (up to 86% ee). This asymmetric hydrogenation step could be utilised to efficiently furnish (+)-CP-99994, a non-peptide antagonist of the NK1 receptor (Scheme 22).
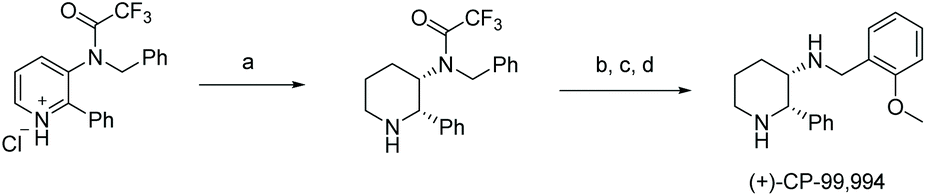 | ||
| Scheme 22 Synthetic application of Ir-catalysed hydrogenation. (a) [Ir(H)[L12]2(μ-Cl)3]Cl (5.0 mol%), (+)-CSA (100 mol%), 1,4-dioxane, 60 °C, H2 (30 bar), 18 h, then basic work-up; (b) K2CO3, MeOH/H2O (3/1), 65 °C, 12 h; (c) Pd(OH)2/C, HCl (ether solution), MeOH, 50 °C, H2 (15 bar), 18 h; (d) o-anisaldehyde, NaBH(OAc)3, i-PrOAc, rt, 3 h. | ||
|
|
||||
|---|---|---|---|---|
| Entry | R1/R2/R3 | Temp. (°C) | Yield (%) | ee % |
| Reaction conditions: pyridinium salt (0.15 mmol), L12 (7.5 mmol), (−)-CSA (0.15 mmol), and 1,4-dioxane (3 ml). dr = >95:5. |
||||
| 1 | Ph/Ph/CF3 | 60 | 82 | 84 (−) |
| 2 | 4-MeOC6H5/Ph/CF3 | 80 | 88 | 80 (−) |
| 3 | 4-CF3C6H5/Ph/CF3 | 60 | 75 | 70 (−) |
| 4 | 4-COOMeC6H5/Ph/CF3 | 60 | 94 | 81 (−) |
| 5 | 2-MeC6H5/Ph/CF3 | 80 | 69 | 77 (−) |
| 6 | 2-Thienyl/Ph/CF3 | 80 | 38 | 70 (+) |
| 7 | 2-Naphthyl/Ph/CF3 | 60 | 73 | 76 (−) |
| 8 | H/Ph/CF3 | 80 | Trace | NA |
| 9 | Ph/Ph/Ph | 60 | 75 | 86 (−) |
| 10 | Ph/Ph/3,5-bis(CF3)C6H3 | 60 | Trace | NA |

Around the same time, Lefort et al. described a mixed-ligand approach for iridium-catalysed asymmetric hydrogenation of N-benzyl-2-arylpyridinium salts and proposed a mechanism based on experimental evidence.72 This EH of pyridinium salts was performed using an Ir complex which was generated in situ from two monodentate ligands (a chiral phosphoramidite and an achiral phosphine) with [Ir(cod)Cl]2. A range of ligands was tested along with modification of the ratios of chiral ligand/achiral ligand and iridium. The optimised ratio was found to be 1:1:1 [Ir(cod)Cl]2/L13/P(p-CF3C6H4)3 under which enantioselectivities of up to 82% were obtained (Scheme 23). The protocol was found to be efficient for a range of 2-arylpyridinium salts. An initial 1,4-hydride addition to the substrate was proposed, leading to the partially reduced N-benzyl-2-phenyl-1,4-dihydropyridine. This species was further hydrogenated to the enamine via the iminium intermediate. Tautomerisation of the enamine to the imine allowed an enantioselective 1,2-hydride addition, providing the final chiral piperidine (Scheme 24).
 | ||
| Scheme 23 EH of pyridinium salt using a mixed-ligand approach. | ||
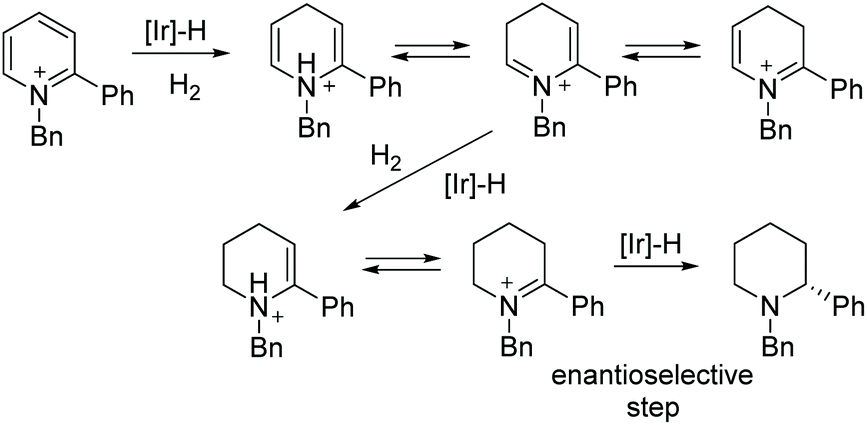 | ||
| Scheme 24 Proposed mechanism of the Ir-catalysed EH of pyridines. | ||
In 2018, Kozlowski and co-workers reported the EH of α-(hetero)aryl pyridinium salts to the corresponding piperidines and investigated the reaction mechanism.73 The EH of pyridinium salts with a heteroaryl functionality has not been widely reported. Previous reports have mainly focussed on α-phenyl derivatives without such a functionality. An [Ir(cod)Cl]2/L14 catalyst system was used in the EH (Scheme 25). Through DFT calculations a detailed mechanism for pyridinium reduction was elucidated. Interestingly, the calculation indicated that protonation of the final enamine intermediate determines the enantioselection of the EH, rather than hydride transfer to the iminium intermediate.
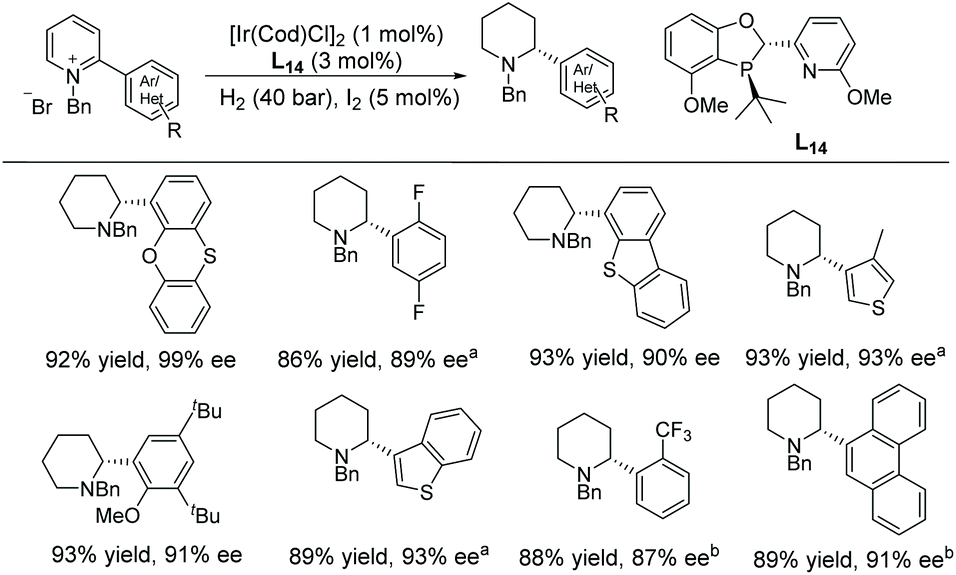 | ||
| Scheme 25 Synthesis of α-(hetero)aryl piperidines via EH of pyridinium salts. Reactions were run at 40 °C for 24 h. a20 °C, b50 °C. | ||
In 2021, Zhang et al. established the synthesis of chiral indolizidine by EH of cyclic pyridinium salts using an Ir–L15 catalytic system.74 Initially, cyclic pyridinium salts were accessed by reducing 2-(2-acylphenyl)pyridines using NaBH4 followed by the cyclization using HBr/HCl. The counteranion exchange reaction was carried out with the corresponding silver salts to access other counterions of cyclic pyridinium salts. In the initial screening, Ir–L15 showed excellent reactivity and acetone as solvent improved the ee. The scope and limitation of this reaction were explored on various EDG and EWG on the benzene ring, which was found to give excellent yield with moderate enantioselectivity and excellent diastereoselectivity (Scheme 26).
 | ||
| Scheme 26 Ir-Catalysed EH of various cyclic pyridinium salts. Reaction conditions: substrate (0.2 mmol), acetone (2 mL). aThe reaction was conducted at 40 °C. | ||
Our group recently reported an asymmetric transfer hydrogenation (ATH) of 2-substituted pyridinium salts with formic acid using a chiral cyclometalated iridium complex to yield chiral piperidines in up to 77% ee.75 Initially various chiral iridacycles were examined for the ATH of N-benzyl-2-phenylpyridinium bromide using IPA as solvent and it was found that oxazoline-containing iridacycles provided a lower selectivity than imidazoline-containing iridacycles. Under this condition a few pyridinium salts were hydrogenated to provide chiral 2-substituted piperidines with moderate enantioselectivities (Scheme 27).
 | ||
| Scheme 27 ATH of selected 2-substituted pyridinium salts. | ||
 | ||
| Scheme 28 Piperazine-based natural products. | ||
 | ||
| Scheme 29 Piperazine in approved drugs. | ||
Despite the prevalent use of the piperazine scaffold, piperazines in drug molecules are generally only substituted on one or both nitrogen atoms. Only 12% of piperazine-based drugs have one or more substitutions at the carbon position (Scheme 30). Piperazine appears to be a generally under-utilised structure, in part due to the lack of efficient reactions to produce carbon substituted derivatives.
 | ||
| Scheme 30 Comparison of piperazine substitution patterns. | ||
More diverse piperazine structures can be found in drugs; however, their occurrence is much rarer than N-substitutions. Vestipitant was developed by GSK and is used to treat tinnitus and insomnia. Pyrrolopyridine quinolone carboxylate is amongst a series of compounds synthesised that were found to exhibit potent antibacterial properties.78 Indinavir, developed by Merk, is a protease inhibitor and has been used to treat HIV/AIDS (Scheme 31). C-Substituted piperazines have great potential. Substitution on carbon in the ring would introduce chirality and diversify piperazine's 3-D space. In addition, these substitutions could also provide an extra chemical handle, allowing for increased complexity in a potential drug molecule. Efforts to form C-substituted piperazine rings are therefore of great interest to both synthetic and medicinal chemists.
 | ||
| Scheme 31 C-Substituted piperazine drugs. | ||
In 1998, Fuchs et al. first established the synthesis of piperazine via EH in 78% ee.79 Later in 2014, Zhou et al. developed highly enantioselective hydrogenation of pyrrolo[1,2-a]pyrazinium salts, providing direct access to the corresponding 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazine derivatives with up to 95% ee, using an Ir catalyst (Table 13).80 Initial attempts during optimisation gave disappointingly low ee values. Previous literature had indicated that racemisation could occur in structurally similar molecules through enamine/iminium isomerisation in acidic conditions. To probe this further, the hydrogenated product was mixed with 1 equivalent of HCl and stirred. The ee of the product was indeed found to drop over time. As HBr is created in situ during the reaction, several different organic and inorganic bases were tested to see if substantial racemisation could be eliminated. The addition of Cs2CO3 to the reaction was found to be the most efficient, allowing high ees to be achieved.
|
|
|||
|---|---|---|---|
| Entry | R | Yield (%) | ee % |
| Reaction conditions: substrate (0.20 mmol), Cs2CO3 (0.12 mmol), THF (3 mL), 24 h.a Cs2CO3 (0.10 mmol) was used, 36 h.b L8 was used as the ligand. | |||
| 1 | Ph | 97 | 84 (−) |
| 2 | 3-MeOC6H4 | 90 | 70 (−) |
| 3a | 4-tBuC6H4 | 91 | 77 (−) |
| 4 | 4-FC6H4 | 93 | 70 (+) |
| 5 | 4-ClC6H4 | 94 | 76 (−) |
| 6 | 3,5-F2C6H3 | 97 | 76 (−) |
| 7 | 4-CF3C6H4 | 97 | 76 (−) |
| 8 | 4-NCC6H4 | 94 | 76 (−) |
| 9a | 4-PhC6H4 | 95 | 76 (−) |
| 10 | 2-Naphthyl | 74 | 76 (−) |
| 11b | Me | 97 | 76 (−) |
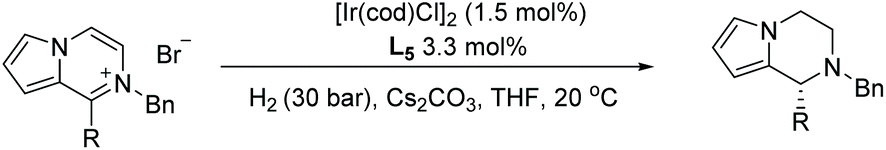
Building on this, Zhou et al. reported Ir-catalysed hydrogenation of pyrazines activated by alkyl halides. Pyrazines are often hard to hydrogenate due to their strong aromaticity, and because they have two nitrogen atoms, each of which could potentially coordinate to the catalyst and poison it. A strategy was devised where one nitrogen on the pyrazine ring was alkylated and the second from the product could form a salt from the in situ generated acid. This reduces substrate coordination to the catalyst and facilitates reduction. Initially, solvents and catalysts were screened to find optimal conditions using 2-phenyl-pyrazinium salt as a model substrate. On this basis, a wide range of chiral cis-piperazines was accessed, including not only 3-substituted but also 2,3- and 3,5-disubstituted piperazines with up to 96% ee (Table 14).81 Deuterium labelling experiments suggested that the mechanism involves an initial 1,4-hydride addition followed by an enamine-iminium tautomerisation. It was suggested that the EH of the iminium salt leads to the chirality in the final product.
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Yield (%) | ee (%) |
| Pyrazines (0.2 mmol), [Ir(cod)Cl]2 (1.0 mol%), L16 (2.2 mol%). dr >20:1. |
||||
| 1 | Me | Ph | 94 | 91 (3R,5S) |
| 2 | Me | 4-MeC6H4 | 95 | 88 (−) |
| 3 | Me | 4-MeOC6H4 | 95 | 84 (−) |
| 4 | Me | 4-FC6H4 | 93 | 92 (+) |
| 5 | Me | 4-CF3C6H4 | 94 | 93 (−) |
| 6 | Et | Ph | 96 | 80 (+) |
| 7 | n Pr | Ph | 90 | 77 (+) |
| 8 | n Pr | Ph | 92 | 86 (+) |
| 9 | n Pr | Ph | 86 | 76 (+) |
| 10 | Cyclopropyl | Ph | 90 | 83 (+) |

