
Mechanistic insights into the pressure-induced polymerization of aryl/perfluoroaryl co-crystals†
Margaret C.
Gerthoffer
 a,
Bohan
Xu
b,
Sikai
Wu
a,
Jordan
Cox
c,
Steven
Huss
a,
Shalisa M.
Oburn
a,
Steven A.
Lopez
c,
Vincent H.
Crespi
abd,
John V.
Badding
abd and
Elizabeth
Elacqua
*a
a,
Bohan
Xu
b,
Sikai
Wu
a,
Jordan
Cox
c,
Steven
Huss
a,
Shalisa M.
Oburn
a,
Steven A.
Lopez
c,
Vincent H.
Crespi
abd,
John V.
Badding
abd and
Elizabeth
Elacqua
*a
aDepartment of Chemistry, The Pennsylvania State University, University Park, PA 16802, USA
bDepartment of Physics, The Pennsylvania State University, University Park, PA 16802, USA
cDepartment of Chemistry and Chemical Biology, Northeastern University, Boston, MA 02115, USA
dDepartment of Materials Science and Engineering, The Pennsylvania State University, University Park, PA 16802, USA
First published on 3rd February 2022
Abstract
Recently discovered diamond nanothreads offer a stiff, sp3-hybridized backbone unachievable in conventional polymer synthesis that is formed through the solid-state pressure-induced polymerization of simple aromatics. This method enables monomeric A-B alternation to fully translate from co-crystal design to polymer backbone in a sequence-defined manner. Here, we report the compression of aryl:perfluoroaryl (Ar/ArF) co-crystals containing –OH and –CHO functional groups. We analyze the tolerance of these functional groups to polymerization, explore the possibility of keto–enol tautomerization, and compare the reaction outcomes of targeted solid-state Ar/ArF design on nanothread formation. Two new co-crystals comprising phenol:pentafluorobenzaldehyde (ArOH:ArFCHO) and benzaldehdye:pentafluorophenol (ArCHO:ArFOH) were synthesized through slow solvent evaporation. Analysis of the single-crystal structures revealed different hydrogen bonding patterns between the –OH and –CHO in each solid (tape and orthogonal dimers, respectively), in addition to markedly different π–π stacking distances within the Ar/ArF synthons. In situ Raman spectroscopy was used to monitor the compression of each co-crystal to 21 GPa and illustrated peak shifts for the –OH and –CHO stretching regions during compression. Photoluminescence corresponding to polymerization appeared at a lower pressure for the co-crystal with the smallest π–π stacking distance. Nevertheless, the recovered solid with the larger centroid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :centroid and centroid:plane π–π stacking distances featured a diffraction ring consistent with the anticipated dimensions of a co-crystal-derived nanothread packing, indicating that both functional group interactions and parallel stacking affect the pressure-induced polymerization to form nanothreads. IR spectroscopy of the recovered samples revealed large shifts in the –OH & –CHO stretching regions, particularly noticable for ArCHO:ArFOH, which may reflect geometrical constraints associated with forming a rigid thread backbone under pressure. Simulation suggests that hydrogen bonding networks may affect the relative compressibility of the co-crystal along a thread-forming axis to modulate the propensity for nanothread formation.
:centroid and centroid:plane π–π stacking distances featured a diffraction ring consistent with the anticipated dimensions of a co-crystal-derived nanothread packing, indicating that both functional group interactions and parallel stacking affect the pressure-induced polymerization to form nanothreads. IR spectroscopy of the recovered samples revealed large shifts in the –OH & –CHO stretching regions, particularly noticable for ArCHO:ArFOH, which may reflect geometrical constraints associated with forming a rigid thread backbone under pressure. Simulation suggests that hydrogen bonding networks may affect the relative compressibility of the co-crystal along a thread-forming axis to modulate the propensity for nanothread formation.
Introduction
Nature uses complex machinery that promotes supramolecular self-assembly toward the design of precise sequence-defined architectures.1 Recent efforts in synthetic polymer chemistry have focused on using noncovalent interactions in monomer design to expand the range of sequence-defined architectures accessible.2,3 Such methods offer opportunities toward new donor–acceptor polymers as push–pull chromophores, sequence-defined structures, and structure–function tunable support systems.4–6 Yet the design of monomers (and/or intricate methods) presents a trade-off between resultant polymer properties, molecular weights, sequential defect sites, and complex design methodologies.Methods to achieve sequence definition often focus on a prescribed monomer order that directly translates into a given polymer sequence, utilizing electronics to either iteratively add monomers in a stepwise fashion7 or through step-growth polymerization.8 Other tactics using ring-opening metathesis polymerization (ROMP) can promote controlled ring-opening, attaining high degrees of sequence control.9,10 Entropy-driven ROMP, for instance, can be utilized to polymerize cyclic macromonomers with living character using enthalpic control.11 Meanwhile, selective installation of a conjugated sequence has been accomplished using electronically governed ROMP, wherein the polymerization of donor–acceptor-based monomers has afforded sequence control mediated by electronic and steric effects.9 Using a similar electronic design, supramolecular strategies have realized sequence-controlled electrosynthesis to form organometallic polymers12 and the growth of discrete precursors from orthogonal coordination-driven self-assembly and hydrogen bonding.2,13 Thus, supramolecular polymerization offers concise monomer design that prudently plans directional routes for polymerization through noncovalent interactions.14
Supramolecularly guided methods for sequence definition have recently been applied in the synthesis of diamond nanothreads. Nanothreads are 1D polymers synthesized through the pressure-induced polymerization of small aromatic molecules (e.g. benzene),15,16 along a stack of molecules in the solid state. These polymers, theorized to form through a series of [4 + 2] cycloadditions,17,18 have been reported from several monomers, including pyridine,19,20 furan,21,22 thiophene,23 aniline,24 and azobenzene.25 Notably, the compression of benzene had been performed for decades prior to nanothread discovery, but primarily lead to amorphous carbon as a result of multiple kinetically favorable reaction pathways opening upon quick collapse of the crystal.26–29 Upon the discovery of a concise method to reproducibly form nanothreads from benzene requiring slow compression,15,16 interest in these polymer threads has been fueled by their potential use as high tensile strength materials,30,31 along with possible applications envisioned as novel semiconductors32 or catalyst-anchoring platforms, with highly sensitive properties dependent on monomer composition.33
Alternating copolymer nanothreads have been reported through the compression of co-crystals, in which two monomers crystallize in a singular lattice, generally supported by strong noncovalent interactions (e.g., hydrogen bonds and quadrupole forces).34 Aryl:perfluoroaryl interactions (Ar/ArF)35–44 in co-crystals imbue supramolecularly guided sequence definition through alternating electron-rich and electron-poor monomers and have been utilized in many systems including synthetic foldamers,39,40 and liquid crystals.43 Compression of octafluoronaphthalene co-crystals with naphthalene (i.e, C10H8:C10F8)45,46 or anthracene46 form nanothreads without the need for slow kinetically controlled compression, likely owing to preferential shrinkage of the Ar/ArF stacking distance upon pressure application and a more geometrically robust stacking in these larger, non-substituted aromatics. Similar discoveries have been illustrated for para-substituted benzenes with cyano, ethynyl, or nitro functionalities in which co-crystals with the smallest stacking distances require less extreme synthetic conditions.47 Pressure-induced phase changes may also affect reaction outcomes by disrupting stacking geometries favorable to 1D pressure-induced polymerization. A co-crystal of C6H6:C6F6, which illustrates close π–π stacking at ambient pressure, undergoes a high-pressure phase change that enables closer contacts between adjacent π-stacks as opposed to within the Ar/ArF stack, yielding a graphane-like structure.48 In contrast, a phenol:pentafluorophenol co-crystal features pressure-induced phase changes, yet affords alternating copolymer nanothreads.49
Given nanothreads are synthesized from diverse monomers that vary in electrostatics, crystal structures, intermolecular interactions, and degrees of aromaticity – and polymerize using different conditions – an underlying criterion is likely guiding the mechanism toward reaction. Whereas comparisons of π-stacked para-disubstituted benzenes,47 including co-crystals, have revealed a trend between the slippage angle and the necessity for slow compression and/or heat, the direct effect of external functionalities remains largely unexplored.
Here, we report the structures of two new Ar/ArF co-crystals ((i) phenol:pentafluorobenzaldehyde and (ii) benzaldehyde:pentafluorophenol (coined ArOH:ArFCHO and ArCHO:ArFOH, respectively)) and detail their pressure-induced polymerizations toward saturated sequence-defined polymeric architectures. The resultant polymers, obtained from the compression of individual co-crystals, feature pendant –OH and –CHO functionalities. Our design interrogates functional group tolerance from pressure-induced reactions, including the potential for keto–enol tautomerization.49 We observe that the ArOH:ArFCHO co-crystal reacts at a lower pressure than the ArCHO:ArFOH co-crystal, likely owing to a lower reaction barrier. IR spectroscopy of the recovered polymers illustrates functional group preservation, while the ArCHO:ArFOH-derived product's X-ray diffraction features a ring at a d-spacing range that suggests successful polymerization. This complementary pair of co-crystals therefore offers a strategic view into the effects of geometry through hydrogen bonding and Ar/ArF electronics on precursor design for synthesizing sequence-defined polymeric nanothreads.
Experimental
Co-crystal synthesis
Co-crystals selected for this study focused on opposing electronically activating/withdrawing groups (–OH/–CHO, respectively) to analyze the effects of ambient-pressure crystal structure on the pressure-induced reactivity. Phenol (≥99%, reagent grade, solid, Sigma Aldrich), pentafluorophenol (>99%, reagent grade, low-melting solid, Sigma Aldrich), benzaldehyde (>99%, reagent grade, liquid, Alfa Aesar), pentafluorobenzaldehyde (>98%, reagent grade, low-melting solid, Oakwood Chemicals) were used as purchased for co-crystal growth. Co-crystallization of ArOH:ArFCHO (MP = 53.4–54.0) and ArCHO:ArFOH (MP = 38.0–42.1) was achieved by mixing each of the two desired components in a 1:1 equimolar ratio within a scintillation vial containing hexane. The solvent was slowly evaporated over the course of three days to reveal clear solid crystals. Data collection parameters, key intermolecular interactions, the experimental specs for instrumentation (Tables S1 and S2†) and figures of the molecular structures with details provided on stacks in the unit cell (Fig. S1and S2†) are provided in the ESI.†
High-pressure synthesis and in situ Raman spectroscopy
To achieve high pressures, a symmetric diamond anvil cell (DAC) equipped with 400 μm diameter culets of Type IIA diamonds was used to compress each co-crystal to 21 GPa. T301 stainless steel gaskets were pre-indented between 45 and 55 μm and subsequently drilled with an 80 μm hole (using an electric discharge machine or a laser-drilling system50) to serve as a sample chamber. Each co-crystal was loaded in the DAC by scooping a small amount of crystal over the pre-indented drilled hole in the gasket when placed on the bottom diamond with a ruby chip without a pressure transmitting media.51 The DAC was closed and pressurized to circa 2–3 GPa to form a powder sample. For compressions of polycrystals, the DAC was heated externally with a heat gun. The pressure was then released manually using the DAC screws to melt the heated and pressurized co-crystal until small seed crystals remained. Pressure was then slowly reapplied to grow the sample into an array of crystals using the in situ seeds. Retention of co-crystallinity under pressure was evaluated from concise shifts in individual precursor peaks by Raman spectroscopy.52Raman spectroscopy during compression monitored for potential chemical changes during reaction, including modifications related to external functional groups. Slow compression was applied using a double-membrane Druck gas controller at a rate of 0.08–0.10 GPa min−1 from 2 to 10 GPa, 0.05–0.07 GPa min−1 from 10 to 15 GPa, 0.03–0.04 GPa min−1 from 15 to 18 GPa, and 0.01–0.02 GPa min−1 from 18 to 21 GPa, resulting in a ∼24 hours compression overall, including the collection time for Raman spectra. Slower compression rates were initiated upon the observation of photoluminescence and continued up to the maximum pressure to facilitate production of nanothread products as previously reported for benzene-type systems.16 Mirrored rates were followed for decompression.
Results and discussion
Ambient pressure co-crystal X-ray diffraction
X-ray diffraction (XRD) was collected for the ArOH:ArFCHO and ArCHO:ArFOH co-crystals to discern the Ar/ArF stacking distance and hydrogen-bonding pattern variances for the complementary co-crystals (Fig. 1). When the electron-withdrawing group is on the fluorinated ring, as for ArOH:ArFCHO, the π–π stacking distance (centroid:centroid, defined as dc) is 3.53–3.58 Å with a modest slippage angle ranging from 17.4° to 21.2° along the stacks. The centroid:plane distance (dp) ranges from 3.30–3.46 Å. The varied ranges of the slippage angle and dp arises from non-perfectly parallel planes in the Ar/ArF co-crystal. In contrast, placement of the electron donating –OH group on the electron deficient perfluorinated ring as in ArCHO:ArFOH yields a larger dc π–π stacking distance of 3.96–3.98 Å and dp of 3.36–3.44 Å with an angle of 29.8–32.2°. Lessons from threads with only core carbon rings (e.g., from benzene and C10H8:C10F8) suggest that stacking geometry controls whether slow compression is needed (benzene)15,16 or not (C10H8:C10F8)45,46 to obtain crystalline polymers. This line of reasoning may suggest that nanothread formation is more plausible from ArOH:ArFCHO than ArCHO:ArFOH owing to the larger slippage angle and Ar/ArF distance between the centroids and non-perfectly parallel planes in the ArCHO:ArFOH co-crystal. However, benzene and naphthalene derivatives do not contain external functionalities, which exhibit the potential to both crosslink and modulate potential high-pressure phase transitions due to steric bulk, lower-symmetry packing, and competing intermolecular interactions. Diethynylbenzene:dicyanobenzene and dinitrobenzene:dicyanobenzene co-crystals, although not Ar/ArF systems, exhibit close π-stacking, with a dp of 3.49 and 3.54 Å respectively (dc for both = 3.78 Å). Both co-crystals feature preferred packing orientation after pressure-induced polymerization;47 however, no hydrogen bonding is present in these co-crystals, though their functional groups present a higher propensity for cross-linking than aldehydes or alcohols. In the ArOH:ArFCHO co-crystal, the functional groups at ambient pressure hydrogen bond between the –OH and –CHO in a continuous tape pattern with the functional groups within a stack eclipsed to one another. In contrast, hydrogen bonding between –OH and –CHO in the ArCHO:ArFOH co-crystal does not extend along the stack but forms inter-stack dimers that have a gauche orientation along a stack. In engineering the Ar/ArF stacks, no direct control was sought over the functional group interaction, yet an interesting contrast was obtained in this aspect as well. The ArCHO:ArFOH/ArOH:ArFCHO comparison thus provides opportunity to extract lessons towards nanothread synthesis not only regarding Ar/ArF distance and slippage angle, but also in the geometry of intermolecular functional group interactions.