To show the practical use of the developed reaction, the authors developed a concise synthesis of the drug Vestipitant and a key intermediate for the drug (S)-Mirtarzapine. EH of the pyrazine produced the corresponding piperazine in a fair ee. Urea formation with the optically pure amine allowed separation of the resulting diastereoisomers, providing the protected vestipitant in 99% ee. Removal of the benzyl protecting group yielded Vestipitant in a total of 4 steps from the pyrazine (Scheme 32). It takes more steps to synthesise such pyrazines with other methods.
 | ||
| Scheme 32 Synthesis of Vestipitant from an EH product. | ||
To synthesise Mirtazapine, the pyrazine was subjected to EH, affording the corresponding piperazine with high ee (90%). A one-pot reaction was developed which allowed the Boc group to be installed in the same step with high yield. Removal of the benzyl protecting group allowed N-methylation through reductive amination. Finally, removal of the Boc protecting group furnished the advanced intermediate, which can be elaborated to the anti-depressant drug (S)-Mirtazapine (Scheme 33).81,82 Mirtazapine is usually used as the racemate; however, only the S enantiomer is under investigation for the treatment of insomnia.83 The development of further reactions to product chirally pure C-substituted piperazines will help develop unsearched areas of chemical space. These reactions will benefit the synthesis of new compounds of pharmaceutical relevance or provide more efficient routes to known compounds.
 | ||
| Scheme 33 Formal synthesis of (S)-Mirtazapine. | ||
 | ||
| Scheme 34 Examples of tetrahydro-2-pyrimidinone drugs. | ||
Kuwano et al. reported in 2015 a highly EH of pyrimidines by iridium catalysis.86 A broad range of pyrimidines were converted into the corresponding 1,4,5,6-tetrahydropyrimidines with high ees using a catalyst derived from [Ir(cod)Cl]2/Josiphos/I2 in the presence of Yb(OTf)3. Later Zhou et al.87 developed an efficient palladium-catalysed EH of 2-hydroxypyrimidines; however, the reaction required harsh conditions, such as high pressure and high catalyst loading. Recently in 2018, the same group established the EH of 4,6-disubstituted-2–hydroxypyrimidines using an Ir catalyst.88 Halogenide additives were used to increase the reactivity of the Ir-catalysed hydrogenation through oxidising Ir(I) to Ir(III) and also the in situ generation of hydrogen halide as a substrate activator. Using 4-methyl-6-phenylpyrimidin-2-ol as a model substrate, various additives and conditions were tested and the optimal conditions were found to be [Ir(cod)Cl]2/L11/H2 (55 bar)/TCCA (10 mol%). This highly enantioselective catalytic system was explored on a broad range of substrates (Scheme 35). Both electron-donating and electron-withdrawing groups were well tolerated, affording the chiral ureas with good yields and high ees.
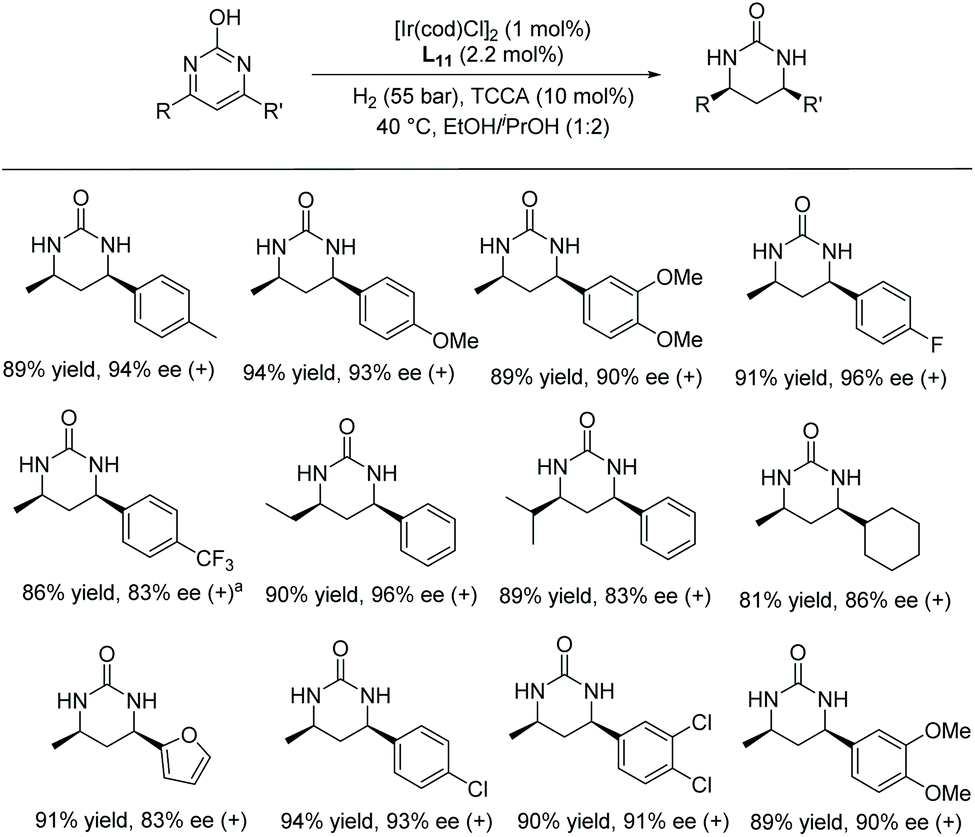 | ||
| Scheme 35 EH of hydroxypyrimidines with an Ir–Binaphane catalyst. Reaction conditions: pyrimidines (0.30 mmol), L11 (2.2 mol%), 24 h. aPerformed at 80 °C. | ||
 | ||
| Scheme 36 Examples of THQ-based natural products with bioactive properties. | ||
The prevalence and properties of these natural products have helped highlight the importance of the THQ core. As such, this has been frequently studied as a privileged scaffold in drug discovery (Scheme 37). Torcetrapib was found to inhibit a cholesteryl ester transfer protein.95 Androgen receptor binder S-40503 was developed as a potential treatment for osteoporosis. THQs have also been found in molecules that have been developed as opioid receptor antagonists96 or neurotoxins.97 However, despite the extensive research on the THQ core, very few drugs have been approved. Examples of current drugs include aragotroban and oxamniquine (Scheme 37).
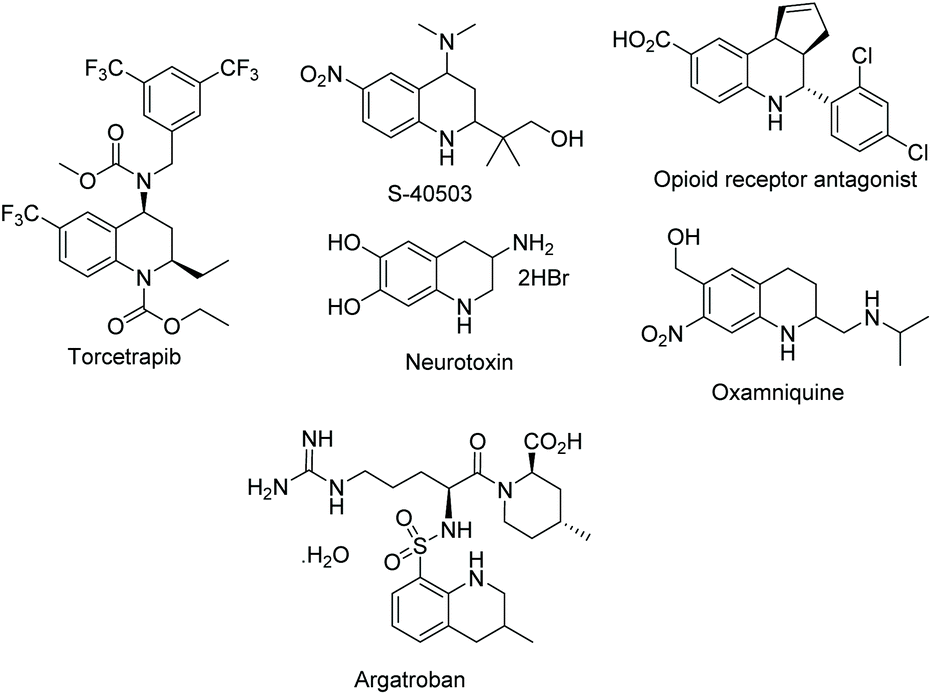 | ||
| Scheme 37 Examples of non-natural THQs under investigation and approved THQ drugs. | ||
In 2013, Agbossou-Niedercorn et al. reported the asymmetric hydrogenation of substituted quinolines for the synthesis of optically enriched 2-functionalised-1,2,3,4-tetrahydroquinoline derivatives using [Ir(cod)Cl]2, a bisphosphine ligand, and iodine (Table 15).98 With reaction conditions already found, several different ligands were screened on various substrates. Quinoline derivatives, such as quinoline carboxylates, hydroxymethylene quinoline, bromoquinoline, and amino-substituted quinolines, were shown to give high reactivity and varying degrees of selectivity. To demonstrate the practicality of the Ir–L19 system, a gram scale reaction was performed and achieved the same level of reactivity and enantioselectivity as the small-scale reactions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Ligand | Conv. (%) | Yield (%) | ee (%) |
| Reaction condition: 1 mmol substrate, substrate/Ir/ligand/I2 = 100/1/1.1/10; toluene 7 ml.a Solvent: toluene/iPrOH: 7/1, 8 mL.b 2-Methyl-1,2,3,4-tetrahydroquinoline was observed. | |||||
| 1 | COOH | L17 | 90 | 82 | 41 |
| 2 | COOMe | L17 | 100 | 88 | 66 |
| 3 | COOMe | L4 | 100 | 95 | 74 |
| 4 | COOEt | L17 | 94 | 89 | 58 |
| 5 | COOEt | (R)-Binap | 100 | 100 | 64 |
| 6 | COOEt | L6 | 100 | 100 | 64 |
| 7 | COOEt | L4 | 99 | 94 | 94 |
| 8 | COOEt | L18 | 88 | 88 | 46 |
| 9 | COOEt | L21 | 14 | 12 | 3 |
| 10 | COOnPr | L4 | 100 | 100 | 90 |
| 11 | COOiPr | L4 | 100 | 100 | 75 |
| 12 | COOiBu | L19 | 100 | 98 | 94 |
| 13 | COOiBu | L4 | 97 | 97 | 96 |
| 14a | CH2OH | L6 | 100 | 100 | 80 |
| 15a | CH2OH | L4 | 97 | 97 | 84 |
| 16a | CH2OH | L20 | 100 | 100 | 70 |
| 17a,b | CH2Cl | L19 | Mixture of products | — | — |
| 18a,b | CH2Br | L6 | 100 | 44 | 79 |
| 19b | CH2Br | L4 | 100 | 65 | 69 |
| 20 | CH2Br | L19 | 100 | 100 | 81 |

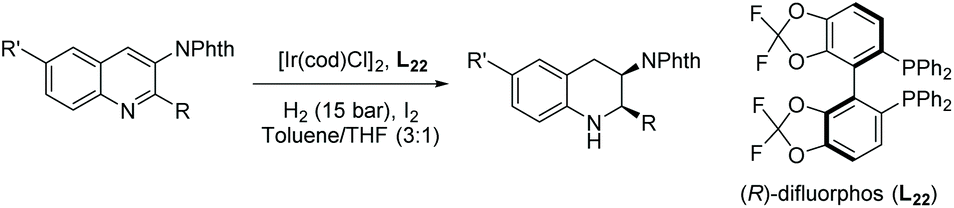
Later, Zhou et al. developed the diastereoselective and enantioselective hydrogenation of quinolin-3-amines to synthesise chiral exocyclic amines with up to 94% ee.99 Such chiral exocyclic amines appear in many biologically important compounds, such as sumanirole, and they also serve as functional handles for further elaboration of the compound. To eliminate the inhibition effect caused by the substrate and the product, a phthaloyl moiety was introduced to the amino group. This also activated the aromatic ring and improved diastereoselectivity. The reported EH of quinolin-3-amines provided the corresponding exocyclic amines with high yields, diastereoselectivities, and enantioselectivities. Although several amino protecting groups were tested, the N-phth-protecting group gave high yield and diastereoselectivity despite the lower ees compared to other protecting groups. The optimal conditions for this reaction were found to be [Ir(cod)Cl]2/L22/H2/I2 (Table 16). Key to this reaction was the introduction of the phthaloyl group which allowed access to the valuable quinoline substrates. The EH of simple exocyclic enamines and imines by transition-metal catalysts with high ee is rarely reported, in part due to the highly coordinating nature of the substrate and product.
|
|
||||
|---|---|---|---|---|
| Entry | R′/R | T (°C) | Yield (%) | ee (%) |
| Reaction condition: quinoline (0.1 mmol), [Ir(cod)Cl]2 (2.0 mol%), L22 (4.4 mol%), I2 (5.0 mol%), 18 h. In all cases the d.r. >20:1.a The conjugated double bond was also hydrogenated.b H2 (3 bar). |
||||
| 1 | H/nBu | 25 | 99 | 93 (R,R) |
| 2 | H/Me | 70 | 97 | 81 (+) |
| 3 | H/Et | 45 | 97 | 90 (+) |
| 4 | H/nPr | 45 | 94 | 92 (+) |
| 5 | H/iBu | 45 | 99 | 94 (+) |
| 6 | H/iPentyl | 45 | 97 | 88 (+) |
| 7 | H/nHexyl | 25 | 97 | 92 (+) |
| 8 | H/Phenethyl | 25 | 99 | 93 (+) |
| 9 | H/(E)-Styryla | 25 | 97 | 90 (+) |
| 10b | H/Ph | 70 | 97 | 40 (+) |
| 11 | MeO/nBn | 45 | 97 | 87 (+) |
More recently, Hu et al. reported the highly enantio- and cis-diastereoselective hydrogenation of unfunctionalised 2,3-disubstituted quinolines (Table 17). The report focussed on the EH of 3-alkyl-2-arylquinolines using an iridium catalyst with a structurally fine-tuned phosphine-phosphoramidite ligand L23.100 Previous success in the hydrogenation of sterically hindered N-arylimines using the Ir–phosphine–phosphoramidite catalyst encouraged the authors to explore EH of quinoline substrates. Initial screening led to the optimal conditions using [Ir(cod)Cl]2/L23/H2. With this catalytic system, the scope of the EH of 3-alkyl-2-arylquinolines was examined (Table 17). Products were obtained in high enantioselectivity and cis-diastereoselectivity in general, regardless of the various substitutions.
| Reaction conditions: substrate (0.4 mmol), 1,4-dioxane (4 mL), 25 °C. In all cases, dr >20:1. |
|---|

|
2.2. Ru-Catalysed enantioselective hydrogenation
Many ruthenium complexes are highly active homogenous catalysts for hydrogenation reactions. For instance, transfer hydrogenation with chiral ruthenium catalysts has been utilised for various enantioselective hydrogenations including ketones, aldehydes, and imines.101–106 Chiral ruthenium complexes for asymmetric reduction were pioneered by Noyori et al.107 These chiral ruthenium complexes have been further developed to reduce prochiral unsaturated compounds which were then utilised for the asymmetric hydrogenation of N-heteroarenes.5,108 Following are the recent reports on the EH of N-heteroaromatic compounds.|
|
|||
|---|---|---|---|
| Entry | R1/R2/R3 | Yield (%) | ee (%) |
| Reaction conditions: substrates (0.2 mmol), RT for 24 h. | |||
| 1 | Me/H/H | 94 | 96 (R) |
| 2 | n Pr/H/H | 88 | 96 (R) |
| 3 | n Bu/H/H | 92 | 97 (R) |
| 4 | n Pentyl/H/H | 89 | 96 (R) |
| 5 | C6H5CH2/H/H | 93 | 97 (R) |
| 6 | 4-FC6H4CH2/H/H | 92 | 97 (R) |
| 7 | 4-MeC6H4CH2/H/H | 90 | 96 (R) |
| 8 | 3-MeC6H4CH2/H/H | 93 | 97 (R) |
| 9 | Me/H/Me | 93 | 95 (R) |
| 10 | Me/H/OMe | 93 | 95 (R) |
| 11 | Me/H/F | 94 | 94 (R) |
| 12 | (CH2)3/H | 92 | 89 (R,R) |
| 13 | (CH2)4/H | 91 | 95 (R,R) |
| 14 | (CH2)4/CH3 | 94 | 90 (R,R) |
| 15 | (CH2)4/F | 92 | 94 (R,R) |
| 16 | (CH2)5/H | 73 | 99 (R,R) |
| 17 | Me/Me/H | 95 | 97 (R,R) |
| 18 | H/Me/Me | 85 | 40 (R) |
| 19 | Ph/H/H | 53 | 42 (S) |

A similar catalytic system was explored by the Arai group on a wide range of halogenated indoles and indoles with acid-sensitive protective groups (Scheme 38).112 The substrates were well tolerated to afford the corresponding chiral indolines with excellent yields and ees. The functional group tolerability of this catalytic system is of high importance to medicinal chemistry, as these groups could be found in molecules of interest in pharmacology, such as the example shown in Scheme 38 which possesses anti-inflammatory properties.113 They also serve as functional handles, allowing for further elaboration of the molecule.
 | ||
| Scheme 38 EH of functionalised indoles. | ||
This method was also used to produce a chiral indoline with a branched-chain ending in an acetal.112 In one step, a tricyclic core could be produced, which is found in a compound under investigation for antitumor properties (Scheme 39).114
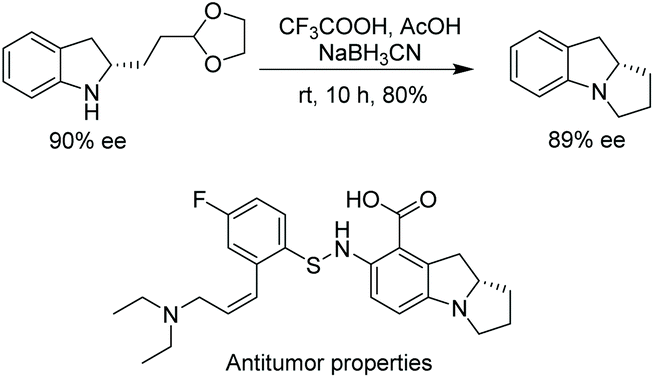 | ||
| Scheme 39 Synthesis of chiral tricyclic indoline. | ||
:4) selectivity in the case of 2,2′-bisquinoline.116 Cationic chiral η6-arene-Ru-diamine catalysts were explored for the EH of bisquinoline and bisquinoxaline derivatives (Scheme 40). The optimal conditions for this hydrogenation were found to be the Ru-(R,R)-1 complex as catalyst (2.0 mol%), H2 (50 bar) and room temperature in iPrOH. Under these optimised conditions, a variety of 2,2′-bisquinoline derivatives were shown to produce the desired diamines with excellent enantioselectivity in most cases, regardless of steric hindrance from alkyl side chain at the 6,6′-, 7,7′-, or 5,5′-positions. A notable decrease in diastereoselectivity was observed, however, when introducing alkyl groups into the 8,8′-positions of 2,2′-bisquinoline. In addition, substrates bearing methoxy and CF3 groups at the 6,6′-positions demonstrated low reactivity, diastereoselectivity, and enantioselectivity. The synthesised chiral vicinal diamines were employed for the preparation of chiral benzimidazolium salts, a precursor to chiral NHC ligands, which can be difficult to access through other methods.
 | ||
| Scheme 40 EH of 2,2′-bisquinoline derivatives using a chiral Ru–diamine catalyst. | ||
More recently, Fan and co-workers demonstrated the asymmetric hydrogenation of terpyridine type N-heteroarenes using a similar Ru–diamine complex as a catalyst. Partially reduced chiral pyridine-amine type products were obtained in high yield with excellent diastereo- and enantioselectivity (Scheme 41).117
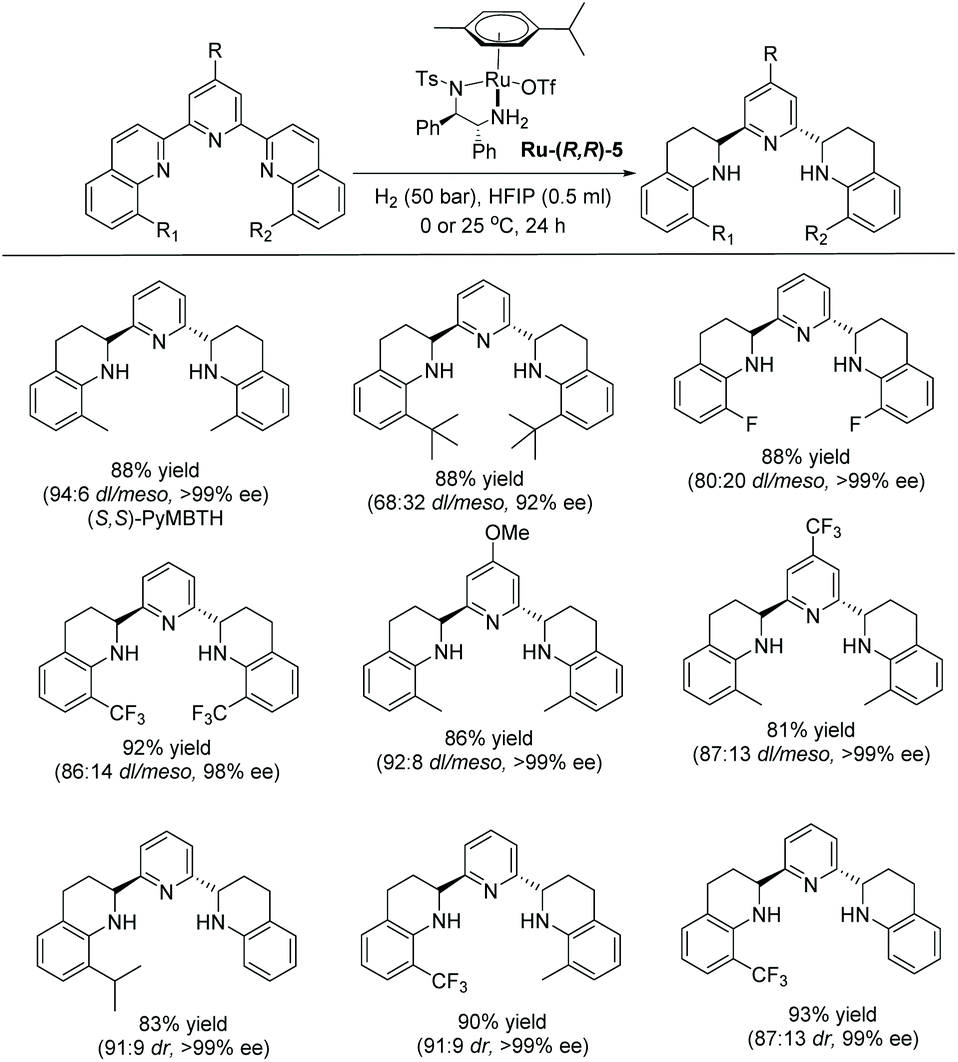 | ||
| Scheme 41 EH of terpyridine type N-heteroarenes using a Ru–diamine catalyst. | ||
This catalytic system worked well on a broad range of 2,6-bis(quinolinyl) pyridines, providing the corresponding chiral 2,6-bis(tetrahydroquinolin-2-yl)pyridines. The substituents at the 8,8′-positions of the quinolinyl rings influenced the stereoselectivity. Thus, substitution with small groups, such as Me and CF3, showed high diastereoselectivities and enantioselectivities, whereas the sterically hindered t-butyl group led to lower diastereoselectivity and enantioselectivity. The resulting pyridine-amine-type compounds from this new method could serve as novel, chiral tridentate nitrogen ligands. Ligands of this nature are difficult to access with other approaches.
The potential of the compounds as ligands were showcased with a copper-catalysed asymmetric Friedel–Crafts type alkylation reaction of indoles with nitroalkenes, resulting in high ees.117 Initial optimisation of the asymmetric alkylation reaction showed that the Cu(OTf)2-(S,S)-PyMBTH (see Scheme 41 for the ligand) catalyst was effective in the presence of HFIP (2.0 eq.) at 0 °C. Under the optimised conditions all substrates underwent alkylation efficiently with excellent reactivity and selectivity (Scheme 42).
 | ||
| Scheme 42 Asymmetric Friedel–Crafts alkylation using a Cu-(S,S)-PyMBTH catalyst. a10 mol% catalyst (Cu(OTf)2/(S,S)-PyMBTH = 1:1.1); double alkylation occurred. | ||
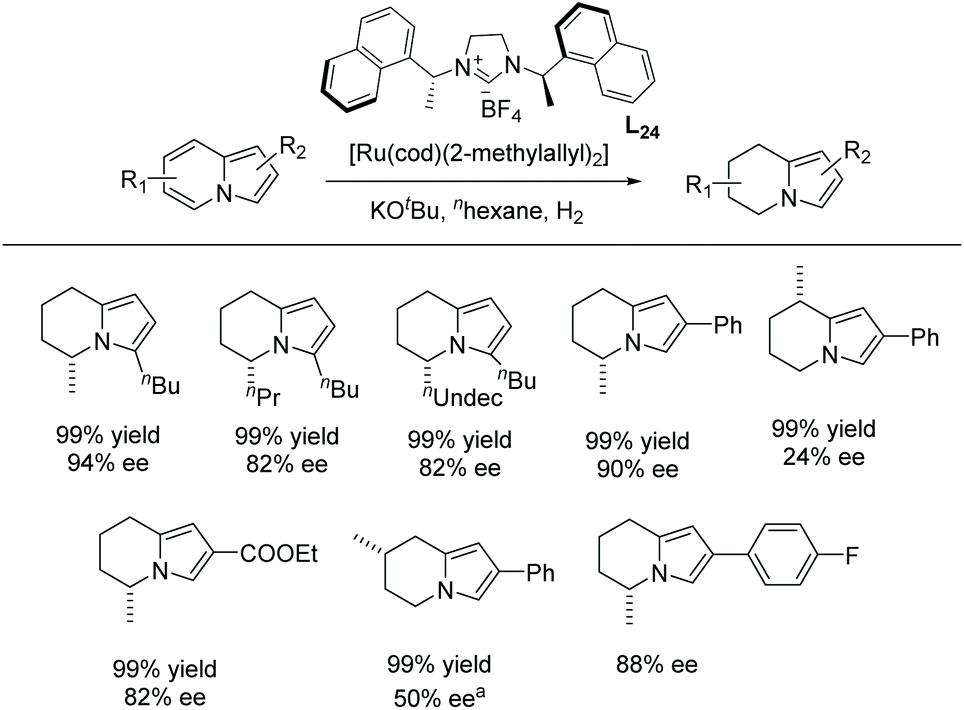 | ||
| Scheme 43 EH of indolizines using a chiral Ru–NHC complex. [Ru(cod)(2-methylallyl)2] (0.015 mmol), KOtBu (0.045 mmol), and L24 (0.03 mmol) were stirred at 70 °C in n-hexane (2 mL) for 12 h; later substrate (0.30 mmol) was added, H2 (100 bar), rt, 24 h. a40 °C. | ||
The scope of the reaction was extended to the EH of 1,2,3-triazolo-[1,5-a]pyridines. Under the same conditions these substrates also showed significant reactivity (Scheme 44). The enantioselectivity decreased slightly when the length of the alkyl substitution was increased at C7. Finally, the product was explored by further hydrogenation, allowing access to the enantiomer of the natural product monomorine, as shown in Scheme 45.
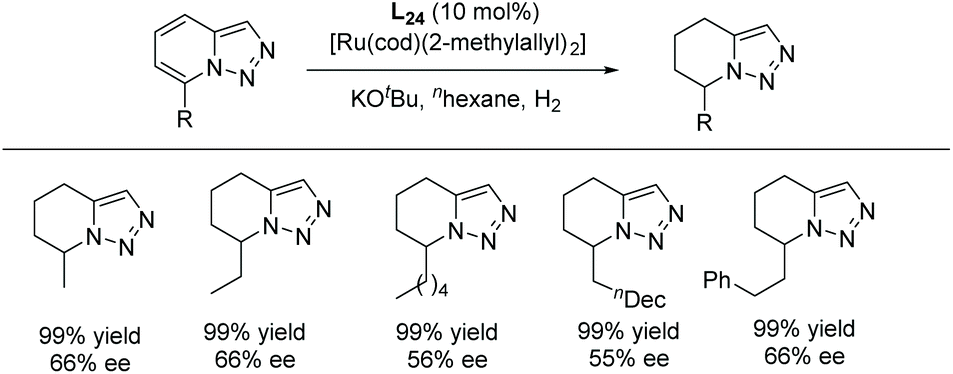 | ||
| Scheme 44 EH of 1,2,3-triazolo-[1,5-a]pyridines using a chiral Ru–NHC complex. [Ru(cod)(2-methylallyl)2] (0.015 mmol), KOtBu (0.045 mmol), and L24 were stirred at 70 °C in n-hexane (2 mL) for 12 h; later substrate (0.30 mmol) was added, H2 (100 bar), rt, 24 h. | ||
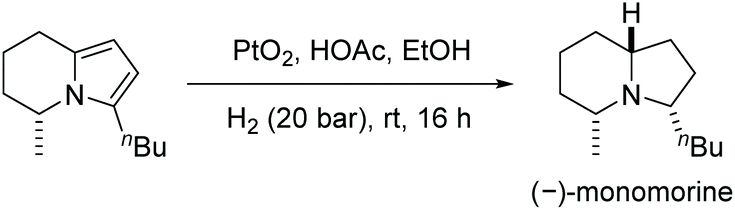 | ||
| Scheme 45 Synthesis of unnatural (−)-monomorine. | ||
Later in 2016 Glorius and co-workers described a structural investigation of the ruthenium–NHC catalysts that had been established for the EH of heteroarenes.119 To examine the catalytic activity and selectivity, several chiral and achiral Ru–NHC complexes were tested on the hydrogenation of 2-methylbenzofuran. The investigation revealed the formation of a structurally novel complex (Ru–L24) arising from 2 eq. of the homochiral carbene precursor L24 reacting with [Ru(cod)(2-methylallyl)2] in the presence of KOtBu. The molecular structure of Ru–L24 features a unique tridentate ligand as a result of deprotonation of the methyl and naphthyl groups from one of the usually monodentate carbene ligand. The study also shows that ligand hydrogenation plays a key role in forming the active catalyst. Ru–L24 showed high activity and selectivity in the hydrogenation of 2-methylbenzofuran (Scheme 46). Very recently, a detailed mechanistic study of a related asymmetric hydrogenation reaction catalysed Ru–L24 has been reported.120
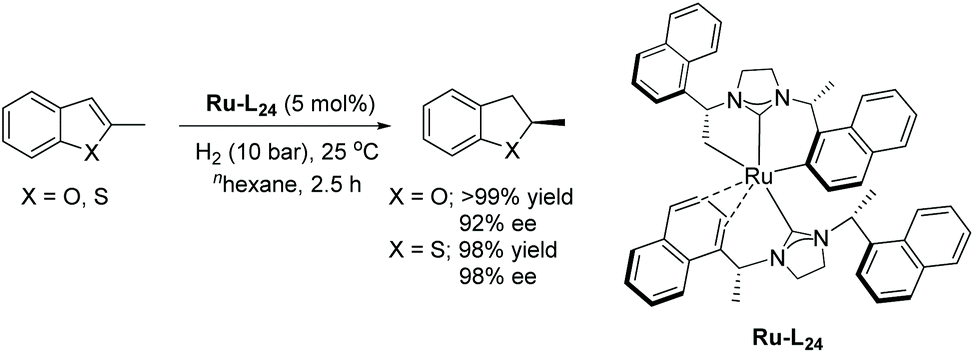 | ||
| Scheme 46 EH of 1,2,3-triazolo-[1,5-a]pyridines using a chiral Ru–NHC complex. | ||
Kuwano et al. in 2016 reported the EH of azaindoles using a chiral catalyst prepared from [Ru(η3-methallyl)2(cod)] and a trans-chelating bis-phosphine ligand (L25) (Table 19).121 The catalytic system was established to access partially reduced azaindoles, i.e. azaindolines. Selected examples were further hydrogenated with Pt/C to provide the chiral products as fully saturated heterocycles.
|
|
||||
|---|---|---|---|---|
| Entry | R1/R2/R3 | Conv (%) | Yield (%) | ee |
| Reactions condition: 0.20 mmol scale in 1.0 mL of EtOAc.a Without Et3N.b In toluene at 40 °C for 48 h.c H2 (100 bar). | ||||
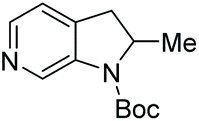
| 1a | Me/H/H | 100 | 98 | 88 (+) |
| 2 | n Hex/H/H | 100 | 98 | 76 (+) |
| 3 | iBu/H/H | 80 | 71 | 54 (+) |
| 4 | n Hex/H/H | <5 | — | — |
| 5 | Ph/H/H | 17 | 10 | 92 |
| 6b,c | Ph/H/H | 84 | 77 | 94 (−) |
| 7b | 4-MeOC6H4/H/H | 84 | 77 | 94 (−) |
| 8b | 4-CF3C6H4/H/H | 60 | 56 | 82 (−) |
| 9a | COOEt/H/H | 100 | 96 | 94 (−) |
| 10a | Me/Me/H | 87 | 81 | 82 (+) |
| 11a | Me/F/H | 100 | 93 | 68 (+) |
| 12a | Me/CF3/H | 88 | 79 | 62 (+) |
| 13a | Me/H/Me | 100 | 85 | 86 (+) |
Initial attempts at this catalytic EH of azaindoles were performed using a protected 2-methyl-7-azaindole as a model substrate. Whilst a [Rh(nbd)2]+/PhTRAP (L25) catalyst exclusively reduced the pyrrole ring, a remarkable improvement in the yield and enantioselectivity was observed when a catalyst derived from [Ru(η3-methallyl)2(cod)]/L25 was used for the hydrogenation. Under the optimised conditions, a broad range of 2-substituted 7-azaindoles underwent the EH, and 6-, 5-, and 4-azaindoles were also well-tolerated to provide high yields and high enantioselectivity (Tables 19 and 20).
|
|
||||
|---|---|---|---|---|
| Entry | Product | Conv (%) | Yield (%) | ee |
| Reaction conditions: 0.20 mmol scale in 1.0 mL of EtOAc.a Without Et3N.b In toluene at 40 °C for 48 h.c At 100 °C. | ||||
| 1a |
|
100 | 98 | 82 (−) |
| 2b |

|
90 | 86 | 88 (−) |
| 3 |

|
100 | 90 | 82 (−) |
| 4 |

|
100 | 94 | 90 (−) |
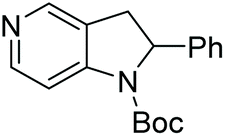
| 5b |
|
87 | 81 | 84 (−) |
| 6 |

|
100 | 82 | 84 (−) |
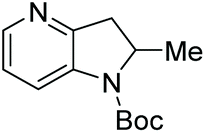
| 7a |
|
100 | 99 | 86 (−) |
| 8 |
|
100 | 99 | 58 (−) |
| 9 |
|
100 | 89 | 54 (−) |
| 10 |
|
<5 | — | — |
| 11b |