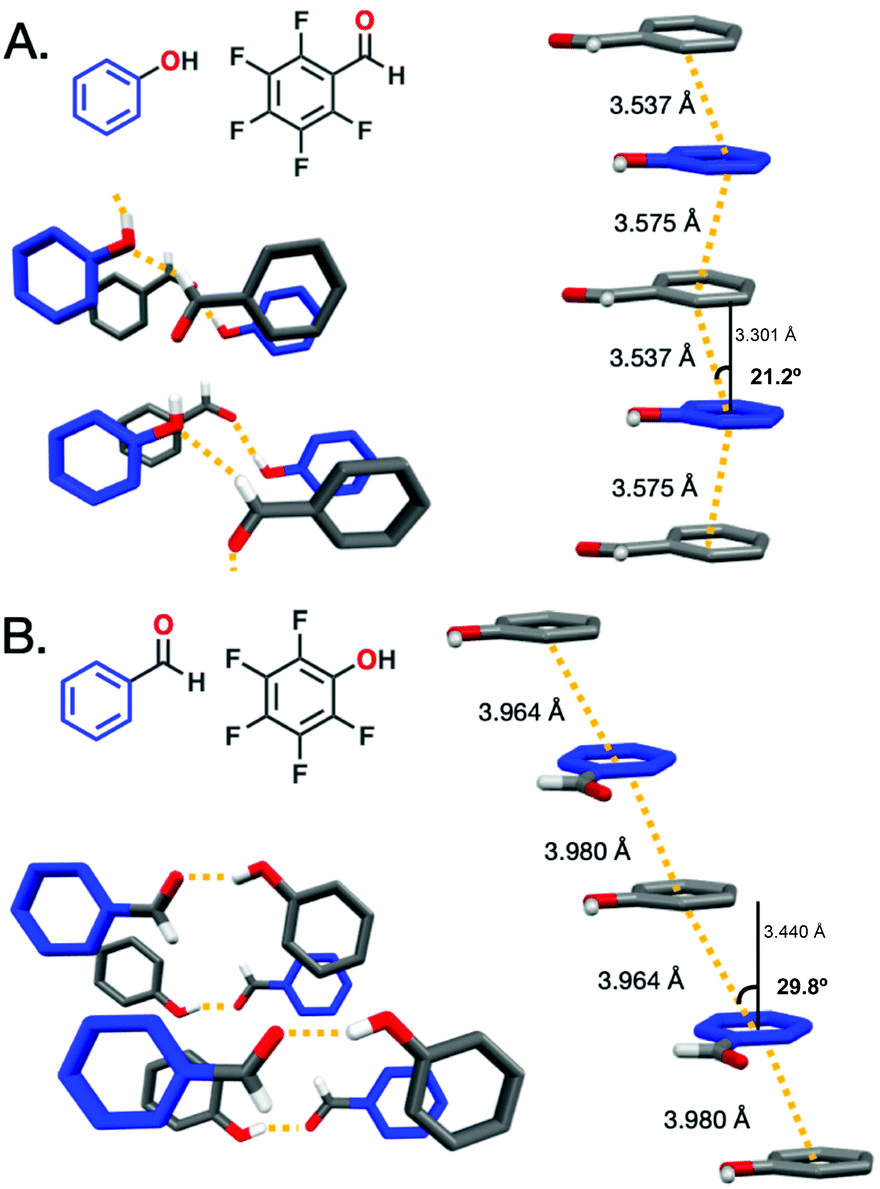 | ||
| Fig. 1 View of each perspective co-crystal along both the aryl:perfluoroaryl stacks and the hydrogen-bonding pattern. Shorter stacking distances and “tape-like” hydrogen-bonding patterns are observed for ArOH:ArFCHO (A) while longer stacking distances and dimer-like hydrogen-bonding patterns are observed for ArCHO:ArFOH (B). | ||
Simulation of co-crystal overcompression
Noting these two potential influences on reaction outcomes, simulations were performed to monitor (i) Ar/ArF centroid:centroid separations, (ii) centroid:plane separations, (iii) slippage angle, and (iv) functional group relationships during hydrostatic compression. Each experimentally obtained co-crystal structure was relaxed by first-principles density functional theory with the Perdew–Burke–Ernzerh of exchange correlation functional53,54 and D3(BJ) dispersion correction55 at pressures from 0 to 22 GPa in 2 GPa increments, starting from the experimental co-crystal structure at ambient pressure. Various intermolecular contacts were monitored at each pressure point, including dc, dp, and (phenol) O–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (aldehyde) hydrogen bonding distances for each respective co-crystal (Fig. 2). As pressure is applied, both the dc and dp begin to collapse to shorter distances within each co-crystal monomer. Due to alternating distances of dc and dp in each co-crystal stack as illustrated in Fig. 1, we present the theoretical collapse as a range of potential distances. For both monomer pairs, reduction of the Ar/ArF centroid:centroid separation was more rapid than that between the hydrogen-bonded aldehyde/alcohol, especially below 5 GPa. However, the rate of collapse along the Ar/ArF stack was faster for the ArCHO:ArFOH co-crystal that exhibited longer stacking distances initially at ambient pressure. We observe general shortening of hydrogen bonding up to 22 GPa, maintaining the same geometry of functional group pairings for both co-crystals. The ArOH:ArFCHO co-crystal hydrogen-bond separation is longer at ambient pressure but shrinks at a faster rate than for the ArCHO:ArFOH co-crystal.
C (aldehyde) hydrogen bonding distances for each respective co-crystal (Fig. 2). As pressure is applied, both the dc and dp begin to collapse to shorter distances within each co-crystal monomer. Due to alternating distances of dc and dp in each co-crystal stack as illustrated in Fig. 1, we present the theoretical collapse as a range of potential distances. For both monomer pairs, reduction of the Ar/ArF centroid:centroid separation was more rapid than that between the hydrogen-bonded aldehyde/alcohol, especially below 5 GPa. However, the rate of collapse along the Ar/ArF stack was faster for the ArCHO:ArFOH co-crystal that exhibited longer stacking distances initially at ambient pressure. We observe general shortening of hydrogen bonding up to 22 GPa, maintaining the same geometry of functional group pairings for both co-crystals. The ArOH:ArFCHO co-crystal hydrogen-bond separation is longer at ambient pressure but shrinks at a faster rate than for the ArCHO:ArFOH co-crystal.
 | ||
| Fig. 2 Measured intermolecular interactions of simulated compressed co-crystals up to 22 GPa. Distances include centroid:centroid dc (purple), centroid:plane dp (black), and intra-stack O⋯O hydrogen bonding (red) of each co-crystal ArOH:ArFCHO (top) and ArCHO:ArFOH (bottom). | ||
The interplane distances (dp) are ∼2.8 Å between 14–16 GPa (Fig. 2), similar to the interplane distances for co-crystals of octafluoronaphthalene with naphthalene or anthracene at initation.45,46 Since –CHO and –F functionalities are both electron withdrawing while –OH groups are electron donating, we expect ArOH:ArFCHO to maintain a shorter interplane distance than ArCHO:ArFOH due to the stronger attraction between paired rings. However, the simulations suggest that the interplane distances of the two co-crystals are similar in intermolecular separation throughout their compression.
The aldehyde and hydroxyl groups of the ArCHO:ArFOH co-crystal dimerize through hydrogen bonding (Fig. 1 and 2). The hydrogen bonded dimers form layers of parallelly aligned planes of molecules. Moreover, the hydrogen bond dimer is orthogonal to the Ar/ArF stacking direction. In contrast, ArCHO:ArFOH's functional groups form an extended linear tape rather than dimers between eclipsed Ar/ArF pairs due to the closest proximity of nearest neighbor –OH and –CHO. This hydrogen bonding pattern does not illustrate the previously mentioned layering behavior and the direction of the hydrogen bond is not orthogonal to the stacks. The change in slippage angle upon pressure induction also differs between the co-crystals (Fig. S7†). For ArOH:ArFCHO, the slippage angle increases upon pressure induction consistently for all non-symmetric stacks. For ArCHO:ArFOH, the slippage angle decreases along one stack and increases along another stack, suggesting that the hydrogen bonding pattern is regulating the ability of the stack to collapse. The tendency of ArCHO:ArFOH to form layers and exhibit direct hydrogen-bonding orthogonality to the Ar/ArF axis may guide the inter-stack hydrogen-bond distance to be relatively maintained. Summation of the inter-stack and intra-stack interactions and their relative alignment to the thread-formation axis suggests that an unknown balance of hydrogen bonding and Ar/ArF interactions could have the potential to govern the experimental outcome for these co-crystal pairs.
Dimer energetic calculations
With the potential for multiple intermolecular interactions to affect polymerization, we thus investigated gas-phase barrier calculations to elucidate the respective [4 + 2] dimer energies necessary for polymerization. Calculations revealed gas-phase dimer energies for the ArOH:ArFCHO and ArCHO:ArFOH co-crystals were 55.3 and 62.4 kcal mol−1 respectively, which is similar to that reported for benzene-derived threads at 68 kcal mol−1 and ArOH:ArFOH-derived threads at 60.3 kcal mol−1.49 The relative energy necessary to make the dimer for each co-crystal is thus on par with prior reported threads. Collapse of both co-crystals along the centroids provides intuition towards valid precursors for controlled polymerizations under pressure.High pressure in situ Raman spectroscopy
With viable candidates to form nanothreads, each co-crystal was individually loaded into a diamond anvil cell (DAC) as a polycrystal for polymerization without a pressure transmitting medium. Perfect uniaxial stress is thus not achieved while monitoring in situ Raman spectroscopy, allowing for collapse of the polycrystalline and powder samples in all directions, resulting in additional texture grains from the images illustrated in Fig. 3 upon pressure application. In situ Raman spectroscopy (Fig. 3) performed under pressure for both compressed powder and polycrystalline samples of each co-crystal provides information on possible structural phase transitions and the initiation of polymerization reactions. Compression of ArOH:ArFCHO up to 21 GPa reveals multiple changes in Raman vibrational bands (Fig. 3, top), including the disappearance of a broad band at 3250 cm−1 attributed to aromatic sp2 C–H stretching. Continuously throughout compression, the aldehyde CO peak at 1700 cm−1 broadens, accompanied by an increase in photoluminescent background indicative of further chemical reaction connected to a reduction in band gap. This steady broadening implies that the (phenol) O–H⋯OC (aldehyde) hydrogen bonded tape non-orthogonal to the Ar/ArF centroid:centroid stacks is maintained, but obtains disorder as a result of pressure. Further evidence toward structural complexity under pressure is provided by C–C aromatic ring vibrations around 1400–1500 cm−1 that begin to split between 5–9 GPa and become two distinct peaks by 11.1 GPa. Such peak splits can follow a pressure-induced symmetry breaking within an aromatic ring that more strongly distinguishes previously symmetry-equivalent (or near-equivalent) carbon atoms within the ring.56–58 These two structural changes may suggest an increased diversity of reaction pathways becoming available to the ArOH:ArFCHO co-crystal under pressure. Further peak broadening of C–F vibrations around 550 cm−1 from 11 GPa upwards suggests changes in halogen bonding between adjacent Ar/ArF stacks that could potentially facilitate inter-stack crosslinking. This may emerge from crystal cracking under non-perfect uniaxial strain as featured in prior nanothread syntheses.19,21,49 Upon further compression the broad photoluminescent background rises, masking further spectral changes; this is a common observation in pressure-induced polymerization.59 We note that the emergence of the photoluminescent background above 11 GPa is at a lower pressure than that reported for the pressure-induced polymerization of ArOH:ArFOH threads at 15.7 GPa,49 an Ar/ArF co-crystal that exhibits a larger average centroid:centroid stacking distance than that of ArOH:ArFCHO. Peak broadening also occurs at a far lower pressure (14 GPa) as compared to the ArOH:ArFOH co-crystal,49 indicating that crystal cracking, amorphous material formation, or polymerization could be occuring.
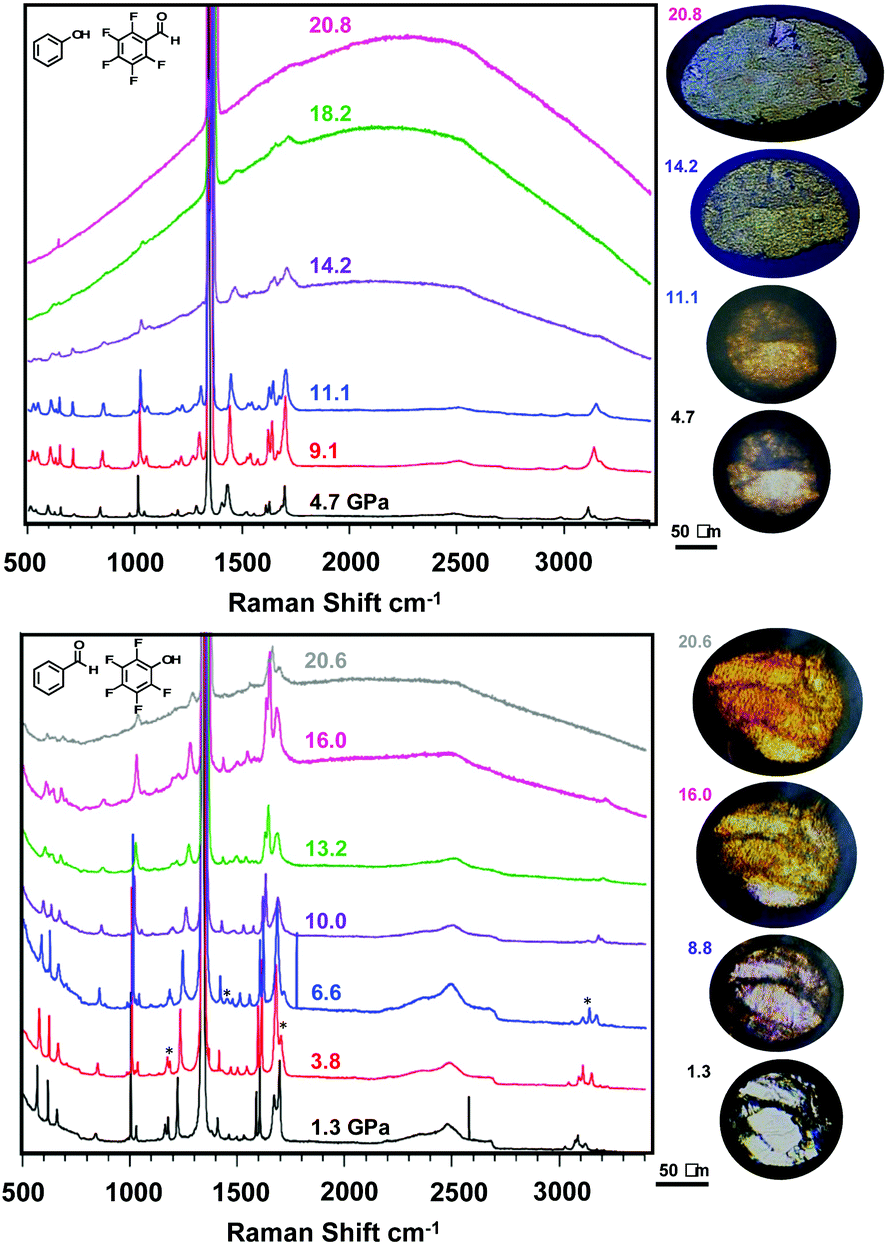 | ||
| Fig. 3 In situ Raman spectroscopy collected during polymerization of ArOH:ArFCHO (top) and ArCHO:ArFOH (bottom), with respective images during polymerization featured to the right. For the ArCHO:ArFOH, bands featuring the onset of the suspected phase transition at 3.8 GPa, continuing to 6.6 GPa are highlighted by an asterisk (*). | ||
Raman spectroscopy during the compression of ArCHO:ArFOH to similar pressures (Fig. 3, bottom) reveals a broad photoluminescent background that rises at 7 to 10 GPa, a lower pressure than for ArOH:ArFCHO. The ArCHO:ArFOH co-crystal underwent at least one structural phase transition between initiating at 3.8 GPa that was not observed by theory, as evidenced by changes in the C–H stretching region around 3000 cm−1, CO stretching around 1700 cm−1, aryl ring vibration around 1200 cm−1, and the emergence of a new band below 1000 cm−1. This structural phase transition (like the co-crystal itself) is not reported and would be difficult to solve to completeness at these pressures. We can infer, however, that the phase change likely altered the intermolecular interactions of the benzaldehyde component in the co-crystal, as all bands that changed are singularly present in benzaldehyde. For example, the peaks associated with both CC and CO stretching continue to broaden up to 10 GPa, indicating the accumulation of disorder. The experimental Raman spectra of the two co-crystals, the heightened photoluminescent background in ArCHO:ArFOH, the symmetry lowering in ArOH:ArFCHO from the tape hydrogen bonding pattern, and the hints of possible inter-stack close contacts developing under pressure in ArOH:ArFCHO, may suggest a higher propensity for columnar polymerization into nanothreads in ArCHO:ArFOH than in ArOH:ArFCHO.