|
75 | 68b | 66 (−) |
| 12c |

|
100 | 85 | 50 (−) |

In 2017, Glorius et al. established the first EH of imidazo[1,2-a]pyridines using the same Ru–NHC catalyst.122 In the initial screening, the hydrogenation was performed on 5-methylimidazo[1,2-a]pyridine as model substrate in the presence of in situ prepared Ru–L24 in various solvents. Under the optimised conditions, a series of mono and disubstituted imidazo[1,2-a]pyridines were hydrogenated providing products in high yields and enantioselectivities. The reaction was found to tolerate multiple functional groups well, including amines, amides, esters, silyl ethers, pyridines and halides which could be utilised for further transformations (Scheme 47).
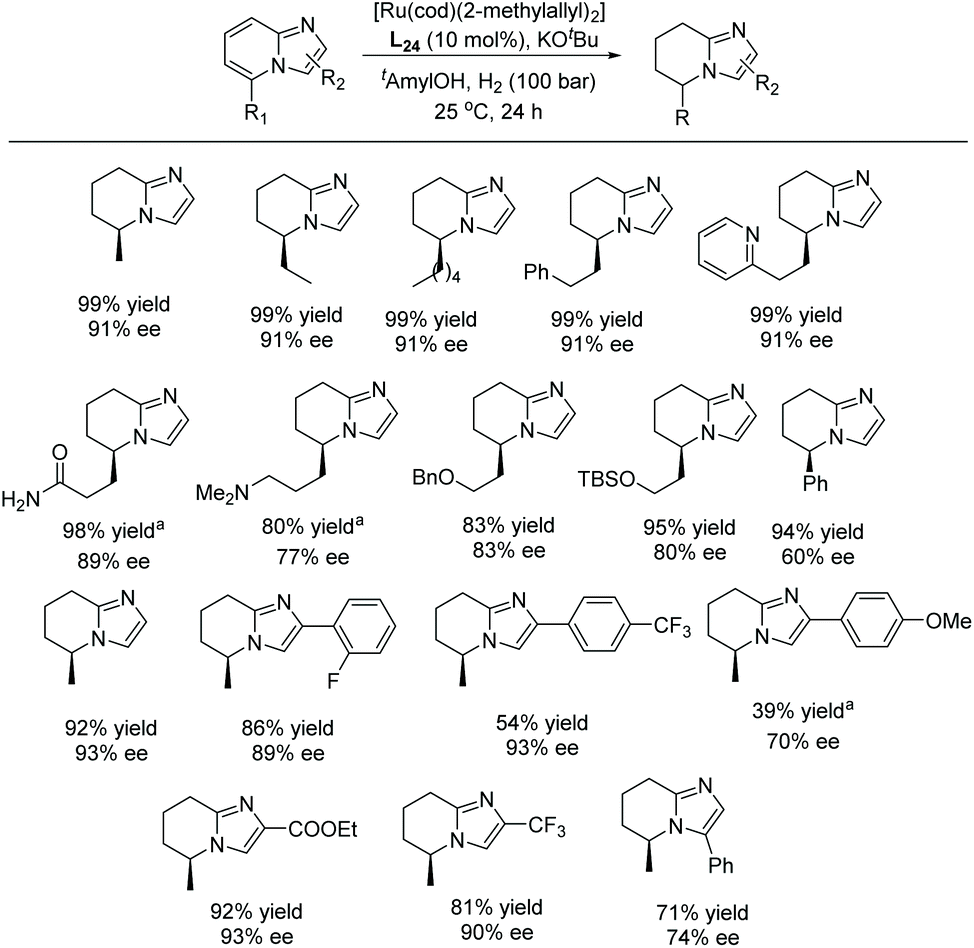 | ||
| Scheme 47 EH of mono- & di-substituted imidazo[1,2-a]pyridines. [Ru(cod)(2-methylallyl)2] (5 mol%), KOtBu (10 mol%), tAmylOH (1 mL). aH2 (150 bar). | ||
To showcase the synthetic utility of this newly developed reaction, a product from the EH was further functionalised into a bioactive molecule (Scheme 48). The product was deprotected and oxidized under Jones-oxidation conditions to provide 5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-5-carboxylic acid. This molecule could further undergo an Ugi reaction to provide an IDH1 mutant inhibitor. Furthermore, a new chiral NHC ligand was synthesised, derived from an imidazo[1,5-a]pyridine.
 | ||
| Scheme 48 Application of the tetrahydroimidazopyridine scaffold. | ||
More recently, the Glorius group explored the EH of 2-quinolones to access chiral 3,4-dihydro-2-quinolones.123 Initial experiments were conducted using their previously reported method, with Ru–L24 as catalyst under 70 bar H2 in hexane at room temperature. In an earlier report,124 no desired product was obtained for unprotected quinolones. The reaction was tested on various alkylated quinolones under the optimised conditions, showing excellent yield and moderate to high enantioselectivity. The catalytic system tolerated various substitutions, such as alkyl, methoxy, aryl, and fluorine on the 2-quinolones (Scheme 49).
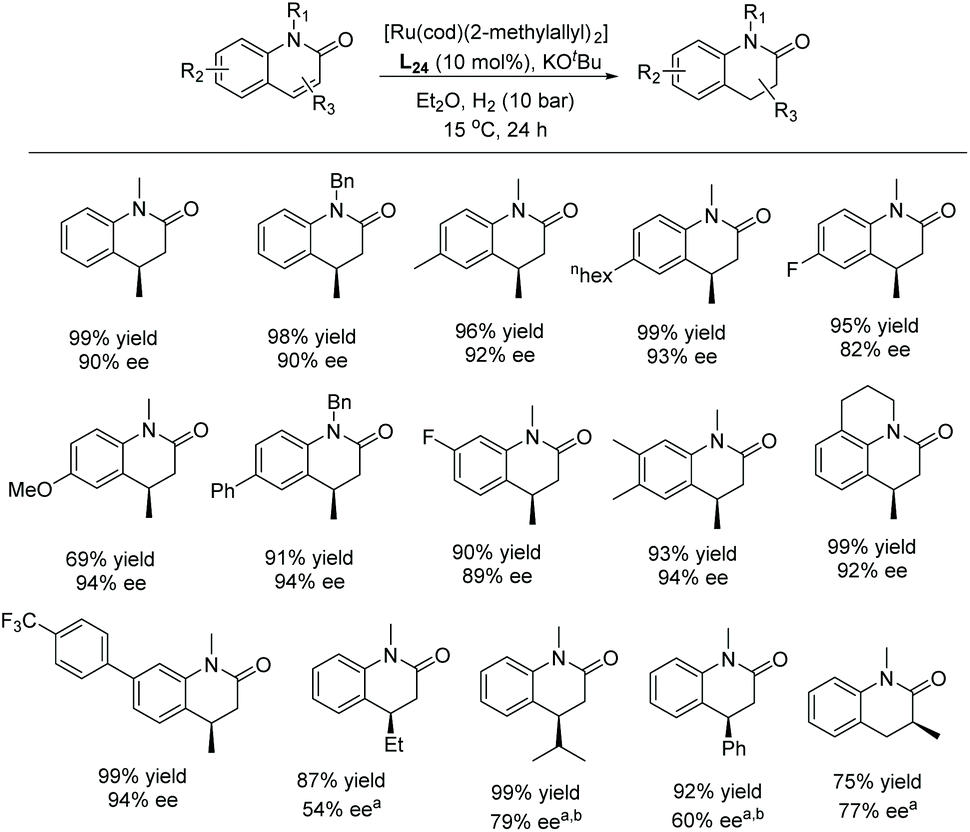 | ||
| Scheme 49 EH of substituted 2-quinolones using Ru–NHC catalyst. [Ru(cod)(2-methylallyl)2] (5 mol%), KOtBu (12.5 mol%), Et2O (1 mL). aHexane (1 mL) and H2 (70 bar). b30 °C, 20% [Ru] catalyst and H2 (70 bar) for 48 h. | ||
The same group also reported the EH of pyrido-pyrimidones to form multiple stereocenters in adjacent rings leading to structurally complex motifs.125 The previously reported conditions122,123 were adopted for the hydrogenation, which showed complete chemoselectivity, with moderate to excellent diastereo-, and enantioselectivity (Scheme 50).
 | ||
| Scheme 50 EH of substituted pyrido[1,2-a]pyrimidinones using a Ru–NHC catalyst. aH2 (80 bar). | ||
The reaction could be stopped to yield tetrahydropyrido-pyrimidinone as a sole intermediate. EH of the isolated intermediate led to opposite diastereomers with excellent diastereo- and enantioselectivity depending on the chirality of the catalyst used (Scheme 51).
 | ||
| Scheme 51 EH of a tetrahydropyrido–pyrimidinone intermediate. | ||
2.3. Rh-Catalysed enantioselective hydrogenation
Rhodium catalysis has been widely used for hydrogenation since the discovery of Wilkinson's catalyst. The catalyst is one of the most widely used transition metal catalysts for the hydrogenation of various substrates such as olefins, imines and later arenes and heteroaromatic compounds. In 2008, our group disclosed the first examples of EH of dihydroisoquinolines and analogues with a chiral cationic Cp*Rh–diamine catalyst, which afforded tetrahydroisoquinolines and tetrahydro-β-carbolines with up to >99% ee.126 We subsequently reported achiral transfer hydrogenation of various N-heteroaromatic compounds, such as pyridines and quinolines, using a Cp*Rh(III) catalyst promoted by iodide.127,128 Thereafter several groups have exploited chiral rhodium complexes to reduce various N-heteroaromatic compounds. | ||
| Scheme 52 EH of indoles using an Rh–L26 catalyst. | ||
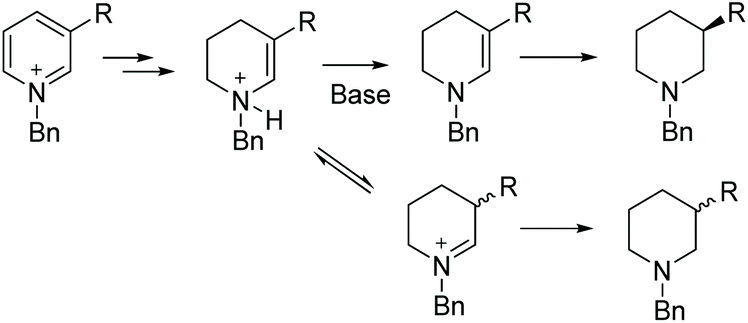 | ||
| Scheme 53 Proposed mechanism for base-promoted EH of 3-substituted pyridines. | ||
In the initial screening, various bases were examined. It was found that the use of basic amines like DIPEA, DMAP and Et3N gave the best ee. The optimised conditions for this transformation were found to be [Rh(cod)2]OTf /L27/H2. Under these optimised conditions, a range of substrates was hydrogenated with various ees observed (Table 21). The EH was carried out both in the presence and absence of Et3N to verify the importance of the base. The ee of the corresponding piperidines in the presence of Et3N was found to be moderate to good compared to the poor value found in its absence.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | With Et3N | Without Et3N | ||
| Yield (%) | ee (%) | Yield (%) | ee (%) | ||
| Reaction conditions: Rh–L27 (2.2 mol%), Et3N (5 eq.), 20 h. | |||||
| 1 | Ph | 50 | 90 (S) | 5 | 23 (S) |
| 2 | Ph | 57 | 84 (S) | — | — |
| 3 | 4-CF3C6H4 | 20 | 83 (−) | 12 | 14 (−) |
| 4 | 2-MeC6H4 | 50 | 75 (−) | 7 | 30 (−) |
| 5 | 4-MeOC6H4 | 52 | 90 (−) | 8 | 40 (−) |
| 6 | 2-naphthyl | 42 | 86 (−) | 21 | 20 (−) |
| 7 | Me | 36 | 57 (R) | <1 | n.d. |
| 8 | COOEt | 2 | 33 | 3 | −17 |
| 9 | NHBoc | 24 | 55 (R) | 25 | 27 (R) |
| 10 | CF3 | 2 | 41 | 2 | 11 |
| 11 | n Bu | 43 | 32 (−) | <1 | n.d. |
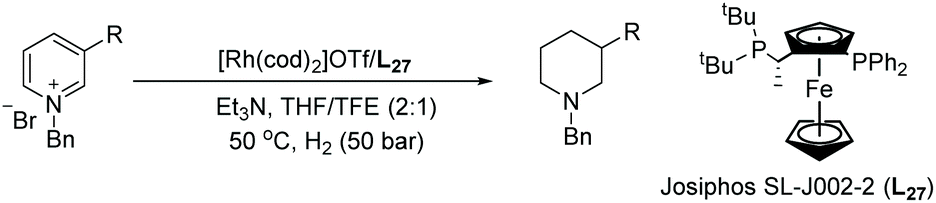
In 2019, Glorius et al. established a straightforward route to access the fluorinated cis-piperidines via hydrogenation using a dearomatization strategy.131 This reaction overcomes the limitation of the hydrogenation of fluorinated pyridines, avoiding both the catalyst deactivation and the hydrodefluorination side reactions. In the initial screening, a borane reagent was introduced to co-ordinate to the pyridine, not only causing dearomatization but also preventing poisoning of the catalyst in following hydrogenation. A known additive 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (HBpin) was used for the dearomatization step and various Rh precursors were tested for the hydrogenation. The Rh–L28 complex was found to show high activity as well as selectivity (Scheme 54).
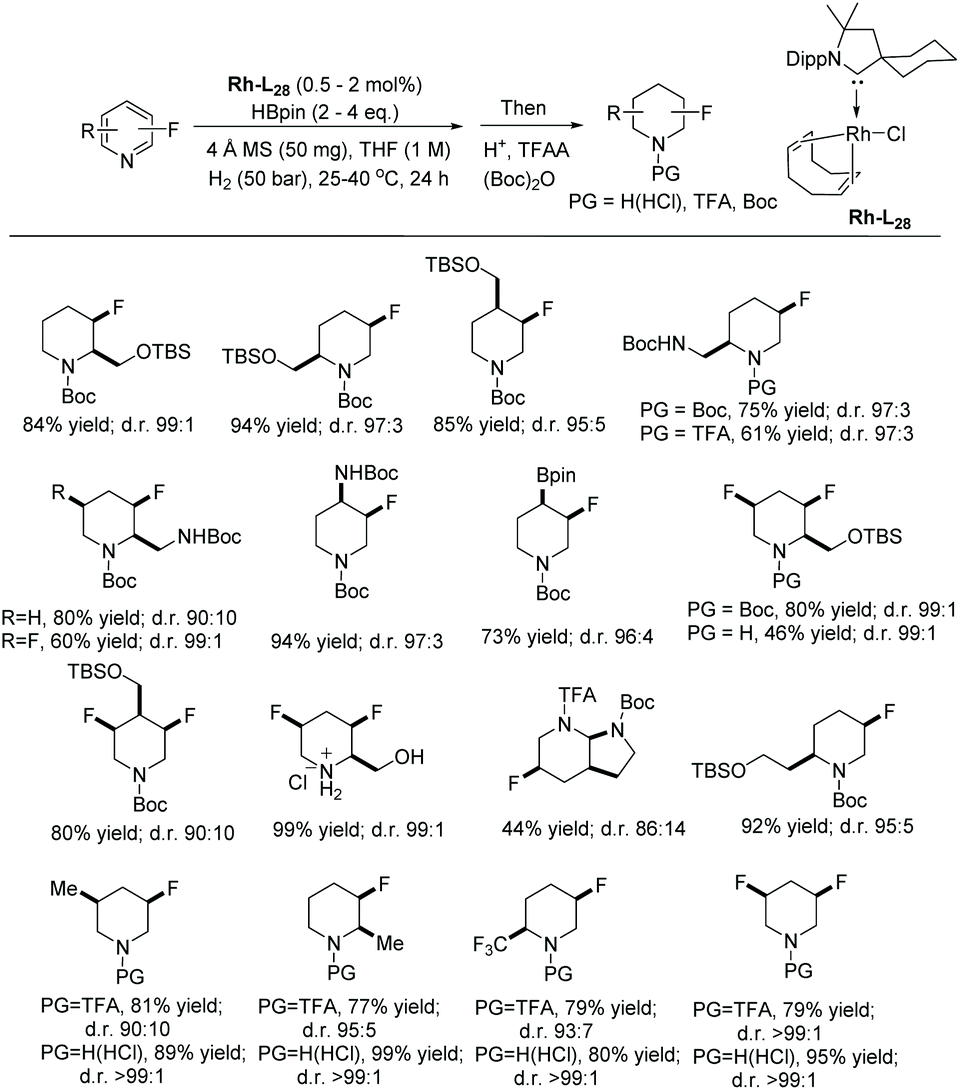 | ||
| Scheme 54 Synthesis of cis-(multi)fluorinated piperidine building blocks. | ||
Under the optimised conditions identified, various di and multi-substituted fluoropyridines were hydrogenated to the corresponding fluoropiperidines in high yields and moderate to excellent diastereoselectivities. Various functional group, such as silyl ethers, amines and pinacol boronic esters, were tolerated. To prevent the loss of the volatile fluorinated piperidine, the product was trapped after the completion of the reaction using trifluoroacetic anhydride. The TFA analogues were then deprotected to provide fluoropiperidine hydrochlorides. The reaction was also scalable, affording good yields and excellent cis-selectivities.
To demonstrate the utility of this method, several fluoropiperidine products were derivatised to commercial drugs. Applications towards the synthesis of a Pipphos ligand and ionic liquids were also demonstrated (Scheme 55).
 | ||
| Scheme 55 Synthesis of fluorinated analogues of commercial drugs, ligand and ionic liquids. | ||
Fluorinated scaffolds, particularly those based on piperidines, are of great interest to medicinal chemists due to the modification of the chemical and physical properties of a molecule.132 For example, during an investigation into 3-piperidinylindole antipsychotic drugs, fluorination was found to decrease the basicity of the amine (Scheme 56). This increases the bioavailability and affinity to 5-HT2 receptors, which has been suggested to decrease some negative side effects.133
 | ||
| Scheme 56 pKa value of fluorinated and non-fluorinated piperidines. | ||
 | ||
| Scheme 57 EH of quinolines and isoquinolines using a Rh–thiourea phosphine complex. Reaction conditions: substrate (0.2 mmol) in 1.2 mL solvent, substrate/[Rh(COD)Cl]2/L29 ratio = 100/0.5/1. | ||
2.4. Pd-Catalysed enantioselective hydrogenation
 | ||
| Scheme 58 Pd-Catalysed EH of indole to yield a tricyclic indoline derivative. | ||
Later in 2018, the same group expanded on the Pd-catalysed synthesis of chiral indoles.136 A one-pot process was reported, where substituted anilines could be cyclised to the corresponding indoles and hydrogenated asymmetrically to chiral indolines (Table 22). Such a reaction could pose several challenges. Firstly, conditions for the formation and asymmetric hydrogenation of indoles must be compatible; secondly, by-products such as water may affect the catalysis and thirdly, the carbonyl group may undergo hydrogenation before the formation of the indole. A strong Brønsted acid played a crucial for both the formation of the indoles as well as their asymmetric hydrogenation. The optimal conditions for this one-pot reaction were found to be Pd(OCOCF3)2/L30/H2.
|
|
|||
|---|---|---|---|
| Entry | R/R′ | Yield (%) | ee (%) |
| Reactions conditions: substrate (0.25 mmol), Pd(OCOCF3)2/L30 (2.5 mol%), EtSO3H (0.50 mmol). | |||
| 1 | Bn/H | 98 | 95 (+) |
| 2 | 2-MeC6H4CH2/H | 91 | 94 (+) |
| 3 | 3-MeC6H4CH2/H | 90 | 95 (+) |
| 4 | 4-MeC6H4CH2/H | 93 | 95 (+) |
| 5 | Me/H | 96 | 90 (+) |
| 6 | Et/H | 82 | 94 (+) |
| 7 | n Pr/H | 84 | 94 (+) |
| 8 | iPr/H | 94 | 96 (+) |
| 9 | n Bu/H | 98 | 94 (+) |
| 10 | n Pentyl/H | 97 | 93 (+) |
| 11 | Me/2-Me | 94 | 96 (+) |
| 12 | Me/2-MeO | 91 | 80 (+) |
| 13 | Me/4-MeO | 81 | 84 (+) |
| 14 | Me/2,4-Me2 | 90 | 94 (+) |
| 15 | Ph/H | 55 | 68 (−) |
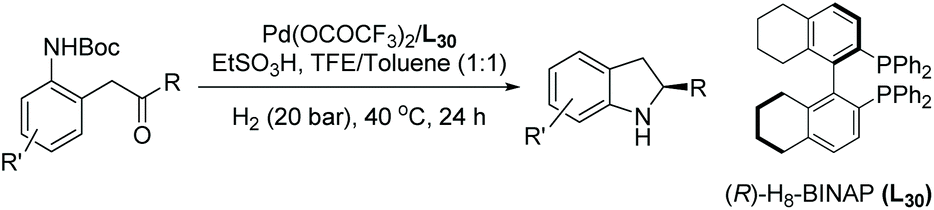
Under the conditions established, various aniline ketones underwent cyclization and reduction in a one-pot fashion, furnishing indolines with up to 96% ee (Table 22). The length of the alkyl chain (R) of the ketone moiety had only little influence on the ee values. To further demonstrate the practical usability of the catalytic system, a gram scale experiment was carried out. Loss of activity or enantioselectivity was not observed during this reaction.
|
|
||||
|---|---|---|---|---|
| Entry | H(or)F/R1 | Temp (°C) | Yield (%) | ee (%) |
| Reaction conditions: substrate (0.10 mmol), Pd(OCOCF3)2 (5.0 mol%) and L31 (6.0 mol%), TFA (60 mol%), CH2Cl2 (4 mL), H2 (70 bar), and 18 h. | ||||
| 1 | H/nBu | 70 | 91 | 90 (S,S) |
| 2 | H/Me | 70 | 86 | 81 (−) |
| 3 | H/Et | 80 | 93 | 85 (−) |
| 4 | H/nPr | 80 | 97 | 87 (−) |
| 5 | H/iPr | 80 | 72 | 80 (−) |
| 6 | H/iBu | 80 | 94 | 90 (−) |
| 7 | H/iPentyl | 80 | 91 | 90 (−) |
| 8 | H/nhexyl | 70 | 86 | 90 (−) |
| 9 | H/phenethyl | 80 | 95 | 90 (−) |
| 10 | F/nBu | 80 | 97 | 79 (−) |
| 11 | H/Ph | 70 | 83 | 14 (−) |
| 12 | H/(E)-styryl | 80 | 99 | 90 (−) |
| 13 | H/(E)-4-fluorostyryl | 80 | 86 | 88 (−) |