Recovered X-ray diffraction
X-ray diffraction collected on the recovered sample from the co-crystal with the shorter π–π stacking distance (i.e., ArOH:ArFCHO) reveals primarily amorphous scattering upon polymerization, while the ArCHO:ArFOH-derived polymer shows a broad diffraction ring from d = 6.0–7.2 Å (maximum intensity at d = 7.07 Å; Fig. 4). While the origin for these differences isn't fully understood, we suspect that the unusual increase of slippage angle in ArOH:ArFCHO upon increasing pressure and the difference in hydrogen bonding patterns that guide layers of Ar/ArF stacks allows for the ArCHO:ArFOH to retain better order during nanothread formation. | ||
| Fig. 4 (A and B) X-ray diffraction and recovered microscopy images with (top) and without cross-polarizers (bottom) of polymers formed from ArOH:ArFCHO (gray) and ArCHO:ArFOH (red). (C) Comparison of experimentally recovered diffraction of ArCHO:ArFOH to theoretical packings, and (D) Thread 1. | ||
Although weaker than the diffractive features of most crystalline inorganic materials, the diffraction ring derived from ArCHO:ArFOH is much sharper than is typically seen in organic polymers and thus reflects an usually high degree of polymeric order. Under polarized light, both polymer products appear to exhibit birefringent characteristics, but the ArCHO:ArFOH co-crystal product is brighter, potentially reflecting an optical anisotropy consistent with a more thread-like mesoscale morphology (Fig. 4).
Unlike nanothreads derived from benzene,15 thiophene,23 furan,21 pyridine,19 naphthalene:octafluoronaphthalene,45,46 and others24,45,49 which show the six diffraction spots of a quasi-hexagonal packing,16 the diffraction ring formed by polymerization of ArCHO:ArFOH indicates the presence of domains with multiple azimuthal orientations around the local thread axis (in addition to wandering of the thread axis about the axis of compression). That this result is obtained regardless of whether the monomer is loaded as a powder or polycrystal – in contrast to the more orientationally ordered outcomes obtained for the aforementioned precursors – may suggest a relation to the distinct character of functionalization in pressure-polymerized ArCHO:ArFOH. The relatively sparse interthread hydrogen bonding between –OH and –CHO functionalities affords a diversity of interthread separations in an ideal packing (Fig. 4C); any azimuthal irregularities in the placement of these functionalities around the threads’ axes in a real crystal would then cause the orientation of the domain to wander about the thread axis as the “tight-packed” and “loose-packed” directions vary from place to place. The outcome would be a diffraction ring aligned generally to the axis of compression and with a width corresponding to the span from the shortest to the longest d-spacings of the ideal packing.
To interrogate this notion, Fig. 4C shows the radial diffraction profile of compressed ArCHO:ArFOH compared to simulated diffraction from both the molecular co-crystal and from well-ordered packings of candidate thread structures that follow the two most likely [4 + 2] cycloaddition reaction outcomes based on the closest carbon–carbon contact distances (Fig. S13†), denoted Thread 1 and 2. Given the large number of isomers for nanothreads, it is difficult to determine if two threads are the exact experimental product. For the purpose of this work, we are using two thread candidates to estimate the diffraction ring's d-spacing, which mostly depend on the approximate positioning of functional groups, rather than exact structure. All simulations were relaxed at the same level of convergence with an empirical 2% thermal expansion applied to these otherwise T = 0 K calculations. The three major peaks correspond to the three diffraction planes of a quasi-hexagonal packing (shown in Fig. 4D for the packing of Thread 1), showing the role of functional group interaction in modulating the d-spacing. This range in d-spacing corresponds reasonably well to the experimental ring width, lending a measure of credence to this model. The co-crystal, in contrast, has a prominent peak above 7.5 Å. The presence of a ring rather than spots for diffraction from threads derived from ArCHO:ArFOH would not be surprising.
While the exact structure and its packing are still to be determined, the simulated d-spacing corresponds reasonably well to the experimental ring width. This diffraction ring is distinct from the diffraction formed from nanoribbons60,61 or the graphane-like structure produced by the compression of a C6H6:C6F6 co-crystal.48 Furthermore, the diffraction ring is also not from the residual co-crystal as the prominent peak for the co-crystal occurs above 7.5 Å. While the experiment and simulation suggest that the product is likely to be nanothread, the confirmation of thread formation still requires further data, such as solid-state NMR spectroscopy and TEM. However, we note that compared to the naphthalene:octafluoronaphthalene45 and anthracene:octafluoronaphthalene co-crystals that exhibit sharp diffraction peaks,45,46 the co-crystals studied in this paper are weakly diffracting to other 1D nanoarchitectures formed under pressure. This is perhaps due to the thinner aromatic cores of the singularly-substituted benzene-rings and weak hydrogen-bonding over the polycyclic arenes. Yet, a weaker diffraction is unsurprising and not discouraging, as 1D rods with weaker affinity to pack tightly (combined with the additional utility of commodity functional groups) would be easier to pull apart for practical material use in solvation, suspensions, interchelations, and post-polymerization functionalization.