3. Heterogeneous catalytic hydrogenation
Heterogeneous asymmetric catalysis presents great opportunities for the production of high-value compounds, including enantio-enriched heterocycles. The advantages provided include easy recycling of the usually expensive catalysts, facile purification of products, and the possibility of the continuous production of chiral compounds with a fixed-bed reactor. Despite the great impact of homogeneous asymmetric catalysis in the pharmaceutical and fine chemical industries and research laboratories, heterogeneously enantio-catalysed reactions have been underdeveloped.139,140 A notable contribution was made by Glorius and co-workers in 2004, who reported the EH of pyridines substituted with a chiral oxazolidinone moiety using a heterogenous catalyst.141 The chiral auxiliary on the C2 position of pyridines was found to induce chirality onto the piperidines with heterogeneous Pd, Pt or Rh catalysts in an acidic medium. This was the first report showing highly selective EH of multisubstituted pyridines. To induce enantioselectivity with solid metal catalysts, the simplest and currently most promising strategy is the modification of the metal surfaces with chiral organic compounds.142 Whilst there are reports on the non-asymmetric hydrogenation of N-heteroaromatic compounds catalysed by metal nanoparticles, little work has been reported on the asymmetric version. Wu et al. in 2017 explored the application of binaphthyl-stabilised palladium nanoparticles (BIN-PdNPs) containing a chiral modifier as a chiral nanocatalyst in the asymmetric hydrogenation of indole and quinoline derivatives.143 The Bin-PdNPs nanocatalyst comprised Pd(0) and Pd(II), prepared by in situ reduction of palladium acetate in the presence of 1,1′-binaphthyl-2,2′-bis(diazonium-tetrafluoroborate) providing a size distribution of 2.5 ± 0.5 nm. The optimal ratio for the chiral modifier H8-BINAP to Bin-PdNPs was found to be 1.2:1, with the chiral ligand occupying 34% of the surface of the nanoparticles.
The screening results on solvents, additives, hydrogen pressure, and reaction temperature showed that the optimal conditions for this EH were Bin-PdNPs/(R)-H8-BINAP/H2/L-CSA additive. With these optimised conditions in hand, the scope of mainly 2-substituted indoles was explored, displaying good to excellent yields of hydrogenated products with moderate ees (Table 24). Furthermore, the reactions were diastereoselective; cis-products were detected for the 2,3-substituted indoles. The protocol also worked for the EH of quinolines, with the optimum chiral modifier being BINAP (Table 25). Previous work had already established a series of metal–carbon bond stabilized palladium nanoparticles (MCBS-PdNPs) as catalysts for the hydrogenation and dehydrogenation of N-heterocycles.144
|
|
|||
|---|---|---|---|
| Entry | R1/R2/R3 | Yield (%) | ee (%) |
| Reaction condition: indole (0.25 mmol), Bin-PdNPs/(R)-H8-BINAP (2 mol%/2.4 mol%), 1 equiv. of L-CSA, 24 h. | |||
| 1 | H/Me/H | 85 | 64 (R) |
| 2 | 5-F/Me/H | 81 | 50 (R) |
| 3 | 5-Me/Me/H | 92 | 47 (R) |
| 4 | H/Me/4-F-Benzyl | 83 | 48 (R) |
| 5 | H/4-Me-Benzyl/H | 81 | 55 (R) |
| 6 | H/3-Me-Benzyl /H | 86 | 53 (R) |
| 7 | H/Benzyl /H | 93 | 54 (R) |
| 8 | H/Phenyl/H | 76 | 31 (S) |
| 9 | H/p-F-C6H4 /H | 51 | 35 (S) |
| 10 | H/p-MeO-C6H4/H | 86 | 22 (S) |
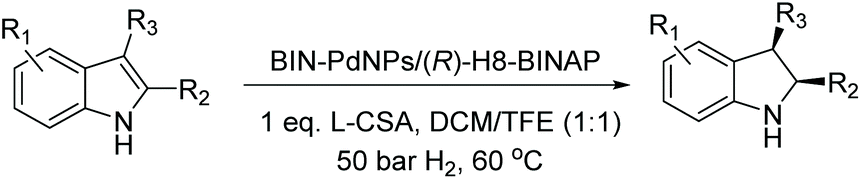
|
|
|||
|---|---|---|---|
| Entry | R1/R2 | Yield (%) | ee (%) |
| Reaction condition: quinolines (0.25 mmol), Bin-PdNPs/(R)-BINAP (2 mol%/2.4 mol%), 1 equiv. of L-CSA, 24 h. | |||
| 1 | H/Me | 93 | 70 (R) |
| 2 | 6-Me/Me | 95 | 62 (R) |
| 3 | 6-MeO/Me | 95 | 46 (R) |
| 4 | 6-F/Me | 71 | 54 (R) |
| 5 | 6-COOMe/Me | 63 | 59 (R) |
| 6 | H/Et | 86 | 66 (R) |
| 7 | H/Phenyl | 91 | 56 (S) |
| 8 | H/o-Tolyl | 84 | 46 (S) |
| 9 | H/2,4-dimethylphenyl | 82 | 52 (S) |

In 2014, the Fan group described a dendronised chiral Ir/Ru bimetallic polymer catalyst, which was generated using a chiral polymer derived from flourene and (S)-BINAP. The dendronised polymer was used in the asymmetric hydrogenation of quinaldine with good reactivity and moderate selectivity (70–73% ee) as shown in Scheme 59.145
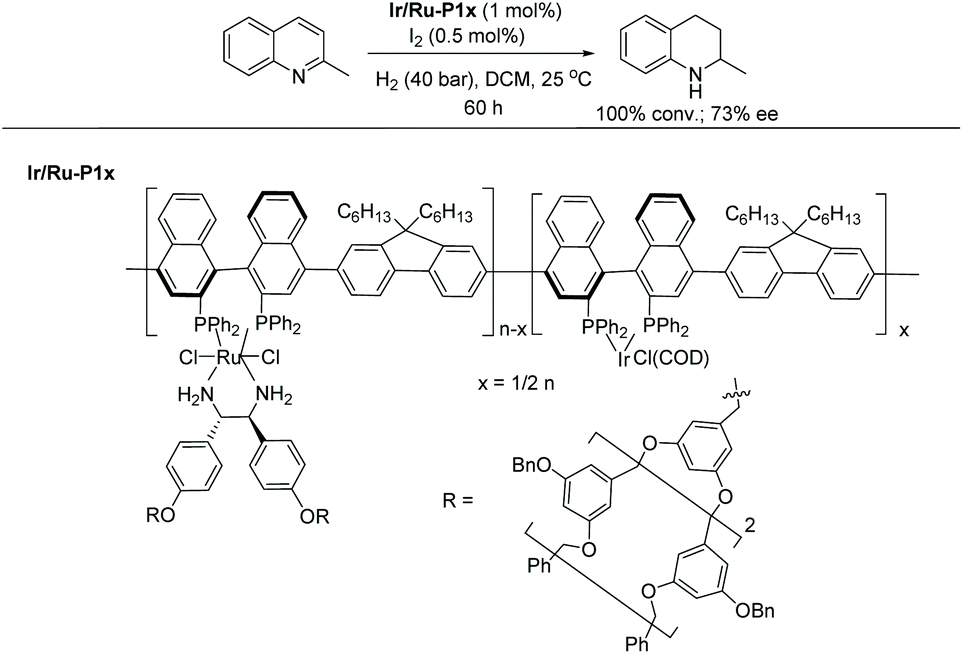 | ||
| Scheme 59 EH of quinaldine using dendronised polymer catalyst. | ||
Liu, Li and co-workers developed in 2015 a conjugated microporous polymer (CMP) embedded with a chiral Ir–BINAP complex as a heterogeneous catalyst. The EH of a range of quinolines proceeded with excellent yield and moderate ees.146 Previous reports on the asymmetric hydrogenation of quinolines showed excellent selectivities using various Ir complexes;147–149 but the catalytic systems have the limitation of high catalyst consumption due to the formation of an irreversible iridium dimer and trimer during the reaction. To overcome this limitation, Fan developed the EH of quinolines with chiral dendritic catalysts bearing BINAP.150 In the work of Li and co-workers, a series of CMPs were synthesized by embedding the chiral BINAP ligand into a CMP network. Using the optimised conditions, a range of substituted quinolines were reduced with a low catalyst loading (Table 26).
|
|
||||
|---|---|---|---|---|
| Entry | R1/R2 | Conv. (%) | ee (%) | Config. |
| Reaction conditions: [Ir(COD)Cl]2 0.00125 mmol, I2 0.05 mmol.a Reprinted from ref 146 with permission from Elsevier. | ||||
| 1 | Me/H | 99 | 70 | R |
| 2 | Et/H | 99 | 77 | R |
| 3 | iPr/H | 83 | 79 | R |
| 4 | Me/F | 99 | 63 | S |
| 5 | Me/OMe | 97 | 70 | R |

More recently, Yang and co-workers reported polymer/CNT composites as a heterogeneous catalyst for the asymmetric hydrogenation of 2-methyl quinoline to give over 90% ee.151 In 2020, this work was developed further for the asymmetric hydrogenation of quinolines (Table 27). A chiral porous polymer was integrated with chiral (1R,2R)-N-(4-vinylbenzenesulfonyl)-1,2-diphenylethane-1,2-diamine(VDPEN)-Ru-OTs sites as well as substrate activation sites (TsOH).152 Initially various catalysts at different temperatures were screened. The bifunctional porous polymer (P–C–AX)–Ru–OTs catalyst, where C denotes the chiral site, A the acidic site and X refers to the molar ratio between A and C, provided 99% yield of the corresponding tetrahydroquinoline product in 12 h when x > 0. The activity of the catalyst was drastically increased by the incorporation of TsOH into the polymer network. With a similar H+/Ru ratio, P–C–A1.0-Ru–OTs was found to be much more active than P–C–A0–Ru–OTs in the presence of 1 equiv. of TsOH, showing that the pre-organised close contact of acidic sites and chiral centres has a synergistic effect on the EH. TsOH incorporation in the polymer network also improved the stability, allowing the catalyst P–C–A1.0–RuOTs to be recycled 5 times without a drop in ee and with only a small drop in activity. In addition to the EH reaction, benzo-quinolizidines were synthesised using the bifunctional polymer catalyst via a cascade asymmetric hydrogenation/reductive amination reactions from 5-(quinolin-2-yl)pentan-2-one.
|
|
||||
|---|---|---|---|---|
| Entry | R1/R2 | Temp (°C) | Yield (%) | ee (%) |
| Reaction conditions: S/C = 100, 0.20 mmol of the substrate, 1 mL of MeOH.a Reprinted from ref 152 with permission from American Chemical Society.b 2 mL of MeOH. | ||||
| 1 | H/nBu | 20 | 96 | 88 |
| 2 | Me/Me | 20 | 95 | 92 |
| 3 | F/Me | 20 | 94 | 90 |
| 4 | Br/Me | 20 | 96 | 88 |
| 5 | H/Ph | 40 | 87 | 72 |
| 6 | H/4-MeOC6H4 | 40 | 83 | 71 |
| 7b | H/2-naphthyl | 40 | 99 | 50 |

Very recently, Glorius et al. reported a simple, cis-selective hydrogenation of multi-substituted fluorinated pyridine derivatives using a heterogeneous Pd catalyst (Scheme 60).153 Initially, the hydrogenation of 3-fluoropyridine was studied using various heterogeneous catalysts in a variety of organic solvents. In these conditions, the catalyst was not found to be active enough. Several modifications to the reaction conditions, such as pressure, temperature, catalyst, and the addition of acid, were tested. The optimal conditions were found to be 5–10 mol% Pd(OH)2/C/H2/aq. HCl (or aq. H2SO4). Other heterogeneous catalysts could lead to defluorination, and the absence of acid resulted in low yields of hydrogenated product. Purification of the unprotected fluorinated piperidines was found to be challenging due to their volatility. The piperidines were therefore trapped with different protecting groups. Fmoc and sulfonamide-protected fluorinated piperidines showed good yields with excellent diastereoselectivities. Selected examples are seen in Scheme 60.
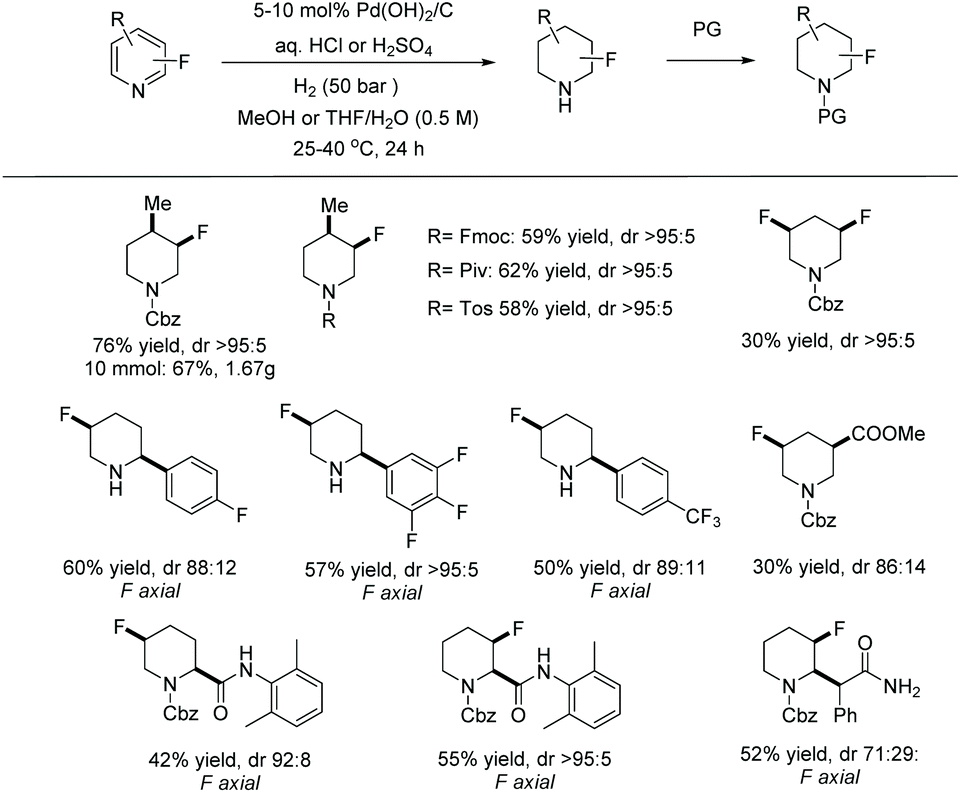 | ||
| Scheme 60 Diastereoselective hydrogenation of fluoropyridines with Pd(OH)2/C. | ||
To showcase the utility of the method, several fluorinated drug derivates were prepared. As shown in Scheme 61, fluorinated derivatives of methylphenidate, bupivacaine and ropivacaine were synthesised from the corresponding fluorinated piperidines. In the presence of a chiral auxiliary, the hydrogenation allowed enantioenriched fluorinated piperidine to be synthesised.
 | ||
| Scheme 61 Synthesis of fluorinated piperidine and drug derivatives. | ||
The auxiliary strategy was further advanced by Glorius et al. in 2021, who reported the enantio- and diastereoselective hydrogenation of oxazolidinone-substituted pyridines using Pd/C as catalyst to access enantioenriched δ-lactams.154 Initial screening showed that the use of a strong Brønsted acid in combination with H2O was critical to the formation of the lactams. Under the optimised conditions, a series of oxazolidinone-substituted pyridines were reduced, affording alkylated δ-lactams in high yield and excellent enatioselctivities as shown in Scheme 62. Various functional groups could be tolerated without a considerable decrease in yield or ee. This method could also be used to form multiple stereocenters in high selectivity. The utility of this reaction was further demonstrated on a 2 g scale.
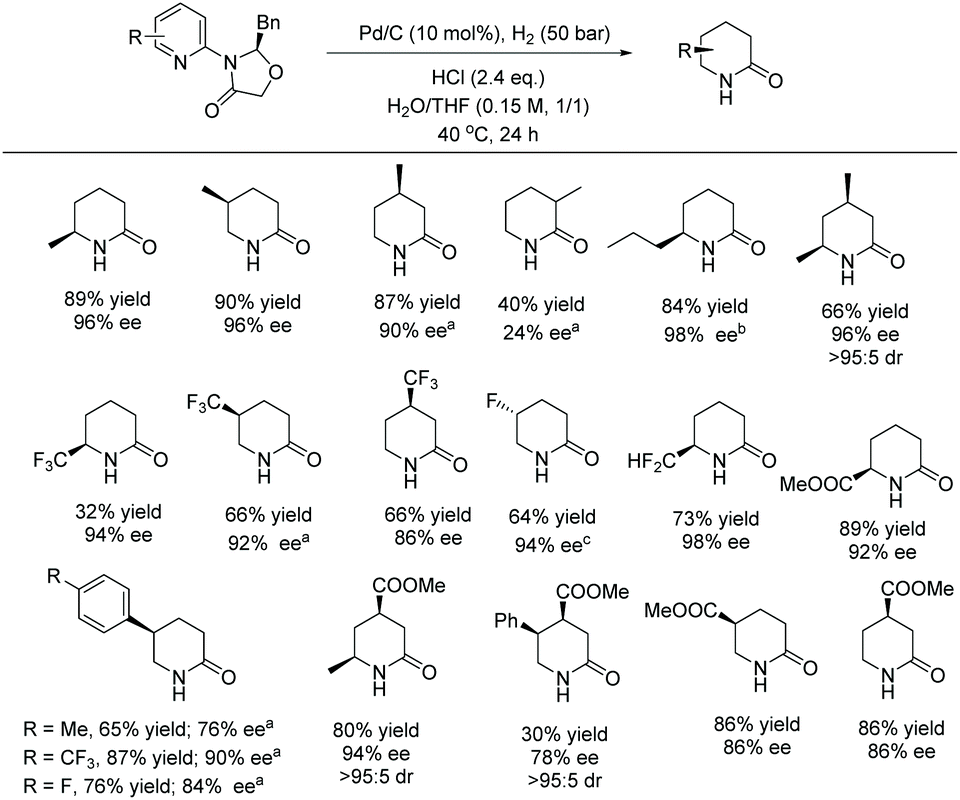 | ||
| Scheme 62 Synthesis of 2-piperidones via palladium catalysed hydrogenation. Chiral auxiliary: a4-isopropyloxazolidinone, b4-tert-Butyloxazolidinone, cEpimeric (R)-isopropyloxazolidinone. | ||
A proposed mechanism for the hydrogenation is seen in Scheme 63. Initial hydrogenation occurs by the addition of H2 to form an oxazolidinone-substituted iminium I. Nucleophilic substitution with water forms the product II while releasing the chiral oxazolidinone, and tautomeriztion of II yields the final product. Due to the interruption by water, the hydrogenation of I to give the normal hydrogenation product III is suppressed.141,155
 | ||
| Scheme 63 Proposed mechanism for the hydrogenation of pyridines to form lactams. | ||
4. Organocatalytic and other miscellaneous asymmetric hydrogenation
As is clear, the asymmetric hydrogenation of N-heteroaromatic compounds has been widely explored with various transition metal catalysts. This often requires harsh reaction conditions and/or specific substrates. In addition, the use of expensive and toxic noble metals poses a potential problem for practical applications. Recently, an organocatalytic transfer hydrogenation strategy has been explored for the asymmetric hydrogenation of N-heteroaromatic compounds. This metal-free approach to hydrogenation is of great interest and an underdeveloped field in the EH of N-heteroaromatic compounds.In 2014, Zhou et al. developed an efficient ATH of quinolin-3-amines with a phosphoric acid as catalyst to access chiral exocyclic amines with high diastereo- and enantioselectivities (Table 28).156 Previous successful applications of chiral phosphoric acids (CPAs) in the ATH of CC, CN, and CO double bonds with Hantzsch esters as the hydrogen source led to further development of the catalytic system for N-heteroaromatic compounds.157–163 Initial screening established the use of a sterically demanding CPA catalyst (5 mol%) and Hantzsch esters (2.4 eq.) as the hydrogen source in 1,4-dioxane/CH2Cl2. With these optimised conditions, the substrate scope and limitation of this strategy were explored. All the 2-aryl substituted substrates were converted to the corresponding products in high yields with high ees, regardless of the electronic properties of the C2 substitution.
|
|
|||
|---|---|---|---|
| Entry | R1/R2 | Yield (%) | ee (%) |
| Reaction conditions: substrate (0.125 mmol), Hantzsch ester (0.300 mmol) in 1,4-dioxane/CH2Cl2 (2:1, 3.0 mL), C1 (5 mol%), 24 h. |
|||
| 1 | H/C6H5 | 94 | 95 (S,S) |
| 2 | H/3-MeC6H4 | 96 | 97 (−) |
| 3 | H/4-MeC6H4 | 98 | 91 (−) |
| 4 | H/4-nBuC6H4 | 93 | 94 (−) |
| 5 | H/4-MeOC6H4 | 96 | 99 (−) |
| 6 | H/4-ClC6H4 | 97 | 95 (−) |
| 7 | H/4-BrC6H4 | 99 | 96 (−) |
| 8 | H/4-FC6H4 | 93 | 98 (−) |
| 9 | H/4-CF3C6H4 | 99 | 98 (−) |
| 10 | H/2-naphthyl | 91 | 83 (−) |
| 11 | F/C6H5 | 94 | 73 (−) |
| 12 | H/3-pyridinyl | 70 | 97 (−) |
Two possible mechanistic routes were postulated for the reaction (Scheme 64). In path a, the partially reduced intermediate I isomerises to exocyclic imine II; in path b, I isomerises to endocyclic imine III. A deuterium labelling experiment illustrated that the reaction mainly proceeds via the endocyclic imine intermediate III (path b), and a kinetic dynamic resolution process is involved to afford the enantio-enriched product.
 | ||
| Scheme 64 Proposed mechanism for the hydrogenation of quinolin-3-amines. | ||
The discovery of frustrated Lewis pair (FLP) chemistry was a breakthrough in the emerging metal-free hydrogenation using molecular H2.164,165 Initial studies in FLP catalysis was conducted on a wide range of unsaturated compounds, such as olefins, alkynes, imines, oxime ethers etc.166 Stephan et al. in 2010 first described the hydrogenation of N-heteroaromatic compounds using the Lewis acid B(C6F5)3.167 Later in 2013, Du et al. established the cis-selective hydrogenation of simple pyridines using a borane catalyst (Table 29).168
|
|
|||
|---|---|---|---|
| Entry | R/Ar | Yield (%) | dr (%) |
| Reactions condition: substrate (0.25 mmol) in toluene (2.0 mL). | |||
| 1 | Me/C6H5 | 96 | 95:5 |
| 2 | Me/4-MeOC6H4 | 98 | 96:4 |
| 3 | Me/4-PhC6H4 | 96 | 96:4 |
| 4 | Me/4-CF3C6H4 | 86 | 97:3 |
| 5 | Me/4-ClC6H4 | 88 | 96:4 |
| 6 | Me/3-MeOC6H4 | 96 | 96:4 |
| 7 | Me/2-naphthyl | 99 | 96:4 |
| 8 | Me/4-allyloxyC6H4 | 80 | 96:4 |
| 9 | R = Ar = Ph | 98 | 98:2 |
| 10 | R = Ar = 4-MeC6H4 | 97 | 98:2 |
| 11 | R = Ar = 4-MeOC6H4 | 99 | 98:2 |
| 12 | R = Ar = 2-furyl | 93 | 90:10 |
| 13 | 4-FC6H4/4-MeOC6H4 | 92 | 99:1 |
Reaction conditions were initially screened for the hydrogenation of 2,6-diphenylpyridine. A Piers’ borane catalyst with H2 (50 bar) in toluene at 100 °C was chosen as the most efficient condition. In situ generation of boranes by hydroboration of an alkene with HB(C6F5)2 led to a more active catalyst, and electron-deficient alkenes were found to be more effective. With these conditions in hand, the substrate scope was explored. Various 2,6-disubstituted pyridines were reduced, affording high to excellent yield of the corresponding cis-piperidines (Table 29). This method was used to synthesise the natural product isosolenopsin A169 in racemic form (Scheme 65). Good yield and diastereoselectivity were achieved.168 Isosolenopsin A displays various biological activities, such as antibacterial properties.170
 | ||
| Scheme 65 Synthesis of natural product isosolenopsin A. | ||
In further studies, Du et al. expanded on the previously reported work by developing an enantioselective and cis-selective metal-free hydrogenation of quinoxalines (Scheme 66).171 Previous results on novel FLP catalytic systems for the asymmetric hydrogenation of imines and silyl enol ethers encouraged them to expand the utility of FLP on the EH of other N-heterocycles. Initial optimisation of the reaction allowed for the hydrogenation of 2,3-disubstituted quinoxalines giving cis-products. An asymmetric version was then developed using a chiral diene–borane catalyst, with the diene based on a chiral binaphthyl framework (C2). The borane catalyst was generated by the in situ hydroboration of the chiral diene with HB(C6F5)2 under mild reaction conditions The catalyst allowed for the formation of tetrahydroquinoxalines in moderate to high enantioselectivities.
 | ||
| Scheme 66 Diastereo and enantioselective reduction of quinoxalines with a borane catalyst. aCatalyst: B(C6F5)3 (10 mol%) or B(p-HC6F4)3 (5 mol%) was used. bCatalyst: chiral diene C2 (10 mol%) was used. | ||
The ATH of 2,3-disubstituted quinoxalines was also realised using the combination of HB(C6F5)2 and (R)-tert-butylsulfinamide as a chiral FLP catalyst (Scheme 67). Ammonia borane was used as the hydrogen source.172 The ATH of 2-alkyl-3-arylquinoxalines provided high cis-selectivities with up to 86% ee. However, when 2,3-dialkylquinoxalines were used as substrates, the desired products were obtained with lower dr values (trans:cis) but higher ees.
 | ||
| Scheme 67 ATH of quinoxalines using a butylsulfinamide derived borane catalyst. Reaction condition: quinoxaline (0.30 mmol), HB(C6F5)2 (0.06 mmol), (R)-tert-butylsulfinamide (0.09 mmol), and ammonia borane (0.60 mmol) in a solvent (3.0 mL) at 30 °C. aSolvent = C6H5Br/n-hexane (3:7). bSolvent = CH2Cl2. | ||
Following their work on quinoxalines, Du et al. reported the metal-free EH of 2,4- and 2,3-disubstituted quinolines using a similar chiral diene-derived borane catalyst to yield highly diastereoselective tetrahydroquinolines with good to excellent ees (Table 30).173 Various chiral dienes were initially investigated for this hydrogenation. On finding a chiral diene that provided high yields and selectivities, reaction conditions were further optimised. Lowering the temperature to 15 °C was found to give 90% ee and a catalyst loading of 10 mol% provided good to excellent yields without loss of diastereoselectivity or enantioselectivity. The EH tolerated aryl substituents containing both EWGs and EDGs at the 2- and 4-positions of quinolines, showing high levels of diastereoselectivities and enantioselectivities (Table 30). Furans, thiophenes, and alkene substituents were all well tolerated in this FLP catalytic system. However, alkyl substituents at 2- or 4-position of quinoline gave only low to moderate ees.
|
|
||||
|---|---|---|---|---|
| Entry | Product | Yield (%) | cis/trans | ee (%) |
| Reaction condition: quinoline (0.40 mmol), HB(C6F5)2 (0.04 mmol), chiral diene (0.02 mmol) in toluene (0.8 mL). | ||||
| 1 |

|
91 | 97:3 |
91 |
| 2 | R = 2-Br | 86 | 99:1 |
97 |
| 3 | R = 4-CF3 | 75 | 95:5 |
87 |
|
||||
| 4 | Ar = 2-furyl | 80 | 98:2 |
87 |
| 5 | Ar = 2-thienyl | 93 | 98:2 |
89 |
| 6 |
|
89 | 96:4 |
96 |
| 7 | R=Me | 90 | 98:2 |
90 |
| 8 |