Recovered IR spectroscopy
IR spectroscopy of the recovered samples after compression revealed sp3-hybridized carbon (2900 cm−1) in both polymer products (Fig. 5, 6 and S8–11†), as anticipated. Remaining, unreacted co-crystal and/or partially saturated portions of threads are still observed in both samples as illustrated by remaining CC and sp2-hybridized C–H stretching. Moreover, O–H (3400 cm−1) and aldehyde CO peaks (1700 cm−1) were observed in both recovered samples and shifted significantly post-compression, suggesting their tolerance to polymerization. After compression of ArOH:ArFCHO, the O–H stretching mode broadens and shifts to higher frequency, a change commonly associated with changes in hydrogen bonding or hybridization compared to the co-crystal precursor (Fig. 5).
 | ||
| Fig. 5 IR spectrum of precursor ArOH:ArFCHO co-crystal prior to compression (black, bottom) and polymer after compression (top, red), featuring the shift to higher wavenumbers of both the –OH and CO stretching. | ||
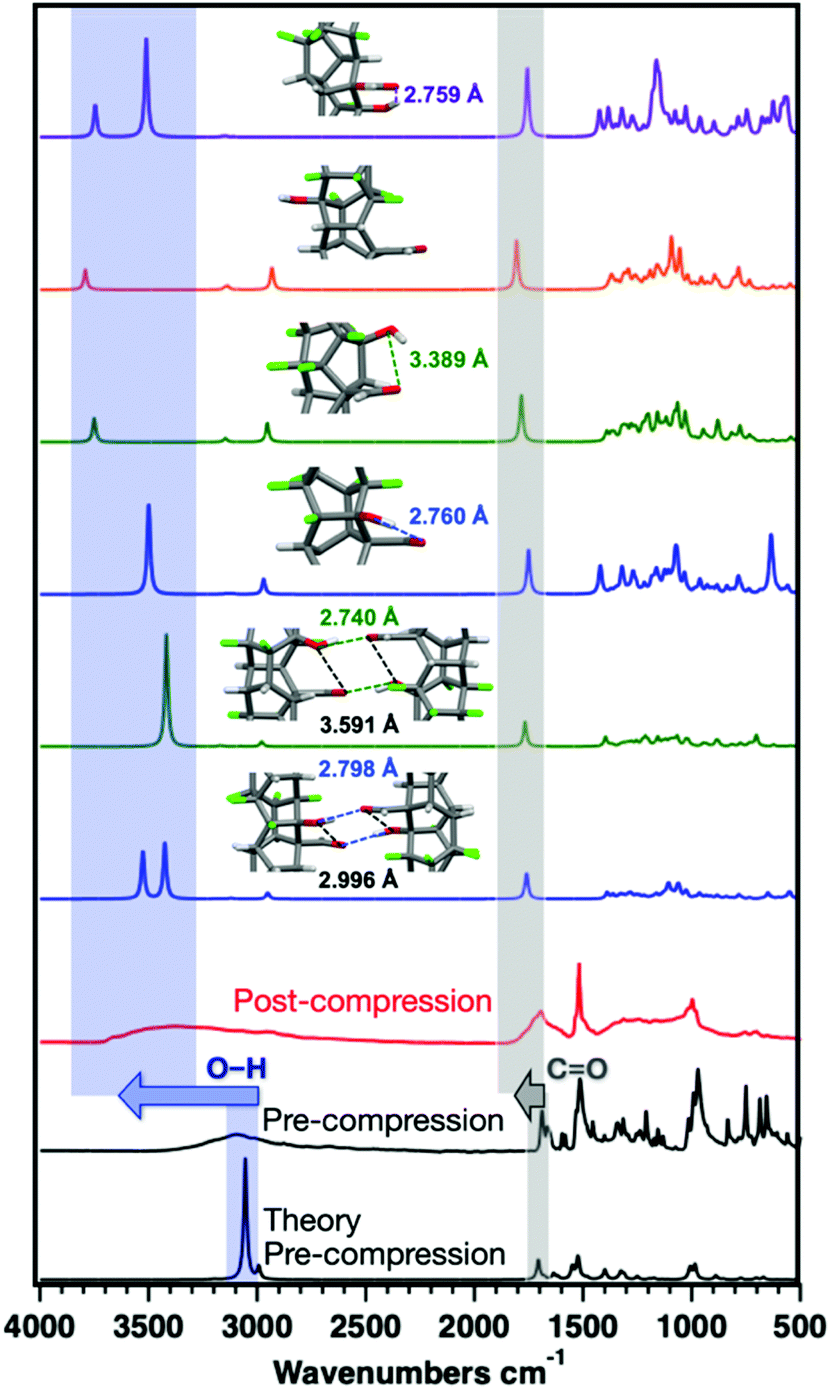 | ||
| Fig. 6 IR spectrum of ArCHO:ArFOH co-crystal from bottom to top as follows: theoretical co-crystal simulation (black), experimental co-crystal prior to compression (black), polymer after compression (red), the following simulated threads with respective packings of Thread 1 (blue) and Thread 2 (green), and simulations of isolated individual threads: Thread 1 (blue), Thread 2 (green), Thread 3 (orange), and Thread 4 (purple). | ||
Much larger O–H stretching shifts are observed for the ArCHO:ArFOH-derived polymer that yielded a more well-ordered structure. These frequency shifts could arise from changes in hybridization on the polymer backbone, electron delocalization around the O–H group in the perfluoroaryl ring, or functional group interactions between threads or along a given thread. We thus simulated the IR spectra of the Thread 1 and Thread 2 candidates for an ordered ArCHO:ArFOH-derived polymer (Fig. 6) as both isolated threads and crystal-packed threads, where Thread 2 has significantly weaker intra-thread hydrogen bonding than Thread 1 (Fig. S16†). We also examined a backbone that eliminates hydrogen bonding entirely by placing the functional groups para to one another (Thread 3), and another that features carboxylic acids in place of the aldehydes (Thread 4), to further elucidate effects of functionality on vibrational shifts.
The difference of the –OH stretching frequency between packed and isolated threads (Thread 1 and 2) can be as large as 300 cm−1 due to the presence of interthread hydrogen-bonding when packed, suggesting that interthread interactions can play an important role in the observed upshifts (Fig. 6). Moreover, simulated threads with stronger intra-thread H-bonding have lowered O–H stretching frequencies. Thus, the increase in frequency of the O–H and CO vibrations after compression can be attributed to constrained intermolecular interactions as a result of the formation of a stiff, multiply connected sp3 backbone. This stiff backbone prevents the free rotation of functional groups to find the most enthalpically favorable interaction, as would be available in solution-phase co-crystal synthesis. Moreover, simulations of Thread 3, in which hydrogen bonding is eliminated, produce maximal upshifts in the O–H and CO frequencies as compared to the isolated precursor, suggesting that the upshifts observed for other threads are not due to the direct change in hybridization of the carbon atoms involved.