|
91 | >99:1 |
69 |
| 9 | R = 3-Br | 99 | >99:1 |
66 |
| 10 | R = 4-CF3 | 92 | >99:1 |
70 |

More recently, Song et al. described a new Brønsted acid catalyst for the asymmetric reduction of indoles with a hydroborane (Scheme 68). The chiral Brønsted acid was generated in situ from a chiral phosphoric acid (C4) boron complex (CPAB) by interaction with water.174 TsOH, a strong acid, had been previously used to protonate the C3 position of 2-alkyl-indoles forming an iminium salt.175,176 Treatment with a suitable reducing agent allows for the conversion of the iminium into the corresponding indoline. DFT calculation demonstrated that the newly designed CPAB-derived acid catalyst is more acidic than TsOH and possesses higher reactivity, higher selectivity, and better versatility. The Brønsted acid activates the indole and facilitates its stereoselective reduction to form the chiral indoline. Initial optimisation showed that the CPAB–H2O catalysed reaction proceeded at −50 °C in toluene. Under the optimised conditions, a range of substituted indoles was reduced with up to 96% ee. The results show that the substitution on the indole moiety has no major effect on the reactivity or selectivity. Potential functional handles such as BPin, Br, COOMe, CN and OMe were tolerated. To demonstrate the practical usability of the catalytic system, a gram scale asymmetric reduction was performed. No detrimental effect on reactivity or enantioselectivity was observed.
 | ||
| Scheme 68 EH of indoles using a chiral phosphoric acid–boron complex. Reaction conditions: substrate (0.1 mmol), catecholborane (0.3 mmol), H2O (0.3 mmol), C4 (10 mol%), toluene (4 mL), −50 °C. | ||
The importance of the resulting indolines was further demonstrated by elaboration of the phenyl substituted indoline into two different potential drug molecules, each with differing biological targets. The methodology was also used to synthesise a chiral ligand (Scheme 69).
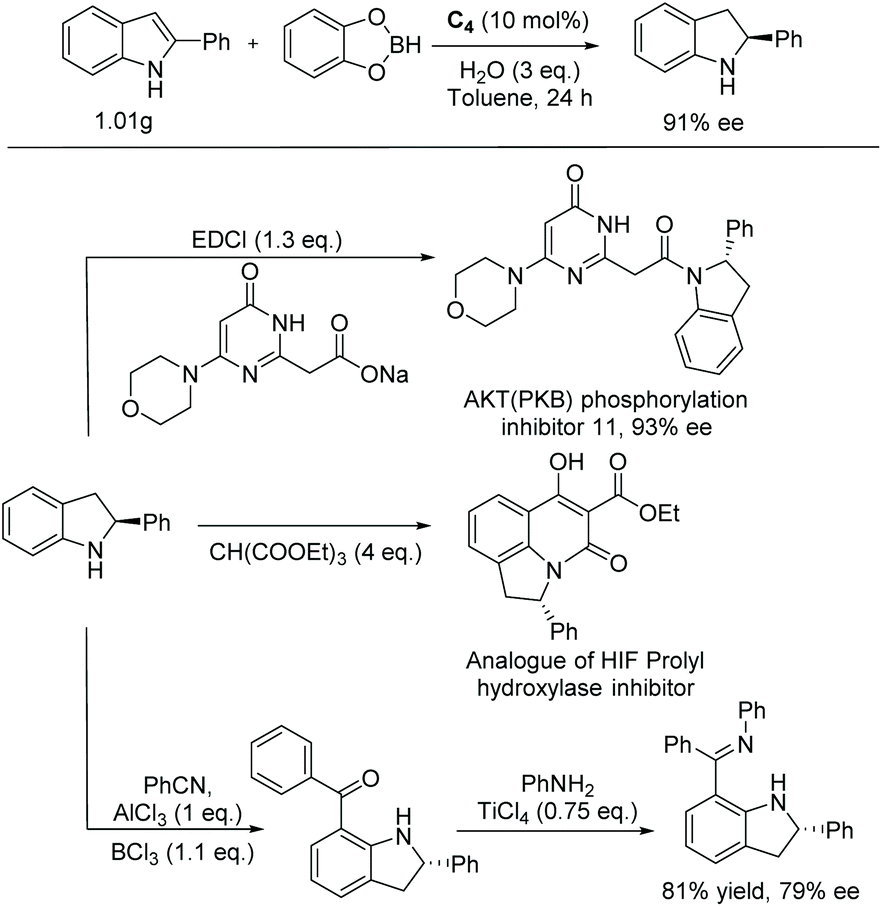 | ||
| Scheme 69 Synthetic applications of the chiral phosphoric acid–boron complex. | ||
5. Conclusion
This review shows the scope of a number of catalytic systems for the diastereo- and enantioselective hydrogenation of various N-heterocycles that have been developed over the past decade. While great progress has been made in transition metal catalysis, recent developments in organocatalysis and heterogeneous asymmetric catalysis have shown new promise. However, despite the advances made, there remain several challenges. For most of the catalysts reported, the substrate scope is narrow and often, a different type of substrate requires a different set of conditions. The field is also dominated by metal catalysts based on a few noble metals, Ir, Rh and Ru, which are not only expensive but also toxic, making their complete removal and reuse necessary. In addition, the reaction conditions tend to be harsh, with high pressure usually necessary when using H2. Consequently, the reaction is yet to be widely utilised in industrial processes. In comparison, the asymmetric reduction of olefins has seen many successful examples at an industrial scale. Clearly, much remains to be done to provide more efficient, greener and more economic catalytic systems for the asymmetric hydrogenation of N-heteroaromatic compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Innovate UK – Knowledge Transfer Partnerships and Liverpool ChiroChem (LCC) for the financial support (R. G.) and Drs Paul Colbon & Jiwu Ruan for discussions & technical assistance.Notes and references
- K. C. Majumdar and S. K. Chattopadhyay, Heterocycles in Natural Product Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- C. T. Walsh, Tetrahedron Lett., 2015, 56, 3075–3081 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- F. Glorius, Org. Biomol. Chem., 2005, 3, 4171–4175 RSC.
- Y. G. Zhou, Acc. Chem. Res., 2007, 40, 1357–1366 CrossRef CAS PubMed.
- D. S. Wang, Q. A. Chen, S. M. Lu and Y. G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed.
- Z. Yu, W. Jin and Q. Jiang, Angew. Chem., Int. Ed., 2012, 51, 6060–6072 CrossRef CAS PubMed.
- R. N. Guo, X. F. Cai, L. Shi, Z. S. Ye, M. W. Chen and Y. G. Zhou, Chem. Commun., 2013, 49, 8537–8539 RSC.
- B. Balakrishna, J. L. Núñez-Rico and A. Vidal-Ferran, Eur. J. Org. Chem., 2015, 2015, 5293–5303 CrossRef CAS.
- Z. P. Chen and Y. G. Zhou, Synthesis, 2016, 48, 1769–1781 CrossRef CAS.
- K. Mashima, K. Higashida, A. Iimuro, H. Nagae and Y. Kita, Chem. Rec., 2016, 16, 2585–2594 CrossRef CAS PubMed.
- Z. X. Giustra, J. S. A. Ishibashi and S. Y. Liu, Coord. Chem. Rev., 2016, 314, 134–181 CrossRef CAS.
- M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10460–10476 CrossRef CAS PubMed.
- J. Cheng and T. J. Deming, Pept. Mater., 2011, 310, 1–26 Search PubMed.
- A. N. Kim and B. M. Stoltz, ACS Catal., 2020, 10, 13834–13851 CrossRef CAS PubMed.
- S. Murata, T. Sugimoto and S. Matsuura, Heterocycles, 1987, 26, 763 CrossRef CAS.
- T. Ohta, T. Miyake, N. Seido, H. Kumobayashi and H. Takaya, J. Org. Chem., 1995, 60, 357–363 CrossRef CAS.
- C. Bianchini, P. Barbaro, G. Scapacci, E. Farnetti and M. Graziani, Organometallics, 1998, 17, 3308–3310 CrossRef CAS.
- R. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS.
- A. Lightfoot, P. Schnider and A. Pfaltz, Angew. Chem., Int. Ed., 1998, 37, 2897–2899 CrossRef CAS PubMed.
- S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402–1411 CrossRef CAS PubMed.
- S. J. Roseblade and A. Pfaltz, C. R. Chim., 2007, 10, 178–187 CrossRef CAS.
- A. Baeza and A. Pfaltz, Chem. – Eur. J., 2010, 16, 4003–4009 CrossRef CAS PubMed.
- L. Massaro, J. Zheng, C. Margarita and P. G. Andersson, Chem. Soc. Rev., 2020, 49, 2504–2522 RSC.
- A. L. Nelson, W. J. Brehm and R. B. Woodward, J. Am. Chem. Soc., 1947, 69, 2250 CrossRef PubMed.
- W. G. Kim, J. P. Kim, H. Koshino, K. Shin-Ya, H. Seto and I. D. Yoo, Tetrahedron, 1997, 53, 4309–4316 CrossRef CAS.
- N. H. Greig, X. F. Pei, T. T. Soncrant, D. K. Ingram and A. Brossi, Med. Res. Rev., 1995, 15, 3–31 CrossRef CAS PubMed.
- P. W. Moore, J. J. Rasimas and J. W. Donovan, J. Med. Toxicol., 2015, 11, 159–160 CrossRef PubMed.
- D. Hayoz, G. Bizzini, B. Noël, M. Depairon, M. Burnier, C. Fauveau, A. Rouillon, R. Brouard and H. R. Brunner, Rheumatology, 2000, 39, 1132–1138 CrossRef CAS PubMed.
- M. Odabas-Geldiay, H. Shields, L. F. Berro, K. C. Rice and L. L. Howell, Drug Alcohol Depend., 2019, 194, 252–256 CrossRef CAS PubMed.
- R. Kuwano, K. Sato, T. Kurokawa, D. Karube and Y. Ito, J. Am. Chem. Soc., 2000, 122, 7614–7615 CrossRef CAS.
- R. Kuwano, K. Kaneda, T. Ito, K. Sato, T. Kurokawa and Y. Ito, Org. Lett., 2004, 6, 2213–2215 CrossRef CAS PubMed.
- A. Baeza and A. Pfaltz, Chem. – Eur. J., 2010, 16, 2036–2039 CrossRef CAS PubMed.
- H. Fernández-Pérez, M. A. Pericàs and A. Vidal-Ferran, Adv. Synth. Catal., 2008, 350, 1984–1990 CrossRef.
- H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119–2176 CrossRef PubMed.
- J. L. Núñez-Rico and A. Vidal-Ferran, Org. Lett., 2013, 15, 2066–2069 CrossRef PubMed.
- J. L. Núñez-Rico, H. Fernández-Pérez and A. Vidal-Ferran, Green Chem., 2014, 16, 1153–1157 RSC.
- B. Balakrishna, A. Bauzá, A. Frontera and A. Vidal-Ferran, Chem. – Eur. J., 2016, 22, 10607–10613 CrossRef CAS PubMed.
- D. V. Ozolin, S. E. Lyubimov and V. A. Davankov, Russ. Chem. Bull. Int. Ed., 2014, 63, 2399–2401 CrossRef CAS.
- S. E. Lyubimov, D. V. Ozolin and V. A. Davankov, Tetrahedron Lett., 2014, 55, 3613–3614 CrossRef CAS.
- T. C. Gray and J. Halton, Br. Med. J., 1946, 2, 293–295 CrossRef PubMed.
- N. Iwatsuki, Y. Hashimoto, K. Amaha, S. Obara and K. Iwatsuki, Anesth. Analg., 1980, 59, 717–721 CAS.
- A. D. Pechulis, J. P. Beck, M. A. Curry, M. A. Wolf, A. E. Harms, N. Xi, C. Opalka, M. P. Sweet, Z. Yang, A. S. Vellekoop, A. M. Klos, P. J. Crocker, C. Hassler, M. Laws, D. B. Kitchen, M. A. Smith, R. E. Olson, S. Liu and B. F. Molino, Bioorg. Med. Chem. Lett., 2012, 22, 7219–7222 CrossRef CAS PubMed.
- M. Dean and V. W. Sung, Drug Des., Dev. Ther., 2018, 12, 313–319 CrossRef CAS PubMed.
- M. B. Jiménez-Díaz, D. Ebert, Y. Salinas, A. Pradhan, A. M. Lehane, M.-E. Myrand-Lapierre, K. G. O'Loughlin, D. M. Shackleford, M. Justino de Almeida, A. K. Carrillo, J. A. Clark, A. S. M. Dennis, J. Diep, X. Deng, S. Duffy, A. N. Endsley, G. Fedewa, W. A. Guiguemde, M. G. Gómez, G. Holbrook, J. Horst, C. C. Kim, J. Liu, M. C. S. Lee, A. Matheny, M. S. Martínez, G. Miller, A. Rodríguez-Alejandre, L. Sanz, M. Sigal, N. J. Spillman, P. D. Stein, Z. Wang, F. Zhu, D. Waterson, S. Knapp, A. Shelat, V. M. Avery, D. A. Fidock, F.-J. Gamo, S. A. Charman, J. C. Mirsalis, H. Ma, S. Ferrer, K. Kirk, I. Angulo-Barturen, D. E. Kyle, J. L. DeRisi, D. M. Floyd and R. K. Guy, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5455–E5462 CrossRef PubMed.
- M. O. Duffey, D. England, S. Freeze, Z. Hu, S. P. Langston, C. Mcintyre, H. Mizutani, K. Ono and H. Xu, US-9683003-B2, 2016.
- S. M. Lu, Y. Q. Wang, X. W. Han and Y. G. Zhou, Angew. Chem., Int. Ed., 2006, 45, 2260–2263 CrossRef CAS PubMed.
- A. Iimuro, K. Yamaji, S. Kandula, T. Nagano, Y. Kita and K. Mashima, Angew. Chem., Int. Ed., 2013, 52, 2046–2050 CrossRef CAS PubMed.
- L. Shi, Z. S. Ye, L. L. Cao, R. N. Guo, Y. Hu and Y. G. Zhou, Angew. Chem., Int. Ed., 2012, 51, 8286–8289 CrossRef CAS PubMed.
- L. Shi, Z. S. Ye, L. L. Cao, R. N. Guo, Y. Hu and Y. G. Zhou, Angew. Chem., Int. Ed., 2012, 51, 8286–8289 CrossRef CAS PubMed.
- T. Nagano, A. Iimuro, R. Schwenk, T. Ohshima, Y. Kita, A. Togni and K. Mashima, Chem. – Eur. J., 2012, 18, 11578–11592 CrossRef CAS PubMed.
- Z. S. Ye, R. N. Guo, X. F. Cai, M. W. Chen, L. Shi and Y. G. Zhou, Angew. Chem., Int. Ed., 2013, 52, 3685–3689 CrossRef CAS PubMed.
- M. W. Chen, Y. Ji, J. Wang, Q. A. Chen, L. Shi and Y. G. Zhou, Org. Lett., 2017, 19, 4988–4991 CrossRef CAS PubMed.
- A. N. Kim, A. Ngamnithiporn, E. R. Welin, M. T. Daiger, C. U. Grünanger, M. D. Bartberger, S. C. Virgil and B. M. Stoltz, ACS Catal., 2020, 10, 3241–3248 CrossRef CAS PubMed.
- E. R. Welin, A. Ngamnithiporn, M. Klatte, G. Lapointe, G. M. Pototschnig, M. S. J. McDermott, D. Conklin, C. D. Gilmore, P. M. Tadross, C. K. Haley, K. Negoro, E. Glibstrup, C. U. Grünanger, K. M. Allan, S. C. Virgil, D. J. Slamon and B. M. Stoltz, Science, 2019, 363, 270–275 CrossRef CAS PubMed.
- B. J. Petek and R. L. Jones, Molecules, 2014, 19, 12328–12335 CrossRef PubMed.
- P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS PubMed.
- M. Studer, C. Wedemeyer-Exl, F. Spindler and H. U. Blaser, Monatsh. Chem., 2000, 131, 1335–1343 CrossRef CAS.
- C. Y. Legault and A. B. Charette, J. Am. Chem. Soc., 2005, 127, 8966–8967 CrossRef CAS PubMed.
- X. B. Wang, W. Zeng and Y. G. Zhou, Tetrahedron Lett., 2008, 49, 4922–4924 CrossRef CAS.
- Z. S. Ye, M. W. Chen, Q. A. Chen, L. Shi, Y. Duan and Y. G. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10181–10184 CrossRef CAS PubMed.
- N. S. Sheikh, D. Leonori, G. Barker, J. D. Firth, K. R. Campos, A. J. H. M. Meijer, P. O'Brien and I. Coldham, J. Am. Chem. Soc., 2012, 134, 5300–5308 CrossRef CAS PubMed.
- D. Xiao, B. J. Lavey, A. Palani, C. Wang, R. G. Aslanian, J. A. Kozlowski, N. Y. Shih, A. T. McPhail, G. P. Randolph, J. E. Lachowicz and R. A. Duffy, Tetrahedron Lett., 2005, 46, 7653–7656 CrossRef CAS.
- W. Tang, Y. Sun, L. Xu, T. Wang, Q. Fan, K. H. Lam and A. S. C. Chan, Org. Biomol. Chem., 2010, 8, 3464–3471 RSC.
- W. J. Tang, J. Tan, L. J. Xu, K. H. Lam, Q. H. Fan and A. S. C. Chan, Adv. Synth. Catal., 2010, 352, 1055–1062 CrossRef CAS.
- A. Cadu, P. K. Upadhyay and P. G. Andersson, Asian J. Org. Chem., 2013, 2, 1061–1065 CrossRef CAS.
- M. Chang, Y. Huang, S. Liu, Y. Chen, S. W. Krska, I. W. Davies and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 12761–12764 CrossRef CAS PubMed.
- Y. Kita, A. Iimuro, S. Hida and K. Mashima, Chem. Lett., 2014, 43, 284–286 CrossRef CAS.
- M.-W. Chen, Z.-S. Ye, Z.-P. Chen, B. Wu and Y.-G. Zhou, Org. Chem. Front., 2015, 2, 586–589 RSC.