Recovered X-ray photoelectron spectroscopy
Both recovered polymers were analysed using XPS to gain further insights on their overall compositions (Tables S7 & S8†). The high-resolution carbon spectrum of the ArCHO:ArFOH-derived polymer reveals a small fraction of CF2 which can be attributed to keto–enol tautomerization, akin to that observed in nanothreads derived from ArOH:ArFOH.49 This is also supported by the much higher-than-anticipated prevalence of C–O. XPS of this polymer product confirms that a large amount of precursor monomer is recovered, as components supporting oxidation of the aldehyde into a carboxylic acid are observed (i.e., COO). According to the high-resolution oxygen spectrum, O–CF was also confirmed for the ArCHO:ArFOH-derived polymer, suggesting that detachment of fluorine atoms is possible during polymerization. If either of the functional groups crosslink with one another, an ether-like functionality could arise, resulting in consumption of some CO. However, since O–C–O, OC, and C–F are indistinguishable in XPS, the relative amounts of crosslinking, fluorinated carbons, and the remaining aldehyde cannot be deconvoluted and quantified. For the ArOH:ArFCHO-derived polymer, no CF2 or COO is observed in the high-resolution carbon spectrum. Rather, an unusually high amount of C–O relative to CO is present, which would be expected if keto–enol tautomerization readily occurred.
Conclusions
This work has explored the impact of competing intermolecular interactions on the ability of a well-stacked Ar/ArF monomer to be consumed under pressure into an ordered thread-like product. We compressed two monomer pairs – whose preorganization was fixed through aryl/perfluoraryl (Ar/ArF) interactions – of the same chemical composition to witness remarkable functional group tolerance under pressure en route to forming saturated architectures. The two new Ar/ArF co-crystals, ArOH:ArFCHO and ArCHO:ArFOH, exhibit contrasting solid-state frameworks, with ArOH:ArFCHO possessing closely eclipsed stacks maintained by a tape-like hydrogen bonding pattern offset from the Ar/ArF stacks while ArCHO:ArFOH witnesses slipped Ar/ArF stacks with orthogonal hydrogen bond dimers perpendicular to the polymerization axis, enabling more facile stack collapse. Although solid-state favorability towards columnar polymerization was originally anticipated to be governed by centroid separation at ambient pressure, solids recovered from putatively non-ideal stacks in ArCHO:ArFOH exhibited the greatest experimental support of polymerizing in a singular direction by yielding a crystalline sample with a diffraction ring on par with anticipated nanothread packings. Moreover, simulations of the IR spectra of the ArCHO:ArFOH nanothread product support candidate thread structures, relating shifts in both the O–H and CO aldehyde vibrations that lend toward alterations in hydrogen bonding as a result of backbone formation.
Our evidence, combined with insights from prior studies, points to a summation of intermolecular interactions as key to the design of pressure-induced stack collapse. Future work aims at further elucidating the control necessary to dictate high-pressure phases using external stimuli (e.g., heat), and/or varied compression rates to maximize the crystalline output for each co-crystal. Further co-crystal monomers possessing strong intermolecular interactions directly perpendicular to the stacking axis may lend toward achieving anew nanothread architectures with designed functional group supports. The success of forming threads from precursors compressed herein thus offer design opportunities to target dual functional sequence-controlled nanothreads.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The funding for this work was provided by the Center for Nanothread Chemistry, a National Science Foundation (NSF) Center for Chemical Innovation (CHE-1832471). We would like to thank the Argonne National Lab and the Advanced Photon Source (APS). In particular, we would like to send our sincere thanks to the HPCAT staff, in particular Dr Changyong Park, for the use of the beamline 16-BM-D. At Pennsylvania State University, we would like to thank Dr Hemant Yennawar for his assistance with single-crystal X-ray diffraction (CHE-0131112), along with Dr Jeffrey Shallenberger for his assistance with XPS. We extend our sincere thanks to Jihyeong Wu, Vincent Torres, Dr Hee Jeung Oh, and Dr Robert Hickey for collecting DSC. We finally thank the Materials Characterization Laboratory in the Materials Research Institute at the Pennsylvania State University for the use of infrared spectroscopy and XPS.Notes and references
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed
.
- X. Yan, S. Li, J. B. Pollock, T. R. Cook, J. Chen, Y. Zhang, X. Ji, Y. Yu, F. Huang and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1558515590 Search PubMed
- J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC
- K. Müllen and W. Pisula, J. Am. Chem. Soc., 2015, 137, 9503–9505 CrossRef PubMed
- J. Babi, L. Zhu, A. Lin, A. Uva, H. El-Haddad, A. Peloewetse and H. Tran, J. Polym. Sci., 2021, 59, 2378–2404 CrossRef CAS
- Z.-A. Lan, M. Wu, Z. Fang, X. Chi, X. Chen, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 16355–16359 CrossRef CAS PubMed
- S. Zhang, N. E. Bauer, I. Y. Kanal, W. You, G. R. Hutchison and T. Y. Meyer, Macromolecules, 2017, 50, 151–161 CrossRef CAS
- A. D. Todd and C. W. Bielawski, ACS Macro Lett., 2015, 4, 1254–1258 CrossRef CAS
- E. Elacqua and M. Gregor, Angew. Chem., Int. Ed., 2019, 58, 9527–9532 CrossRef CAS PubMed
- W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2015, 137, 8038–8041 CrossRef CAS PubMed
- J. A. Nowalk, C. Fang, A. L. Short, R. M. Weiss, J. H. Swisher, P. Liu and T. Y. Meyer, J. Am. Chem. Soc., 2019, 141, 5741–5752 CrossRef CAS PubMed
- J. Zhang, J. Wang, C. Wei, Y. Wang, G. Xie, Y. Li and M. Li, Nat. Commun., 2020, 11, 2530 CrossRef CAS PubMed
- S.-H. Ahn, J. W. Grate and E. F. Darve, J. Chem. Phys., 2018, 149, 072330 CrossRef PubMed
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed
- T. C. Fitzgibbons, M. Guthrie, E.-s. Xu, V. H. Crespi, S. K. Davidowski, G. D. Cody, N. Alem and J. V. Badding, Nat. Mater., 2015, 14, 43–47 CrossRef CAS PubMed
- X. Li, M. Baldini, T. Wang, B. Chen, E.-s. Xu, B. Vermilyea, V. H. Crespi, R. Hoffmann, J. J. Molaison, C. A. Tulk, M. Guthrie, S. Sinogeikin and J. V. Badding, J. Am. Chem. Soc., 2017, 139, 16343–16349 CrossRef CAS PubMed
- P. Duan, X. Li, T. Wang, B. Chen, S. J. Juhl, D. Koeplinger, V. H. Crespi, J. V. Badding and K. Schmidt-Rohr, J. Am. Chem. Soc., 2018, 140, 7658–7666 CrossRef CAS PubMed
- B. Chen, R. Hoffmann, N. W. Ashcroft, J. Badding, E. Xu and V. Crespi, J. Am. Chem. Soc., 2015, 137, 14373–14386 CrossRef CAS PubMed
- X. Li, T. Wang, P. Duan, M. Baldini, H.-T. Huang, B. Chen, S. J. Juhl, D. Koeplinger, V. H. Crespi, K. Schmidt-Rohr, R. Hoffmann, N. Alem, M. Guthrie, X. Zhang and J. V. Badding, J. Am. Chem. Soc., 2018, 140, 4969–4972 CrossRef CAS PubMed
- S. Fanetti, M. Santoro, F. Alabarse, B. Enrico and R. Bini, Nanoscale, 2020, 12, 5233–5242 RSC
- S. Huss, S. Wu, B. Chen, T. Wang, M. C. Gerthoffer, D. J. Ryan, S. E. Smith, V. H. Crespi, J. V. Badding and E. Elacqua, ACS Nano, 2021, 15, 4134–4143 CrossRef CAS PubMed
- B. S. Matsuura, S. Huss, Z. Zheng, S. Yuan, T. Wang, B. Chen, J. V. Badding, D. Trauner, E. Elacqua, A. C. T. van Duin, V. H. Crespi and K. Schmidt-Rohr, J. Am. Chem. Soc., 2021, 143, 9529–9542 CrossRef CAS PubMed
- A. Biswas, M. D. Ward, T. Wang, L. Zhu, H.-T. Huang, J. V. Badding, V. H. Crespi and T. A. Strobel, J. Phys. Chem. Lett., 2019, 10, 7164–7171 CrossRef CAS PubMed
- M. M. Nobrega, E. Teixeira-Neto, A. B. Cairns, M. L. A. Temperini and R. Bini, Chem. Sci., 2018, 9, 254–260 RSC
- S. Romi, S. Fanetti, F. Alabarse, A. M. Mio and R. Bini, Chem. Sci., 2021, 12, 7048–7057 RSC
- L. Ciabini, M. Santoro, F. A. Gorelli, R. Bini, V. Schettino and S. Raugei, Nat. Mater., 2007, 6, 39–43 CrossRef CAS PubMed
- M. Citroni, R. Bini, P. Foggi and V. Schettino, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7658 CrossRef CAS PubMed
- M. M. Thiéry, J. M. Besson and J. L. Bribes, J. Chem. Phys., 1992, 96, 2633–2654 CrossRef
- L. Ciabini, F. A. Gorelli, M. Santoro, R. Bini, V. Schettino and M. Mezouar, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 094108 CrossRef
- H. Zhan, G. Zhang, V. B. C. Tan and Y. Gu, Nat. Commun., 2017, 8, 14863 CrossRef CAS PubMed
- H. Zhan, G. Zhang, J. M. Bell, V. B. C. Tan and Y. Gu, Nat. Commun., 2020, 11, 1905 CrossRef CAS PubMed
- R. E. Roman, K. Kwan and S. W. Cranford, Nano Lett., 2015, 15, 1585–1590 CrossRef CAS PubMed
- P. G. Demingos and A. R. Muniz, J. Phys. Chem. C, 2019, 123, 3886–3891 CrossRef CAS
- C. A. Gunawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047–14060 RSC
- S. Meejoo, B. M. Kariuki and K. D. Harris, ChemPhysChem, 2003, 4, 766–769 CrossRef CAS PubMed
- G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 15, 3641–3649 CrossRef
- C. R. Patrick and G. S. Prosser, Nature, 1960, 187, 1021 CrossRef CAS
- M. A. Sinnwell, J. Baltrusaitis and L. R. MacGillivray, Cryst. Growth Des., 2015, 15, 538–541 CrossRef CAS
- J. Lu, N. ten Brummelhuis and M. Weck, Chem. Commun., 2014, 50, 6225–6227 RSC
- E. Elacqua, G. T. Geberth, D. A. Vanden Bout and M. Weck, Chem. Sci., 2019, 10, 2144–2152 RSC
- A. S. Batsanov, J. C. Collings and T. B. Marder, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m229–m231 CrossRef PubMed
- C. E. Smith, P. S. Smith, R. L. Thomas, E. G. Robins, J. C. Collings, C. Dai, A. J. Scott, S. Borwick, A. S. Batsanov, S. W. Watt, S. J. Clark, C. Viney, J. A. K. Howard, W. Clegg and T. B. Marder, J. Mater. Chem., 2004, 14, 413–420 RSC
- M. Weck, A. R. Dunn, K. Matsumoto, G. W. Coates, E. B. Lobkovsky and R. H. Grubbs, Angew. Chem., Int. Ed., 1999, 38, 2741–2745 CrossRef CAS PubMed
- A. Schwarzer and E. Weber, Cryst. Growth Des., 2008, 8, 2862–2874 CrossRef CAS
- M. D. Ward, W. S. Tang, L. Zhu, D. Popov, G. D. Cody and T. A. Strobel, Macromolecules, 2019, 52, 7557–7563 CrossRef CAS
- A. Friedrich, I. E. Collings, K. F. Dziubek, S. Fanetti, K. Radacki, J. Ruiz-Fuertes, J. Pellicer-Porres, M. Hanfland, D. Sieh, R. Bini, S. J. Clark and T. B. Marder, J. Am. Chem. Soc., 2020, 142, 18907–18923 CrossRef CAS PubMed
- W. S. Tang and T. A. Strobel, J. Phys. Chem. C, 2020, 124, 25062–25070 CrossRef CAS
- Y. Wang, X. Dong, X. Tang, H. Zheng, K. Li, X. Lin, L. Fang, G. A. Sun, X. Chen, L. Xie, C. L. Bull, N. P. Funnell, T. Hattori, A. Sano-Furukawa, J. Chen, D. K. Hensley, G. D. Cody, Y. Ren, H. H. Lee and H.-k. Mao, Angew. Chem., Int. Ed., 2019, 58, 1468–1473 CrossRef CAS PubMed
- M. C. Gerthoffer, S. Wu, B. Chen, T. Wang, S. Huss, S. M. Oburn, V. H. Crespi, J. V. Badding and E. Elacqua, Chem. Sci., 2020, 11, 11419–11424 RSC
- R. Hrubiak, S. Sinogeikin, E. Rod and G. Shen, Rev. Sci. Instrum., 2015, 86, 072202 CrossRef PubMed
- G. Shen, Y. Wang, A. Dewaele, C. Wu, D. E. Fratanduono, J. Eggert, S. Klotz, K. F. Dziubek, P. Loubeyre, O. V. Fat'yanov, P. D. Asimow, T. Mashimo and R. M. M. Wentzcovitch, High Pressure Res., 2020, 40, 299–314 CrossRef
- M. A. Elbagerma, H. G. M. Edwards, T. Munshi, M. D. Hargreaves, P. Matousek and I. J. Scowen, Cryst. Growth Des., 2010, 10, 2360–2371 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS
- L. He, F. Liu, G. Hautier, M. J. T. Oliveira, M. A. L. Marques, F. D. Vila, J. J. Rehr, G. M. Rignanese and A. Zhou, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 064305 CrossRef
- A. F. Goncharov, Int. J. Spectrosc., 2012, 2012, 617528 Search PubMed
- M. Fisch, A. Lanza, E. Boldyreva, P. Macchi and N. Casati, J. Phys. Chem. C, 2015, 119, 18611–18617 CrossRef CAS
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2018, 140, 6361–6373 CrossRef CAS PubMed
- S. B. Dierker and M. C. Aronson, Rev. Sci. Instrum., 2018, 89, 053902 CrossRef CAS PubMed
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS PubMed
- E. Castillo-Martínez, J. Carretero-González, J. Sovich and M. D. Lima, J. Mater. Chem. A, 2014, 2, 221–228 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2098283 and 2098284. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1py01387d |
| This journal is © The Royal Society of Chemistry 2022 |