- W. X. Huang, C. B. Yu, Y. Ji, L. J. Liu and Y. G. Zhou, ACS Catal., 2016, 6, 2368–2371 CrossRef CAS.
- A. Iimuro, K. Higashida, Y. Kita and K. Mashima, Adv. Synth. Catal., 2016, 358, 1929–1933 CrossRef CAS.
- M. Renom-Carrasco, P. Gajewski, L. Pignataro, J. G. de Vries, U. Piarulli, C. Gennari and L. Lefort, Adv. Synth. Catal., 2016, 358, 2589–2593 CrossRef CAS.
- B. Qu, H. P. R. Mangunuru, S. Tcyrulnikov, D. Rivalti, O. V. Zatolochnaya, D. Kurouski, S. Radomkit, S. Biswas, S. Karyakarte, K. R. Fandrick, J. D. Sieber, S. Rodriguez, J. N. Desrosiers, N. Haddad, K. McKellop, S. Pennino, H. Lee, N. K. Yee, J. J. Song, M. C. Kozlowski and C. H. Senanayake, Org. Lett., 2018, 20, 1333–1337 CrossRef CAS PubMed.
- W. Li, S. Zhang, X. Yu, X. Feng, Y. Yamamoto and M. Bao, J. Org. Chem., 2021, 86, 10773–10781 CrossRef CAS PubMed.
- J. Smith, A. Kacmaz, C. Wang, B. Villa-Marcos and J. Xiao, Org. Biomol. Chem., 2021, 19, 279–284 RSC.
- S. Kohmoto, Y. Kashman, O. J. McConnell, K. L. Rinehart, A. Wright and F. Koehn, J. Org. Chem., 1988, 53, 3116–3118 CrossRef CAS.
- S. TAMAI, M. KANEDA and S. NAKAMURA, J. Antibiot., 1982, 35, 1130–1136 CrossRef CAS PubMed.
- B. Zhu, B. A. Marinelli, R. Goldschmidt, B. D. Foleno, J. J. Hilliard, K. Bush and M. J. Macielag, Bioorg. Med. Chem. Lett., 2009, 19, 4933–4936 CrossRef CAS PubMed.
- R. Fuchs, Chem. Abstr., 1998, 128, 803502 Search PubMed.
- W. X. Huang, C. Bin Yu, L. Shi and Y. G. Zhou, Org. Lett., 2014, 16, 3324–3327 CrossRef CAS PubMed.
- W. X. Huang, L. J. Liu, B. Wu, G. S. Feng, B. Wang and Y. G. Zhou, Org. Lett., 2016, 18, 3082–3085 CrossRef CAS PubMed.
- M. Van Der Linden, J. Borsboom, F. Kaspersen and G. Kemperman, Eur. J. Org. Chem., 2008, 2989–2997 CrossRef CAS.
- N. Ivgy-May, F. Ruwe, A. Krystal and T. Roth, Sleep Med., 2015, 16, 838–844 CrossRef PubMed.
- C. Harrison, Nat. Biotechnol., 2020, 38, 379–381 CrossRef PubMed.
- J. C. Barrow, P. G. Nantermet, H. G. Selnick, K. L. Glass, K. E. Rittle, K. F. Gilbert, T. G. Steele, C. F. Homnick, R. M. Freidinger, R. W. Ransom, P. Kling, D. Reiss, T. P. Broten, T. W. Schorn, R. S. L. Chang, S. S. O'Malley, T. V. Olah, J. D. Ellis, A. Barrish, K. Kassahun, P. Leppert, D. Nagarathnam and C. Forray, J. Med. Chem., 2000, 43, 2703–2718 CrossRef CAS PubMed.
- R. Kuwano, Y. Hashiguchi, R. Ikeda and K. Ishizuka, Angew. Chem., Int. Ed., 2015, 54, 2393–2396 CrossRef CAS PubMed.
- G. S. Feng, M. W. Chen, L. Shi and Y. G. Zhou, Angew. Chem., Int. Ed., 2018, 57, 5853–5857 CrossRef CAS PubMed.
- G. S. Feng, L. Shi, F. J. Meng, M. W. Chen and Y. G. Zhou, Org. Lett., 2018, 20, 6415–6419 CrossRef CAS PubMed.
- V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS PubMed.
- I. Muthukrishnan, V. Sridharan and J. C. Menéndez, Chem. Rev., 2019, 119, 5057–5191 CrossRef CAS PubMed.
- J. G. Lee, I. D. Yoo and W. G. Kim, Biol. Pharm. Bull., 2007, 30, 795–797 CrossRef CAS PubMed.
- J. H. Rakotoson, N. Fabre, I. Jacquemond-Collet, S. Hannedouche, I. Fourasté and C. Moulis, Planta Med., 1998, 64, 762–763 CrossRef CAS PubMed.
- I. Jacquemond-Collet, F. Benoit-Vical, Mustofa, M. A. Valentin, E. Stanislas, M. Mallié and I. Fourasté, Planta Med., 2002, 68, 68–69 CrossRef CAS PubMed.
- K. M. Witherup, W. C. Lumma, S. M. Pitzenberger, S. L. Varga, R. W. Ransom, A. C. Graham, A. M. Bernard, M. J. Salvatore and P. S. Anderson, J. Am. Chem. Soc., 1995, 117, 6682–6685 CrossRef CAS.
- R. W. Clark, T. A. Sutfin, R. B. Ruggeri, A. T. Willauer, E. D. Sugarman, G. Magnus-Aryitey, P. G. Cosgrove, T. M. Sand, R. T. Wester, J. A. Williams, M. E. Perlman and M. J. Bamberger, Arterioscler. Thromb. Vasc. Biol., 2004, 24, 490–497 CrossRef CAS PubMed.
- C. Zhang, S. M. Westaway, J. D. Speake, M. J. Bishop, A. S. Goetz, L. H. Carballo, M. Hu and A. H. Epperly, Bioorg. Med. Chem. Lett., 2011, 21, 670–676 CrossRef CAS PubMed.
- R. J. Lewis, C. A. Francis, R. E. Lehr and C. L. Blank, Tetrahedron, 2000, 56, 5345–5352 CrossRef CAS.
- A. M. Maj, I. Suisse, C. Hardouin and F. Agbossou-Niedercorn, Tetrahedron, 2013, 69, 9322–9328 CrossRef CAS.
- X. F. Cai, R. N. Guo, M. W. Chen, L. Shi and Y. G. Zhou, Chem. – Eur. J., 2014, 20, 7245–7248 CrossRef CAS PubMed.
- X. H. Hu and X. P. Hu, Org. Lett., 2019, 21, 10003–10006 CrossRef CAS PubMed.
- A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS.
- W. Zuo and R. H. Morris, Nat. Protoc., 2015, 10, 241–257 CrossRef CAS PubMed.
- A. A. Mishra and B. M. Bhanage, Chirality, 2021, 33, 337–378 CrossRef CAS PubMed.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- H. G. Nedden, A. Zanotti-Gerosa and M. Wills, Chem. Rec., 2016, 16, 2619–2639 CrossRef CAS PubMed.
- J. Barrios-Rivera, Y. Xu, M. Wills and V. K. Vyas, Org. Chem. Front., 2020, 7, 3312–3342 RSC.
- R. Noyori, T. Ohkuma, M. Kitamura, H. Takaya, N. Sayo, H. Kumobayashi and S. Akutagawa, J. Am. Chem. Soc., 1987, 109, 5856–5858 CrossRef CAS.
- P. Etayo and A. Vidal-Ferran, Chem. Soc. Rev., 2013, 42, 728–754 RSC.
- Z. Yang, F. Chen, Y. He, N. Yang and Q. H. Fan, Angew. Chem., Int. Ed., 2016, 55, 13863–13866 CrossRef CAS PubMed.
- T. Wang, L. G. Zhuo, Z. Li, F. Chen, Z. Ding, Y. He, Q. H. Fan, J. Xiang, Z. X. Yu and A. S. C. Chan, J. Am. Chem. Soc., 2011, 133, 9878–9891 CrossRef CAS PubMed.
- J. Qin, F. Chen, Z. Ding, Y. M. He, L. Xu and Q. H. Fan, Org. Lett., 2011, 13, 6568–6571 CrossRef CAS PubMed.
- T. Touge and T. Arai, J. Am. Chem. Soc., 2016, 138, 11299–11305 CrossRef CAS PubMed.
- R. M. Jones, S. Han and J. V. Moody, WO/2009/151626, 2008.
- S. M. Cramp, H. J. Dyke, T. D. Pallin and R. Zahler, WO2012/154678, 2012.
- W. Ma, J. Zhang, C. Xu, F. Chen, Y. M. He and Q. H. Fan, Angew. Chem., Int. Ed., 2016, 55, 12891–12894 CrossRef CAS PubMed.
- J. Wu, J. H. Barnard, Y. Zhang, D. Talwar, C. M. Robertson and J. Xiao, Chem. Commun., 2013, 49, 7052–7054 RSC.
- C. Li, Y. Pan, Y. Feng, Y. M. He, Y. Liu and Q. H. Fan, Org. Lett., 2020, 22, 6452–6457 CrossRef CAS PubMed.
- N. Ortega, D. T. D. Tang, S. Urban, D. Zhao and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 9500–9503 CrossRef CAS PubMed.
- D. Paul, B. Beiring, M. Plois, N. Ortega, S. Kock, D. Schlüns, J. Neugebauer, R. Wolf and F. Glorius, Organometallics, 2016, 35, 3641–3646 CrossRef CAS.
- A. Hamza, D. Moock, C. Schlepphorst, J. Schneidewind, W. Baumann and F. Glorius, Chem. Sci., 2022, 13, 985–995 RSC.
- Y. Makida, M. Saita, T. Kuramoto, K. Ishizuka and R. Kuwano, Angew. Chem., Int. Ed., 2016, 55, 11859–11862 CrossRef CAS PubMed.
- C. Schlepphorst, M. P. Wiesenfeldt and F. Glorius, Chem. – Eur. J., 2018, 24, 356–359 CrossRef CAS PubMed.
- T. Hu, L. Lückemeier, C. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 23193–23196 CrossRef CAS PubMed.
- J. Wysocki, C. Schlepphorst and F. Glorius, Synlett, 2015, 26, 1557–1562 CrossRef CAS.
- M. P. Wiesenfeldt, D. Moock, D. Paul and F. Glorius, Chem. Sci., 2021, 12, 5611–5615 RSC.
- C. Li and J. Xiao, J. Am. Chem. Soc., 2008, 130, 13208–13209 CrossRef CAS PubMed.
- J. Wu, C. Wang, W. Tang, A. Pettman and J. Xiao, Chem. – Eur. J., 2012, 18, 9525–9529 CrossRef CAS PubMed.
- J. Wu, W. Tang, A. Pettman and J. Xiao, Adv. Synth. Catal., 2013, 355, 35–40 CrossRef CAS.
- J. Wen, X. Fan, R. Tan, H. C. Chien, Q. Zhou, L. W. Chung and X. Zhang, Org. Lett., 2018, 20, 2143–2147 CrossRef CAS PubMed.
- M. Renom-Carrasco, P. Gajewski, L. Pignataro, J. G. de Vries, U. Piarulli, C. Gennari and L. Lefort, Chem. – Eur. J., 2016, 22, 9528–9532 CrossRef CAS PubMed.
- Z. Nairoukh, M. Wollenburg, C. Schlepphorst, K. Bergander and F. Glorius, Nat. Chem., 2019, 11, 264–270 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- M. Rowley, D. J. Hallett, S. Goodacre, C. Moyes, J. Crawforth, T. J. Sparey, S. Patel, R. Marwood, S. Patel, S. Thomas, L. Hitzel, D. O'Connor, N. Szeto, J. L. Castro, P. H. Hutson and A. M. Macleod, J. Med. Chem., 2001, 44, 1603–1614 CrossRef CAS PubMed.
- J. Wen, R. Tan, S. Liu, Q. Zhao and X. Zhang, Chem. Sci., 2016, 7, 3047–3051 RSC.
- Y. Duan, L. Li, M. W. Chen, C. Bin Yu, H. J. Fan and Y. G. Zhou, J. Am. Chem. Soc., 2014, 136, 7688–7700 CrossRef CAS PubMed.
- C. Bin Yu, J. Wang and Y. G. Zhou, Org. Chem. Front., 2018, 5, 2805–2809 RSC.
- W. B. Wang, S. M. Lu, P. Y. Yang, X. W. Han and Y. G. Zhou, J. Am. Chem. Soc., 2003, 125, 10536–10537 CrossRef CAS PubMed.
- X. F. Cai, W. X. Huang, Z. P. Chen and Y. G. Zhou, Chem. Commun., 2014, 50, 9588–9590 RSC.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed.
- F. Glorius, N. Spielkamp, S. Holle, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2004, 43, 2850–2852 CrossRef CAS PubMed.
- F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522–11569 CrossRef CAS PubMed.
- Y. T. Xia, J. Ma, X. D. Wang, L. Yang and L. Wu, Catal. Sci. Technol., 2017, 7, 5515–5520 RSC.
- Y. Zhang, J. Zhu, Y. T. Xia, X. T. Sun and L. Wu, Adv. Synth. Catal., 2016, 358, 3039–3045 CrossRef CAS.
- Y. Y. Huang, X. Yang, Y. Feng, F. Verpoort and Q. H. Fan, J. Mol. Catal. A: Chem., 2014, 393, 150–155 CrossRef CAS.
- X. Wang, J. Li, S. Lu, Y. Liu and C. Li, Chin. J. Catal., 2015, 36, 1170–1174 CrossRef.
- S.-M. Lu, X.-W. Han and Y.-G. Zhou, Adv. Synth. Catal., 2004, 346, 909–912 CrossRef CAS.
- X. Wang and Y. Zhou, J. Org. Chem., 2008, 73, 5640–5642 CrossRef CAS PubMed.
- K. H. Lam, L. Xu, L. Feng, Q.-H. Fan, F. L. Lam, W. Lo and A. S. C. Chan, Adv. Synth. Catal., 2005, 347, 1755–1758 CrossRef CAS.
- Z. J. Wang, G. J. Deng, Y. Li, Y. M. He, W. J. Tang and Q. H. Fan, Org. Lett., 2007, 9, 1243–1246 CrossRef CAS PubMed.
- L. Tao, C. Li, Y. Ren, H. Li, J. Chen and Q. Yang, Chin. J. Catal., 2019, 40, 1548–1556 CrossRef CAS.
- L. Tao, Y. Ren, C. Li, H. Li, X. Chen, L. Liu and Q. Yang, ACS Catal., 2020, 10, 1783–1791 CrossRef CAS.
- T. Wagener, A. Heusler, Z. Nairoukh, K. Bergander, C. G. Daniliuc and F. Glorius, ACS Catal., 2020, 10, 12052–12057 CrossRef CAS PubMed.
- T. Wagener, L. Lückemeier, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 6425–6429 CrossRef CAS PubMed.
- D. Moock, M. P. Wiesenfeldt, M. Freitag, S. Muratsugu, S. Ikemoto, R. Knitsch, J. Schneidewind, W. Baumann, A. H. Schäfer, A. Timmer, M. Tada, M. R. Hansen and F. Glorius, ACS Catal., 2020, 10, 6309–6317 CrossRef CAS PubMed.
- X. F. Cai, R. N. Guo, G. S. Feng, B. Wu and Y. G. Zhou, Org. Lett., 2014, 16, 2680–2683 CrossRef CAS PubMed.
- S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef CAS PubMed.
- C. Zheng and S. L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC.
- M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781–3783 CrossRef CAS PubMed.
- S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424–7427 CrossRef CAS PubMed.
- R. I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84–86 CrossRef CAS PubMed.
- D. Šterk, M. Stephan and B. Mohar, Org. Lett., 2006, 8, 5935–5938 CrossRef PubMed.
- C. Rampalakos and W. D. Wulff, Adv. Synth. Catal., 2008, 350, 1785–1790 CrossRef CAS PubMed.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC.
- S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884–4886 RSC.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 12968–12971 CrossRef CAS PubMed.
- T. H. Jones, M. S. Blum and H. M. Fales, Tetrahedron, 1982, 38, 1949–1958 CrossRef CAS.
- D. C. Sullivan, H. Flowers, R. Rockhold, H. M. T. B. Herath and N. P. D. Nanayakkara, Am. J. Med. Sci., 2009, 338, 287–291 CrossRef PubMed.
- Z. Zhang and H. Du, Angew. Chem., Int. Ed., 2015, 54, 623–626 CAS.
- S. Li, W. Meng and H. Du, Org. Lett., 2017, 19, 2604–2606 CrossRef CAS PubMed.
- Z. Zhang and H. Du, Org. Lett., 2015, 17, 6266–6269 CrossRef CAS PubMed.
- K. Yang, Y. Lou, C. Wang, L. W. Qi, T. Fang, F. Zhang, H. Xu, L. Zhou, W. Li, G. Zhang, P. Yu and Q. Song, Angew. Chem., Int. Ed., 2020, 59, 3294–3299 CrossRef CAS PubMed.
- D. S. Wang, Q. A. Chen, W. Li, C. Bin Yu, Y. G. Zhou and X. Zhang, J. Am. Chem. Soc., 2010, 132, 8909–8911 CrossRef CAS PubMed.
- Y. Duan, L. Li, M. W. Chen, C. Bin Yu, H. J. Fan and Y. G. Zhou, J. Am. Chem. Soc., 2014, 136, 7688–7700 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |